
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced functional DNA biosensor for distance-based read-by-eye quantification of various analytes based on starch-hydrolysis-adjusted wettability change in paper devices†
Yijing Chen‡
,
Lang Zhang‡,
Jinkun Huang‡,
Zihao Deng,
Yali Yuan *,
Jianmei Zou,
Jinfang Nie* and
Yun Zhang*
*,
Jianmei Zou,
Jinfang Nie* and
Yun Zhang*
College of Chemistry and Bioengineering, Guilin University of Technology, Guilin 541004, P. R. China. E-mail: zy@glut.edu.cn; thankSIN2013@163.com; Niejinfang@glut.edu.cn; Fax: +86 773 5896839; Tel: +86 773 5896453
First published on 27th July 2020
Abstract
Low-cost, equipment-free and quantitative detection of a wide range of analytes of interest at home and in the field holds the potential to revolutionize disease diagnosis, environmental pollution monitoring, and food safety analysis. Herein, we describe a functional DNA biosensor for the first time that integrates analyte-directed assembly of enzyme-coated microbead probes for robust yet efficient signal amplification with a simple quantitative detection motif of distance measurement on portable paper devices based on starch-hydrolysis-adjusted wettability change of paper. Its utility is well demonstrated with highly sensitive and specific detection of model analytes ranging from adenosine (an important small biomolecule; 1.6 μM detection limit) to interferon-γ (a protein marker; 0.3 nM detection limit) and Pb2+ (a highly toxic metal ion; 0.5 nM detection limit) by simply using an inexpensive, ubiquitous ruler. The developed general method with the distance-measuring readout should be easily tailored for the portable, read-by-eye, quantitative detection of many other types of analytical targets by taking advantage of their specific functional DNA partners like aptamers and DNAzymes.
Introduction
DNA has been known as a very special and vital class of biological polymers for the storage and propagation of genetic information. Since the early 1990s, however, certain DNA molecules have been unexpectedly found to show catalytic activity (called DNAzymes) like protein enzymes1 and/or binding affinity to target molecules (called aptamers).2 The DNAzymes, aptamers and their combination (called aptazymes) are now collectively called functional DNA and can be selected by means of a process known as in vitro selection3 or systematic evolution of ligands by exponential enrichment (SELEX).4,5 In comparison with the most-widely applied recognition biomolecules such as antibodies, these functional DNA molecules can offer a great number of competitive advantages including simpler production with better reproducibility, higher chemical stability, lower cost, easier storage, and an especially wider range of recognizable targets.6,7 As a result, extensive efforts have been devoted to the development of functional DNA sensors for various analytical applications in medical diagnosis, environmental analysis, and food safety testing.8–11 The functional DNA biosensors typically transduce the recognition chemistry using electrochemical, absorbance, and fluorescence readers.12,13 They have been proven to allow for highly sensitive and specific detection of toxic ions (e.g., Hg2+ and UO22+),14,15 small molecules such as aflatoxin B1 (ref. 16) and adenosine 5′-triphosphate,17 microRNA,18 protein biomarkers like thrombin,19 cancer cells,20 and bacteria such as Escherichia coli21 and Fusarium proliferatum.22 Nevertheless, most of these measurements may suffer from the requirements of expensive yet bulky analytical instruments that can only be operated by well-trained personnel to obtain final results, which make these useful biosensors difficult to be accessed in less-industrialized areas, in field-based analysis, in emergency situations, or in-home healthcare services.The reports of emerging paper microfluidics are a positive step towards developing promising alternative analytical devices for use in such resource-limited environments because of its many attractive properties.23–25 The paper-based devices are extremely cheap, easy to create/store, portable and disposable, typically consume several microliters of reagent or sample per assay, and can function just via capillary-action-driven fluid flow.26,27 Paper microfluidics has its genesis in the lateral flow test strip technique, the most representative example of which is the home pregnancy test kit that was commercially available as early as 1988. In 2007, the Whitesides' group patterned hydrophobic materials into hydrophilic paper body to upgrade the basic lateral flow design for providing more functionality.28 This family of portable assay platforms is now known as microfluidic paper-based analytical devices (μPADs).29,30 Recently, a few of colorimetric functional DNA sensors have been successfully established using lateral flow strips31,32 or μPADs.33–36 Such biosensors enabled rapid qualitative and/or semi-quantitative naked-eye analysis of exosomes,31 cortisol,32 Helicobacter pylori,33 platelet-derived growth factor,34 pan malaria and P. falciparum species36 or multiple cancer cells.35 Nevertheless, quantitative measurements are necessary in many cases, such as disease stage diagnosis, therapeutic efficacy monitoring, etc. Alternatively, some efforts have been dedicated to designing μPAD-based functional DNA sensing systems for direct quantification of analytes of interest including K+,37 Pb2+,38 Hg2+,39 adenosine40–44 and cocaine42,43 by just using a timer, a blood glucose meter, a digital multimeter, or even a ruler. In particular, the distance-based detection strategies with the ruler have been well-recognized to be capable of achieving the ultimate economic goal of quantifying targets without the use of any electronic readers for applications especially in resource-poor settings in either the developing world or developed countries.45–49
Herein, we respond to this challenge by initially describing the proof-of-concept of a new functional DNA biosensor for equipment-free, read-by-eye quantification of adenosine, interferon-γ and Pb2+ ion as three types of model analytical targets. This sensor integrates enzyme-coated microbeads for efficient signal amplification with distance measurement on μPADs based on starch-hydrolysis-adjusted wettability change of paper body for portable quantitative readout. Its working principle is illustrated in Fig. 1. For each type of analyte, super-paramagnetic microbead (SPM) is covalently modified with a terminally biotinylated DNA strand for anchoring an analyte-specific aptamer or DNAzyme. After the functional DNA probe recognizes the analyte in a sample, the biotin is thus exposed to capture a streptavidin-labeled SiO2 microbead that is loaded with lots of glucoamylase tags (via biotin–streptavidin interaction). The glucoamylase is then adopted to further catalyze the hydrolysis of starch polymer with low solubility to produce glucose molecules with high solubility. After adding the reaction mixture onto a strip-like μPAD fabricated by laser cutting,50 the unhydrolyzed starch polymer can enhance the hydrophobicity of the paper body to inversely adjust flowing distance of a colored reagent (red ink) pre-loaded in the paper device. In other words, the ink's flowing distance is positively proportional to the target concentration in the sample. The results show that our method requires only a ubiquitous, cheap ruler and the ability to see the color to sensitively, selectively quantify the level of the chosen model analytes in artificial buffer samples as well as real complex samples (e.g., undiluted human serum, tap water, pond water, etc.). To the best of our knowledge, this may be the first report of applying the simple distance-measuring quantitative detection motif on the basis of the starch-hydrolysis-adjusted wettability change in μPADs to design general, equipment-free functional DNA biosensors.
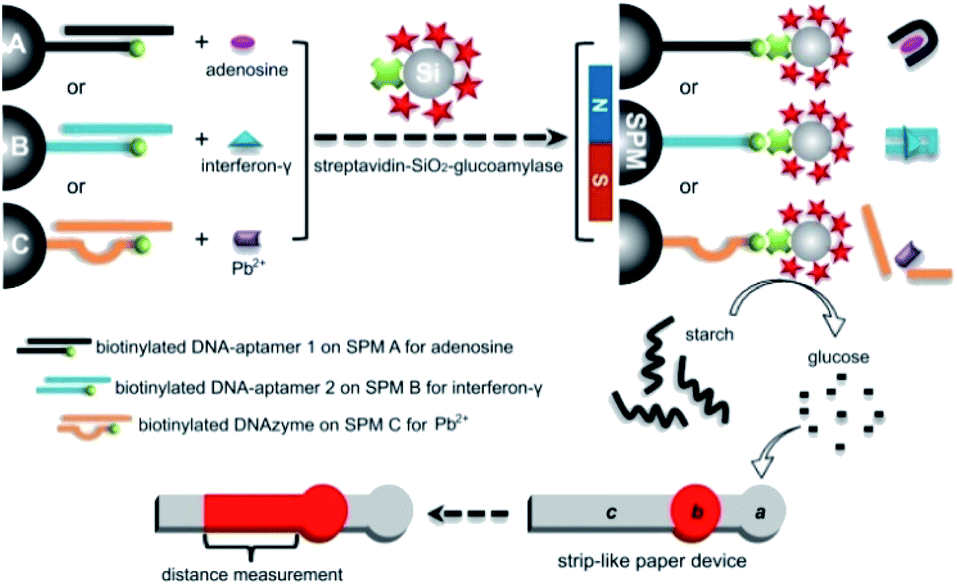 | ||
| Fig. 1 Schematic working principle of the developed functional DNA biosensors that use SPMs A, B and C loaded with aptamers or DNAzymes and enzyme-coated SiO2 microbeads for read-by-eye distance-measuring quantification of adenosine, interferon-γ, and Pb2+ (as three model analytes) based on starch-hydrolysis-adjusted wettability change in strip-like μPADs, respectively. | ||
Experimental
Materials and apparatus
All of the used oligonucleotides were synthesized from Takara Biotechnology Co., Ltd. (Dalian, China). Their thermodynamic parameters were calculated by using the bioinformatics software (http://www.bioinfo.rpi.edu/applications/). Their sequences (from 5′ to 3′) are as follows: biotinylated capture DNA strand for adenosine, biotin-CCC AGG TCA GTG GAG-(CH2)6-NH2; adenosine aptamer, CAC TGA CCT GGG GGA GTA TTG CGG AGG AAG GT (the sequence for adenosine binding is italic);51 biotinylated capture DNA strand for interferon-γ, NH2-(CH2)6-A12-AAC ACA ACC AAC C-biotin; interferon-γ aptamer, TGG GGT TGG TTG TGT TGG GTG TTG TGT;52 biotinylated capture DNA strand for Pb2+, NH2-(CH2)6-A3-AAT CAT CTC TGA AGT AGC GCC GCC GTA GTG-biotin (the sequence of DNAzyme is italic); substrate strand, AAA CAC TrA GGA AGA GAT GAT T (the italic “rA” represents adenosine ribonucleotide).53Streptavidin (from Streptomyces avidinii, >17 U mg−1) and glutaraldehyde were purchased from Sigma-Aldrich. Adenosine, cytidine, uridine, and thymidine were products of Jiehui Biotechnology Co., Ltd. Recombinant human interferon-γ, human serum albumin, human IgG, glucose oxidase, glucoamylase (>20 U mg−1), bovine serum albumin, lysine, and polyethylene glycol (molecular weight range 7000–9000) were provided by Shanghai Sangon Biotechnology Co., Ltd. (Shanghai, China). Soluble starch was obtained from Xilong Chemical Co., Ltd. (Shanghai, China). Amine-coated super-paramagnetic microbeads (SPMs, 1 μm in average diameter) and amine-coated SiO2 microbeads (0.2 μm in average diameter) were from Tianjin BaseLine Chrom Tech Research Centre (Tianjin, China). All other chemicals of analytical grade were used as received. Human serum was collected from healthy volunteers. Real water samples include commercially-available drinking water (a product of Hangzhou Wahaha Co., Ltd., Hangzhou, China), tap water obtained from our lab, and pond water collected from a pond on our campus. All of these real water samples were filtrated with polycarbonate nanoporous membranes (Whatman, 0.2 μM in average pore size) before analysis. Unless specially stated, ultrapure water (with a resistivity of ∼18.2 MΩ cm) was used to prepare stock solutions and buffer. The deionized water instrument was gained from Chengdu Yuechun Technology Co., Ltd. (Chengdu, China). The used buffers include: 10 mM phosphate-buffered saline (PBS, pH 6 or 7.4) that contains 100 mM NaCl and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.4) containing 50 mM NaCl and 5 mM MgCl2.
Quantitative filter paper (thickness ∼195 μm, ash ≤0.009%, pore size 1–3 μM, basis weight of ∼100 g m−2) was purchased from Hangzhou Xinhua Paper Industry Co., Ltd. (Hangzhou, China). The filter paper was chosen herein as one example of practice for fabricating paper devices, but other types of paper substrates such as chromatography paper are also suitable. A mini-type CO2 laser machine (XB-3020) was a product of Shandong Xinbang Laser Equipment Co., Ltd. (Shandong, China).
Preparation of functional DNA–SPM conjugates
In brief, biotinylated capture DNA strands and aptamer or substrate strands in PBS (pH 7.4, 0.5 μM each) were mixed, heated to 90 °C, incubated for 10 min, and finally allowed to cool down slowly to room temperature (∼2 h). Duplex DNA strands could be formed through hybridization reactions. Meanwhile, 1 mL of a 1 mg mL−1 SPM suspension in PBS (pH 7.4) was incubated with 5 mL of a glutaraldehyde solution (5%, w/v) in PBS (pH 7.4) for 3 h at room temperature. The resultant aldehyde-modified SPMs were isolated magnetically and washed with the buffer (3 times), and then dispersed in 1 mL of the 0.5 μM duplex DNA solution above. The mixture was incubated under continuous gentle stirring at 4 °C overnight. Moreover, 1 mL of a 0.1 M lysine solution was further utilized to block residuary aldehyde on SPMs, followed by magnetic segregation and thorough washings. The as-prepared DNA–SPM bioconjugates for the assays of adenosine, interferon-γ, and Pb2+ were re-suspended in 1 mL of PBS, HEPES, and HEPES buffers (pH 7.4, containing 1.5% (w/v) polyethylene glycol), respectively, and stored at 4 °C for further use.Preparation of glucoamylase–SiO2–streptavidin conjugates
Briefly, 1 mL of a 1 mg mL−1 SiO2 microbead suspension in PBS (pH 7.4) was mixed with 5 mL of a glutaraldehyde solution (5%, w/v) in PBS (pH 7.4). Three hours later, excess glutaraldehyde was removed from the mixture by centrifuging and washing these microbeads 3 times with water, followed by re-dispersion in 5 mL of PBS buffer (pH 7.4). Then, 1 mL of a streptavidin solution (1 mg mL−1) and 1 mL of a 10 mg mL−1 glucoamylase solution were added into the suspension of aldehyde-activated microbeads and were incubated under continuous gentle stirring at 4 °C overnight. After centrifugal separation and washing, the resultant biofunctionalized SiO2 microbeads were re-suspended in 6 mL of a solution of bovine serum albumin (10 mg mL−1, containing 1.5% (w/v) polyethylene glycol) in PBS buffer (pH 6) and finally stored at 4 °C in a refrigerator.Fabrication of strip-like μPADs via laser cutting
The used strip-like μPADs were fabricated with a laser cutting method.50 In brief, each device mainly, sequentially consists of a circular region a (4.6 mm in diameter) for reaction mixture addition, a circular region b (3.5 mm in diameter) for colored reagent (red ink) immobilization, and a 2 mm × 25 mm rectangular region c for distance-measuring readout (as depicted in Fig. 1). The pattern and size (as black lines on a white background) were designed using CorelDrawX6 on a PC. After a piece of filter paper was placed at the working platform in the laser machine, the laser with a spot size of ∼0.3 mm cut the paper according to the designed pattern at the applied current of 4 mA, cutting rate of 20 mm s−1, and laser power of 40 W. The patterned paper devices could be produced one by one from the main paper substrate every short period of time less than 10 s. For each μPAD, ∼0.5 μL of red ink was added onto its region b and then allowed to air dry at room temperature (∼25 °C). After that, the two regions b and c were sandwiched with two pieces of transparent adhesive tape. Each freshly prepared μPAD was singly loaded in a vacuum-packed storage bag and finally stored in a dark place until used.Analytical procedures for adenosine detection
In a typical assay, 10 μL of an artificial sample in PBS (pH 7.4) or analyte-spiked undiluted human serum, 10 μL of SPMs immobilized with the adenosine-specific aptamer, and 10 μL of a biofunctionalized SiO2 microbead suspension were mixed and incubated at room temperature (∼25 °C) for 1 h. The complex product was separated magnetically and rinsed with PBS (pH 6) for three times, followed by reacting with 100 μL of a 0.5 wt% starch solution in PBS (pH 6) for 40 min. Then, 5 μL aliquot of the final reaction mixture was dropped onto the circular region a of a μPAD; the liquid would flow into the circular region b via capillary action. As a result, the red ink sediment in the region b would be redissolved and in turn, would flow in the rectangular region c. After the flowing of the colored reagent stopped (within in 1 min), its flowing distance (FD) was then measured with a ruler. The FD value is positively proportional to the level of adenosine target in the sample. The corresponding signal of FD change (ΔFD) was defined as ΔFD = FDadenosine − FDblank, in which FDadenosine and FDblank were the FD values measured for an adenosine sample and a blank PBS sample, respectively. Specificity experiments were also performed in the same manner but using cytidine, uridine or thymidine instead of adenosine.Analytical procedures for interferon-γ detection
Except for the SPMs coated with interferon-γ-specific aptamer, the interferon-γ assay was conducted according to the same procedures for the adenosine analysis at room temperature (∼25 °C). The measured FD signals positively relied on the tested protein levels in artificial samples in HEPES buffer or analyte-spiked undiluted human serum samples. The ΔFD value for each interferon-γ sample was defined as ΔFD = FDinterferon-γ − FDblank, in which FDinterferon-γ and FDblank were the FD values measured for an interferon-γ sample and a blank buffer sample, respectively. Specificity experiments were conducted in the same manner but using human serum albumin, human IgG or glucose oxidase instead of interferon-γ.Analytical procedures for Pb2+detection
Except for the SPMs functionalized with Pb2+-specific DNAzyme, the ion analysis was performed through the same procedures for the adenosine detection at room temperature (∼25 °C). The measured FD signals were positively proportional to the tested Pb2+ levels in artificial samples in HEPES buffer or analyte-spiked real water samples (i.e., drinking water, tap water, and pond water). The ΔFD value for each Pb2+ sample was defined as ΔFD = FDPb2+ − FDblank, in which FDPb2+ and FDblank were the FD values measured for a Pb2+ sample and a blank buffer sample, respectively. Specificity experiments were carried out in the same manner but using K+, Na+, Ag+, Ni2+, Co2+, Cu2+, Hg2+, Ca2+, Cd2+, Cr3+, Al3+ or Fe3+ instead of Pb2+.Results and discussion
As aforementioned, external electronic readers are typically required for most of the current functional DNA sensors to perform quantitative analysis. Herein, we describe a new functional DNA sensor in which the equipment-free distance readout in the strip-like μPADs is simply adopted to quantify the level of three types of model analytes (as depicted in Fig. 1). In addition, the method combines SPMs loaded with aptamer or DNAzyme for highly specific target recognition (binding) with enzymatic reactions based on enzyme-immobilized SiO2 microbeads for highly efficient signal amplification. Each paper device consists of a bare circular region a (for the addition of the final reaction mixture containing unhydrolyzed starch polymer), a circular region b deposited with red ink, and a rectangular measurement region c. As the level of starch polymer with low solubility can adjust the paper's hydrophobicity of the region a, the volume of the reaction mixture solution which will flow into the region b (to redissolve the red ink sediment) and region c (where red staining can be produced) can be adjusted subsequently. The flowing (staining) distance (FD) of the red ink in the region c positively relies on the starch level that is positively related to the target concentration in the sample. The red ink is chosen as the reporting reagent for color development in the paper device because it is quite cheap and commercially available in daily life.The key concept of our method is the transition of the target detection into the read-by-eye measurement of the FD value of red ink. Thus, the starch level-dependant ink flowing in the strip-like μPAD was first investigated. Fig. 2 shows the images of the ink's FD results obtained from several starch solutions with different levels. Obviously, in the absence of starch (i.e., 0 wt% in the solution), a long ink staining (with an FD value up to ∼13.8 mm) was observed in the region c in the used paper device (Fig. 2a), directly indicating the excellent (original) hydrophilicity of the paper body in its region a. As expected, on the other hand, a relatively shorter ink staining (with an FD value of ∼11.3 mm) was obtained from the 0.12 wt% starch solution (Fig. 2b). This phenomenon might be explained in that the starch with low solubility could enhance the hydrophobicity of the region a in the corresponding μPAD by physically blocking the paper's porous microstructures. As a result, only a partial solution could flow into the region b to dissolve a partial amount of red ink sediment for creating relatively shorter staining in the region c via the capillary action. Moreover, higher starch levels led to shorter ink staining results. For example, two FD values of ∼9.3 (Fig. 2c) and 4.5 (Fig. 2d) mm were produced from 0.25 and 0.5 wt% starch solutions, respectively. The ink's FD values were inversely yet linearly associated with the starch levels in a proper range, though too high a starch level could totally block the paper's porous microstructures resulting in no ink flowing in the paper devices (Fig. S1 in ESI†). However, after a 10 μg mL−1 glucoamylase solution was incubated with the 0.5 wt% starch solution, a long ink staining with a large FD value of ∼13 mm was interestedly realized in the involved μPAD (Fig. 2e), presumably due to the fact that the starch of low solubility had been hydrolyzed by the enzyme to small molecule glucose with high solubility which hardly changed the paper's original wettability. In other words, it should be expected to adopt the FD value to indirectly measure the concentration of glucoamylase probe capable of adjusting the starch level. Moreover, it was also experimentally found that the encapsulation of the μPADs with transparent adhesive tape benefited stable (repeatable) ink flowing in the paper body (Fig. S2 in ESI†).
 | ||
| Fig. 2 Flowing distance (FD) results of the colored reagent obtained from different solutions in the strip-like μPADs. The scale bar is 3.5 mm. | ||
After demonstrating the starch-controlled ink flowing in the μPADs that could be adjusted by the glucoamylase, the feasibility of the proposed DNA biosensor with aptamer-loaded SPMs was studied for a small molecule model analyte, i.e., adenosine, a vital cofactor in many biological processes such as kidney function.51
Assays of a 50 μM adenosine sample and a blank sample (PBS buffer without the analyte) were performed according to the schematic procedures shown in Fig. 1, compared with a background starch solution (control). The FD values on the red ink-loaded μPADs used were measured. Fig. 3A displays that no significative differences are observed in small FD values obtained from the PBS buffer (∼4.7) and the control (∼4.5), while the FD measured for the adenosine is as large as ∼10.8. Such a dramatically increased red ink's FD proved that after the adenosine targets had been captured by their aptamers, the recognition events could be further traced by the SiO2 microbeads immobilized with lots of glucoamylases that subsequently catalyzed the hydrolysis of starch molecules capable of enhancing the hydrophobicity of the paper devices. Moreover, the detection specificity of the sensor was also tested by carrying out the analysis of the other three types of small molecules belonging to the nucleosides family, i.e., 100 μM cytidine, thymidine, and uridine. The corresponding FD results were also provided in Fig. 3A. As clearly shown in Fig. 3A, although their concentrations were far higher than the adenosine level, all of the FD values obtained from the three non-specific small molecules are only around 4.8, which is quite similar to that recorded from the blank buffer (∼4.7). This comparison study confirmed that only the adenosine target could be recognized and bound specifically by its aptamer on the SPM to trigger the glucoamylase-catalyzed starch hydrolysis.
 | ||
| Fig. 3 (A) Flowing distance (FD) results obtained from a starch solution (0.5 wt%), a blank sample (PBS buffer without the analyte), 50 μM adenosine, and three other sorts of small molecules (100 μM each). (B) Calibration curve that describes the relationships between the FD change (ΔFD = FDadenosine − FDblank) and the logarithm values of the adenosine concentrations (log[adenosine]) in the buffer samples. The linear regression equation is y = 2.7651x + 1.8551 (R = 0.9914). Each error bar represents a standard deviation across three replicate experiments. | ||
Then, our new biosensor was used to detect a series of buffer samples containing varying adenosine levels under the optimized experimental conditions for assessing its equipment-free analytical performance (Fig. S3–S5 in ESI†). As presented in Fig. 3B that the ΔFD value (FDadenosine − FDblank) increased as the analyte level increased. Fig. 3B further showed that the ΔFD value was linear in a detection range of 1.7–62.5 μM adenosine, realizing a limit of detection (LOD) of ∼1.6 μM (3σ). It should be noteworthy that this LOD was achieved just with simple distance readout in paper microfluidics but was comparable to or even lower than that obtained using some recent instrumental methods with fluorescence, electrochemistry, surface plasmon resonance, surface-enhanced Raman scattering, electromagnetic piezoelectric acoustic platform, and absorbance or distance measurements (Table S1 in ESI†). The satisfactory sensitivity was mainly attributed to the use of enzyme-coated microbead probes for significantly amplifying the signal per each adenosine–aptamer binding event. Moreover, its analytical repeatability was proved with the small relative standard deviations (RSDs) of ∼3.1 and 4.4% (n = 5) obtained from the assays of 1.7 and 15 μM adenosine samples, respectively (Fig. S6 and S7 in ESI†). In addition, to verify the sensor's accuracy for the adenosine detection, several undiluted human serum samples spiked with fixed analyte levels were assayed according to the general procedures. The calculated recovery results were from 97.26 to 101.07% (Table S2 in ESI†), which indicated excellent practicality of the developed approach. In particular, the data proved that the aptamer still kept its specific molecular recognition capability to the adenosine even in the real serum. The SPM platform could additionally minimize undesirable effects that possibly existed in such a complex matrix.
Next, to further evaluate the versatility of this distance-measuring- based functional DNA sensor, interferon-γ was assayed as a protein analyte using SPMs immobilized with its corresponding aptamer under the optimal experimental conditions (Fig. S5 and S8 in ESI†). Abnormal interferon-γ levels are clinically associated with many pathogen-caused responses of the human immune system as well as infectious diseases.52 The sensor's specificity for the interferon-γ analysis was studied by assaying human serum albumin, human IgG, glucose oxidase, and blank HEPES buffer (negative control). It is obviously presented in Fig. 4A that only the interferon-γ target gave a large FD value of ∼11.5, while all of the other non-specific proteins (10 μM) gave small signals (∼4.7) quite similar to that obtained from the control buffer sample (∼4.6). A set of interferon-γ samples (in HEPES buffer containing 0–64 nM analyte) were analyzed. The calculated ΔFD results (FDinterferon-γ − FDblank) which were positively proportional to the analyte level were displayed in Fig. 4B. This figure illustrates a linear relationship of the ΔFD value and the interferon-γ concentration in a range from 0.5 to 32 nM. The LOD was estimated to be as low as ∼0.3 nM (3σ), thus making our instrument-free approach one of the most sensitive interferon-γ methods in comparison with many other typical instrumental techniques (Table S3 in ESI†). Furthermore, the RSDs obtained from the analysis of 0.5 and 32 nM interferon-γ samples were ∼2.7 and 4.6% (n = 5), respectively, suggesting good detection stability of the μPADs used (Fig. S9 and S10 in ESI†). These results of the adenosine and interferon-γ assays clearly proved that our equipment-free functional DNA sensor should be highly applicable to the aptamer's diversity for realizing sensitive, specific, and reliable detection of small molecules or macromolecules like proteins of interest.
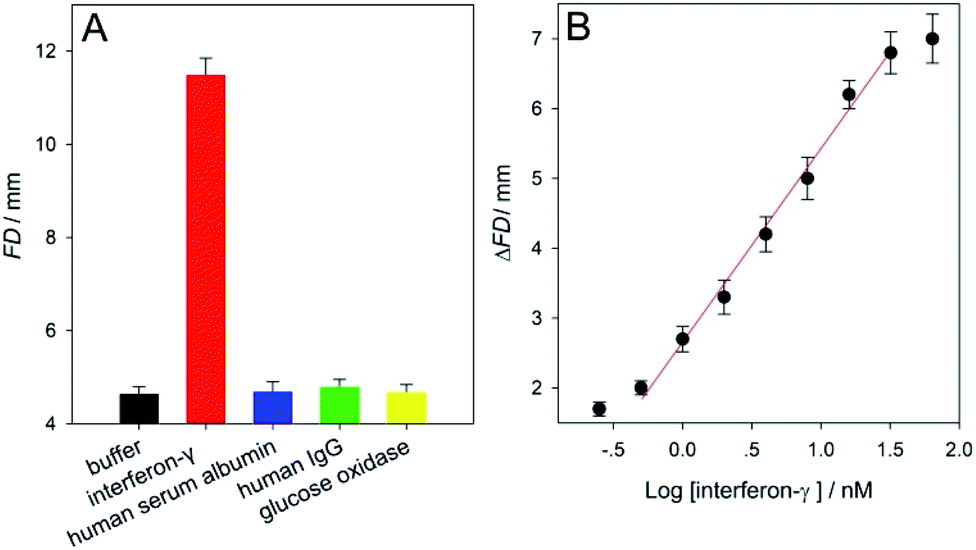 | ||
| Fig. 4 (A) Flowing distance (FD) results obtained from a blank sample (HEPES buffer without the analyte), 30 nM interferon-γ, and three other sorts of proteins (10 μM each). (B) Calibration curve that describes the relationships between the FD change (ΔFD = FDinterferon-γ − FDblank) and the logarithm values of the interferon-γ concentrations (log[interferon-γ]) in the buffer samples. The linear regression equation is y = 2.7406x + 2.6643 (R = 0.9961). Each error bar represents a standard deviation across three replicate experiments. | ||
Apart from the aptamer, DNAzyme was also used to design (extend) the biosensor with distance-measuring readout developed herein to detect Pb2+ ions (that belong to the highest toxicity class of heavy metal pollutants54) using SPMs coated with Pb2+-specific DNAzymes. The working principle of the DNAzyme sensor was also depicted in Fig. 1. The related main experimental conditions had been optimized (Fig. S5 and S11 in ESI†). The feasibility and specificity results were summarized in Fig. 5A. One can find from Fig. 5A that, for either a blank sample (HEPES buffer without the analyte), or 1 mM K+, Na+, Ag+, Ni2+, Co2+, Cu2+, Hg2+, Ca2+, Cd2+, Cr3+, Al3+ and Fe3+, the recorded small FD values that could not be clearly distinguished from each other were about 4.6. On the contrast, the detection of 50 nM Pb2+ led to a large FD up to ∼11, indirectly reflecting that only in this case efficient glucoamylase-catalyzed starch hydrolysis occurred successfully after the target ions had been specifically recognized by their DNAzymes functionalized on the SPMs. Then, our biosensor was utilized to analyze a series of Pb2+ samples in the buffer. The results of ΔFD (FDPb2+ − FDblank) were shown in Fig. 5B, from which a linear detection range of 0.75–50 nM Pb2+ was determined. The LOD for the analyte ion was estimated to be ∼0.5 nM (3σ); it was about 100 times lower than the maximum level (∼48 nM) of Pb2+ in drinking water permitted by the U.S. Environmental Protection Agency.54 Compared with some recently-reported representative fluorescent, electrochemical, colorimetric or distance-measuring methods, this new approach achieved comparable or even better detection sensitivity (Table S4 in ESI†). Moreover, both of 3 and 12 nM Pb2+ samples were assayed five times in parallel and the calculated RSDs were ∼1.9 and 2.7% (n = 5), respectively (Fig. S12 and S13 in ESI†). Additionally, the recovery results for Pb2+ obtained from several commercially-available drinking water samples, tap water samples, and pond water samples were between 98.5–101.6%, 97.1–104.2%, and 95.4–109.6%, respectively (Table S5 in ESI†). The data confirmed that our method possessed good detection repeatability as well as accuracy.
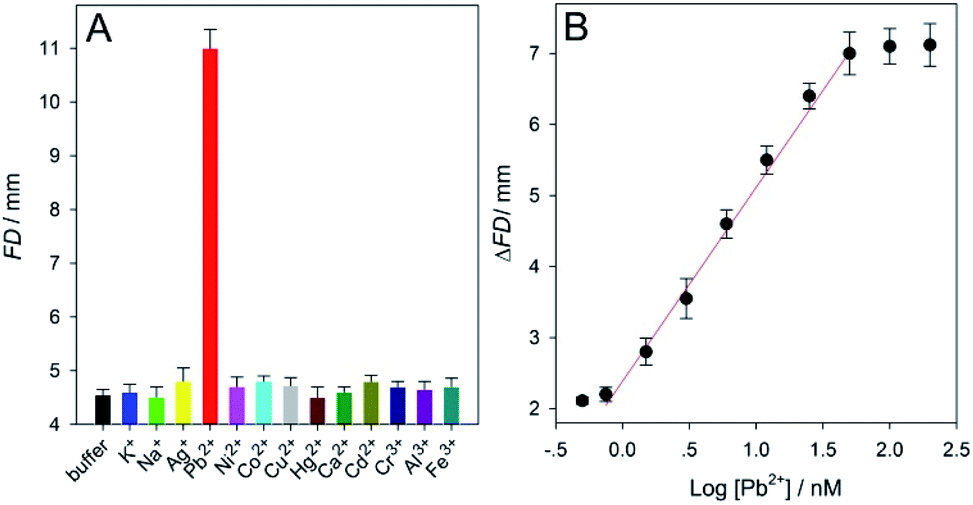 | ||
| Fig. 5 (A) Flowing distance (FD) results obtained from a blank sample (HEPES buffer without the analyte), 50 nM Pb2+, and twelve other sorts of metal ions (1 mM each). (B) Calibration curve that describes the relationships between the FD change (ΔFD = FDPb2+ − FDblank) and the logarithm values of the Pb2+ concentrations (log[Pb2+]) in the buffer samples. The linear regression equation is y = 2.7252x + 2.3799 (R = 0.9973). Each error bar represents a standard deviation across three replicate experiments. | ||
Conclusions
We have developed an equipment-free functional DNA sensor with a simple quantitative detection motif of distance measurement based on starch-hydrolysis-adjusted wettability change in μPADs. The SiO2 microbead-enhanced enzymatic reactions were adopted for highly efficient signal amplification. The results demonstrate that only a ubiquitous ruler is required to realize the low-cost, read-by-eye detection of trace levels of three model targets, namely the adenosine (∼1.6 μM LOD), interferon-γ (∼0.3 nM LOD), and Pb2+ (∼0.5 nM LOD). With the diversity of available functional DNAs like aptamers and DNAzymes, our general strategy can be easily tailored to achieve simple quantification of many other targets of interest ranging from ions to small molecules and macromolecules. It thus holds great potential as a sustainable technique to especially facilitate novel paradigms of personalized medical diagnosis, environmental monitoring in the field and food safety evaluation at home. Although, in its current state, the instrument-free biosensor designed is not yet available for practical point-of-care testing uses, it can be further simplified through more professional engineering in terms of reaction format and data analysis, on which some research work is now underway in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from National Natural Science Foundation of China (21874032, 21765007, and 21765005), Guangxi Key Research Project (GuikeAB17129003), Guangxi Natural Science Foundation (2018GXNSFAA138047), Guangxi Science Fund for Distinguished Young Scholars (2018GXNSFFA281002), Guangxi Scholarship Fund of Guangxi Education Department (Guijiaoren-2018-18), Fund of Guangxi Education Office (2018KY0260), and Fund of Guangxi Key Laboratory of Electrochemical and Magneto-chemical Functional Materials (EMFM20161202).References
- R. R. Breaker, Nat. Biotechnol., 1997, 15, 427 CrossRef CAS PubMed.
- D. S. Wilson and J. W. Szostak, Annu. Rev. Biochem., 1999, 68, 611 CrossRef CAS PubMed.
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344, 467 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505 CrossRef CAS PubMed.
- L. Gong, Y. F. Lv, H. Liang, S. Y. Huan, X. B. Zhang and W. J. Zhang, Rev. Anal. Chem., 2014, 33, 201 CAS.
- J. Li, L. T. Mo, C. H. Lu, T. Fu, H. H. Yang and W. H. Tan, Chem. Soc. Rev., 2016, 45, 1410 RSC.
- S. A. Li, X. T. Zhao, X. X. Yu, Y. Q. Wan, M. Y. Yin, W. W. Zhang, B. Q. Cao and H. Wang, Anal. Chem., 2019, 91, 14737 CrossRef CAS PubMed.
- M. Majdinasab, A. Hayat and J. L. Marty, TrAC, Trends Anal. Chem., 2018, 107, 60 CrossRef CAS.
- K. Mao, H. Zhang, Z. L. Wang, H. R. Cao, K. K. Zhang, X. Q. Li and Z. G. Yang, Biosens. Bioelectron., 2020, 148, 111785 CrossRef CAS PubMed.
- H. M. Meng, H. Liu, H. L. Kuai, R. Z. Peng, L. T. Mo and X. B. Zhang, Chem. Soc. Rev., 2016, 45, 2583 RSC.
- G. C. Han, X. Z. Feng and Z. C. Chen, Int. J. Electrochem. Sci., 2015, 10, 3897 CAS.
- Q. Yuan, D. Q. Lu, X. B. Zhang, Z. Chen and W. H. Tan, TrAC, Trends Anal. Chem., 2012, 39, 72 CrossRef CAS.
- D. Tan, Y. He, X. Xing, Y. Zhao, H. Tang and D. Pang, Talanta, 2013, 113, 26 CrossRef CAS PubMed.
- Y. Xiang and Y. Lu, Nat. Chem., 2011, 3, 697 CrossRef CAS PubMed.
- F. S. Sabet, M. Hosseini, H. Khabbaz, M. Dadmehr and M. R. Ganjali, Food Chem., 2017, 220, 527 CrossRef CAS PubMed.
- F. L. Gao, J. Wu, Y. Yao, Y. Zhang, X. J. Liao, D. Q. Geng and D. Q. Tang, RSC Adv., 2018, 8, 28161 RSC.
- Y. J. Yang, J. Huang, X. H. Yang, X. X. He, K. Quan, N. L. Xie, M. Ou and K. M. Wang, Anal. Chem., 2017, 89, 5851 Search PubMed.
- J. M. Yang, B. T. Dou, R. Yuan and Y. Xiang, Anal. Chem., 2016, 88, 8218 CrossRef CAS PubMed.
- X. Chang, C. Zhang, C. Lv, Y. Sun, M. Z. Zhang, Y. M. Zhao, L. L. Yang, D. Han and W. H. Tan, J. Am. Chem. Soc., 2019, 141(32), 12738 CrossRef CAS PubMed.
- Y. N. Guo, Y. Wang, S. Liu, J. H. Yu, H. Z. Wang, Y. L. Wang and J. D. Huang, Biosens. Bioelectron., 2016, 75, 315 CrossRef CAS PubMed.
- Y. Wang, X. D. Li, D. M. Xia and X. Q. Wang, RSC Adv., 2019, 9, 37144 RSC.
- D. M. Cate, J. A. Adkins, J. Mettakoonpitak and C. S. Henry, Anal. Chem., 2015, 87, 19 CrossRef CAS PubMed.
- S. G. Ge, L. N. Zhang, Y. Zhang, F. F. Lan, M. Yan and J. H. Yu, Nanoscale, 2017, 9, 4366 RSC.
- K. Yamada, T. G. Henares, K. Suzuki and D. Citterio, Angew. Chem., Int. Ed., 2015, 54, 5294 CrossRef CAS PubMed.
- J. F. Nie, Y. Zhang, L. W. Lin, C. B. Zhou, S. H. Li, L. M. Zhang and J. P. Li, Anal. Chem., 2012, 84, 6331 CrossRef CAS PubMed.
- Y. Zhang, C. B. Zhou, J. F. Nie, S. W. Le, Q. Qin, F. Liu, Y. P. Li and J. P. Li, Anal. Chem., 2014, 86, 2005 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3 CrossRef CAS PubMed.
- Y. Y. Yang, E. Noviana, M. P. Nguyen, B. J. Geiss, D. S. Dandy and C. S. Henry, Anal. Chem., 2017, 89, 71 CrossRef CAS PubMed.
- N. Cheng, Y. Song, Q. R. Shi, D. Du, D. Liu, Y. B. Lu, W. T. Xu and Y. H. Lin, Anal. Chem., 2019, 91, 13986 CrossRef CAS PubMed.
- S. Dalirirad and A. J. Steckl, Sens. Actuators, B, 2019, 83, 79 CrossRef.
- M. M. Ali, M. Wolfe, K. Tram, J. Gu, C. D. M. Filipe, Y. F. Li and J. D. Brennan, Angew. Chem., Int. Ed., 2019, 58, 9907 CrossRef CAS PubMed.
- X. Li, X. He, Q. Zhang, Y. Y. Chang and M. Liu, Anal. Methods, 2019, 11, 4328 RSC.
- L. L. Liang, M. Su, L. Li, F. F. Lan, G. X. Yang, S. G. Ge, J. H. Yu and X. R. Song, Sens. Actuators, B, 2016, 229, 347 CrossRef CAS.
- N. K. Singh, P. Jain, S. Das and P. Goswami, Anal. Chem., 2019, 91, 4213 CrossRef CAS PubMed.
- Y. Zhang, J. L. Fan, J. L. Nie, S. W. Le, W. Y. Zhu, D. Gao, J. N. Yang, S. B. Zhang and J. P. Li, Biosens. Bioelectron., 2015, 73, 13 CrossRef CAS PubMed.
- Y. Zhang, J. M. Xu, S. Zhou, L. Zhu, X. Lv, J. Zhang, L. N. Zhang, P. H. Zhu and J. H. Yu, Anal. Chem., 2020, 92, 3874 CrossRef CAS PubMed.
- T. Y. Dong, G. A. Wang, M. W. Li and F. Li, Anal. Methods, 2019, 11, 5376 RSC.
- H. Y. Fu, J. H. Yang, L. Guo, J. F. Nie, Q. B. Yin, L. Zhang and Y. Zhang, Biosens. Bioelectron., 2017, 96, 194 CrossRef CAS PubMed.
- H. Liu, Y. Xiang, Y. Lu and R. M. Crooks, Angew. Chem., Int. Ed., 2012, 51, 6925 CrossRef CAS PubMed.
- X. F. Wei, T. Tian, S. S. Jia, Z. Zhu, Y. L. Ma, J. J. Sun, Z. Y. Lin and C. J. Yang, Anal. Chem., 2016, 88, 2345 CrossRef CAS PubMed.
- T. Tian, Y. An, Y. P. Wu, Y. L. Song, Z. Zhu and C. Y. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 30480 CrossRef CAS PubMed.
- Y. Zhang, D. Gao, J. L. Fan, J. F. Nie, S. W. Le, W. Y. Zhu, J. N. Yang and J. P. Li, Biosens. Bioelectron., 2016, 78, 538 CrossRef CAS PubMed.
- X. F. Wei, T. Tian, S. S. Jia, Z. Zhu, Y. L. Ma, J. J. Sun, Z. Y. Lin and C. J. Yang, Anal. Chem., 2016, 88, 2345 CrossRef CAS PubMed.
- Y. Zhang, D. Gao, J. L. Fan, J. F. Nie, S. W. Le, W. Y. Zhu, J. N. Yang and J. P. Li, Biosens. Bioelectron., 2016, 78, 538 CrossRef CAS PubMed.
- Y. Zhang, J. M. Xu, S. Zhou, L. Zhu, X. Lv, J. Zhang, L. N. Zhang, P. H. Zhu and J. H. Yu, Anal. Chem., 2020, 92, 3874 CrossRef CAS PubMed.
- S. Chung, C. M. Jennings and J. Yoon, Chem.–Eur. J., 2019, 25, 13070 CrossRef CAS PubMed.
- B. Alsaeed and F. R. Mansour, Microchem. J., 2020, 155, 104664 CrossRef.
- J. F. Nie, Y. Z. Liang, S. B. Zhang, S. W. Le, D. N. Li and Y. Zhang, Analyst, 2013, 138, 671 RSC.
- H. L. Wang, Y. Zhang, R. X. Li, J. F. Nie, A. H. El-Sagheer, T. Brown, Z. Y. Liu and W. C. Xiao, Chem. Commun., 2017, 53, 8407 RSC.
- W. C. Xiao, Z. H. Deng, J. K. Huang, Z. H. Huang, M. M. Zhuang, Y. L. Yuan, J. F. Nie and Y. Zhang, Anal. Chem., 2019, 91, 15114 CrossRef CAS PubMed.
- X. H. Zhao, R. M. Kong, X. B. Zhang, H. M. Meng, W. N. Liu, W. H. Tan, G. L. Shen and R. Q. Yu, Anal. Chem., 2011, 83, 5062 CrossRef CAS PubMed.
- Z. Y. Liu, W. Y. Jin, F. X. Wang, T. C. Li, J. F. Nie, W. C. Xiao, Q. Zhang and Y. Zhang, Sens. Actuators, B, 2019, 296, 126698 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04619a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |