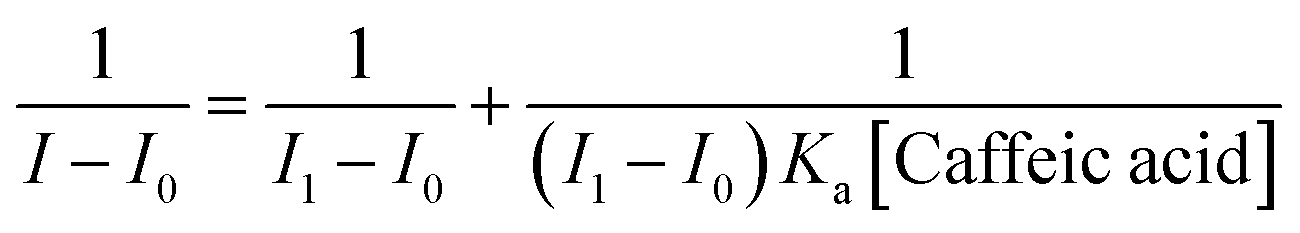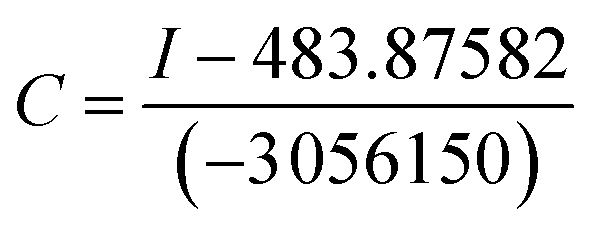DOI:
10.1039/D0RA00980F
(Paper)
RSC Adv., 2020,
10, 28148-28156
A water-soluble boronic acid sensor for caffeic acid based on double sites recognition†
Received
1st February 2020
, Accepted 14th July 2020
First published on 27th July 2020
Abstract
Due to reversibly and covalently binding with Lewis bases and polyols, boronic acid compounds as fluorescent sensors have been widely reported to recognize carbohydrates, ions, hydrogen peroxide, and so on. However, boronic acid sensors for highly selective recognition of caffeic acid rather than catechol or catechol derivatives have not been reported yet. Herein a novel water-soluble sensor 5c with double recognition sites based on a boronic acid was reported. When 2.3 × 10−4 M of caffeic acid was added, the fluorescence intensity of sensor 5c decreased by 99.6% via inner filter effect (IFE) because its excitation spectrum well overlaps with the absorption spectrum of caffeic acid under neutral condition, while the fluorescence increased or did not change obviously after binding with other analytes including carbohydrates and other catechol derivatives. In addition, the response time to caffeic acid is fast at room temperature, and a high binding constant (9245.7 ± 348.3 M−1) and low LOD (1.81 × 10−6 M) was calculated. Moreover, determination of caffeic acid content in caffeic acid tablets was studied, and the recovery rate is sufficient. Therefore, sensor 5c can be used as a potential tool for detecting biologically significant caffeic acid in real samples.
Introduction
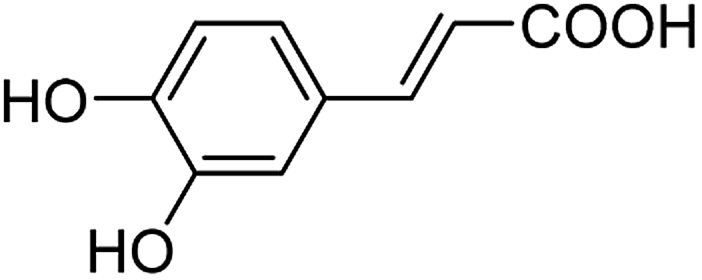
As a natural and ubiquitous substance in many people's daily diet, caffeic acid (its structure is shown in Fig. 1) has exhibited excellent pharmacological effects in terms of healthcare and clinical treatment over the past few decades.1–4 It was reported that 3 μg of caffeic acid can completely inhibit 20 μg phosphodiesterase which is the main composition of snake poison, and 100 μM of caffeic acid can protect venous endothelial cells from the induction of cell apoptosis. In addition, diseases such as upper respiratory infections in children and obesity problems can be prevented by caffeic acid according to the latest researches.5,6 Caffeic acid preparations in modern medicine including caffeic acid tablets have been widely used clinically for the treatment of hemostasis, leukopenia, and thrombocytopenia etc.7 Moreover, the chemoprotective effect on cancers has been confirmed.8,9 However, excessive amounts of caffeic acid lead to carcinogenic effects. For instance, studies reported by Hirose and Takesada et al. showed that phenolic compounds (including caffeic acid, catechol, sesamol, BHA and 4-methoxyphenol etc.) have an additive/synergistic effect on cancer development even at low dose levels compared to control groups.10 Therefore, quantitative detection of caffeic acid is of great significance for comprehend our daily diet and early diagnosis of diseases.11
 |
| | Fig. 1 The structure of caffeic acid. | |
To date, several analytical methods have been reported to determine caffeic acid, including chromatography (HPLC, GC),12–15 capillary electrophoresis,16 voltammetry,17–19 electrochemical methods,20–23 and UV-Visible spectrophotometry,24,25 etc. However, there are still some limitations, such as needing expensive instruments, complex sample pretreatment, lengthy analysis time and high cost, etc. Therefore, a new platform with simple, sensitive and efficient detection technology is still urgently needed. Fluorescence techniques have been most widely explored due to their high sensitivity, feasibility, and easy-to-operation.26 However, at present, there are few reports on the detection of caffeic acid utilizing fluorescence sensors, and most of them are detected by quantum dots or enzymatic methods.23,27–29 For example, a novel caffeic acid fluorescence detection method based on molecularly imprinted polymers (CDs@MIPs) coated with silane functional carbon dots was proposed by Xu et al.28 Although CD has excellent photoluminescence performance and high biocompatibility, the selectivity of a single carbon dot to the target compound is low, and it needs to be used in combination with other substances, so the synthesis and assembly process is tedious and difficult. Moreover, expensive instruments are needed to characterize the carbon dots. In addition, based on the peroxidase-mimicking activities of the G-quadruplex/hemin DNAzyme, a fluorometric assay platform for fluorescent detection of caffeic acid was designed and reported by Cai et al.23 The experimental operation is complicated and requires dangerous reagents such as H2SO4, HCl, HNO3, and DMSO, etc. Moreover, the activity of a DNAzyme is closely related to its configuration, and it is susceptible to environmental influences.
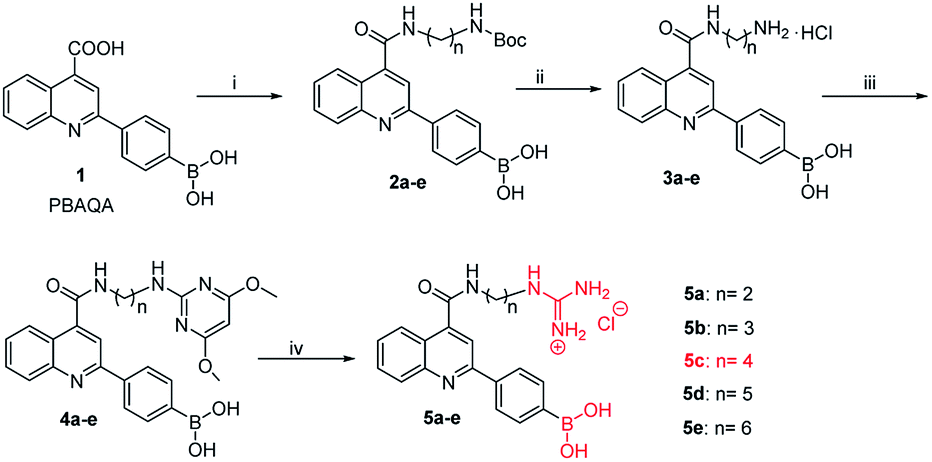
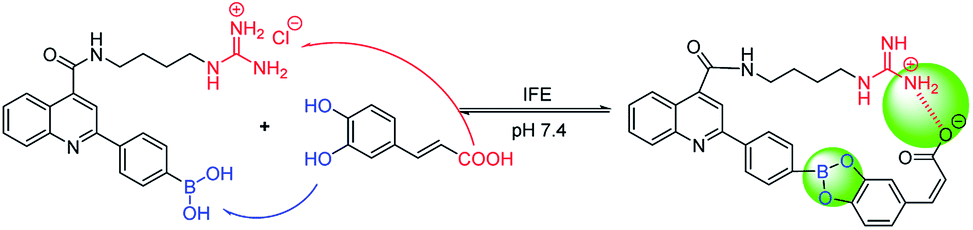
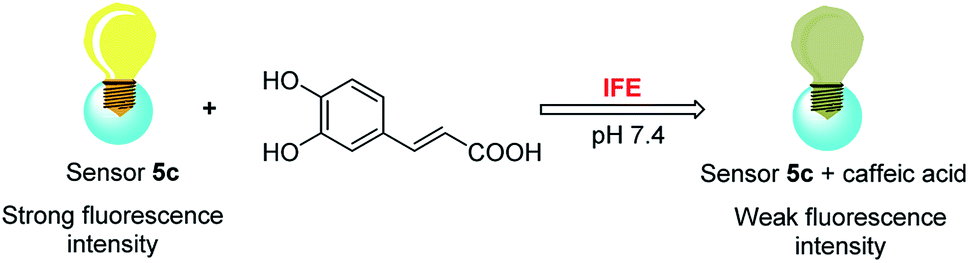
Owing to boronic acid has the unique ability to reversibly bind 1,2- or 1,3-diols in aqueous media, boronic acid-based fluorescent sensors can provide some of the advantages of detecting diols, including high sensitivity and fast response.30–33 Boronic acid derivatives and their utility in diols sensing including: (1) in clinical practice, some complications such as diabetes and cancer, etc. are early diagnosed by detecting certain components in body fluids, saliva or blood;34 (2) it can be used to test the content of certain ingredients in drug or food products;35 (3) in the future, fluorescence guided surgery (FGS) can also help doctors determine tumor boundaries and find metastases in the clinic, and guide surgeons to accurately remove tumors. In addition, the structure of 2-aryl-quinoline-4-carboxy derivatives has a large aromatic conjugated system, it has good fluorescent properties.36,37 Our research group has reported the synthesis of 2-(4-boranephenyl)-quinoline-4-carboxylic acid and its diboronic acid derivatives and studied their fluorescent properties.30 Sensors have been reported for recognizing sorbitol,35 ribose,30 dopamine,38 levodopa,36 catechol37 and Fe3+,39 respectively. Herein, we reported novel water soluble boronic acid compounds with double recognition sites (5a–e) for selective detection of caffeic acid based on inner filter effect (IFE).40,41 As illustrated in Scheme 1, compounds 5 are synthesized through an optimized route and used as fluorescent sensors to detect caffeic acid. Take the representative sensor 5c for instance, in addition to boronic acid group binding with the o-dihydroxy group of caffeic acid, the guanidino group combined with the carboxyl group of caffeic acid to achieve a double recognition effect, which significantly improved the selectivity and affinity, as shown in Fig. 2. The main reason for the interaction between the guanidino group of the sensor 5 and the carboxyl group of caffeic acid is electrostatic attraction. Moreover, the fluorescence of sensor 5c can be quenched by caffeic acid via IFE because its excitation spectrum (328 nm) well overlaps with the absorption spectrum (maximum absorption wavelength at 323 nm) of caffeic acid under neutral condition, as shown in Fig. 3. Due to the simple synthesis, good water solubility, sensitivity, and selectivity, sensor 5c can be used as a potential tool to detect the caffeic acid content in drug or food products, and even to detect the complications of diabetes early by detecting the caffeic acid content in body fluids.
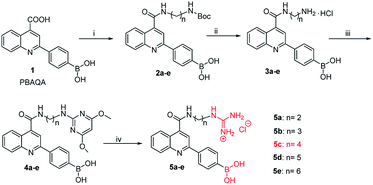 |
| | Scheme 1 Synthetic route of sensor 5: (i) diamine, CH3OH, DMT-MM, N-methylmorpholine, rt, 20 h, 2a: 75%, 2b: 81%, 2c: 73%, 2d: 79%, 2e: 78%. (ii) EtOAC, HCl, rt, 18 h, 3a: 70%, 3b: 75%, 3c: 72%, 3d: 85%, 3e: 71%. (iii) 2-Chloro-4,6-dimethoxypyrimidine, i-PrOH, NEt3, reflux, 120 °C, 12 h, 4a: 35%, 4b: 52%, 4c: 47%, 4d: 46%, 4e: 48%. (iv) 4 M HCl AcOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), reflux, 100 °C, 10 h, 5a: 88%, 5b: 82%, 5c: 88%, 5d: 83%, 5e: 81%. :1, v/v), reflux, 100 °C, 10 h, 5a: 88%, 5b: 82%, 5c: 88%, 5d: 83%, 5e: 81%. | |
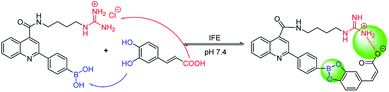 |
| | Fig. 2 The process of sensor 5c recognizes caffeic acid. | |
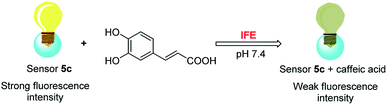 |
| | Fig. 3 Mechanism of sensor 5c recognizes caffeic acid. | |
Experimental
Materials and methods
All materials for synthesis were commercially available without further purification. All solvents used were of analytical reagent grade and all aqueous solutions were prepared using pure water. The caffeic acid tablets were purchased from Dezhou Deyao Pharmaceutical Co., Ltd. The 1H NMR spectra and 13C NMR spectra were recorded on a Bruker AM-600 spectrometer (Billerica, MA), and chemical shift (δ) were given in parts per million relative to tetramethylsilane (TMS). High-resolution mass spectrometry (HRMS) spectra were recorded on an Agilent 1290LC-6540 Accurate-Mass Q-TOF by using electrospray ionization (ESI). Chromatographic datas were recorded on a Waters high-performance liquid chromatograph (Waters Corporation, USA), and the separated compounds were collected using a SHIMADZU LC-20AR preparative liquid chromatograph (Shimadzu, Japan). Ultraviolet absorption datas were collected on a HITACHI U-2910 UV-Visible spectrophotometer (Hitachi, Japan). Fluorescence datas were collected on an RF5301PC Fluorescence Spectrophotometer (Shimadzu, Japan).
Synthesis
Owing to compound 1 (PBAQA, 2-(4-boronophenyl)quinoline-4-carboxylic acid) is a known compound and the synthesis process is complicated. Therefore, compound 1 used in our experiments was purchased from a commercial supplier.36,37,42 As shown in Scheme 1, previously, our research group has reported the synthesis method of compound 3.30 In addition, in the synthesis of compound 5, thiourea trioxide was used as the raw material, and the by-products were more difficult to purify and produced environmentally unfriendly sulfur dioxide after the reaction was complete. So, 2-chloro-4,6-dimethoxypyrimidine was used instead of thiourea trioxide, the reaction was easy to proceed and produced less by-products and waste liquid. Moreover, the experimental operations were relatively simple and the reagents used in the experiment were less toxic.
(4-(4-((2-((tert-butoxycarbonyl)amino)ethyl)carbamoyl)quinolin-2-yl)phenyl)boronic acid (2a)
Compound 1 (0.5 g, 1.7 × 10−3 mol) was added to a round bottom flask and methanol (50 mL) was added to dissolve the compound 1. DMT-MM (0.5 g, 1.9 × 10−3 mol) and N-Boc-ethylenediamine (520 μL, 1.9 × 10−3 mol) were then added and completely dissolved by ultrasound, followed by 2 drops of N-methylmorpholine. Stirring for 20 h at room temperature, the reaction was stopped, and TLC was used to detect whether the reaction was complete. The reaction solution was concentrated under reduced pressure to at least the amount, and then the reaction solution was slowly added dropwise to 150 mL of ice water with continuous stirring, and a pale yellow precipitate gradually precipitated, followed by filtering. It was then recrystallized from methanol and filtered to give a white powder compound 2a (0.5549 g, 75%).30 Compounds 2b, 2c, 2d, and 2e were synthesized in the same manner as 2a. The yields of compound 2 as follows: 2a: 75%, 2b: 81%, 2c: 73%, 2d: 79%, 2e: 78%, respectively.
(The diamine in reaction (i) includes N-Boc-ethylene diamine, N-Boc-1,3-propane diamine, N-Boc-1,4-butane diamine, N-Boc-1,5-diaminopentane, N-Boc-1,6-hexane diamine).
(4-(4-((2-aminoethyl)carbamoyl)quinolin-2-yl)phenyl)boronic acid hydrochloride (3a)
The product 2a (0.5549 g, 1.3 × 10−3 mol) obtained in the previous step was dissolved in a round bottom flask using ethyl acetate (150 mL). Subsequently, 5 mL of hydrochloric acid was slowly added and stirred at room temperature for 18 h. The reaction solution changed from a yellow clear state to a yellow turbid state. After the reaction was completed, the reaction was stopped, and a yellow solid was obtained by suction filtration. It was then washed three times with ethyl acetate and suction filtered, and dried under vacuum to obtain the pale yellow powder compound 3a (0.3382 g, 70%).35 Compounds 3b, 3c, 3d, and 3e were synthesized in the same manner as 3a. The yields of each compound as follows: 3a: 70%, 3b: 75%, 3c: 72%, 3d: 85%, 3e: 71%, respectively.
(4-(4-((2-((4,6-dimethoxypyrimidin-2-yl)amino)ethyl)carbamoyl)quinolin-2-yl)phenyl)boronic acid (4a)
The compound 3a (0.9034 g, 2.4 × 10−3 mol) was added to a round bottom flask, followed by 12 mL i-PrOH, 1152 μL NEt3, and the 3a was completely dissolved by ultrasound. Then, 2-chloro-4, 6-dimethoxypyrimidine (1.0475 g, 6.0 × 10−3 mol) was added and heated to reflux at 120 °C. The reaction was left at this temperature until deemed complete by TLC (CH2Cl:CH3OH = 10:1) analysis, typically 12 h. After the reaction was complete, the reaction was then allowed cool to room temperature and then 10 mL of EtOAc and 5 mL of distilled water were added to the reaction liquid to extract and separate the two phases. The aqueous phase was further washed twice with 5 mL of EtOAc and the two phases were separated. The separated EtOAc phases were combined, washed with a saturated NaCl solution (10 mL), and the two phases were separated. Then anhydrous Na2SO4 was used to dry the EtOAc phase, and the precipitate was removed by filtration. The filtrate was reduced by rotary evaporator and concentrated to dry to obtain the off-white powder. Purification was performed by column chromatography (CH2Cl:CH3OH = 100:0 → 95:5). The collected components were concentrated under reduced pressure to give a white powder compound 4a (0.3951 g, 35%).43,44 Compounds 4b, 4c, 4d, and 4e were synthesized and post processed in the same manner as 4a. The yields of each compound as follows: 4a: 35%, 4b: 52%, 4c: 47%, 4d: 46%, 4e: 48%, respectively.
4a: 1H NMR (600 MHz, CD3OD) δ (ppm) (Fig. S8, ESI†): 8.94 (d, J = 5.1 Hz, 1H), 8.25 (d, J = 8.2 Hz, 1H), 8.23–8.15 (m, 2H), 8.16–8.06 (m, 1H), 7.99 (d, J = 8.1 Hz, 1H), 7.88–7.75 (m, 1H), 7.69–7.54 (m, 1H), 7.28 (t, J = 5.3 Hz, 1H), 5.44–5.31 (m, 1H), 3.79 (s, 4H), 3.57 (t, J = 7.0 Hz, 2H), 3.34 (s, 3H), 2.56–2.45 (m, 1H). HRMS (ESI) (Fig. S9, ESI†): calculated for C24H25BN5O5+ [M + H]+: 474.1943 found 474.1937.
4c: HRMS (ESI) (Fig. S10, ESI†): calculated for C26H28BN5O5+ [M + H]+: 502.2256 found 502.2271.
Synthesis of compound 5
(4-(4-((2-guanidinoethyl)carbamoyl)quinolin-2-yl)phenyl)boronic acid hydrochloride (5a). The synthesized compound 4a (0.3949 g, 0.8 × 10−3 mol) was added to the round-bottom flask, followed by 3.5 mL AcOH, 3.5 mL distilled water and 18 mL 4 M HCl were added in this order, and 4a was completely dissolved by ultrasound and refluxed at 100 °C for 10 h. After the reaction was detected to be complete by TLC, the reaction was stopped and cooled to room temperature. 20 mL of EtOAc was added to the reaction solution, and the two phases were separated by extraction. The aqueous phase was further washed with EtOAc (2 × 20 mL), and then the aqueous phase was washed with 10 mL CH2Cl2:CH3OH = 8.5:1.5 and the two phases were separated. The separated aqueous phase was concentrated to dryness under reduced pressure by a rotary evaporator, and a yellow oily solid was obtained after vacuum drying. The previously obtained yellow oily solid was separated by liquid preparative chromatography (chromatographic methanol with a mobile phase of 35%), and the collected components were concentrated under reduced pressure and dried under vacuum to obtain a yellow powder compound 5a (0.2921 g, 88%).43,45 1H NMR (600 MHz, DMSO) δ (ppm) (Fig. S11, ESI†): 9.15 (t, J = 5.5 Hz, 1H), 8.35 (d, J = 7.4 Hz, 2H), 8.25 (d, J = 8.3 Hz, 1H), 8.19 (d, J = 8.3 Hz, 1H), 8.07 (t, J = 5.9 Hz, 1H), 8.01 (d, J = 8.2 Hz, 1H), 7.88–7.84 (m, 1H), 7.70–7.66 (m, 1H), 3.53 (dd, J = 11.4, 5.6 Hz, 2H), 3.46 (dd, J = 11.5, 5.7 Hz, 2H). 13C NMR (151 MHz, DMSO) δ (ppm) (Fig. S12, ESI†): δ 168.25, 158.58, 157.04, 148.62, 144.47, 140.22, 137.37, 135.95, 131.77, 130.2, 128.65, 127.77, 127.48, 126.87, 124.72, 118.57, 100.00, 41.45, 39.92. HRMS (ESI) (Fig. S13, ESI†): calculated for C19H21BN5O3+ [M + H]+: 378.1732 found 378.1715.
(4-(4-((3-guanidinopropyl)carbamoyl)quinolin-2-yl)phenyl)boronic acid hydrochloride (5b). The compound synthesis operation was the same as that of 5a. Compound 4b (0.1901 g, 0.39 × 10−3 mol) was used instead of 4a to obtain a yellow powder compound 5b (0.1360 g, 82%). 1H NMR (600 MHz, MeOD) δ (ppm) (Fig. S14, ESI†): 8.25–8.12 (m, 4H), 8.11 (s, 1H), 7.95 (s, 1H), 7.90–7.75 (m, 2H), 7.71–7.64 (m, 1H), 3.60 (t, J = 6.9 Hz, 2H), 3.36 (t, J = 6.9 Hz, 2H), 1.99 (p, J = 6.9 Hz, 2H). 13C NMR (151 MHz, MeOD) δ (ppm) (Fig. S15, ESI†): δ 168.58, 157.42, 157.19, 147.42, 143.83, 139.23, 134.19, 130.66, 127.87, 127.36–127.23, 126.58, 125.01, 123.57, 117.31, 38.74, 36.73, 28.46. HRMS (ESI) (Fig. S16, ESI†): calculated for C20H23BN5O3+ [M + H]+: 392.1888 found 392.1850.
(4-(4-((4-guanidinobutyl)carbamoyl)quinolin-2-yl)phenyl)boronic acid hydrochloride (5c). The compound synthesis operation was the same as that of 5a. Compound 4c (0.5679 g, 1.1 × 10−3 mol) was used instead of 4a to obtain a yellow powder compound 5c (0.4258 g, 88%). 1H NMR (600 MHz, DMSO) δ (ppm) (Fig. S17, ESI†): 9.02 (t, J = 5.5 Hz, 1H), 8.30 (d, J = 8.2 Hz, 2H), 8.20 (d, J = 8.7 Hz, 3H), 8.01 (d, J = 8.1 Hz, 2H), 7.87 (dd, J = 13.2, 6.7 Hz, 2H), 7.69 (t, J = 7.7 Hz, 1H), 3.43–3.38 (m, 2H), 3.22–3.16 (m, 2H), 1.70–1.57 (m, 4H). 13C NMR (151 MHz, MeOD) δ (ppm) (Fig. S18, ESIb†): δ 167.21, 162.26, 209.14–136.26, 134.41, 134.34, 135.29–127.72, 127.19, 126.23, 66.08. HRMS (ESI) (Fig. S19, ESI†): calculated for C21H25BN5O3+ [M + H]+: 406.2045 found 406.2087.
(4-(4-((5-guanidinopentyl)carbamoyl)quinolin-2-yl)phenyl)boronic acid hydrochloride (5d). The compound synthesis operation was the same as that of 5a. Compound 4d (0.2100 g, 0.41 × 10−3 mol) was used instead of 4a to obtain a yellow powder compound 5d (0.1556 g, 83%). 1H NMR (600 MHz, MeOD) δ (ppm) (Fig. S20, ESI†): 8.17 (dd, J = 12.7, 11.8 Hz, 4H), 8.05 (s, 1H), 7.95 (s, 1H), 7.84 (ddd, J = 8.2, 7.0, 1.2 Hz, 2H), 7.69–7.63 (m, 1H), 3.52 (t, J = 7.1 Hz, 2H), 3.22 (t, J = 7.1 Hz, 2H), 1.79–1.72 (m, 2H), 1.69 (dt, J = 15.0, 7.4 Hz, 2H), 1.57–1.49 (m, 2H). 13C NMR (151 MHz, MeOD) δ (ppm) (Fig. S21, ESI†): δ 168.35, 157.16, 147.61, 144.04, 139.44, 132.37, 130.39–130.13, 128.43, 127.36, 126.53, 124.97, 123.60, 117.14, 41.03, 39.37, 28.62, 28.12, 23.74. HRMS (ESI) (Fig. S22, ESI†): calculated for C22H27BN5O3+ [M + H]+: 420.2201 found 420.2173.
(4-(4-((6-guanidinohexyl)carbamoyl)quinolin-2-yl)phenyl)boronic acid hydrochloride (5e). The compound synthesis operation was the same as that of 5a. Compound 4e (0.1300 g, 0.25 × 10−3 mol) was used instead of 4a to obtain a yellow powder compound 5e (0.0760 g, 81%). 1H NMR (600 MHz, MeOD) δ (ppm) (Fig. S23, ESI†): 8.19 (ddd, J = 13.1, 7.1, 6.3 Hz, 4H), 8.06 (s, 1H), 7.97 (s, 1H), 7.90–7.75 (m, 2H), 7.67 (ddd, J = 8.2, 7.0, 1.0 Hz, 1H), 3.53 (t, J = 7.2 Hz, 2H), 3.21 (t, J = 7.1 Hz, 2H), 1.82–1.70 (m, 2H), 1.70–1.60 (m, 2H), 1.60–1.45 (m, 4H). 13C NMR (151 MHz, MeOD) δ (ppm) (Fig. S24, ESI†): δ 168.34, 157.22, 157.06, 147.66, 144.02, 134.18, 130.48, 128.51, 127.30, 126.51, 124.94, 123.59, 117.06, 41.04, 39.47, 28.86, 28.44, 26.27, 25.92. HRMS (ESI) (Fig. S25, ESI†): calculated for C23H29BN5O3+ [M + H]+: 434.2358 found 434.2325.
Results and discussion
A sensor stock solution (1 × 10−3 M) was prepared in water, and 100 μL stock solution was diluted to 10 mL using 0.1 M phosphate buffer (PBS, pH 7.4) to obtain a blank sensor solution (1 × 10−5 M). Then, the 0.1 M phosphate buffer solution (pH 7.4) was used to prepare analytes stock solution, such as saccharide analytes stock solution (8 × 10−1 M) and acid analytes stock solution (8 × 10−3 M). And a blank sensor solution (2 × 10−5 M) was mixed with the saccharide and acid analytes stock solution 1:1 to prepare saccharide analytes stock solution (4 × 10−1 M) and acid analytes stock solution (4 × 10−3 M), respectively. By reducing different volumes of saccharide or acid analytes sensor stock solutions and adding different volumes of sensor stock solutions (1 × 10−5 M), obtaining 1 mL sensor (1 × 10−5 M) with different analyte concentrations (0 to 4 × 10−1 M or 4 × 10−3 M), and the fluorescence spectrum was recorded while testing. The UV-Vis absorption spectrum of the sensor 1 was recorded in DMSO/PBS (1:99, v/v) and the sensor 5c was recorded in PBS (Fig. S1, ESI†). The excitation wavelength of the boronic acid sensor 5c was set to 328 nm (slit: 5 nm/5 nm), while the maximum fluorescence emission intensity of the boronic acid sensor 5c was approximately 395 nm, as shown in Fig. S4 (ESI†).
Fluorescence properties
To examine the fluorescence binding affinities of compounds 5a–e and various analytes, we conducted a series of fluorescence activity studies. When the concentration of analytes added is 6.7 × 10−4 M. We found that the sensor has a significant fluorescence response to catechol compounds. Especially, sensor 5c is found to have the highest binding constant for caffeic acid (9245.7 ± 348.3 M−1) among the several sensors tested, and the lowest detection limit (LOD) (1.81 × 10−6 M) at pH 7.4, as shown in Fig. S4 (ESI†). Compared with sensor 5, the binding constant of the sensor 1 is significantly lower, as shown in Table 1. The probable reason is that the sensor 1 has only one recognition site (boronic acid group), while the sensor 5 has two recognition sites (boronic acid group and guanidine group). Therefore, the binding affinity and selectivity of the sensor 5 are significantly higher than that of sensor 1. Moreover, the binding constant of the sensor 5c and caffeic acid is higher than that of the sensors 5a, 5b, 5d, and 5e, possibly due to the sensor 5c has a linker of an appropriate length and rigidity, which provides the suitable spatial structure for the sensor 5c.
Table 1 The key information of sensor 1 and 5 combining with caffeic acid (6.7 × 10−4 M)
| Sensors |
(I − I0)/I0 |
LODa (M) |
Keqb (M−1) |
| The value was calculated by 3δ/S (R2 > 0.99). The value was calculated by Benesi–Hildebrand equation based on three times of measurement (R2 > 0.99). |
| 1 |
0.26 |
1.74 × 10−4 |
1179.7 ± 149.0 |
| 5a |
0.96 |
5.11 × 10−6 |
7118.9 ± 281.1 |
| 5b |
0.97 |
2.25 × 10−6 |
7839.4 ± 295.9 |
| 5c |
0.98 |
1.81 × 10−6 |
9245.7 ± 348.3 |
| 5d |
0.95 |
3.85 × 10−6 |
5203.6 ± 92.1 |
| 5e |
0.97 |
5.53 × 10−6 |
5738.6 ± 167.3 |
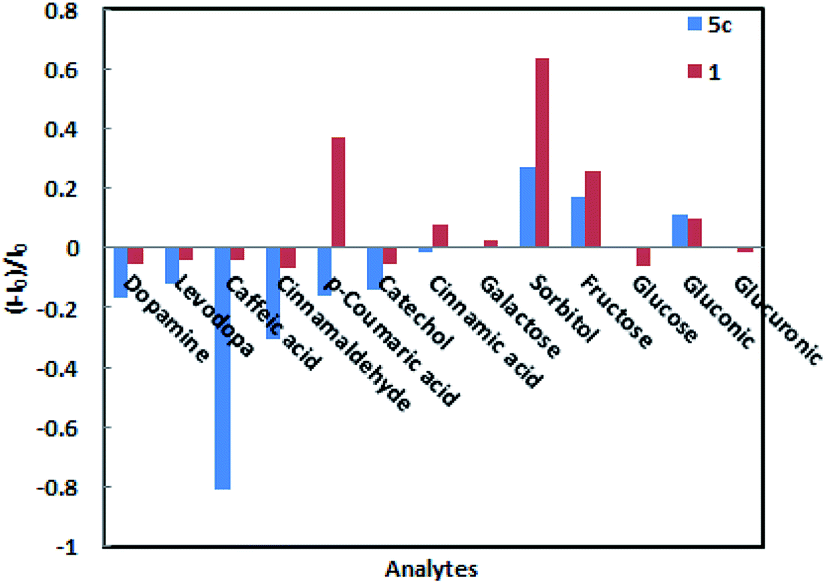
When low concentrations of different analytes (2.3 × 10−4 M) are added, the amplitude changes in the fluorescence intensity of sensors 1 and 5c as shown in Fig. 4. Among them, caffeic acid causes the largest changes in various analytes. The fluorescence intensity of sensor 5c decreased by 99.6% after combined with low-concentration caffeic acid (2.3 × 10−4 M), and its fluorescence is almost completely quenched, followed by dopamine, catechol, and levodopa. However, its fluorescence intensity increased or did not change obviously after binding with other analytes. Especially, when combined with glucose, the fluorescence intensity is virtually zero, indicating that sensor 5c cannot recognize glucose. Although the fluorescence intensity of boronic acid sensor 1 decreased by 93.7% after combined with caffeic acid, the binding constant is low and the linearity is poor. In addition, except sorbitol, the fluorescence intensity of sensor 1 is not change obviously after binding with other analytes. From the above, sensor 5c can selectively recognize low concentrations of caffeic acid in a phosphate buffer solution with a pH of 7.4.
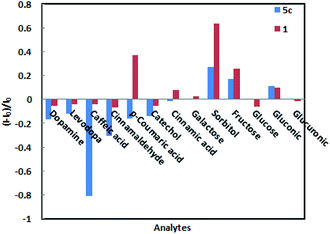 |
| | Fig. 4 Relative fluorescence intensity of sensor 1 and 5c to a low concentration of analytes (2.3 × 10−4 M) in phosphate buffer at pH 7.4. | |
These studies indicate that if there is no hydroxyl group or only one hydroxyl group in the structure of the analytes, such as cinnamaldehyde, p-coumaric acid and cinnamic acid, the fluorescence intensity of the sensor 5c does almost unchanged. Moreover, when it contains only o-dihydroxy group and no carboxyl group or aldehyde group, such as catechol, the fluorescence intensity of the sensor hardly changes. Therefore, when the boronic acid group of the sensor 5c is combined with the o-dihydroxy group of the analytes, the guanidine group is combined with the carboxyl group or the aldehyde group, and the fluorescence intensity changes significantly when the two groups work synergistically.
In addition, under the same conditions, we also studied the optimal binding ability of sensor 5c to other analytes, where the fluorescence intensity reached saturation at the added concentration, as shown in Table 2. When the concentration of the added saccharide analytes is increased from 0 to 4 × 10−1 M, the fluorescence of sorbitol and fructose increased and reached saturation, but the fluorescence intensity of glucose is almost zero. However, when the concentration of the added acid analytes is increased from 0 to 4 × 10−3 M, except for glucuronic acid, the fluorescence is almost quenched after the sensor 5c is combined with the acid analytes.37 Especially, the sensor 5c has the strongest binding capacity to caffeic acid, and its fluorescence is completely quenched (Fig. S7, ESI†).
Table 2 Binding constants (Ka) of sensor 5c with different analytesa
| Analytes |
Ka (M−1) |
| Ka the value was calculated by Benesi–Hildebrand equation based on three times of measurement (R2 > 0.99). |
| Catechol |
795.6 ± 4.2 |
| Dopamine |
893.3 ± 16.3 |
| Levodopa |
746.6 ± 22.1 |
| Caffeic acid |
9245.7 ± 348.3 |
| Galactose |
15.6 ± 0.9 |
| Sorbitol |
347.1 ± 2.4 |
| Fructose |
404.4 ± 4.5 |
| Glucose |
— |
| Gluconic |
135.9 ± 1.0 |
| Glucuronic |
— |
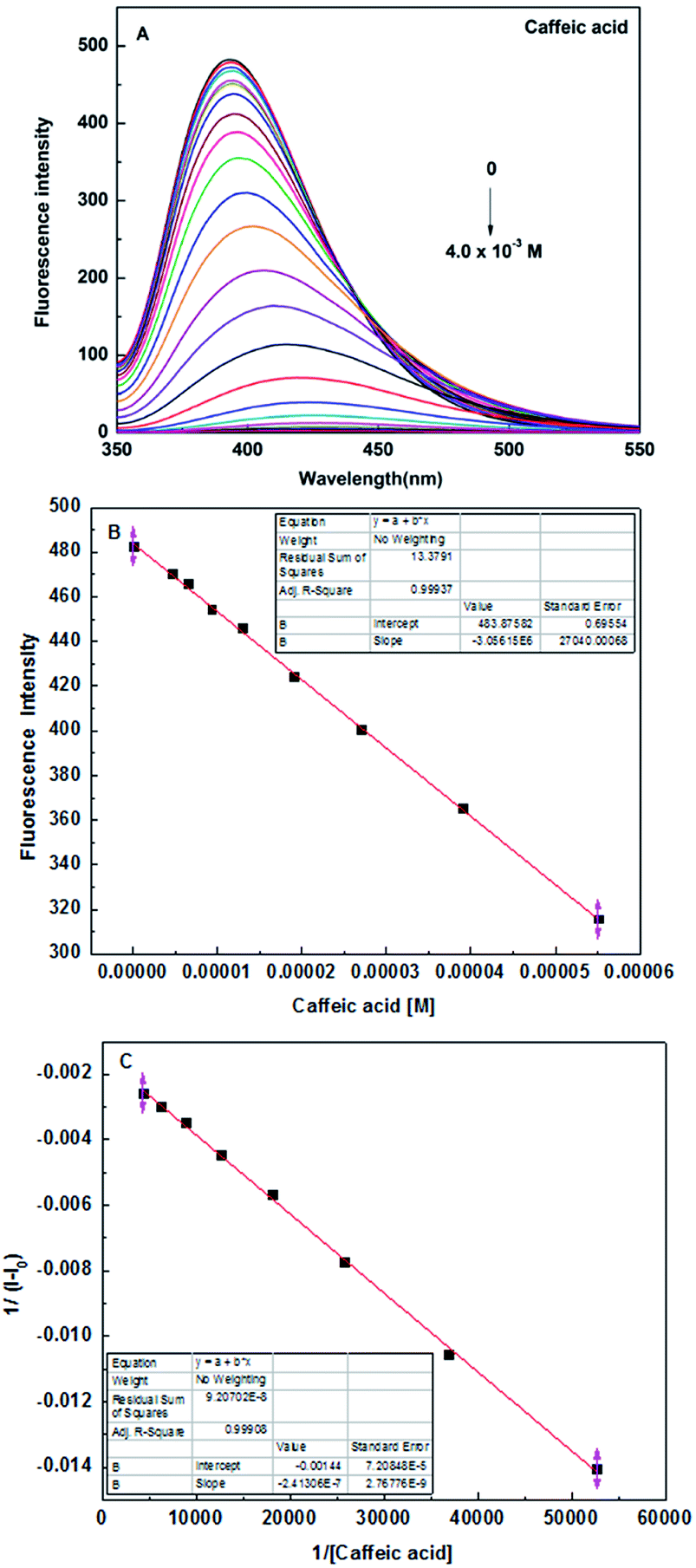
In order to further study the binding ability of sensor 5c to caffeic acid, the fluorescence titration experiment was performed. When the concentration of caffeic acid increased from 0 to 4 × 10−3 M, the fluorescence intensity of sensor 5c decreased by 99.6%, which is almost completely quenched, as shown in Fig. 5A. And the fluorescence titration of sensor 5c with other analytes is shown in Fig. S7 (ESI†). In addition, when the added caffeic acid concentration is in the range of 4.6 × 10−6 M to 5.5 × 10−5 M, there is a good linear relationship between the fluorescence intensity of the sensor 5c and the caffeic acid concentration, and the correlation coefficient is R2 = 0.99937, as shown in Fig. 5B. Therefore, the linear regression equation can be determined from Fig. 5B as I = −3056150c + 483.87582, where c is the concentration of caffeic acid and I is the maximum emission fluorescence intensity at 395 nm. Then the LOD is calculated as 1.81 × 10−6 M by the following equation.46,47
where
δ is the standard deviation of the 5 times blank signal of the sensor, and
S is the slope of the calibration curve.
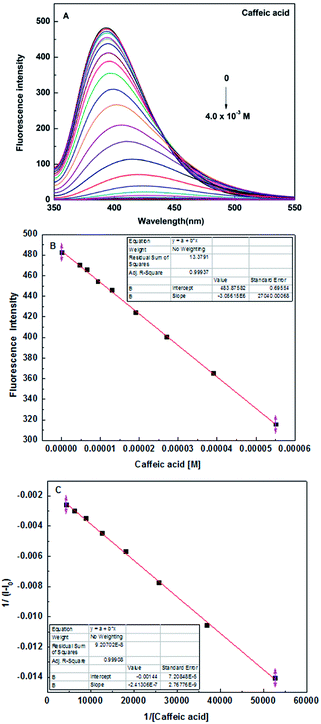 |
| | Fig. 5 (A) Fluorescence spectra of sensor 5c (1 × 10−5 M) in the presence of different concentrations of caffeic acid in PBS (pH 7.4) solution, at room temperature; (B) the photograph of sensor 5c linear range; (C) Benesi–Hildebrand plot of sensor 5c 1/(I − I0) versus 1/[Caffeic acid]. | |
In addition, we found that the reciprocal of the decrease in the fluorescence intensity of the sensor 5c and the reciprocal of the caffeic acid concentration show a good linear relationship, R2 = 0.99908, as shown in Fig. 5C. Benesi–Hildebrand (B–H) equation is a widely used approach for determining the stoichiometry and equilibrium constants of nonbonded interactions, particularly 1:1 and 1:2 interactions.48 Therefore, the titrated fluorescence data were processed using the B–H equation and all titrations were performed three times. The titration curve for caffeic acid is fitted and the binding constant (Ka) is 9245.7 ± 348.3 M−1 according to the following equation:30,35,38,49,50
where
I0 and
I1 are the initial (no caffeic acid) and final fluorescence intensity of the titration curve,
I is the observed fluorescence intensity and [Caffeic acid] is the caffeic acid concentration. Using the curve of (
I1 −
I0)/(
I −
I0)
versus 1/[Caffeic acid],
Ka can be calculated from the intercept/slope. According to the value of
Ka calculated from the B–H equation, it can be found that sensor
5c has a high binding affinity for caffeic acid. And due to
Fig. 5C processed by the B–H equation shows a good linear relationship,
R2 = 0.99908, therefore, it can be determined that the binding ratio of sensor
5c to caffeic acid is 1
:
1.
Response time
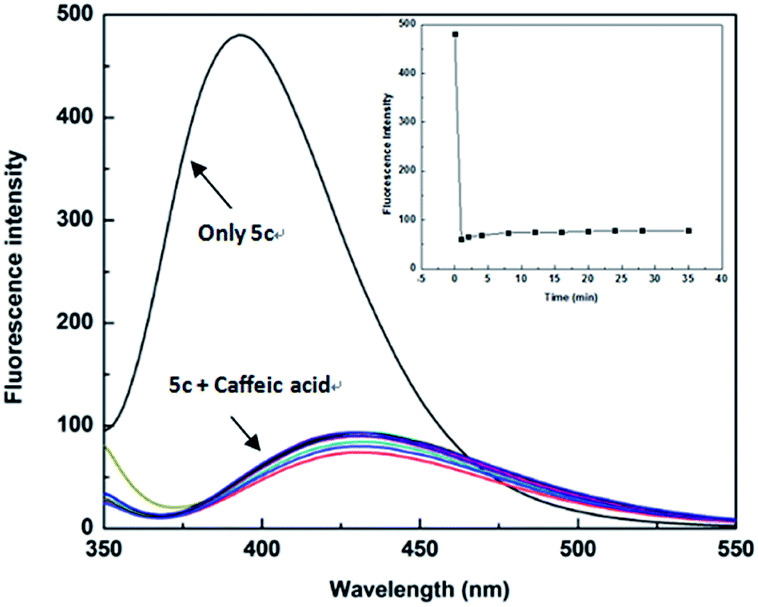
The high sensitivity of the sensor is an important aspect of practical applications. In order to be used for the detection of actual samples, it is necessary to explore the sensitivity of the sensor 5c. Therefore, we performed the response time experiment of the reaction system at room temperature (25 °C). The fluorescence of sensor 5c (1 × 10−5 M) decreased at 395 nm, and its fluorescence intensity is greatly reduced after adding 4.0 × 10−4 M caffeic acid. Fluorescence scans recording requires 0.5 minutes of preparation time. The response time of the test was 0 to 35 minutes, and the recorded time was set to 0, 1, 2, 4, 8, 12, 16, 20, 24, 28, 35 minutes, respectively. Through the response time experiment we found that the fluorescence of the sensor 5c is greatly reduced to a certain intensity of fluorescence emission, and then it showed very weak fluorescence attenuation over time, as shown in Fig. 6. These results indicate that the response time of the sensor 5c for caffeic acid is very fast, and real-time detection can be achieved.
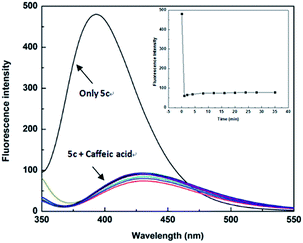 |
| | Fig. 6 Fluorescence spectra of sensor 5c (1.0 × 10−5 M) upon addition of 4.0 × 10−4 M of caffeic acid from 0 to 35 min in PBS (pH 7.4) solution, at room temperature. Inset: plot of the fluorescence intensities at 395 nm over 35 min. | |
pH titration
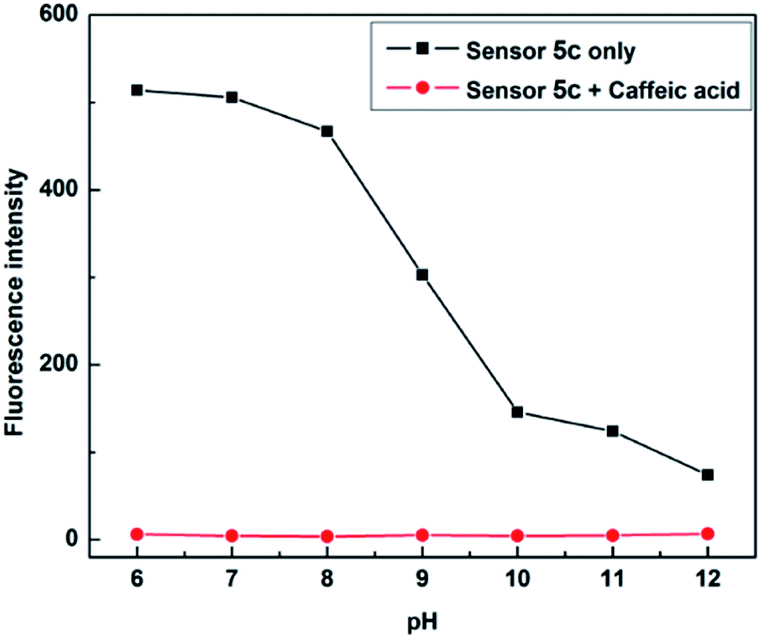
Due to boronic acid shows different binding forms in acidic and alkaline environments, in order to find the appropriate pH conditions, it is necessary to study the recognition process of sensor 5c to caffeic acid. A phosphate buffer (PBS, 0.1 M) was first used as the buffered solvent, followed by the solution of hydrochloric acid and sodium hydroxide to adjust the pH. And the pH was adjusted to 6.0, 7.0, 8.0, 9.0, 10.0, 11.0 and 12.0, respectively, as the pH of the sensor 5c to be studied. Then, different concentrations of caffeic acid were used for fluorescence titration at each pH. The titration curve was made when the concentration of caffeic acid added was 4 × 10−3 M, it could be found that sensor 5c has a large fluorescence response in the range of pH 6 to 8, as shown in Fig. 7. Considering that the detection sample requires a large fluorescence response, to avoid the trouble of pH adjustment of the buffer solution and to meet the requirements of physiological conditions, therefore, pH 7.4 is a suitable condition for detecting caffeic acid, and all experiments are performed at pH 7.4.
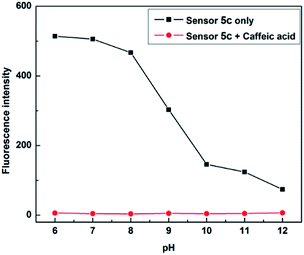 |
| | Fig. 7 Fluorescence responses of sensor 5c (1 × 10−5 M) to caffeic acid (4 × 10−3 M) in phosphate buffer (PBS, 0.1 M) at different pH values. | |
Application of sensor 5c in caffeic acid analysis in real samples
From the above experiments, we know that the sensor 5c has high selectivity to caffeic acid, so we want to know whether the sensor 5c can be used for the detection of actual samples, such as caffeic acid in drugs or food products. So, we choose drug caffeic acid tablets for testing, and recovery studies were carried out by spiking the samples with caffeic acid in different concentrations in the range of the linearity. Since the sensor has a good fluorescence response at pH 7.4, a true sample solution to be measured can be prepared with 0.1 M phosphate buffer (pH 7.4) to measure the fluorescence intensity of the actual sample. Using the linear regression equation obtained in Fig. 3B: I = −3056150c + 483.87582. By processing the linear regression equation of fluorescence intensity to convert a series of actual sample concentrations, the equation for calculating the concentration can be converted as follows:
The recovery and relative standard deviation were calculated by using the calculated concentrations of caffeic acid and spiked caffeic acid. As shown in Table 3, it can be found from the measurement results that caffeic acid has a better recovery rate. Moreover, the active component of caffeic acid tablets is caffeic acid, and there are no pyrocatechol compounds in the pharmaceutical excipients, so the excipients do not interfere with the test results. Therefore, the experimental results show that the sensor 5c can be applied to the analysis of real samples.47,51,52
Table 3 Determination of caffeic acid concentration in caffeic acid tablets
| Sample |
Caffeic acid added (M) |
Caffeic acid found (M) |
Recovery% |
RSDa (%) |
| Relative standard derivation was calculated based on three times of measurements. |
| Caffeic acid tablets |
0 |
1.29 × 10−5 |
— |
2.2 |
| 3.0 × 10−5 |
4.11 × 10−5 |
94 |
1.3 |
| 3.5 × 10−5 |
4.58 × 10−5 |
94 |
1.3 |
| 4.0 × 10−5 |
4.99 × 10−5 |
93 |
1.6 |
| 4.5 × 10−5 |
5.35 × 10−5 |
90 |
1.2 |
Comparison with other detection methods
The fluorescence analysis method for detecting caffeic acid is sensitive, fast and does not require complicated sample pretreatment processes, so it has potential application value in real life. As shown in Table 4, the detection methods of caffeic acid that have been reported are compared with the methods used in this paper. The results show that compared to most previously reported methods, our established method provides a better linear range and LOD.
Table 4 Comparison of linear range and detection limit between the proposed method and other reported detection methods for caffeic acid
| Method |
Linear range (M) |
LOD (M) |
Author |
| UV-Vis spectrometry |
8.8 × 10−4 to 5.6 × 10−1 |
0.3 × 10−3 |
Zitka,53 2011 |
| Liquid chromatography |
2.8 × 10−4 to 5.6 × 10−3 |
0.11 × 10−3 |
Tsai,54 1999 |
| Gas chromatography |
1.6 × 10−3 to 1.2 × 10−1 |
0.53 × 10−3 |
Chu,55 2001 |
| Voltammetric method |
1.0 × 10−5 to 3.5 × 10−1 |
2.4 × 10−6 |
Karikalan,11 2017 |
| Amperometric method |
2.0 × 10−3 to 1.0 × 10−2 |
0.5 × 10−3 |
Demirkol,56 2012 |
| Electrochemical sensor |
5.0 × 10−4 to 6.0 × 10−2 |
0.15 × 10−3 |
Leite,57 2014 |
| 5.0 × 10−4 to 5.0 × 10−2 |
0.05 × 10−3 |
Zhang,26 2013 |
| Electrochemistry |
4.0 × 10−4 to 7.4 × 10−3 |
0.29 × 10−3 |
Radoi,58 2011 |
| 7.4 × 10−4 to 10.5 × 10−3 |
0.15 × 10−3 |
Diaconu,59 2010 |
| Fluorometry |
5.0 × 10−4 to 2.0 × 10−1 |
0.11 × 10−3 |
Xu,16 2018 |
| 1.4 × 10−4 to 1.4 × 10−3 |
0.06 × 10−3 |
Xiang,27 2015 |
| 3.7 × 10−3 to 1.1 × 10−1 |
1.2 × 10−3 |
Fan,29 2011 |
| 2.0 × 10−3 to 3.5 × 10−1 |
0.2 × 10−3 |
Cai,23 2016 |
| 4.6 × 10−6 to 5.5 × 10−5 |
1.81 × 10−6 |
This work |
Conclusions
At present, water-soluble boronic acid sensors for selective identification of caffeic acid have not been reported. We synthesized several water-soluble boronic acid sensors using compound 1 as the building block, and detected changes in their fluorescence properties after binding to the various analytes. Among them, when the concentration of the added analytes is 2.3 × 10−4 M, sensor 5c has the strongest binding capacity to caffeic acid, and its fluorescence intensity is reduced by 99.6%. However, its fluorescence intensity increased or did not change obviously after binding with other analytes. Furthermore, sensor 5c not only has a very fast response time to caffeic acid under mild conditions (at room temperature) but also has a high binding constant (9245.7 ± 348.3 M−1) and low LOD (1.81 × 10−6 M) in phosphate buffer (0.1 M, pH 7.4). This indicates that sensor 5c can selectively recognize caffeic acid via fluorescence quenching. In addition, sensor 5c can be used to detect the caffeic acid content in real samples (such as caffeic acid tablets), and the recovery rate is good. Therefore, the above research shows that the sensor 5c can be used as a potential tool to detect caffeic acid content in drug or food products and even to diagnose diabetic complications early through caffeic acid detection. Finally, we hope to report that this work will encourage other research groups to discovery more potential boronic acid sensors to recognizing biologically meaningful substances, as well as to develop more promising fluorescence navigation sensors tool for clinical use.
Conflicts of interest
The authors confirm that this article content has no conflict of interest.
Acknowledgements
The authors would like to thank the Innovation Project of the Academy of Medical Sciences for financial support. This work was supported by the National Natural Science Foundation of China (Grant No. 21801158), Shandong Academy of Medical Sciences Foundation (Grant No. 2018-17), and Graduate Instructor Guidance Ability Improvement Project University of Jinan (Grant No. JDYY1804).
Notes and references
- J. Shen, G. Wang and J. Zuo, Antiviral Res., 2018, 154, 166–173 CrossRef CAS PubMed.
- X. Zhang, X. He, Q. Chen, J. Lu, S. Rapposelli and R. Pi, Bioorg. Med. Chem., 2018, 26, 543–550 CrossRef CAS PubMed.
- K. M. M. Espindola, R. G. Ferreira, L. E. M. Narvaez, A. C. R. Silva Rosario, A. H. M. da Silva, A. G. B. Silva, A. P. O. Vieira and M. C. Monteiro, Front. Oncol., 2019, 9, 1–10 CrossRef PubMed.
- L. P. Pelinson, C. E. Assmann, T. V. Palma, I. B. M. da Cruz, M. M. Pillat, A. Manica, N. Stefanello, G. C. C. Weis, A. de Oliveira Alves, C. M. de Andrade, H. Ulrich, V. M. M. Morsch, M. R. C. Schetinger and M. D. Bagatini, Mol. Biol. Rep., 2019, 46, 2085–2092 CrossRef CAS PubMed.
- E. Lutfi, P. J. Babin, J. Gutierrez, E. Capilla and I. Navarro, PLoS One, 2017, 12, 1–21 Search PubMed.
- S. Yuksel and S. Akyol, J. Intercult. Ethnopharmacol., 2016, 5, 308–311 CrossRef CAS PubMed.
- M. Zhang, J. Zhou, L. Wang, B. Li, J. Guo, X. Guan, Q. Han and H. Zhang, Biol. Pharm. Bull., 2014, 37, 347–354 CrossRef CAS PubMed.
- J. Min, H. Shen, W. Xi, Q. Wang, L. Yin, Y. Zhang, Y. Yu, Q. Yang and Z. N. Wang, Cell. Physiol. Biochem., 2018, 48, 1433–1442 CrossRef CAS PubMed.
- A. Sharma, M. Kaur, J. K. Katnoria and A. K. Nagpal, Curr. Med. Chem., 2018, 25, 4740–4757 CrossRef CAS PubMed.
- M. Hirose, Y. Takesada, H. Tanaka, S. Tamano, T. Kato and T. Shirai, Carcinogenesis, 1998, 19, 207–212 CrossRef CAS PubMed.
- N. Karikalan, R. Karthik, S. M. Chen and H. A. Chen, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- Ş. Karaman, E. Tütem, K. Sözgen Başkan and R. Apak, Food Chem., 2010, 120, 1201–1209 CrossRef.
- Z. Bartosova, D. Riman, P. Jakubec, V. Halouzka, J. Hrbac and D. Jirovsky, Sci. World J., 2012, 2012, 1–6 CrossRef PubMed.
- E. Mesquita and M. Monteiro, Food Res. Int., 2018, 106, 54–63 CrossRef CAS PubMed.
- H. A. Salman, S. Ramasamy and B. S. Mahmood, J. Intercult. Ethnopharmacol., 2018, 7, 76–81 CAS.
- Y. Shi, H. Xu, J. Wang, S. Li, Z. Xiong, B. Yan, C. Wang and Y. Du, Sens. Actuators, B, 2018, 272, 135–138 CrossRef CAS.
- J. Xue, P. T. Lee and R. G. Compton, Electroanalysis, 2014, 26, 1454–1460 CrossRef CAS.
- Y. Yardim, M. Gulcan and Z. Senturk, Food Chem., 2013, 141, 1821–1827 CrossRef CAS PubMed.
- S. Ramki, P. Balasubramanian, S. M. Chen, T. W. Chen, T. W. Tseng and B. S. Lou, Int. J. Electrochem. Sci., 2018, 13, 1241–1249 CrossRef CAS.
- L. G. Mohtar, P. Aranda, G. A. Messina, M. A. Nazareno, S. V. Pereira, J. Raba and F. A. Bertolino, Microchem. J., 2019, 144, 13–18 CrossRef CAS.
- A. Heras, F. Vulcano, J. Garoz-Ruiz, N. Porcelli, F. Terzi, A. Colina, R. Seeber and C. Zanardi, Sensors, 2019, 19, 1–12 CrossRef PubMed.
- H. Mao, Y. Zhang and G. Chen, Anal. Methods, 2019, 11, 303–308 RSC.
- N. Cai, Y. Li, S. Chen and X. Su, Analyst, 2016, 141, 4456–4462 RSC.
- K. Singh and A. Kumar, Spectrochim. Acta, Part A, 2019, 211, 148–153 CrossRef CAS PubMed.
- P. Wongsa, J. Chaiwarith, J. Voranitikul, J. Chaiwongkhajorn, N. Rattanapanone and R. Lanberg, Chiang Mai J. Sci., 2019, 46, 672–682 CAS.
- J. Song, J. Zhang, F. Lv, Y. Cheng, B. Wang, L. Feng, L. Liu and S. Wang, Angew. Chem., Int. Ed., 2013, 52, 13020–13023 CrossRef CAS PubMed.
- X. Xiang, J. Shi, F. Huang, M. Zheng and Q. Deng, Talanta, 2015, 141, 182–187 CrossRef CAS PubMed.
- X. Xu, G. Xu, F. Wei, Y. Cen, M. Shi, X. Cheng, Y. Chai, M. Sohail and Q. Hu, J. Colloid Interface Sci., 2018, 529, 568–574 CrossRef CAS PubMed.
- X. Fan, S. Liu and Y. He, Colloids Surf., B, 2011, 88, 23–30 CrossRef CAS PubMed.
- H. Wang, G. Fang, H. Wang, J. Dou, Z. Bian, Y. Li, H. Chai, Z. Wu and Q. Yao, New J. Chem., 2019, 43, 4385–4390 RSC.
- X. Zhou, X. Gao, F. Song, C. Wang, F. Chu and S. Wu, Appl. Surf. Sci., 2017, 423, 810–816 CrossRef CAS.
- A. L. Chibac, V. Melinte, T. Buruiana and E. C. Buruiana, Sens. Actuators, B, 2017, 253, 987–998 CrossRef CAS.
- S. K. Munusamy, K. Thirumoorthy, V. P. Muralidharan, U. Balijapalli and S. K. Lyer, Sens. Actuators, B, 2017, 244, 175–181 CrossRef CAS.
- S. Xu, S. Che, P. Ma, F. Zhang, L. Xu, X. Liu, X. Wang, D. Song and Y. Sun, Talanta, 2019, 197, 548–552 CrossRef CAS PubMed.
- G. Fang, Z. Bian, D. Liu, G. Wu, H. Wang, Z. Wu and Q. Yao, New J. Chem., 2019, 43, 13802–13809 RSC.
- Z. Wu, X. Yang, W. Xu, B. Wang and H. Fang, Drug Discoveries Ther., 2012, 6, 238–241 CAS.
- Z. Wu, M. Li, H. Fang and B. Wang, Bioorg. Med. Chem. Lett., 2012, 22, 7179–7182 CrossRef CAS PubMed.
- H. Wang, G. Fang, K. Wang, Z. Wu and Q. Yao, Anal. Lett., 2018, 52, 713–727 CrossRef.
- G. Fang, H. Wang, Z. Bian, M. Guo, Z. Wu and Q. Yao, RSC Adv., 2019, 9, 20306–20313 RSC.
- L. Han, S. G. Liu, J. Y. Liang, Y. J. Ju, N. B. Li and H. Q. Luo, J. Hazard. Mater., 2019, 362, 45–52 CrossRef CAS PubMed.
- Z. R. Zhang J and D. Tang, et al., TrAC, Trends Anal. Chem., 2019, 110, 183–190 CrossRef.
- H. Y. Chen, J. R. Wei, J. X. Pan, W. Zhang, F. Q. Dang, Z. Q. Zhang and J. Zhang, Biosens. Bioelectron., 2017, 91, 328–333 CrossRef CAS PubMed.
- J. W. Shaw, L. Barbance, D. H. Grayson and I. Rozas, Tetrahedron Lett., 2015, 56, 4990–4992 CrossRef CAS.
- K. B. Suhs T, Mini-Rev. Org. Chem., 2006, 3, 315–331 CrossRef.
- D. Hazarika, A. J. Borah and P. Phukan, Chem. Commun., 2019, 55, 1418–1421 RSC.
- R. Hosseinzadeh, M. Mohadjerani, M. Pooryousef, A. Eslami and S. Emami, Spectrochim. Acta, Part A, 2015, 144, 53–60 CrossRef CAS PubMed.
- R. Hosseinzadeh, M. Mohadjerani and M. Pooryousef, Anal. Bioanal. Chem., 2016, 408, 1901–1908 CrossRef CAS PubMed.
- R. Wang and Z. Yu, Acta Phys.-Chim. Sin., 2007, 23, 1353–1359 CrossRef CAS.
- A. A. Bhatti, M. Oguz, S. Memon and M. Yilmaz, J. Fluoresc., 2017, 27, 263–270 CrossRef CAS PubMed.
- Y. Dai, K. Yao, J. Fu, K. Xue, L. Yang and K. Xu, Sens. Actuators, B, 2017, 251, 877–884 CrossRef CAS.
- M. Jamkratoke, G. Tumcharern, T. Tuntulani and B. Tomapatanaget, J. Fluoresc., 2011, 21, 1179–1187 CrossRef CAS PubMed.
- S. Liu, H. Bai, Q. Sun, W. Zhang and J. Qian, RSC Adv., 2015, 5, 2837–2843 RSC.
- O. Zitka, J. Sochor and O. Rop, et al., Molecules, 2011, 16, 2914–2936 CrossRef CAS PubMed.
- T. H. Tsai, Y. F. Chen and I. F. Chen, et al., J. Chromatogr. B: Biomed. Sci. Appl., 1999, 729(2), 119–125 CrossRef CAS.
- T. Y. Chu, C. H. Chang and Y. C. Liao, et al., Talanta, 2001, 54(6), 1163–1171 CrossRef CAS PubMed.
- D. O. Demirkol, B. Gulsunoglu and C. Ozdemir, et al., Food Anal. Methods, 2012, 5(2), 244–249 CrossRef.
- W. J. R. Santos, L. T. Kubota and F. R. F. Leite, Sens. Actuators, B, 2014, 193, 238–246 CrossRef.
- A. Radoi, S. C. Litescu and S. A. V. Eremia, et al., Microchim. Acta, 2011, 175(1–2), 97–104 CrossRef CAS.
- M. Diaconu, S. C. Litescu and G. L. Radu, Sens. Actuators, B, 2010, 145, 800–806 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00980f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence abcd,
Ran Wangabcd,
Dongxue Zhanbcd,
Qingqiang Yaobcd and
Zhongyu Wu
abcd,
Ran Wangabcd,
Dongxue Zhanbcd,
Qingqiang Yaobcd and
Zhongyu Wu