DOI:
10.1039/D0RA04696E
(Paper)
RSC Adv., 2020,
10, 38287-38293
Magnetic thermally sensitive interpenetrating polymer network (IPN) nanogels: IPN-pNIPAm@Fe2O3-SiO2
Received
27th May 2020
, Accepted 8th October 2020
First published on 16th October 2020
Abstract
In this paper, iron oxide-silica@poly(acrylamide-co-N,N-diethylacrylamide)/poly(N,N-diethylacrylamide) interpenetrating polymer network (IPN-pNIPAm@Fe2O3-SiO2) nanogels, possessing both magnetic and thermo-sensitive properties were successfully prepared. The preparation approach involved two steps, consisting of nanoparticle self-assembly and in situ polymerization with monomers. The structural combination of interpenetrating polymer networks (IPNs) with the Fe2O3-SiO2 nanoparticles led to a synergistic property enhancement of both IPNs and nanoparticles, which could increase the mechanical strength of hydrogels and decrease the aggregation of nanoparticles. The synergistic effect was induced by the compatibility of these two individual components. Furthermore, the swelling and shrinking behaviors of the IPN-pNIPAm@Fe2O3-SiO2 nanogels revealed the reversible thermo-responsive properties of IPN nanogels. This fabrication approach for IPN-pNIPAm@Fe2O3-SiO2 nanogels can provide a facile route for manufacturing smart nanocomposites with stability in aqueous solution and reversible swelling/deswelling stimuli-responsive properties to achieve multifunctional tasks in clinical therapy.
Introduction
Recently, intensive research effort has been expended in the smart combination of various micro- and nano-materials to provide multifunctional capabilities for broad nanomedical applications, such as controlled drug delivery and cell detection.1–3 Magnetic nanoparticles have attracted significant interest because of their properties including high saturation magnetization, small coercively and chemical stability.4,5 Besides, they could conjugate with bio-molecules and other functional groups through surface modification for various nanomedical applications.6 However, these functionalized magnetic nanoparticles usually lack sensitivity in response to the changes in the local physiological environment. As a result, the fabrication of functionalized magnetic nanoparticles still needs further optimization in order to achieve a sensitive release strategy.7,8
On the other hand, the smart nanogels which are nanosized hydrogels with three-dimensional polymer networks possessing stimuli-responsive property can act as a promising alternative to deal with this issue. By combining the magnetic nanoparticles with the nanogels, nanocomposites responding to changes in external stimuli,9 including temperature, pH, pressure electronic and magnetic field, can be fabricated. Therefore, the combination of magnetic nanoparticles and nanogels have been extensively explored in biomedical research, for example, drug delivery,10,11 cancer diagnoses,12 and biosensors.13 There are various kinds of polymeric materials with stimuli-responsive property such as hyaluronic acid,14 chitosan,15 dextrin16 and gelatin.17 Among these polymeric materials, the poly(N-isopropylacrylamide) (pNIPAm) has received considerable attention attributed to its discontinuous phase right above the lower critical solution temperature (LCST).18–20 Besides, interpenetrating polymer networks (IPNs) are the polymeric networks consisting of two or more polymers in a network form, entanglements with only occasional covalent bonds between the chains of individual components.21,22 The incorporation of IPN structure can improve the response rate of pNIPAm as well as facilitate therapeutics.23
Motivated by these advantages, researchers have focused on the fabrication of the smart combination of magnetic nanoparticles and nanogels with various stimuli-responsive properties. For instance, Ravi's group synthesized the hydrogels intra-ocular lens applications in a prepared nanoparticle suspension,24 however, this approach based on nanoparticles suspension suffers the drawback of the leaching of nanoparticles out of the hydrogel matrix due to the low density of cross-linking.25 Yu and his colleagues fabricated the nanogels with pH- and temperature-induced gelation of chitosan and ovalbumin based on physical incorporation.26 however, the method still needs to overcome certain problems, including poor stability in the biological environment caused by electrostatic interaction between the nanoparticles and polymers and the low efficiency of fabrication through coprecipitation.27 Therefore, a facile route to fabricate smart composite made of magnetic nanoparticles and nanogels exhibiting both the magnetic and thermo-sensitive properties is very important for its practical applications in biomedicine.
In this work, we established an approach for fabricating the IPN-pNIPAm@Fe2O3-SiO2 nanogels with both magnetic and thermo-sensitive properties via nanoparticles self-assembly and in situ polymerization process. The surface-modified magnetic nanoparticles were adopted as the template while NIPAm and AAm were adopted as the monomer to fabricate the IPN nanogels through polymerization/cross-linking process, which could increase the mechanical strength of hydrogels as well as decrease aggregation of nanoparticles because of the synergistic effect induced by the compatibility of individual components in IPN nanogels.28–30 This fabrication method combining nanoparticle self-assembly and in situ polymerization was demonstrated to be a facile route for manufacturing the smart nanocomposite with stability in aqueous solution and reversible swelling/deswelling stimuli-responsive property. Moreover, the thermo-responsive property and their swelling/shrinking behavior of IPN nanogels were investigated, and the IPN-pNIPAm@Fe2O3-SiO2 nanogels could be applied in protein adsorption and desorption with the thermo-trigger mechanism. This work opened a new pathway for innovative medical therapy applications that make use of stimuli of the external environment.
Experimental
Materials
Iron(III) oxyhydroxide (FeOOH), oleic acid (90%), octadecene (90%), bovine serum albumin (BSA), phosphate buffers (PBS) were purchased from Aldrich. Polyoxyethylene (10) octylphenyl ether (Triton X-100), aqueous ammonia (25%), tetraethyl orthosilicate (TEOS, 98%), ethanol, hexanol, chloroform, cyclohexane, toluene, and 3-aminopropyl triethoxysilane (APTES, 99%) were obtained from Acros. N-Isopropylacrylamide (NIPAm), acrylamide (AAm), sodium dodecyl sulfate (SDS), N,N-methylenebisacrylamide (BIS), and ammonium persulfate (APS) were purchased from Sigma. All chemicals were of analytical grade and could be used without additional purification.
Synthesis of Fe2O3-SiO2 nanoparticles
Fe2O3 magnetic nanoparticles were first obtained by thermal decomposition of iron(III) oxyhydroxide following Augustyn's published procedures.31 The 2 mmol of fine powder (2 mmol), 8 mmol of oleic acid (8 mmol), and 10 mL of octadecene were vigorously stirred in a three neck flask under a flow of nitrogen, the mixture was first heated to 200 °C for 30 min, then heated to 300 °C for 1 hour. Chloroform and acetone at a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 were added to the mixture, and the black precipitate was acquired by centrifugation. The Fe2O3 magnetic nanoparticles were purified three times by adding chloroform and acetone at a volume ratio of 1:3 and the final product was redispersed in chloroform. The yield of Fe2O3 magnetic nanoparticles was collected as high as 92.7%. Subsequently, the as-synthesized Fe2O3 magnetic nanoparticles were modified with silica shell through water-in-oil reverse microemulsion.32 The Fe2O3 magnetic nanoparticles were dissolved in cyclohexane at a concentration of 0.2 mg mL−1. An optimized inverse microemulsion system was formed by mixing Triton-X100 (1.79 g), hexanol (1.6 mL) and aqueous ammonia (107 μL), and stirred at room temperature for 14 hours. After the formation of microemulsion, 25 μL of TEOS was added dropwise, and the mixture was continuously stirred for another 2 hours at room temperature. The final Fe2O3-SiO2 magnetic nanoparticles were obtained by washing and precipitation with ethanol. The yield of Fe2O3-SiO2 magnetic nanoparticles yield is 83.9%. The resulting magnetic nanoparticles reacted with APTES at 100 °C under reflux conditions for 10 hours in order to bind the silane group of APTES to Fe2O3-SiO2 magnetic nanoparticles through the amino functionality. After washing with toluene for three times, the amino-end modified Fe2O3-SiO2 magnetic nanoparticles were acryloylated with acryloyl chloride in toluene solution and vigorously stirring for 12 hours at room temperature under nitrogen. The AAm-Fe2O3-SiO2 magnetic nanoparticles were dried for later use after washing. The synthetic protocol currently yields is 77.6%.
:3 were added to the mixture, and the black precipitate was acquired by centrifugation. The Fe2O3 magnetic nanoparticles were purified three times by adding chloroform and acetone at a volume ratio of 1:3 and the final product was redispersed in chloroform. The yield of Fe2O3 magnetic nanoparticles was collected as high as 92.7%. Subsequently, the as-synthesized Fe2O3 magnetic nanoparticles were modified with silica shell through water-in-oil reverse microemulsion.32 The Fe2O3 magnetic nanoparticles were dissolved in cyclohexane at a concentration of 0.2 mg mL−1. An optimized inverse microemulsion system was formed by mixing Triton-X100 (1.79 g), hexanol (1.6 mL) and aqueous ammonia (107 μL), and stirred at room temperature for 14 hours. After the formation of microemulsion, 25 μL of TEOS was added dropwise, and the mixture was continuously stirred for another 2 hours at room temperature. The final Fe2O3-SiO2 magnetic nanoparticles were obtained by washing and precipitation with ethanol. The yield of Fe2O3-SiO2 magnetic nanoparticles yield is 83.9%. The resulting magnetic nanoparticles reacted with APTES at 100 °C under reflux conditions for 10 hours in order to bind the silane group of APTES to Fe2O3-SiO2 magnetic nanoparticles through the amino functionality. After washing with toluene for three times, the amino-end modified Fe2O3-SiO2 magnetic nanoparticles were acryloylated with acryloyl chloride in toluene solution and vigorously stirring for 12 hours at room temperature under nitrogen. The AAm-Fe2O3-SiO2 magnetic nanoparticles were dried for later use after washing. The synthetic protocol currently yields is 77.6%.
Fabrication of interpenetrating polymer network (IPN) nanogels
The IPN-pNIPAm@Fe2O3-SiO2 nanogels were prepared by in situ polymerization method in deionized water.33 The as-prepared AAm-Fe2O3-SiO2 magnetic nanoparticles was added into an aqueous solution (15 mL) containing N-isopropylacrylamide (NIPAm, 0.20 g), the acrylamide (AAm, 0.014 g), sodium dodecyl sulfate (SDS, 0.025 g) and N,N-methylenebisacrylamide (BIS, 0.003 g), then the mixture was heated to 70 °C for 1 hour to equilibration. In the presence of added ammonium persulfate (APS, 0.0125 g), the mixture was refluxed at 70 °C under nitrogen for 5 hours with in situ polymerization process. Finally, the IPN-pNIPAm@Fe2O3-SiO2 nanogels were obtained through repeated centrifugation and washing with deionized water. The final dried IPN-pNIPAm@Fe2O3-SiO2 nanogels were obtained with 56.8% yield.
Swelling and shrinking behavior of IPN-pNIPAm@Fe2O3-SiO2 nanogels
To measure the absorption mass of the water, the IPN hydrogels were incubated in water at a temperature ranging from 20 to 45 °C. Subsequently, the hydrogels were removed, and their weight was gravimetrically measured after wiping away any surface water with moistened filter paper. The swelling and shrinking behavior of IPN-pNIPAm@Fe2O3-SiO2 nanogels were investigated using the classical gravimetric methods, which measure the equilibrium swelling ratio (ESR) of nanogels.34,35 The equilibrium swelling ratio was calculated by:
where ESR is the equilibrium water absorption, defined as grams of water per gram of sample, the W0 and W1 refer to the weights of sample before and after swelling, respectively.36
Protein adsorption/desorption of IPN-pNIPAm@Fe2O3-SiO2 nanogels
To measure the protein adsorption and desorption, BSA protein experiments with IPN-pNIPAm@Fe2O3-SiO2 nanogels were carried out at different temperatures. 10 mg of IPN-pNIPAm@Fe2O3-SiO2 nanogels was added to the given concentration of BSA protein solution (1 mg mL−1 in PBS). Then, the mixture suspension was incubated for 24 hours with different temperature. The resultant products that adsorbed BSA protein were centrifuged to separate them from the aqueous medium. The supernatant was withdrawn and the amount of protein remaining in the supernatant was obtained by measuring the absorbance at 280 nm using a UV spectrophotometer.34 The amount of BSA protein adsorbed on nanogel was determined from the difference in the concentrations before and after incubation, which were measured based on UV-Vis absorbance spectrum at different temperature.35
Characterization and measurement
Morphologies of the fabricated nanoparticles were characterized using a Philips CM-100 transmission electron microscope (TEM) operated at 100 kV. Structures of the IPN-pNIPAm@Fe2O3-SiO2 nanogels were examined by an LEO 1530 scanning electron microscope (SEM). The Fourier transform infrared (FTIR) spectra were recorded with a Shimadzu FTIR-8300 spectrometer. The thermal property of the IPN-pNIPAm@Fe2O3-SiO2 nanogels was studied using differential scanning calorimetry (DSC) (TA instrument Q20) and thermogravimetric analyses (TGA) (TA instrument Q50), both at a ramp rate of 10 °C min−1 in nitrogen atmosphere. Magnetic properties of nanogels were characterized using a MicroSense EZ vibrating sample magnetometer (VSM) at 300 K. The protein adsorption and desorption were measured by the UV/Visible spectrophotometer (Agilent Cary 60).
Results and discussions
Fabrication mechanism
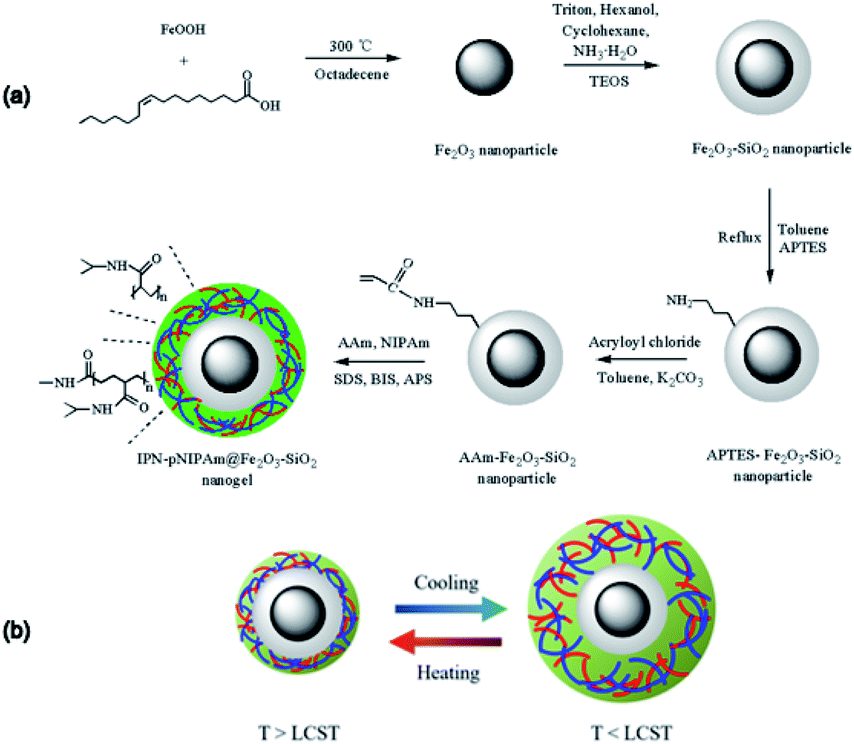
The strategy of preparing IPN-pNIPAm@Fe2O3-SiO2 nanogels involved two steps as schematically displayed in Fig. 1a, mainly the modified AAm-Fe2O3-SiO2 magnetic nanoparticles self-assembly and the in situ polymerization/cross-linking of monomer. The growth of acrylamide monomer on the silica surface of Fe2O3-SiO2 magnetic nanoparticles was modified through aminopropyl modification and acryloylated process. The AAm-capped protection of Fe2O3-SiO2 magnetic nanoparticles could prevent the aggregation of nanoparticles. In the second step, the AAm-Fe2O3-SiO2 magnetic nanoparticles served as the template for in situ formation of nanogels by polymerization of PNIPAm and AAm as the monomer with the crosslinker BIS, the initiator APS and the emulsifier SDS. The formation of IPN rendered the reversible reswelling/deswelling stimuli-responsive property as presented in Fig. 1b. The interpenetration of these two-monomer resulted in a much higher mechanical strength comparing with the homopolymer network. Therefore, the synthesized IPN nanogels exhibited the enhanced stability in aqueous solution.30,37 The structural combination of IPN with the Fe2O3-SiO2 magnetic nanoparticles led to a synergistic property enhancement of both IPN and nanoparticles, including the increased mechanical strength and the decreased aggregation of IPN nanogels.29
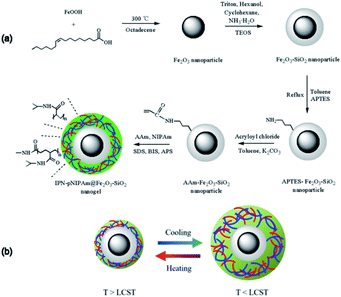 |
| | Fig. 1 (a) Schematic illustration for the preparation of IPN-pNIPAm@Fe2O3-SiO2 nanogels, and (b) the dynamic mechanism of the phase transition of IPN-pNIPAm@Fe2O3-SiO2 nanogels. | |
Characterization of IPN-pNIPAm@Fe2O3-SiO2 nanogels
The and freeze-dried to obtain nanogels NG(P85) with 50% yield by weight. TEM image in Fig. 2a reveals that the as-synthesized Fe2O3 magnetic nanoparticles are in spherical shape with the diameter of 9.8 ± 0.8 nm. The Fe2O3-SiO2 magnetic nanoparticles exhibited the core–shell structure with 28 nm diameter consisting of the 10 nm core diameter and 9 nm shell thickness as shown in Fig. 2b. Through surface modification, the AAm-Fe2O3-SiO2 magnetic nanoparticles possessed stable core–shell structure with ∼30 nm diameter, which is larger than 28 nm because of the acrylamide layer (Fig. 2c). The prepared nanoparticles were used for the polymerization/cross-linking with NIPAm and AAm monomers to fabricate the IPN structure.
 |
| | Fig. 2 TEM images of (a) Fe2O3 magnetic nanoparticles, (b) Fe2O3-SiO2 magnetic nanoparticles, and (c) AAm-Fe2O3-SiO2 magnetic nanoparticles. | |
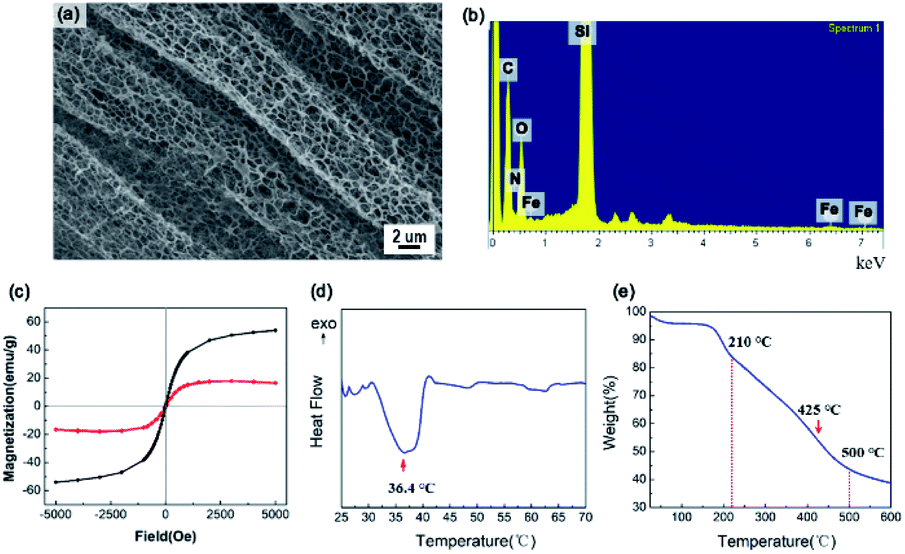
The SEM image in Fig. 3a shows the mesoporous structure of the IPN-pNIPAm@Fe2O3-SiO2 nanogels. The energy-dispersive X-ray spectroscopy (EDX) in Fig. 3b evidences the existence of N, O, and C elements, indicating the modification with amide group. The magnetization curves of the Fe2O3 magnetic nanoparticles and IPN-pNIPAm@Fe2O3-SiO2 nanogels are displayed in Fig. 3c. The coercivity of the IPN nanogels is 17.7 Oe. They exhibited the diamagnetic behavior,38 due to the IPN structure covering on the surface of Fe2O3 magnetic nanoparticles. According to Fig. 3d, the DSC curve shows the exothermic point owing to the phase transition of pNIPAm. The minimum critical point at 36.4 °C is defined as the LCST of the IPN-pNIPAm@Fe2O3-SiO2 nanogels. As displayed in Fig. 3e, the TGA curve of the nanogels shows the weight loss at below 210 °C, which can be attributed to the evaporation of residues organic solvent and water. The weight loss of 56% can be observed in the range 210–500 °C because of the decomposition of the organic polymer on the surface of modified Fe2O3 magnetic nanoparticles. Furthermore, the maximum decomposition rate of the IPN nanogels occurred at 425 °C, which is similar to the decomposition rate of pure pNIPAm.39
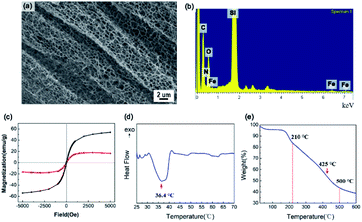 |
| | Fig. 3 (a) SEM image and (b) EDX of IPN-pNIPAm@Fe2O3-SiO2 nanogels, (c) the magnetization curve of Fe2O3 magnetic nanoparticles (black) and IPN-pNIPAm@Fe2O3-SiO2 nanogels (red), (d) DSC curve, and (e) TGA curve of IPN-pNIPAm@Fe2O3-SiO2 nanogels. | |
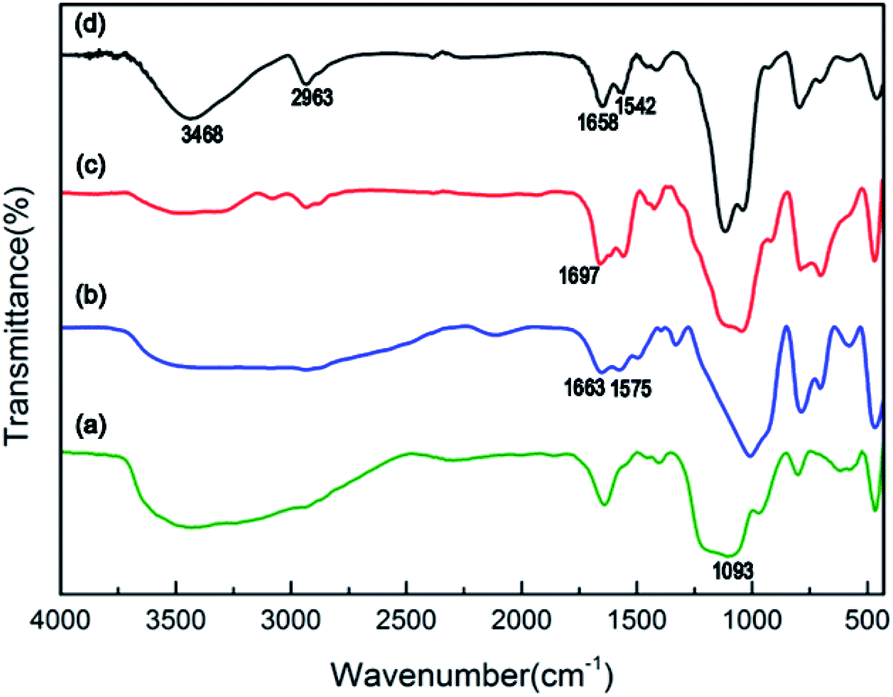
The chemical bonds of the modified magnetic nanoparticles and IPN-pNIPAm@Fe2O3-SiO2 nanogels can be confirmed by FTIR spectrum. Fig. 4a shows the characteristic absorption band of 1093 cm−1 which is assigned to the Si–O–Si band of the silica shell modified on the Fe2O3 magnetic nanoparticles. The peaks at 1663 cm−1 and 1575 cm−1 in Fig. 4b are attributed to the N–H and C–N bonds, indicating the APTES surface modification of the Fe2O3 magnetic nanoparticles. The absorption band at 1697 cm−1 in Fig. 4c can be attributed to the amide group of the acrylamide layer modified on the surface of Fe2O3-SiO2 magnetic nanoparticles.40,41 Two new peaks appear in Fig. 4d, the peak at 3468 cm−1 is attributed to the amide group of pNIPAm, while the stretching band at 2963 cm−1 corresponds to the C–H vibration of –CH2 groups in the NIPAm and AAm. The peaks at 1658 cm−1 and 1542 cm−1 indicate the amide I and amide II, respectively,42 due to the presence of intramolecular hydrogen bonding between –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups and –NH groups of NIPAm and AAm monomers. These results evidence the existence of the IPN-pNIPAm@Fe2O3-SiO2 nanogels formed by polymerization/crosslinking of NIPAm and AAm monomers.
O groups and –NH groups of NIPAm and AAm monomers. These results evidence the existence of the IPN-pNIPAm@Fe2O3-SiO2 nanogels formed by polymerization/crosslinking of NIPAm and AAm monomers.
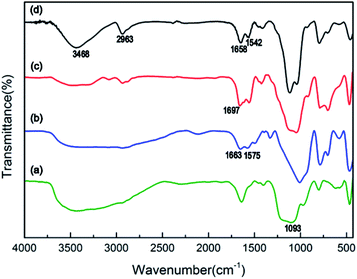 |
| | Fig. 4 FTIR spectrum of (a) Fe2O3-SiO2 magnetic nanoparticles, (b) APTES-Fe2O3-SiO2 magnetic nanoparticles, (c) AAm-Fe2O3-SiO2 magnetic nanoparticles, and (d) IPN-pNIPAm@Fe2O3-SiO2 nanogels. | |
Thermo-responsive property of IPN-pNIPAm@Fe2O3-SiO2 nanogels
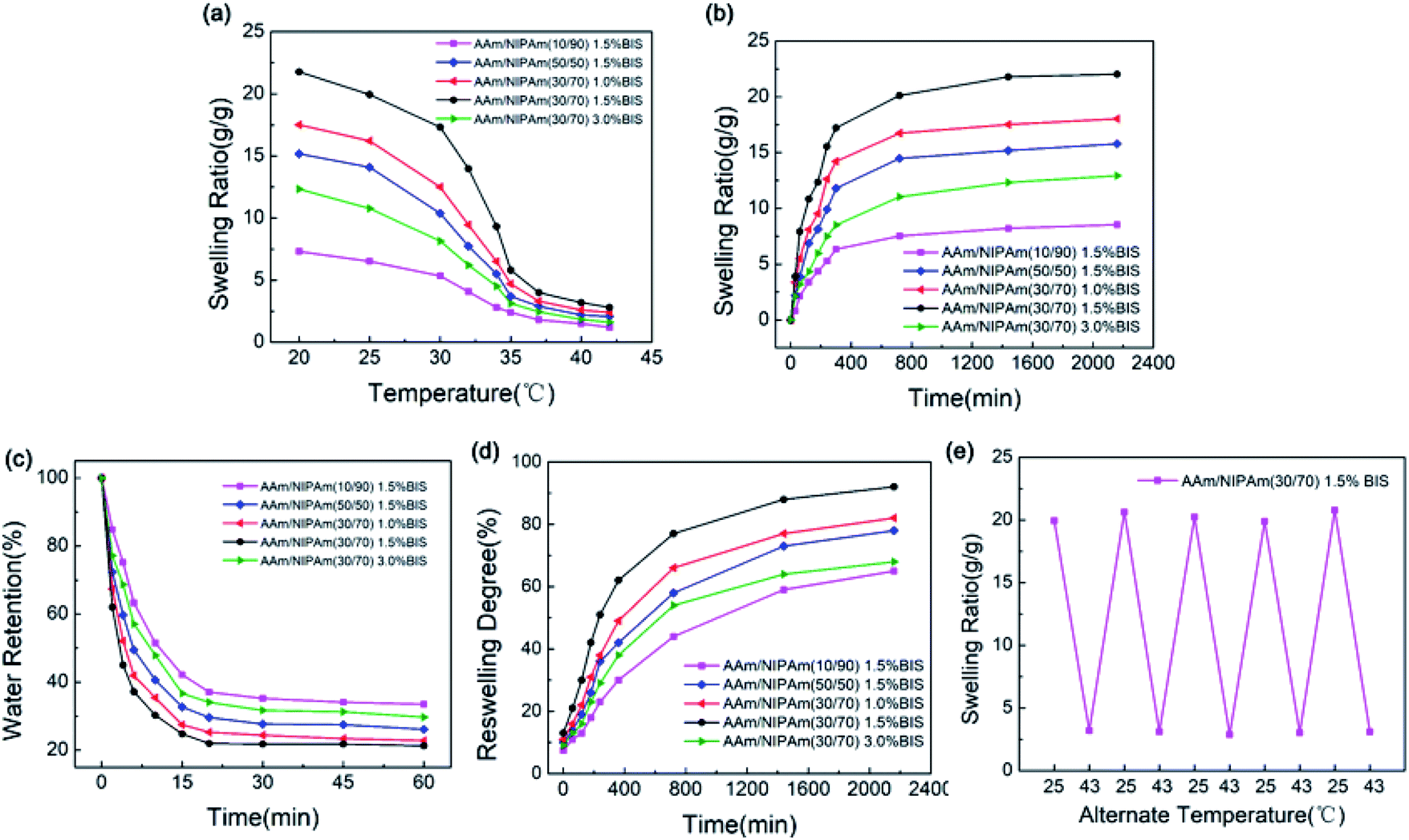
The degree of crosslinking could be tuned by changing the molar ratio of AAm to NIPAm and the concentration of BIS cross-linker (based on the weight of monomer).43 As observed from Fig. 5a–c, the degree of crosslinking in the IPN nanogels became stronger when the molar ratio of AAm to NIPAm was increased from 10/90 to 50/50. Dense and irregular crosslinking network was observed at a molar ratio of 50/50. The degree of crosslinking can be analyzed quantitatively by evaluating the equilibrium swelling ratio (ESR) of IPN nanogels. The nanogel fabricated with 30/70 molar ratio exhibited the highest value of ESR compared with the other groups, revealing that the porosity was enhanced by increasing the molar ratio of AAm. This enhancement effect promoted the hydrophilicity of IPN nanogel, leading to the increased absorption of water in the nanogel.39 However, excessive AAm probably occupied more void spaces within the interpenetrating polymer network, resulting in the decrease of ESR as shown in Fig. 5d.
 |
| | Fig. 5 SEM images of IPN-pNIPAm@Fe2O3-SiO2 nanogels with different molar ratio: (a) AAm/NIPAm (10/90)–1.2%BIS, (b) AAm/NIPAm (30/70)–1.2% BIS, (c) AAm/NIPAm (50/50)–1.2% BIS, and (d) the effect of molar ratio (AAm/NIPAm) on the swelling property of IPN-pNIPAm@Fe2O3-SiO2 nanogels. | |
The IPN nanogel fabricated with the concentration of 1.5% BIS exhibited a more structured microporous morphology compared with the sample with 1.0% BIS as shown in Fig. 6a and b. The IPN nanogel fabricated with 3.0% BIS exhibited the macroporous morphology with thick-walls as shown in Fig. 6c. In additional, the ESR decreased with the increasing BIS concentration from 1.5% to 3.0%. This was because the excessive crosslinks could shorten the distance between the interconnection sites on different polymer chains, leading to the occurrence of shrinking behavior of the nanogels, which was because the excessive crosslinks could shorten the distance between the interconnection sites on different polymer chains, leading to the occurrence of shrinking behavior of the nanogels, the micro-porous size becomes smaller, reducing the water capacity. The 1.5% BIS cross-linker with the highest ESR values was used to fabricate IPN nanogels.
 |
| | Fig. 6 SEM images of IPN-NIPAm@Fe2O3-SiO2 nanogels with different BIS crosslinker concentration: (a) AAm/NIPAm (30/70)–1.0% BIS, (b) AAm/NIPAm (30/70)–1.5% BIS, (c) AAm/NIPAm (30/70)–3.0% BIS, and (d) the effect of BIS crosslinker concentration on the swelling property of IPN-pNIPAm@Fe2O3-SiO2 nanogels. | |
Swelling/shrinking behavior of IPN-pNIPAm@Fe2O3-SiO2 nanogels
The ability to absorb and retain water is one of the essential features of nanogels.44 The thermo-sensitivity of IPN-pNIPAm@Fe2O3-SiO2 nanogels was investigated by measuring the ESR during swelling and shrinking process. As presented in Fig. 7a, the ESR of all samples decreased continuously with the temperature because the hydrophilic parts of the nanogel broke at the temperature over the LCST. According to Fig. 7b, the ESR of all samples first increased rapidly and then tended to stabilize with time. The maximum ESR of the nanogels was reached in 36 hours. The swelling behavior can be explained by the enhancement effect of increased AAm chains on hydrophilicity. According to the previous work,45 the extent of swelling behavior is dependent on the hydrophilicity and the degree of cross-linking of nanogels. Thus, the ESR can be improved by adding the AAm due to its hydrophilicity. These results demonstrated the thermo-sensitive property of the IPN nanogels. Moreover, the deswelling process of all samples was rather short, and it only required 20 minutes to dehydrate, as shown in Fig. 7c. The AAm acted as the water-releasing channels in the deswelling behavior. Therefore, the deswelling rate could increase as the molar ratio of AAm to NIPAm increased, which is similar to the results reported in the previous work.44,46 The deswelling rate of 30/70 molar ratio was faster than that of 10/90 molar ratio which can be observed from Fig. 7d This suggested that the improved response rate can be achieved through increasing the molar ratio of AAm to NIPAm, which could realize a quicker reversible swelling/deswelling process of the IPN nanogels. Fig. 7e presented the reversible swelling/deswelling behaviors of IPN-pNIPAm@Fe2O3-SiO2 nanogels. The alternate temperature was cycled between 25 °C to 43 °C in an aqueous medium. Rapid swelling and deswelling of the gels were observed, and the process was proven to be repeatable, thus the stability (reswelling/deswelling cycle) performances were verified.
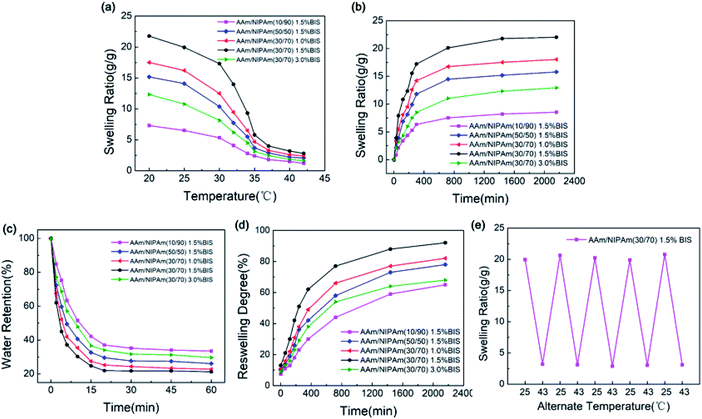 |
| | Fig. 7 Swelling behavior of the IPN-pNIPAm@Fe2O3-SiO2 nanogels: (a) equilibrium swelling ratio of all nanogels at 20–45 °C; (b) equilibrium swelling ratio of all nanogels at 25 °C; (c) deswelling behavior of all nanogels at 40 °C; (d) reswelling behavior of all nanogels at 25 °C, and (e) reversible swelling/deswelling curve of the IPN-pNIPAm@Fe2O3-SiO2 nanogels. | |
Performance of protein adsorption/desorption with thermo-trigger mechanism
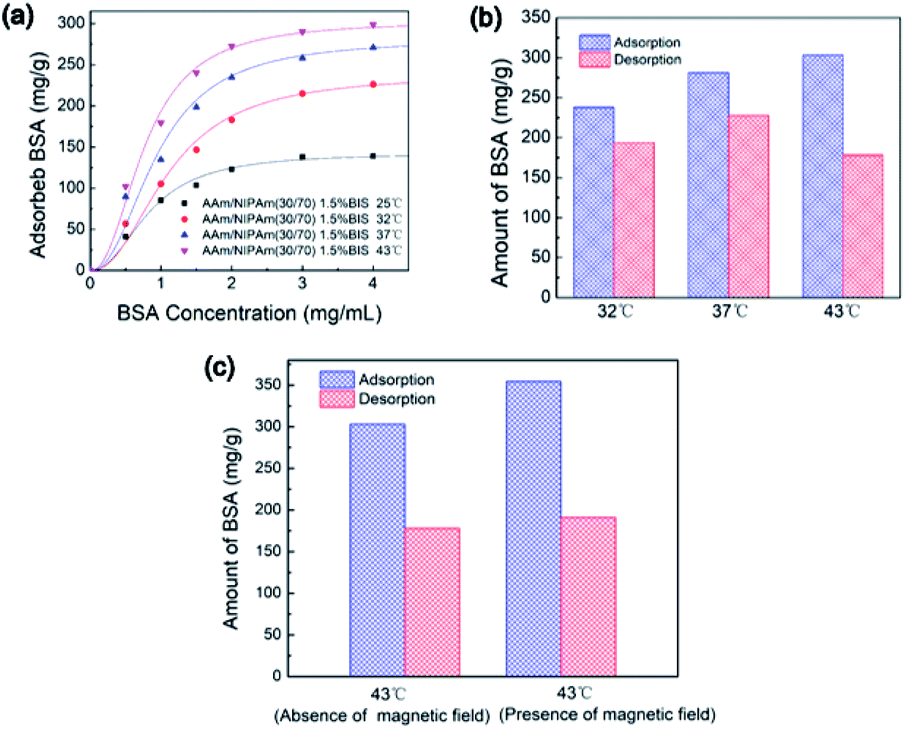
The protein adsorption and desorption experiments were conducted to explore the potential of sensitive release profile of the IPN nanogels in drug delivery. The BSA acted as the model protein. The experimental data in Fig. 8a were fitted well with the Langmuir–Freundlich model. The high R2 values in Table 1 indicated the desirability of adsorption kinetics for the IPN-pNIPAm@Fe2O3-SiO2 nanogels.47 The amount of adsorbed BSA at 43 °C was more than that of the groups operated at lower temperature because of the increased affinity of the protein molecules and IPN nanogels at higher temperature. The maximum protein adsorption capacity was 303.10 mg g−1 (Table 1). Fig. 8b shows the comparison of BSA adsorption and desorption under different temperature, and the detailed amount of adsorbed and desorbed BSA are listed in Table 2. The results showed that more than 80% of BSA could be desorbed from the IPN nanogels when the temperature was below or near the LCST. On the contrary, less than 60% desorption of BSA was observed for the samples operated above the LCST (Table 2). Although the amount of adsorbed BSA increased with the temperature, the deformation of BSA molecules due to their interaction with the polymers at higher temperature resulting in the reduced desorption of BSA.
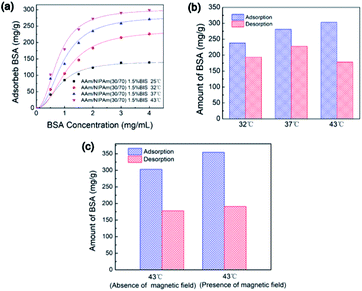 |
| | Fig. 8 (a) The influence of BSA initial concentration on BSA adsorption of IPN-pNIPAm@Fe2O3-SiO2 nanogels, (b) the influence of temperature on BSA adsorption and desorption of IPN-pNIPAm@Fe2O3-SiO2 nanogels, and (c) the influence of applied magnetic field on BSA adsorption of IPN-pNIPAm@Fe2O3-SiO2 nanogels. | |
Table 1 Langmuir–Freundlich parameters for BSA adsorption on IPN-pNIPAm@Fe2O3-SiO2 nanogels at different temperatures
| |
43 °C |
37 °C |
32 °C |
25 °C |
| Cm (mg g−1) |
303.10 |
281.13 |
238.07 |
142.66 |
| K (mg mL−1) |
1.77 |
1.36 |
0.79 |
1.48 |
| R2 |
0.97 |
0.97 |
0.98 |
0.99 |
Table 2 Results of BSA desorption from IPN-pNIPAm@Fe2O3-SiO2 nanogels
| |
Adsorbed BSA (mg g−1) |
Desorbed BSA (mg g−1) |
Desorption percentage (%) |
| AAm/NIPAm(30/70) 1.5% BIS, 32 °C |
238.07 |
193.67 |
81.35% |
| AAm/NIPAm(30/70) 1.5% BIS, 37 °C |
281.13 |
227.60 |
80.96% |
| AAm/NIPAm(30/70) 1.5% BIS, 43 °C |
303.10 |
177.98 |
58.72% |
The influence of external magnetic field on the performance of protein adsorption was investigated. The experiment was operated under an out-of-plane magnetic field for 2 hours. As shown in Fig. 8c, the amount of adsorbed BSA in the presence of magnetic field was more than that of the group carried out in the absence of magnetic field. The maximum protein adsorption capacity reached to 354.37 mg g−1, which was higher than the adsorbed protein in the group under the absence of applied magnetic field. These results suggested that the protein adsorption process was enhanced when external magnetic field was introduced. Such enhancement was presumably related to the aggregation of the magnetic IPN-pNIPAm@Fe2O3-SiO2 nanogels under the guidance of applied magnetic field, which may facilitate the protein adsorption process on nanogels.
Conclusions
To conclude, the IPN-pNIPAm@Fe2O3-SiO2 nanogels with the combined magnetic and thermo-sensitive properties were prepared through the nanoparticle self-assembly and in situ polymerization of the thermo-sensitive polymers on the surface of silica-coated iron-oxide magnetic nanoparticles. The chemically fabricated Fe2O3-SiO2 magnetic nanoparticles were monodisperse and modified with the acylamide-monomer, offering the template for the formation of IPN-pNIPAm@Fe2O3-SiO2 nanogels via the polymerization/cross-linking process in an aqueous solution. The experimental factors including the monomer molar ratio and the concentration of crosslinker as well as the thermo-responsive swelling behavior of the IPN-pNIPAm@Fe2O3-SiO2 nanogels were investigated. The experimental protein adsorption data were fitted well with Langmuir–Freundlich model and it was observed that larger amount of protein was adsorbed at higher temperature above LCST. Besides, over 80% of the protein desorption was released from the IPN-pNIPAm@Fe2O3-SiO2 nanogels at the temperature below LCST. The results of this work suggested that the proposed fabrication approach combining nanoparticle assembly and in situ polymerization can provide a facile route for manufacturing the smart nanocomposite with stability in aqueous solution and reversible swelling/deswelling stimuli-responsive property. It can open a new pathway for innovative medical therapy applications that make use of multifunctional nanomaterials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Seed Funding Program for Basic Research, Seed Funding Program for Applied Research and Small Project Funding Program from the University of Hong Kong, RGC-GRF grant (HKU 17204617). ITF Tier 3 funding (ITS-104/13, ITS-214/14), and University Grants Committee of HK (AoE/P-04/08).
References
- E. Ruiz-Hernandez, A. Baeza and M. Vallet-Regi, ACS Nano, 2011, 5, 1259–1266 CrossRef CAS.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS.
- P. Parhi, C. Mohanty and S. K. Sahoo, Drug Discovery Today, 2012, 17, 1044–1052 CrossRef CAS.
- M. Ma, Y. Wu, J. Zhou, Y. Sun, Y. Zhang and N. Gu, J. Magn. Magn. Mater., 2004, 268, 33–39 CrossRef CAS.
- N. Lee, D. Yoo, D. Ling, M. H. Cho, T. Hyeon and J. Cheon, Chem. Rev., 2015, 115, 10637–10689 CrossRef CAS.
- N. Kohler, C. Sun, J. Wang and M. Zhang, Langmuir, 2005, 21, 8858–8864 CrossRef CAS.
- X. Mou, Z. Ali, S. Li and N. He, J. Nanosci. Nanotechnol., 2015, 15, 54–62 CrossRef CAS.
- C. Fang, F. M. Kievit, O. Veiseh, Z. R. Stephen, T. Z. Wang, D. H. Lee, R. G. Ellenbogen and M. Q. Zhang, J. Controlled Release, 2012, 162, 233–241 CrossRef CAS.
- X. Li, H. Lu, Y. Zhang, F. He, L. Jing and X. He, Appl. Surf. Sci., 2016, 389, 567–577 CrossRef CAS.
- J.-Y. Zhu, Q. Lei, B. Yang, H.-Z. Jia, W.-X. Qiu, X. Wang, X. Zeng, R.-X. Zhuo, J. Feng and X.-Z. Zhang, Biomaterials, 2015, 52, 281–290 CrossRef CAS.
- M. Hayati, G. B. Rezanejade, M. Ramezani, S. S. Hosseini and F. Mizani, Polym. Int., 2020, 69, 156–164 CrossRef CAS.
- R. Ischakov, L. Adler-Abramovich, L. Buzhansky, T. Shekhter and E. Gazit, Bioorg. Med. Chem., 2013, 21, 3517–3522 CrossRef CAS.
- Y. Wang, F. Papadimitrakopoulos and D. J. Burgess, J. Controlled Release, 2013, 169, 341–347 CrossRef CAS.
- A. Fakhari and C. Berkland, Acta Biomater., 2013, 9, 7081–7092 CrossRef CAS.
- B. Anisha, D. Sankar, A. Mohandas, K. Chennazhi, S. V. Nair and R. Jayakumar, Carbohydr. Polym., 2013, 92, 1470–1476 CrossRef CAS.
- S. Manchun, C. R. Dass, K. Cheewatanakornkool and P. Sriamornsak, Carbohydr. Polym., 2015, 126, 222–230 CrossRef CAS.
- K. M. Rao, K. K. Rao, G. Ramanjaneyulu and C.-S. Ha, Int. J. Pharm., 2015, 478, 788–795 CrossRef.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
- K. D. Seo, J. Doh and D. S. Kim, Langmuir, 2013, 29, 15137–15141 CrossRef CAS.
- M. E. Harmon, D. Kuckling, P. Pareek and C. W. Frank, Langmuir, 2003, 19, 10947–10956 CrossRef CAS.
- D. Denmark, R. H. Hyde, C. Gladney, M. H. Phan, K. S. Bisht and S. Witanachchi, Drug Deliv., 2017, 24, 1317–1324 CrossRef CAS.
- K. Frisch, D. Klempner and H. Frisch, Mater. Des., 1983, 4, 821–827 CrossRef.
- L. Klouda and A. G. Mikos, Eur. J. Pharm. Biopharm., 2008, 68, 34–45 CrossRef CAS.
- H. A. Aliyar, P. D. Hamilton and N. Ravi, Biomacromolecules, 2005, 6, 204–211 CrossRef CAS.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- S. Yu, J. Hu, X. Pan, P. Yao and M. Jiang, Langmuir, 2006, 22, 2754–2759 CrossRef CAS.
- C. D. Jones, M. J. Serpe, L. Schroeder and L. A. Lyon, J. Am. Chem. Soc., 2003, 125, 5292–5293 CrossRef CAS.
- K. H. Hsieh, J. L. Han, C. T. Yu and S. C. Fu, Poly, 2001, 42, 2491–2500 CrossRef CAS.
- S. Thomas, D. Grande, U. Cvelbar, K. Raju, S. P. Thomas, R. Narayan and H. Akhina, Micro-and Nano-structured Interpenetrating Polymer Networks: From Design to Applications, John Wiley & Sons, London, 2016 Search PubMed.
- L. H. Sperling, Interpenetrating polymer networks and related materials, Springer Science & Business Media, Berlin, 2012 Search PubMed.
- C. L. Augustyn, T. D. Allston, R. K. Hailstone and K. J. Reed, RSC Adv., 2014, 4, 5228–5235 RSC.
- C. Vogt, M. S. Toprak, M. Muhammed, S. Laurent, J. L. Bridot and R. N. Müller, J. Nanopart. Res., 2010, 12, 1137–1147 CrossRef CAS.
- M. B. Ahmad, Y. Gharayebi, M. S. Salit, M. Z. Hussein and K. Shameli, Int. J. Mol. Sci., 2011, 12, 6040–6050 CrossRef.
- D. Huo, Y. Li, Q. Qian and T. Kobayashi, Colloids Surf., B, 2006, 50, 36–42 CrossRef CAS.
- D. Duracher, R. Veyret, A. Elaïssari and C. Pichot, Polym. Int., 2004, 53, 618–626 CrossRef CAS.
- R. T. Cha, Z. B. He and Y. H. Ni, Carbohydr. Polym., 2012, 88, 713–718 CrossRef CAS.
- J. Zhang and N. A. Peppas, J. Biomater. Sci., Polym. Ed., 2002, 13, 511–525 CrossRef CAS.
- T. Iwamoto, Y. Kitamoto and N. Toshima, Physica B Condens. Matter, 2009, 404, 2080–2085 CrossRef CAS.
- R. G. Sousa, W. F. Magalhães and R. F. S. Freitas, Polym. Degrad. Stab., 1998, 61, 275–281 CrossRef CAS.
- I. A. Rahman, M. Jafarzadeh and C. S. Sipaut, Ceram. Int., 2009, 35, 1883–1888 CrossRef CAS.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS.
- D. E. Owens, Y. Jian, J. E. Fang, B. V. Slaughter, Y. H. Chen and N. A. Peppas, Macromolecules, 2007, 40, 7306–7310 CrossRef CAS.
- H. Lange, Colloid Polym. Sci., 1986, 264, 488–493 CrossRef CAS.
- S. Ekici, J. Mater. Sci., 2011, 46, 2843–2850 CrossRef CAS.
- C. M. A. Lopes and M. I. Felisberti, Biomaterials, 2003, 24, 1279–1284 CrossRef CAS.
- J. T. Zhang, R. Bhat and K. D. Jandt, Acta Biomater., 2009, 5, 488–497 CrossRef CAS.
- A. A. Sarhan, M. A. Aki and K. Razaq, J. Nanomed. Nanotechnol., 2015, 01, 1–9 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Philip W. T. Pong*
and
Philip W. T. Pong*