
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplexation of biologically essential (mono- and divalent) metal cations to cucurbiturils: a DFT/SMD evaluation of the key factors governing the host–guest recognition†
Nikoleta Kirchevaa,
Stefan Dobreva,
Lyubima Dashevab,
Iskra Kolevab,
Valya Nikolovab,
Silvia Angelova *a and
Todor Dudev*b
*a and
Todor Dudev*b
aInstitute of Optical Materials and Technologies “Acad. J. Malinowski”, Bulgarian Academy of Sciences, 1113 Sofia, Bulgaria. E-mail: sea@iomt.bas.bg
bFaculty of Chemistry and Pharmacy, Sofia University “St. Kl. Ohridski”, 1164 Sofia, Bulgaria. E-mail: t.dudev@chem.uni-sofia.bg
First published on 27th July 2020
Abstract
Supramolecular complexes based on classical synthetic macrocyclic host molecules such as cyclodextrins and calixarenes have received much attention recently due to their broad applications as biological and chemical sensors, bioimaging agents, drug delivery carriers, light-emitting materials, etc. Cucurbit[n]urils comprise another group of cavitands known for their high affinity for various guest molecules. Nonetheless, some aspects of their coordination chemistry remain enigmatic. Although they are recognized as potential biomimetic scaffolds, they are still not tested as metalloenzyme models and not much is known about their metal-binding properties. Furthermore, there is no systematic study on the key factors controlling the processes of metal coordination to these systems. In the computational study herein, DFT molecular modeling has been employed in order to investigate the interactions of biologically essential (mono- and divalent) metal cations to cucurbit[n]urils and evaluate the major determinants shaping the process. The thermodynamic descriptors (Gibbs energies in the gas phase and in a water medium) of the corresponding complexation reactions have been estimated. The results obtained shed light on the mechanism of host–guest recognition and disclose which factors more specifically affect the metal binding process.
Introduction
In view of the increasing panic of the globally spreading coronavirus, the urge of developing novel drugs appears even more pressing. Although substantial efforts and resources have been invested worldwide, the list of newly approved pharmaceutical molecules has been declining since the mid-1990s.1 Significant progress has been achieved, however, in developing novel drug delivery technologies resulting in enhanced drug efficacy and specificity, decreased adverse effects, and suppressed unpleasant characteristics of the active substances.2 Various host systems, such as cyclodextrins,3–5 calixarenes6,7 and crown ethers,8–10 able to accommodate diverse guest molecules, have been recognized as efficient drug delivery vehicles and subjected to large-scale investigations. Another group of cavitands with unique properties is that of cucurbit[n]urils11–14 comprising n condensed units formed by glycoluril and formaldehyde fragments (Fig. 1). These are rigid symmetrical macrocycles which, as the name signifies, resemble that of a pumpkin. The geometry of the representatives of the CB[n] family with n = 5–8, namely cucurbit[5]uril (CB[5]), cucurbit[6]uril (CB[6]), cucurbit[7]uril (CB[7]) and cucurbit[8]uril (CB[8]), are given in Fig. 1.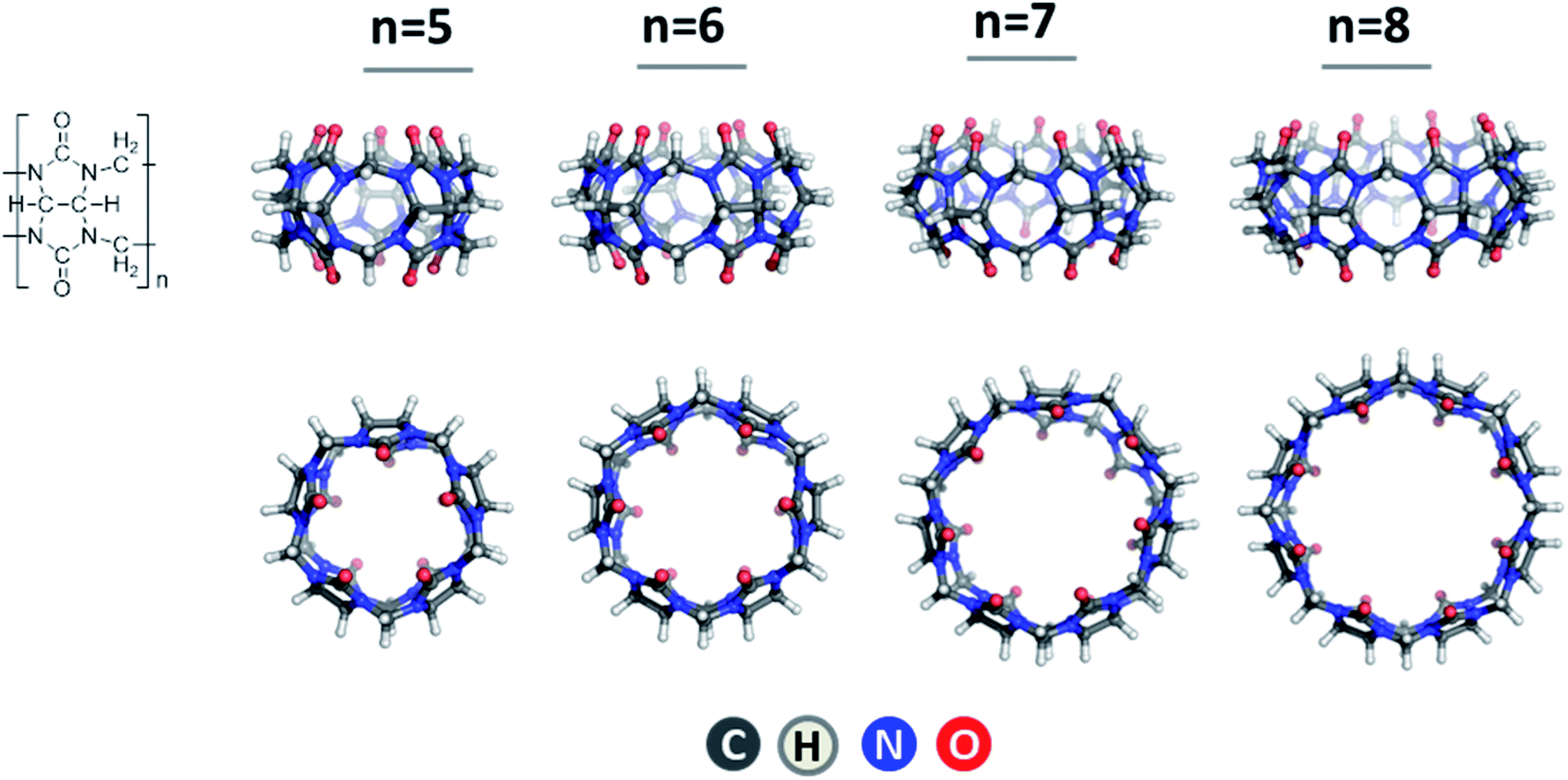 | ||
| Fig. 1 Molecular structures of CB[n], n = 5–8. | ||
As a result of the synthetic work of Kim13 and Day,15 several members of the cucurbituril family with varying pore size comprising 5, 6, 7, 8, 10 and 14 building blocks have been synthesized. Notably, this structural diversity allows CB[n]s to act as host molecules for a great amount of guests, thus finding their way to vast areas of scientific and industrial interest such as catalysis, self-assembled monolayers, drug delivery, photo- and electrochemistry.16–19 Nonetheless, cucurbiturils suffer one main drawback – their insolubility in water environment. Jeon et al.20 suggested a possible way to circumvent this problem as they showed that in aqueous solution of alkali metal salts, in particular sodium sulphate, the cavitands tend to dissolve much more easily. This effect draws from favorable binding of the metal ions to the host macrocycle groups resulting in stable metal–cucurbituril complex formation as revealed in numerous studies.21–29 More importantly, metal–cucurbituril coordination expands the field of their application, as this process results in the formation of various novel constructs such as molecular capsules, tubular polymers and molecular bracelets and necklaces. Consequently, in the newly formed capsules the metal cations act as a “lid”, which can un/block the portal of the cucurbituril under specific conditions, e.g. temperature or acidity, thus allowing the guest-molecule to be specifically released.20,27 Tubular polymers, on the other hand, have gained considerable popularity in the sphere of nanotechnology,30,31 while constructs of the type of molecular bracelets and necklaces are considered as new materials.32–34
A systematic study delineating the key determinants of the metal cation–cucurbituril recognition is, however, lacking. Herein, in an attempt to shed light on the intimate mechanism of the metal binding to cucurbiturils, we systematically study the effect of various factors on the host–guest interactions and unravel the key parameters governing the process. The interaction between CB[n]s (n = 5, 6, 7 and 8) and a series of biologically essential mono- and divalent metal cations (Na+/K+ and Mg2+/Ca2+, respectively) is studied by employing density functional theory (DFT) calculations in conjunction with polarizable continuum model computations (SMD scheme). The influence of different factors on the thermodynamics of the host–guest interactions, such as the size of the host cavitand, metal's nature (cation charge, size and inherent chemical properties), its locality in the complex, the preferable number of ions bound, as well as the presence/absence of a metal hydration shell, is assessed. Notably, this particular approach has proven quite effective in disclosing the basic determinants of host–guest recognition in other macrocyclic systems.35–37
Computational details
The geometries of the host CB[n] systems and their metal complexes were optimized by employing a hybrid M062X Minnesota functional,38 the utilization of which is well known to provide accurate data for thermochemistry and non-covalent interactions.39 Among various basis sets, the 6-31G(d,p) was chosen for the acquisition of the necessary characteristics of the structures using the Gaussian 09 program.40 This specific combination of method/basis set has been recently shown to be largely applicable in modelling the complexes between metal cations and other host-molecules such as cyclodextrins, calixarenes, or lactose.35–37,41Initial geometries of the CB[n] host systems were derived from structural data deposited in the Cambridge Crystallographic Data Centre (CCSD). The necessary constructs were the following: for CB[5] – LOZNIX (CSD Entry from ref. 42); for CB[6] – BATWIE (CSD Entry from ref. 43); for CB[7] – TUHGAG (CSD Entry from ref. 44), and for CB[8] – BATWEA (CSD Entry from ref. 43). The optimization of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CB[n]/metal ion complexes was initiated from a geometry with the metal cation positioned at the center of one of the carbonylated portals of the free CB[n].
:1 CB[n]/metal ion complexes was initiated from a geometry with the metal cation positioned at the center of one of the carbonylated portals of the free CB[n].
The full optimization of each structure was consecutively followed by frequency calculations at the same level of theory, where no negative values were found for the lowest energy configurations. Single point calculations were performed at M062X/6-31+G(d,p) level of theory. Electronic energies obtained at both levels of theory (M062X/6-31G(d,p) and M062X/6-31+G(d,p)//M062X/6-31G(d,p)) were used alongside in the subsequent evaluations. M062X/6-31G(d,p) vibrational frequencies were used to compute the respective thermal energies, Eth, including zero-point energy, and entropy, S, yielding the gas-phase Gibbs energy of the molecule/complex at T = 298.15 K.
| G1 = Eel + Eth − TS | (1) |
The calculations were performed at standard conditions – room temperature (298.15 K) and pressure of 1.0 atm. Changing the temperature and external pressure applied to the system has negligible effect on the evaluated free energies of the complex formation reactions (Table S7, ESI†). Solvation effects were accounted for by employing the SMD (Density-based Solvation Model) method45 as implemented in the Gaussian 09 suite of programs. For each optimized molecule/complex in the gas phase, a single point calculation in water (dielectric constant ε = 78) was performed at both levels of theory: M062X/6-31G(d,p)//M062X/6-31G(d,p) and M062X/6-31+G(d,p)//M062X/6-31G(d,p). Results with the lower basis set are provided in the respective tables of the ESI.† The difference between the gas-phase and SMD calculated energies yields the solvation free energy, ΔGsolv78, of the respective entity: ΔGsolv78 ≈ Eel78 − Eel. Solvation free energies of the products and reactants were used to evaluate the free energy of complex formation, ΔG78, in a water medium:
| ΔG78 = ΔG1 + ΔGsolv78 (products) − ΔGsolv78 (reactants) | (2) |
A positive ΔG78 implies a thermodynamically unfavorable complex formation, whereas a negative value suggests a favorable one. Basis set superposition errors (BSSEs) were accounted for by employing the counterpoise procedure of Boys and Bernardi.46 The artificial stabilization of the complexes (overestimation of the binding energy) was countered for 2 fragments corresponding to the respective host (CB[n]) and guest (bare or hydrated metal cation) in each complex formation reaction. The PyMOL molecular graphics system was used to generate the molecular graphics images.47
Results and discussion
Effect of the cavity size
Complexes between a representative of the metal series, Mg2+ (bare cation), and CB[5], CB[6], CB[7] and CB[8] were optimized and examined (Fig. 2). The thermodynamic data of the reactions of complexation in a gas phase as well as in a water environment are presented in Table 1.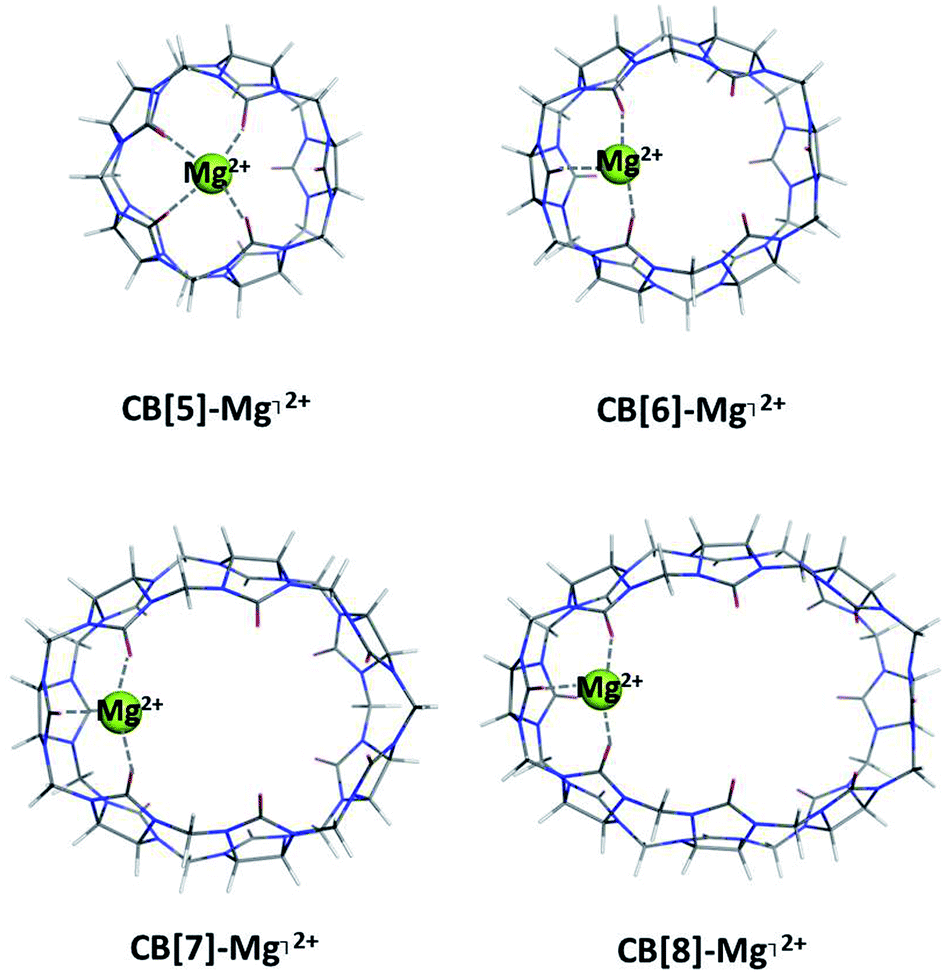 | ||
| Fig. 2 M062X/6-31G(d,p) optimized geometries of the CB[n]–Mg⌉2+-complexes (n = 5–8) in the gas phase. | ||
| Reaction | ΔG1 | ΔG78 |
|---|---|---|
| CB[5] + Mg2+→ CB[5]–Mg⌉2+ | −315.6 | −48.2 |
| CB[6] + Mg2+→ CB[6]–Mg⌉2+ | −293.7 | −41.7 |
| CB[7] + Mg2+→ CB[7]–Mg⌉2+ | −285.8 | −36.2 |
| CB[8] + Mg2+→ CB[8]–Mg⌉2+ | −280.9 | −33.9 |
All calculated Gibbs energies stay firmly on negative ground which corresponds to readily formed CB[n]–Mg⌉2+ complexes. The results obtained show a clear trend: smaller cavitands favor metal complexation in larger extent than their larger homologs (ΔG78 = −48.2 and −41.7 kcal mol−1 for the CB[5] and CB[6], respectively, vs. −36.2 and −33.9 kcal mol−1 for the CB[7] and CB[8], accordingly). A few factors contribute to this outcome. Firstly, as Fig. 2 clearly demonstrates, the number of bonds in the complex between the metal cation and the CB[5] is four due mainly to the smaller radius of the cavity and close proximity of the oxygen-containing residues to the guest metal. As the size of the host molecule increases, the Gibbs energies decrease as a result of the fewer bonds formed (in the CB[6–8]–Mg⌉2+ complexes their number is three). As the bond lengths between the metal and ligating C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups remain almost the same in the series (approximately 1.94 Å), the interactions with the remoter CO groups weaken with enlarging of the cavity, since the distances increase from 3.5 and 4.6 Å in CB[5 and 6] to 5.2 and 5.4 Å in CB[7 and 8], respectively. Another feature that strongly contributes to the de/stabilization of the complexes is the charge transfer. Two schemes were applied, namely the natural population analysis and the Hirshfeld methodology at the same level of theory, and they both show a minor amount of charge transferred from the ligands to the metal cation with increasing the cavity size (e.g. 1.30 e− for the CB[5]-containing complex vs. 1.17 e− for the CB[6–8]-containing structures, Hirshfeld analysis). Thus, the calculations demonstrate that the bare magnesium ion preferentially binds the smaller CB[5] and CB[6] as opposed to their bigger counterparts (CB[7] and CB[8]) due to the better interactions in the decreased cavity size and resulting better charge transfer from the ligating carbonyl groups to the metal cation.
O groups remain almost the same in the series (approximately 1.94 Å), the interactions with the remoter CO groups weaken with enlarging of the cavity, since the distances increase from 3.5 and 4.6 Å in CB[5 and 6] to 5.2 and 5.4 Å in CB[7 and 8], respectively. Another feature that strongly contributes to the de/stabilization of the complexes is the charge transfer. Two schemes were applied, namely the natural population analysis and the Hirshfeld methodology at the same level of theory, and they both show a minor amount of charge transferred from the ligands to the metal cation with increasing the cavity size (e.g. 1.30 e− for the CB[5]-containing complex vs. 1.17 e− for the CB[6–8]-containing structures, Hirshfeld analysis). Thus, the calculations demonstrate that the bare magnesium ion preferentially binds the smaller CB[5] and CB[6] as opposed to their bigger counterparts (CB[7] and CB[8]) due to the better interactions in the decreased cavity size and resulting better charge transfer from the ligating carbonyl groups to the metal cation.
Effect of the cation properties
Complexes between cations from the rest of the series (Na+, K+ and Ca2+) with the middle-size CB[7] were also modelled (Fig. 3) and the effect of the cation characteristics on the thermodynamics of the metal binding was assessed.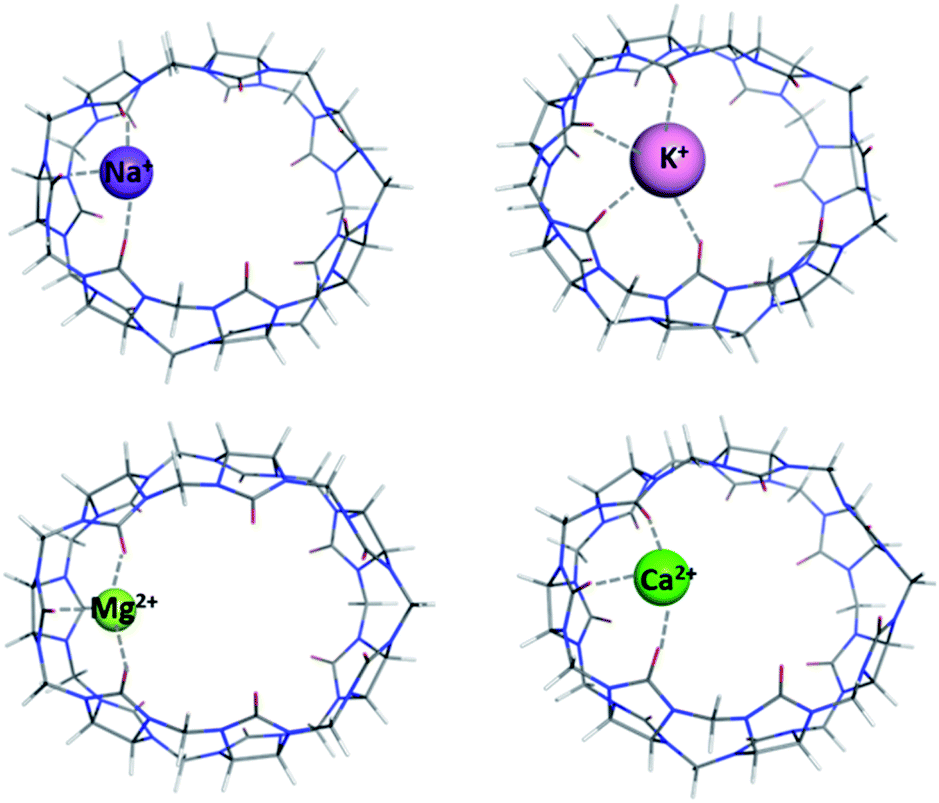 | ||
| Fig. 3 M062X/6-31G(d,p) optimized structures of CB[7]–M⌉+/2+ complexes in the gas phase (M = Na+, K+, Mg2+, Ca2+). | ||
In this particular series of reactions, we have aimed at assessing the role of the complexing metal cation in thermodynamics of complex formation (Tables 2 and 3). The results obtained clearly show the effect of cationic radius and metal charge on the strength of the host–guest interactions: the values of the Gibbs energies indicate a strong preference towards the smallest and doubly charged Mg2+ as its formation ΔG78 is the lowest in the series (−36.2 kcal mol−1) whereas those for the larger Ca2+ and bulkier monovalent alkali metals, Na+ and K+, are 11.9, −17.1 and −13.1 kcal mol−1, respectively (Table 3). Furthermore, with increasing the metal cationic radii in the series, the readiness for complexation subsides as ΔGs increase in absolute value going down group IA and IIA, respectively. The rationale behind this lies primarily in the bond lengths which strongly affect the ion-dipole interactions in the complexes: the sodium cation is 2.26 Å distant from the CO groups (corresponding to ΔG78 of −17.1 kcal mol−1), while the potassium one lies averagely about 2.76 Å away from the surrounding carbonyl groups (ΔG78 is −13.1 kcal mol−1). Similarly, for the group IIA cations, the metal–ligand bond distances are 1.94 Å for the Mg2+ complex and 2.28 Å for its Ca2+ counterpart. Our current findings are in line with previous studies confirming that the electrostatic interactions are of paramount significance for the formation of metal–host systems and that the smaller size of the cation in combination with its greater charge are the driving forces for this type of reactions.49–51 The role of solvation effects should also be pointed out, as the energy loss in aqueous medium is quite substantial, since the metal–cucurbit[n]uril complexes are not as well hydrated as the bare metal. Hence, the differences between the Gibbs energy values in the gas phase and in water medium span the range of 50–60 kcal mol−1 (for the formation of CB[7]–Na/K⌉+ complexes, Table 3) to about 230 – 250 kcal mol−1 (for the Mg–CB[5–8] and Ca–CB[7] complexation, Tables 1 and 3). Note that because of solvation effects, the Gibbs energy for the Ca–CB[7] complex in aqueous solution becomes positive.
| Reaction | ΔG1 | ΔG78 |
|---|---|---|
| CB[7] + Na+→ CB[7]–Na⌉+ | −74.4 | −17.1 |
| CB[7] + K+→ CB[7]–K⌉+ | −61.9 | −13.2 |
| CB[7] + Mg2+→ CB[7]–Mg⌉2+ | −285.8 | −36.2 |
| CB[7] + Ca2+→ CB[7]–Ca⌉2+ | −215.0 | 11.9 |
Effect of the position of a second Mg2+ cation
An interesting feature of the additional metal cations is their applicability as “lids” in the host–guest systems, especially when added to cucurbituril-dyes complexes.52,53 The third component, an alkaline or lanthanide cation, greatly affects the fluorescence of the dye or allow its controlled release.54 Therefore, it is of particular importance for our study to understand where would a second coming metal cation (Mg2+) preferentially bind (designated as position 2, 3 or 4 below) and what could be the factors governing its positioning. Fig. 4 represents the coordination patterns that were studied, namely (1;2), (1;3) and (1;4), while Table 4 summarizes the acquired thermodynamic data.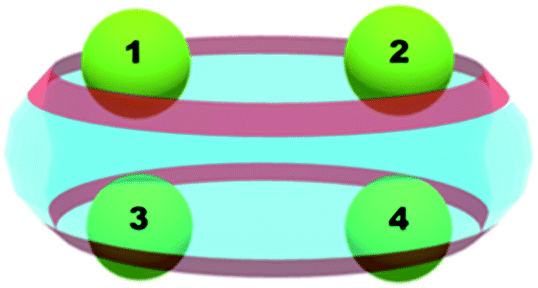 | ||
| Fig. 4 Positions and coordination patterns of two metal cations, possible combinations are (1;2), (1;3) and (1;4). | ||
| Reaction | ΔG1 | ΔG78 |
|---|---|---|
| CB[7] + Mg2+→ CB[7]–Mg⌉2+ | −285.8 | −36.2 |
| CB[7]–Mg⌉2+ + Mg2+→ CB[7]–2*Mg⌉4+ (1;2) | −118.6 | −29.9 |
| CB[7]–Mg⌉2+ + Mg2+→ CB[7]–2*Mg⌉4+ (1;3) | −66.4 | −30.6 |
| CB[7]–Mg⌉2+ + Mg2+→ CB[7]–2*Mg⌉4+ (1;4) | −127.4 | −39.5 |
In accordance with the obtained thermodynamic data, the most preferred position of the second magnesium cation is the one designated as 4, followed by the (1;3) and the (1;2) combinations, where the difference between the last two is negligible and lies within the range of the error of the method (ΔΔG78 = 0.7 kcal mol−1). The analysis of structural data obtained implies that the distances between the metal cations and the carbonyl-groups from the complex play a crucial role as they affect the strength of the ion-dipole interactions. In the (1;2) structure the first Mg2+ ion lies about 1.89 Å from the ligating oxygen atoms, while the other one is 2.06 Å away, which results in an average bond distance for the whole complex of 1.98 Å. The (1;3) complex is entirely symmetrical and both Mg2+ cations form complex bonds of averagely 1.95 Å. Therefore, the second structure appears slightly more stable than the first one in terms of their Gibbs energy values. The third possible combination, (1;4), brings the two magnesium cations even closer to the surrounding carbonyl-groups, as the (C)O–Mg2+ distances decrease to 1.93 Å. The presence of another positively charged particle in the complex, however, inflicts repulsive ion–ion interactions affecting both geometry and thermodynamics of the construct. In the (1;2) and (1;3) structures the distances between the two Mg2+ ions are approximately 6.7 Å, while this gap broadens in the (1;4) complex to 9.2 Å. This reflects on the energetics of the host–guest complexes, as the free energy gain for the (1;4) structure formation is about 10 kcal mol−1 more than that for the other two reactions.
Effect of the metal hydration
The presence/absence of a hydration shell plays a crucial role on the structure and characteristics of a complex, especially when dealing with highly charged species.55 We assessed the effect of the metal hydration by applying a hybrid explicit/implicit solvation model: the first hydration shell around the studied cations has been threated explicitly and the resulting metal/H2O clusters and CB[7]-hydrated metal cations complexes have been surrounded by a dielectric continuum (ε = 78). The optimized structures of the studied CB[7]-hydrated metal complexes as well as the obtained thermodynamic data are given in Fig. 5 and Table 5, respectively. The optimized geometries of the aqua ions of the studied mono- and divalent metals are visualized in Fig. S1, ESI.†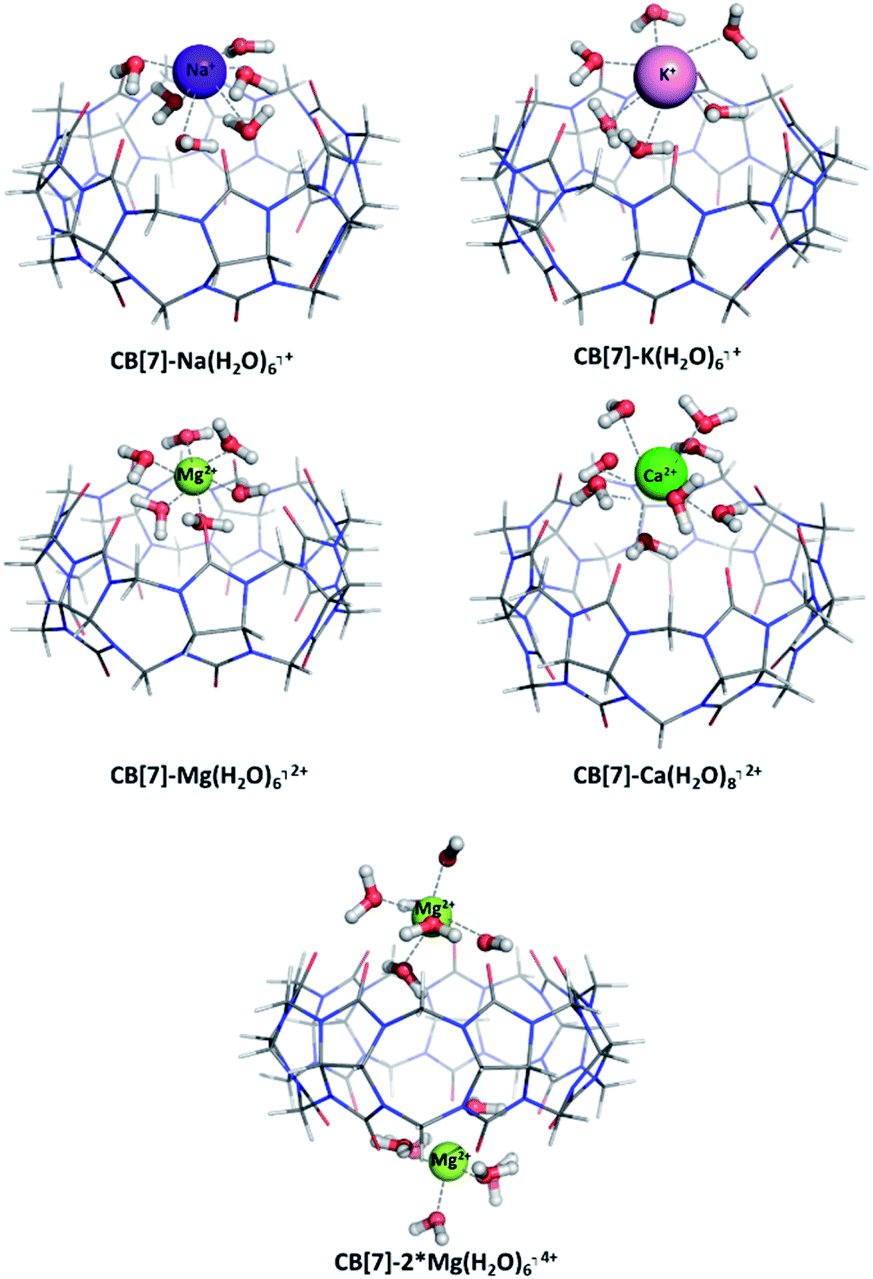 | ||
| Fig. 5 M062X/6-31G(d,p) optimized structures of CB[7]–Na(H2O)6⌉+, CB[7]–K(H2O)6⌉+, CB[7]–Mg(H2O)6⌉2+, CB[7]–Ca(H2O)8⌉2+ and CB[7]–2*Mg(H2O)6⌉4+ complexes in the gas phase. | ||
| Reaction | ΔG1 | ΔG78 |
|---|---|---|
| CB[7] + Na(H2O)6⌉+→ CB[7]–Na(H2O)6⌉+ | −48.6 | −8.6 |
| CB[7] + K(H2O)6⌉+→ CB[7]–K(H2O)6⌉+ | −36.7 | −0.5 |
| CB[7] + Mg(H2O)6⌉2+→ CB[7]–Mg(H2O)6⌉2+ | −128.9 | −33.8 |
| CB[7] + Ca(H2O)8⌉2+→ CB[7]–Ca(H2O)8⌉2+ | −112.4 | −10.1 |
| CB[7]–Mg(H2O)6⌉2+ + Mg(H2O)6⌉2+→ CB[7]–2*Mg(H2O)6⌉4+ | 25.1 | −26.2 |
Except for the well-studied Mg2+, the number of the first-shell water molecules for Na+, K+ and Ca2+ cations varies greatly (between 5 and 7 for Na+, from 6 to 8 for K+ and from 6 to 9 for Ca2+) depending on the environment and experimental/theoretical approach used.56–59 For the purpose of the current study, complexes of CB[7] with Na(H2O)6⌉+, K(H2O)6⌉+, Mg(H2O)6⌉2+, and Ca(H2O)8⌉2+ have been modelled and their Gibbs energies of formation assessed. The results clearly show that the influence of the hydration shell should be taken into account. Although the observed tendencies concerning the effect of the cation properties are still valid (e.g. within a group the preference is toward the smaller cation, in addition to the greatest preference toward the Mg2+), the ΔG78 values decrease in their absolute value. This is mainly due to the lost direct interaction between the metal cation and the carbonyl groups from the host-system and shielding of the metal by a layer of water molecules thus relegating the carbonyl groups from the host macrocycle to the metal second coordination shell. The electron charge transfer from the ligands (now 6–8 water molecules) to the metal in the hydrated structures is: 0.69 e− (Na+), 0.77 e− (K+), 1.59 e− (Mg2+), and 1.84 e− (Ca2+)/Hirshfeld analysis, while in the structures without a hydration shell the observed charge transfer is smaller: 0.48 e− (Na+), 0.50 e− (K+), 1.17 e− (Mg2+), and 1.07 e− (Ca2+). Still, when accounting for the effect of the metal hydration all of the modelled reactions are possible from a thermodynamic point of view (unlike the results obtained for the “naked” metal cations, given in Table 3). This is mainly due to the finely-tuned balance between the great number of intramolecular hydrogen bonds between the water molecules and the cucurbituril, on one hand, and the strong metal/first hydration shell interactions, on the other. This outcome is observed even for the calcium ion, which falls in line with experimental data.31 Thought-provoking is also the fact that the ΔG78 value for the formation of a CB[7]–K(H2O)6⌉+ complex becomes closer to zero as compared to the one for obtaining a CB[7]–K⌉+ (−0.5 kcal mol−1 vs. −13.2 kcal mol−1). The rationale lies in potassium's weak H2O binding ability which results in the formation of a loose hydration shell in aqueous solution.56 All these arguments imply that modelling a hydration shell around a particular metal cation must be considered in accordance with its specific preferences toward the surrounding water molecules.
The effect of the presence of a second metal cation was accounted for by modelling the reaction of formation of a CB[7]–2*Mg(H2O)6⌉4+ complex. The position of the additional Mg2+ is on the lower rim of the cucurbituril in accordance with the tendencies drawn in the previous paragraph.
The formation of the two complexes in water environment is thermodynamically favorable, as the corresponding Gibbs energies stay firmly on negative ground (−33.8 for the mono-Mg2+ and −26.2 kcal mol−1 for the bi-Mg2+ structures, accordingly). Still, these values are less negative in comparison to the ones evaluated for the formation of host–guest structures with bare cations, where the equivalent numbers are −36.2 and −39.5 kcal mol−1 (for the most stable (1;4) combination), as shown in Table 4. The presence of a hydration shell leads to a loss of direct ion-dipole interaction between the metal and the host cucurbituril. This, on the other side, is partially compensated by an additional energy gain arising from the formation of numerous hydrogen bonds between the H2O molecules and the carbonyl groups. Another advantage is that in the structure of CB[7]–Mg(H2O)6⌉2+ the Mg(H2O)6 complex retains its initial geometry, as all angles are in the range of 90° and the bond lengths remain in the diapason of 2.04 to 2.08 Å (in the Mg(H2O)6 alone all distances are 2.06 Å). In the CB[7]–2*Mg(H2O)6 structure, however, the hydration shell around the cation becomes distorted to a certain extent. The valence angles deviate between 80° and 100°, although this tendency does not affect the interatomic distances and they remain up to 2.10 Å. Another feature that is altered is the charge transfer from the water molecules to the magnesium cation: in the Mg(H2O)6 complex its value is 1.36 e−/1.52 e− (natural population analysis/Hirshfeld), while in the host–guest structures it is 1.715; 1.717 e−/1.52; 1.57 e−. Nonetheless, all these results still indicate that the CB[7]-Mg(H2O)6⌉2+ and CB[7]-2*Mg(H2O)6⌉4+ complexes are readily formed.
Conclusions
A systematic study of biologically essential (mono- and divalent) metal cations (Na+, K+, Mg2+, Ca2+) binding thermodynamics to cucurbit[5–8]urils has been performed using density functional theory computations combined with solvation model based on density (SMD) calculations. The effects of the cavity size, as well as the specific metal's physicochemical characteristics such as radius, charge, charge accepting abilities and water hydration have been assessed by modelling the corresponding interaction between the host cavitand and incoming metal species. The results obtained indicate a well-defined preference towards small and highly charged metals, since magnesium is the one that stands out when comparing the Gibbs energy values. The main interactions in the complexes are electrostatic in nature (ion–ion or ion–dipole type), therefore structural characteristics like bond lengths or valence angles play a crucial role. The arising charge transfer from the surrounding carbonyl groups to the metal species is also of great significance. Notably, unlike other host macrocyclic systems (cyclodextrins and calixarenes) the cucurbiturils are quite rigid and less structurally adaptive. This can be seen especially in the CB[7]–2*Mg(H2O)6 structure, where not the molecular container, but the Mg(H2O)6 – construct is the one that undergoes deformation in docking to the pore. Although much about the CB[n]s–metal cations interactions have been revealed in this and other studies, intriguing questions about the basic determinants in CB[n]-complexes with trivalent metals of the p-block or lanthanide series are still awaiting their answers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Bulgarian National Science Fund under grant KP-06-N39/10 (project “BIRDCagE”) and the provided access to the e-infrastructure of the NCHDC – part of the Bulgarian National Roadmap for RIs, with the financial support by the grant no. DO1-271/16.12.2019.References
- T. Takebe, R. Imai and S. Ono, The Current Status of Drug Discovery and Development as Originated in United States Academia: The Influence of Industrial and Academic Collaboration on Drug Discovery and Development, Clin. Transl. Sci., 2018, 11(6), 597–606 CrossRef PubMed.
- R. K. Verma and S. Garg, Current Status of Drug Delivery Technologies and Future Directions, Pharmaceutical Technology On-Line, 2001, 25(2), 1–14 Search PubMed.
- J. Szejtli, Introduction and general overview of cyclodextrin chemistry, Chem. Rev., 1998, 98(5), 1743–1754 CrossRef CAS PubMed.
- M. E. Davis and M. E. Brewster, Cyclodextrin-based pharmaceutics: Past, present and future, Nat. Rev. Drug Discovery, 2004, 3(12), 1023–1035 CrossRef CAS PubMed.
- G. Crini, Review: A history of cyclodextrins, Chem. Rev., 2014, 114(21), 10940–10975 CrossRef CAS PubMed.
- Calixarenes: A Versatile Class of Macrocyclic Compounds, Series: Topics in Inclusion Science, ed. J. Vicens and V. Böhmer, Springer, 1991 Search PubMed.
- D. M. Homden and C. Redshaw, The use of calixarenes in metal-based catalysis, Chem. Rev., 2008, 108(12), 5086–5130 CrossRef CAS PubMed.
- M. Kralj, L. Tušek-Božić and L. Frkanec, Biomedical potentials of crown ethers: Prospective antitumor agents, ChemMedChem, 2008, 3(10), 1478–1492 CrossRef CAS PubMed.
- G. Chehardoli and A. Bahmani, The role of crown ethers in drug delivery, Supramol. Chem., 2019, 31(4), 221–238 CrossRef CAS.
- A. Bukhzam, Nabil Bader, Crown Ethers: Their Complexes and Analytical Applications, J. Appl. Chem., 2017, 3(1), 237–244 Search PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. Del Barrio and O. A. Scherman, Cucurbituril-Based Molecular Recognition, Chem. Rev., 2015, 115(22), 12320–12406 CrossRef CAS PubMed.
- E. Yu. Chernikova, Yu. V. Fedorov and O. A. Fedorova, Cucurbituril as a new “host” of organic molecules in inclusion complexes, Russ. Chem. Bull., 2012, 61, 1363–1390 CrossRef CAS.
- J. W. Lee, S. Samal, N. Selvapalam, H. J. Kim and K. Kim, Cucurbituril homologues and derivatives: New opportunities in supramolecular chemistry, Acc. Chem. Res., 2003, 36(8), 621–630 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Cucurbiturils: From synthesis to high-affinity binding and catalysis, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, Controlling factors in the synthesis of cucurbituril and its homologues, J. Org. Chem., 2001, 66(24), 8094–8100 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, The cucurbit[n]uril family, Angew. Chem., Int. Ed., 2005, 44(31), 4844–4870 CrossRef CAS PubMed.
- O. A. Gerasko, D. G. Samsonenko and V. P. Fedin, Supramolecular chemistry of cucurbiturils, Russ. Chem. Rev., 2002, 71(9), 741–760 CrossRef CAS.
- E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, Cucurbituril Chemistry: A Tale of Supramolecular Success, RSC Adv., 2012, 2, 1213–1247 RSC.
- J. Lü, J. X. Lin, M. N. Cao and R. Cao, Cucurbituril: A promising organic building block for the design of coordination compounds and beyond, Coord. Chem. Rev., 2013, 257(7–8), 1334–1356 CrossRef.
- Y. M. Jeon, J. Kim, D. Whang and K. Kim, Molecular container assembly capable of conrolling binding and release of its guest molecules: Reversible encapsulation of organic molecules in sodium ion complexed cucurbituril, J. Am. Chem. Soc., 1996, 118(40), 9790–9791 CrossRef CAS.
- H. Cong, Q. J. Zhu, S. F. Xue, Z. Tao and G. Wei, Direct coordination of metal ions to cucurbit[n]urils, Chin. Sci. Bull., 2010, 55, 3633–3640 CrossRef CAS.
- X. L. Ni, X. Xiao, H. Cong, L.-L. Liang, K. Cheng, X.-J. Cheng, N.-N. Ji, Q.-J. Zhu, S.-F. Xuea and Z. Tao, Cucurbit[n]uril-based coordination chemistry: From simple coordination complexes to novel poly-dimensional coordination polymers, Chem. Soc. Rev., 2013, 42, 9480–9508 RSC.
- Y. Wu, L. Xu, Y. Shen, Y. Wang, L. Zou, Q. Wang, X. Jiang, J. Liua and H. Tian, The smallest cucurbituril analogue with high affinity for Ag+, Chem. Commun., 2017, 53, 4070–4072 RSC.
- W. M. Nau, M. Florea and K. I. Assaf, Deep inside cucurbiturils: Physical properties and volumes of their inner cavity determine the hydrophobic driving force for host–guest complexation, Isr. J. Chem., 2011, 51(5–6), 559–577 CrossRef CAS.
- L. Mikulu, R. Michalicova, V. Iglesias, M. A. Yawer, A. E. Kaifer, P. Lubal and V. Sindelar, pH Control on the Sequential Uptake and Release of Organic Cations by Cucurbit[7]uril, Chem.–Eur. J., 2017, 23(10), 2350–2355 CrossRef CAS PubMed.
- F. F. Shen, J. L. Zhao, K. Chen, Z.-Y. Hua, M.-D. Chen, Y.-Q. Zhang, Q.-J. Zhub and Z. Tao, Supramolecular coordination assemblies of a symmetrical octamethyl-substituted cucurbituril with alkali metal ions based on the outer-surface interactions of cucurbit[: N] urils, CrystEngComm, 2017, 19, 2464–2474 RSC.
- D. Whang, J. Heo, J. H. Park and K. Kim, A Molecular Bowl with Metal Ion as Bottom: Reversible Inclusion of Organic Molecules in Cesium Ion Complexed Cucurbituril, Angew. Chem., Int. Ed., 1998, 37, 78 CrossRef CAS.
- Y. Q. Yao, K. Chen, Z. Y. Hua, Q. J. Zhu, S. F. Xue and Z. Tao, Cucurbit[n]uril-based host–guest-metal ion chemistry: an emerging branch in cucurbit[n]uril chemistry, J. Inclusion Phenom. Macrocyclic Chem., 2017, 89, 1–14 CrossRef CAS.
- S. Zhang, L. Grimm, Z. Miskolczy, L. Biczók, F. Biedermann and W. M. Nau, Binding affinities of cucurbit[: N] urils with cations, Chem. Commun., 2019, 55, 14131–14134 RSC.
- J. Heo, S. Y. Kim, D. Whang and K. Kim, Shape-induced, hexagonal, open frameworks: Rubidium ion complexed cucurbituril, Angew. Chem., Int. Ed., 1999, 38(5), 641–643 CrossRef CAS PubMed.
- F. Zhang, T. Yajima, Y. Z. Li, G.-Z. Xu, H.-L. Chen, Q.-T. Liu and O. Yamauchi, Iodine-assisted assembly of helical coordination polymers of cucurbituril and asymmetric copper(II) complexes, Angew. Chem., Int. Ed., 2005, 44(22), 3402–3407 CrossRef CAS PubMed.
- K. Kim, Mechanically interlocked molecules incorporating cucurbituril and their supramolecular assemblies, Chem. Soc. Rev., 2002, 31, 96–107 RSC.
- Y. H. Ko, K. Kim, J.-K. Kang, H. Chun, J. W. Lee, S. Sakamoto, K. Yamaguchi, J. C. Fettinger and K. Kim, Designed Self-Assembly of Molecular Necklaces Using Host-Stabilized Charge-Transfer Interactions, J. Am. Chem. Soc., 2004, 126(7), 1932–1933 CrossRef CAS PubMed.
- X.-L. Ni, J.-X. Lin, Y.-Y. Zheng, W.-S. Wu, Y.-Q. Zhang, S.-F. Xue, Q.-J. Zhu, Z. Tao and A. I. Day, Supramolecular bracelets and interlocking rings elaborated through the interrelationship of neighboring chemical environments of alkyl-substitution on cucurbit[5]uril, Cryst. Growth Des., 2008, 8(9), 3446–3450 CrossRef CAS.
- S. Angelova, V. Nikolova, N. Molla and T. Dudev, Factors Governing the Host–Guest Interactions between IIA/IIB Group Metal Cations and α-Cyclodextrin: A DFT/CDM Study, Inorg. Chem., 2017, 56(4), 1981–1987 CrossRef CAS PubMed.
- V. Nikolova, S. Angelova and T. Dudev, IIA/IIB group metal cations hosted by β-cyclodextrin: A DFT study, Bulg. Chem. Commun., 2017, 49, 189–194 Search PubMed.
- V. K. Nikolova, C. V. Kirkova, S. E. Angelova and T. M. Dudev, Host–guest interactions between p-sulfonatocalix[4]arene and p-sulfonatothiacalix[4]arene and group IA, IIA and f-block metal cations: A DFT/SMD study, Beilstein J. Org. Chem., 2019, 15, 1321–1330 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- N. Mardirossian and M. Head-Gordon, How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements?, J. Chem. Theory Comput., 2016, 12(9), 4303–4325 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- S. E. Angelova, V. K. Nikolova and T. M. Dudev, Determinants of the host–guest interactions between α-, β- And γ-cyclodextrins and group IA, IIA and IIIA metal cations: A DFT/PCM study, Phys. Chem. Chem. Phys., 2017, 19, 15129–15136 RSC.
- Y. Miyahara, K. Abe and T. Inazu, “Molecular” molecular sieves: Lid-free decamethylcucurbit[5]uril absorbs and desorbs gases selectively, Angew. Chem., Int. Ed., 2002, 41(16), 3020–3023 CrossRef CAS PubMed.
- D. Bardelang, K. A. Udachin, D. M. Leek, J. C. Margeson, G. Chan, C. I. Ratcliffe and J. A. Ripmeester, Cucurbit[n]urils (n = 5-8): A comprehensive solid state study, Cryst. Growth Des., 2011, 11(12), 5598–5614 CrossRef CAS.
- L. Cao, D. Škalamera, P. Y. Zavalij, J. Hostaš, P. Hobza, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, Influence of hydrophobic residues on the binding of CB[7] toward diammonium ions of common ammonium⋯ammonium distance, Org. Biomol. Chem., 2015, 13, 6249–6254 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- The PyMOL Molecular Graphics System, Version 1.7.6.6, Schrödinger, LLC Search PubMed.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751–767 CrossRef.
- S. J. Angyal, Complexes of Metal Cations with Carbohydrates in Solution, Adv. Carbohydr. Chem. Biochem., 1989, 47, 1–43 CrossRef CAS.
- M. Saladini, L. Menabue and E. Ferrari, Sugar complexes with metal2+ ions: Thermodynamic parameters of associations of Ca2+, Mg2+ and Zn2+ with galactaric acid, Carbohydr. Res., 2001, 336(1), 55–61 CrossRef CAS PubMed.
- J. Yucheng, G. Shiyang, X. Shuping, H. Mancheng, W. Jianji, L. Yan and Z. Kelei, The enthalpy and entropy interaction parameters of cesium chloride with saccharides (D-glucose, D-fructose and sucrose) in water at 298.15 K, Thermochim. Acta, 2003, 400(1–2), 37–42 CrossRef.
- M. Shaikh, J. Mohanty, A. C. Bhasikuttan, V. D. Uzunova, W. M. Nau and H. Pal, Salt-induced guest relocation from a macrocyclic cavity into a biomolecular pocket: Interplay between cucurbit[7]uril and albumin, Chem. Commun., 2008, 31, 3681–3683 RSC.
- Y. Q. Zhang, Q. J. Zhu, S. F. Xue and Z. Tao, Chlorine anion encapsulation by molecular capsules based on cucurbit[5]uril and decamethylcucurbit[5]uril, Molecules, 2007, 12(7), 1325–1333 CrossRef CAS PubMed.
- S. D. Choudhury, J. Mohanty, H. Pal and A. C. Bhasikuttan, Cooperative metal ion binding to a cucurbit[7]uril - Thioflavin T complex: Demonstration of a stimulus-responsive fluorescent supramolecular capsule, J. Am. Chem. Soc., 2010, 132(4), 1395–1401 CrossRef CAS PubMed.
- J. S. Rao, T. C. Dinadayalane, J. Leszczynski and G. N. Sastry, Comprehensive study on the solvation of mono- and divalent metal cations: Li+, Na+, K+, Be2+, Mg2+ and Ca2+, J. Phys. Chem. A, 2008, 112(50), 12944–12953 CrossRef CAS PubMed.
- J. Mähler and I. Persson, A study of the hydration of the alkali metal ions in aqueous solution, Inorg. Chem., 2012, 51, 425–438 CrossRef PubMed.
- M. A. Tahoon, E. A. Gomaa and M. H. A. Suleiman, Aqueous Micro-hydration of Na+(H2O) n=1-7 Clusters: DFT Study, Open Chem., 2019, 17(1), 260–269 CAS.
- R. Mancinelli, A. Botti, F. Bruni, M. A. Ricci and A. K. Soper, Hydration of sodium, potassium, and chloride ions in solution and the concept of structure maker/breaker, J. Phys. Chem. B, 2007, 111, 13570–13577 CrossRef CAS PubMed.
- S. E. Rodriguez-Cruz, R. A. Jockusch and E. R. Williams, Hydration Energies and Structures of Alkaline Earth Metal Ions, M2+(H2O)n , n = 5–7, M = Mg, Ca, Sr, and Ba., J. Am. Chem. Soc., 1999, 121, 8898–8906 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04387g |
| This journal is © The Royal Society of Chemistry 2020 |