
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactions of triosmium and triruthenium clusters with 2-ethynylpyridine: new modes for alkyne C–C bond coupling and C–H bond activation†
Md. Tuhinur R. Joya,
Roknuzzamana,
Md. Emdad Hossaina,
Shishir Ghosh *a,
Derek A. Tocherb,
Michael G. Richmondc and
Shariff E. Kabir*a
*a,
Derek A. Tocherb,
Michael G. Richmondc and
Shariff E. Kabir*a
aDepartment of Chemistry, Jahangirnagar University, Savar, Dhaka 1342, Bangladesh. E-mail: sghosh_006@yahoo.com; skabir_ju@yahoo.com
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
cDepartment of Chemistry, University of North Texas, 1155 Union Circle, Box 305070, Denton, TX 76203, USA
First published on 19th August 2020
Abstract
The reaction of the trimetallic clusters [H2Os3(CO)10] and [Ru3(CO)10L2] (L = CO, MeCN) with 2-ethynylpyridine has been investigated. Treatment of [H2Os3(CO)10] with excess 2-ethynylpyridine affords [HOs3(CO)10(μ-C5H4NCH=CH)] (1), [HOs3(CO)9(μ3-C5H4NC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)] (2), [HOs3(CO)9(μ3-C5H4NCCCO2)] (3), and [HOs3(CO)10(μ-CHCHC5H4N)] (4) formed through either the direct addition of the Os–H bond across the C
CH2)] (2), [HOs3(CO)9(μ3-C5H4NCCCO2)] (3), and [HOs3(CO)10(μ-CHCHC5H4N)] (4) formed through either the direct addition of the Os–H bond across the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond or acetylenic C–H bond activation of the 2-ethynylpyridine substrate. In contrast, the dominant pathway for the reaction between [Ru3(CO)12] and 2-ethynylpyridine is C–C bond coupling of the alkyne moiety to furnish the triruthenium clusters [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CH}] (5) and [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CHCHC(C5H4N)}] (6). Cluster 5 contains a metalated 2-pyridyl-substituted diene while 6 exhibits a metalated 2-pyridyl-substituted triene moiety. The functionalized pyridyl ligands in 5 and 6 derive via the formal C–C bond coupling of two and three 2-ethynylpyridine molecules, respectively, and 5 and 6 provide evidence for facile alkyne insertion at ruthenium clusters. The solid-state structures of 1–3, 5, and 6 have been determined by single-crystal X-ray diffraction analyses, and the bonding in the product clusters has been investigated by DFT. In the case of 1, the computational results reveal a rare thermodynamic preference for a terminal hydride ligand as opposed to a hydride-bridged Os–Os bond (3c,2e Os–Os–H bond).
C bond or acetylenic C–H bond activation of the 2-ethynylpyridine substrate. In contrast, the dominant pathway for the reaction between [Ru3(CO)12] and 2-ethynylpyridine is C–C bond coupling of the alkyne moiety to furnish the triruthenium clusters [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CH}] (5) and [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CHCHC(C5H4N)}] (6). Cluster 5 contains a metalated 2-pyridyl-substituted diene while 6 exhibits a metalated 2-pyridyl-substituted triene moiety. The functionalized pyridyl ligands in 5 and 6 derive via the formal C–C bond coupling of two and three 2-ethynylpyridine molecules, respectively, and 5 and 6 provide evidence for facile alkyne insertion at ruthenium clusters. The solid-state structures of 1–3, 5, and 6 have been determined by single-crystal X-ray diffraction analyses, and the bonding in the product clusters has been investigated by DFT. In the case of 1, the computational results reveal a rare thermodynamic preference for a terminal hydride ligand as opposed to a hydride-bridged Os–Os bond (3c,2e Os–Os–H bond).
1. Introduction
2-Vinylpyridine is well known for its ability to stabilize transition metal–carbon bonds in a chelating coordination mode.1–10 Early investigative work on the coordination chemistry of this ligand at mono- and polynuclear metal compounds has produced many excellent publications from the academic community.1–18 At mononuclear centers, coordination of this ligand occurs through the nitrogen donor of the pyridyl ligand, which in turn derives from the short-lived precursor involving the alkene functionality (π-complex). Once coordinated, the 2-vinylpyridine ligand is activated with respect to competitive C–H bond activation at the β-C–H alkenyl bond.1–10 In contrast, many heterocyclic-substituted polynuclear clusters supporting a non-spectator nitrogen ligand have been isolated and structurally characterized using 2-vinylpyridine as a ligand. As part of our interest in the role played by metal cluster compounds in potential catalytic cycles, we continue to explore the reactivity pathways involving metal clusters and heterocyclic substrates. It should be noted that the majority of examples of metalated polynuclear derivatives derived from 2-vinylpyridine display common coordination modes for the heterocyclic auxiliary. This particular pyridyl ligand has been shown to coordinate up to four metal atoms and function as a 4e or 6e donor (Chart 1).11–18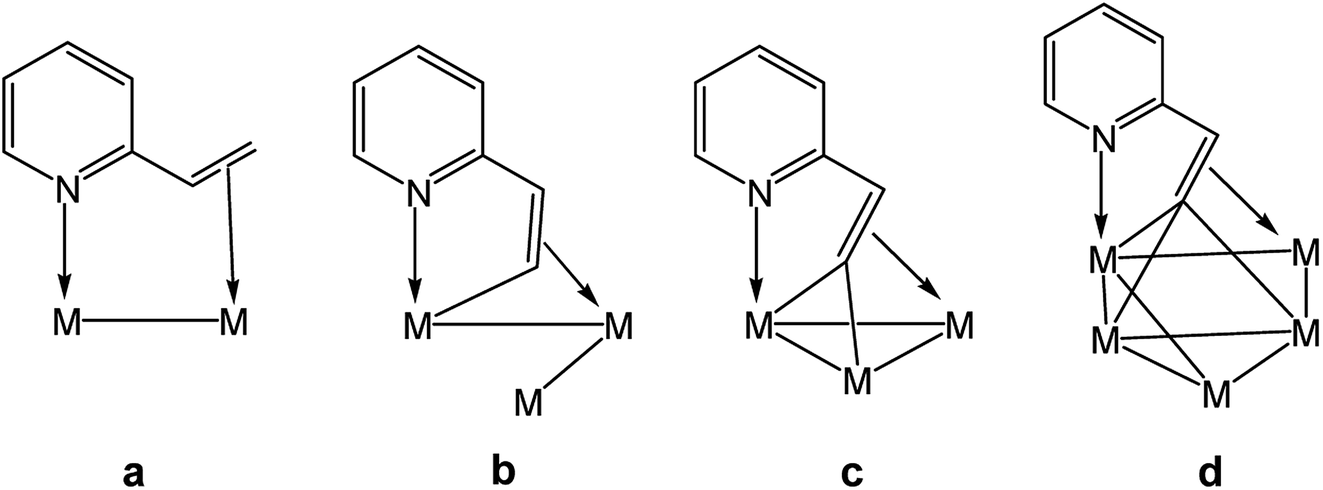 | ||
| Chart 1 Known binding modes of non-metalated (a), mono-metalated (b), di-metalated (c), and tri-metalated (d) 2-vinylpyridine ligands at polynuclear transition metal centers. | ||
In comparison to the large number of reports on the reactivity of 2-vinylpyridine at metal clusters, fewer studies exist on the coordination chemistry of the acetylenic counterpart 2-ethynylpyridine at metal clusters.11,19,20 In 1985, a cooperative investigation on the reaction of [H2Os3(CO)10] with 2-ethynylpyridine was conducted by the groups of Lewis, Hursthouse, and Deeming (LHD). The published report presented data for the existence of two products (Scheme 1), and the structure of one of the products (minor) was unequivocally established by X-ray crystallography. The solid-state structure of the minor product [HOs3(CO)10(μ-C5H4NCHCH)] displays an opened triangular core with the ancillary pyridyl ligand functioning as a 5e donor that includes both nitrogen and (Z)-σ,π-alkenyl contributions.11 The structure of the major isomeric product was formulated to have a dangling (free) pyridyl ligand with an (E)-alkenyl moiety that functions as a traditional σ,π-vinyl donor. The route that affords the major product was ascribed to an addition path involving an Os–H bond and the alkyne unit.
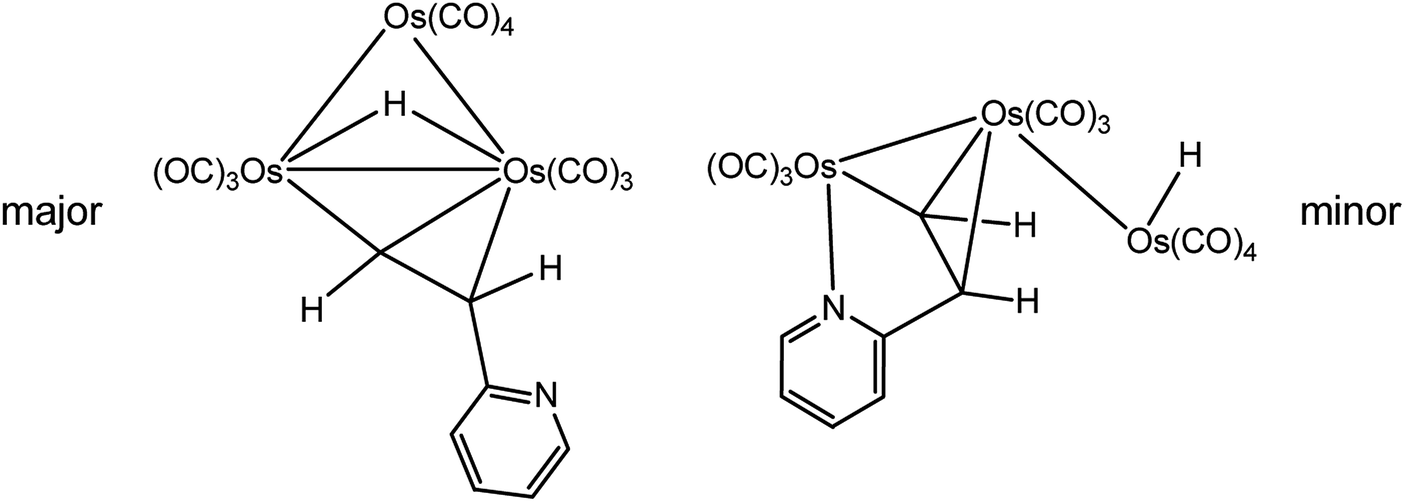 | ||
| Scheme 1 LHD major (left) and minor (right) products isolated from the reaction of [H2Os3(CO)10] with 2-ethynylpyridine.11 | ||
We have investigated the reactivity of different heterocycles containing a vinyl functionality at the ortho position such as 2-vinylpyridine, 2-vinylpyrazine, etc. at polynuclear metal centers over the last few years.16–18,20 Wishing to extend these studies based on alkenyl-substituted heterocyclic ligands, we have investigated the reaction of 2-ethynylpyridine with [Ru3(CO)12], which affords novel Ru3 products based on C–C bond coupling of alkyne carbons of the pyridyl substrate. We have also reinvestigated the reaction of [H2Os3(CO)10] with 2-ethynylpyridine and now report the isolation of two new cluster products 2 and 3 and confirm the unexpected isomerization of the major species found in the LHD study at elevated temperature to the minor isomer having an opened cluster polyhedron.11
2. Experimental section
2.1. General remarks
All reactions were carried out under an inert atmosphere of nitrogen using standard Schlenk techniques unless otherwise stated. Reagent grade solvents were dried by standard methods and were freshly distilled prior to use. Infrared spectra were recorded on a Shimadzu IR Prestige-21 spectrophotometer, while the 1H NMR spectra were recorded on a Bruker Advance III HD (400 MHz) instrument. All chemical shifts are reported in δ units and are referenced to the residual protons of the deuterated solvent. Elemental analyses were performed by the Microanalytical Laboratories of the Wazed Miah Science Research Centre at Jahangirnagar University. [Os3(CO)12] and [Ru3(CO)12] were purchased from Strem Chemical Inc. and used without further purification. 2-Ethynylpyridine was purchased from Acros Organics and used as received. The starting clusters [H2Os3(CO)10]21 and [Ru3(CO)10(NCMe)2]22 were prepared according to the published procedures. All products were separated in the air using preparative TLC plates coated with 0.25 mm of silica gel (HF254-type 60, E. Merck, Germany).2.2. Reaction of [H2Os3(CO)10] with 2-ethynylpyridine
A CH2Cl2 solution (15 mL) of [H2Os3(CO)10] (50 mg, 0.059 mmol) and 2-ethynylpyridine (25 mg, 0.24 mmol) was stirred at room temperature for 15 min. The color of the reaction mixture changed from purple to orange by this time. The solvent was removed under reduced pressure and the residue purified by preparative TLC. Elution with cyclohexane/CH2Cl2 (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) developed four bands which afforded the following compounds, in order of elution, yellow [HOs3(CO)10(μ-C5H4NCHCH)] (1; LHD minor product; 14 mg, 25%), orange [HOs3(CO)9(μ3-C5H4NCCH2)] (2) (8.8 mg, 15%), orange [HOs3(CO)9(μ3-C5H4NCCCO2)] (3) (17 mg, 30%), and yellow [HOs3(CO)10(μ-CHCHC5H4N)] (4; LHD major product; 11 mg, 20%). Data for 2: anal. calc. for C16H7NO9Os3: C, 20.71; H, 0.76; N, 1.51. Found: C, 20.98; H, 0.78; N, 1.55%. IR (ν(CO), CH2Cl2): 2083s, 2051vs, 2028vs, 1995vs, 1983sh, 1970m, 1958m cm−1. 1H NMR (CDCl3): δ 8.56 (d, J 6.0 Hz, 1H), 7.64 (m, 1H), 6.98 (m, 1H), 6.92 (d, J 8.0 Hz, 1H), 4.30 (s, 1H) 2.63 (s, 1H), −17.70 (s, 1H). Data for 3: anal. calc. for C17H5NO11Os3: C, 21.05; H, 0.52; N, 1.44. Found: C, 21.23; H, 0.55; N, 1.51%. IR (ν(CO), CH2Cl2): 2104m, 2079vs, 2060vs, 2025s, 2005m, 1990w, 1693w cm−1. IR (ν(CO), KBr): 2102m, 2075s, 2056s, 2033s, 2018s, 1996vs, 1979s, 1692w cm−1. 1H NMR (CDCl3): δ 8.63 (d, J 6.0 Hz, 1H), 7.88 (m, 1H), 7.76 (d, J 8.0 Hz, 1H), 7.00 (m, 1H), −16.76 (s, 1H).
:3 v/v) developed four bands which afforded the following compounds, in order of elution, yellow [HOs3(CO)10(μ-C5H4NCHCH)] (1; LHD minor product; 14 mg, 25%), orange [HOs3(CO)9(μ3-C5H4NCCH2)] (2) (8.8 mg, 15%), orange [HOs3(CO)9(μ3-C5H4NCCCO2)] (3) (17 mg, 30%), and yellow [HOs3(CO)10(μ-CHCHC5H4N)] (4; LHD major product; 11 mg, 20%). Data for 2: anal. calc. for C16H7NO9Os3: C, 20.71; H, 0.76; N, 1.51. Found: C, 20.98; H, 0.78; N, 1.55%. IR (ν(CO), CH2Cl2): 2083s, 2051vs, 2028vs, 1995vs, 1983sh, 1970m, 1958m cm−1. 1H NMR (CDCl3): δ 8.56 (d, J 6.0 Hz, 1H), 7.64 (m, 1H), 6.98 (m, 1H), 6.92 (d, J 8.0 Hz, 1H), 4.30 (s, 1H) 2.63 (s, 1H), −17.70 (s, 1H). Data for 3: anal. calc. for C17H5NO11Os3: C, 21.05; H, 0.52; N, 1.44. Found: C, 21.23; H, 0.55; N, 1.51%. IR (ν(CO), CH2Cl2): 2104m, 2079vs, 2060vs, 2025s, 2005m, 1990w, 1693w cm−1. IR (ν(CO), KBr): 2102m, 2075s, 2056s, 2033s, 2018s, 1996vs, 1979s, 1692w cm−1. 1H NMR (CDCl3): δ 8.63 (d, J 6.0 Hz, 1H), 7.88 (m, 1H), 7.76 (d, J 8.0 Hz, 1H), 7.00 (m, 1H), −16.76 (s, 1H).
2.3. Isomerization of 4 to 1
To a 5 mm NMR tube was charged cluster 4 (8.0 mg, 0.0080 mmol) and 0.75 mL of CDCl3. The tube was then placed in the NMR probe, and the progress of the isomerization was monitored at 60 °C. Quantitative conversion to 1 was confirmed after 2 h.2.4. Reaction of [Ru3(CO)12] with 2-ethynylpyridine
A thf solution (15 mL) of [Ru3(CO)12] (50 mg, 0.078 mmol) and 2-ethynylpyridine (30 mg, 0.29 mmol) was heated to reflux for 30 min. The solvent was removed under reduced pressure and the residue separated by TLC. Elution with cyclohexane/CH2Cl2 (1:2, v/v) developed five bands. The first band was determined to be unreacted [Ru3(CO)12] (trace). The second and fifth bands afforded [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CH}] (5) (12 mg, 21%) as red crystals and [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CHCHC(C5H4N)}] (6) (10 mg, 15%) as orange crystals after recrystallization from n-hexane/CH2Cl2 at 4 °C. The contents of the other bands were too small for complete characterization. Data for 5: anal. calc. for C22H10N2O8 Ru3: C, 36.02; H, 1.37; N, 3.82. Found: C, 36.22; H, 1.42; N, 3.90%. IR (ν(CO), CH2Cl2): 2068m, 2039vs, 2014s, 1998m, 1965w, 1937w, 1792w, 1719w cm−1. 1H NMR (CD2Cl2): δ 9.49 (d, J 2.4 Hz, 1H), 8.76 (d, J 5.2 Hz, 1H), 8.62 (d, J 5.2 Hz, 1H), 7.77 (t, J 8.0 Hz, 2H), 7.59 (dd, J 12.0, 8.0 Hz, 2H), 7.29 (t, J 6.0 Hz, 1H), 7.21 (t, J 6.0 Hz, 1H), 6.96 (d, J 2.4 Hz, 1H). Data for 6: anal. calc. for C29H15N3O8Ru3: C, 41.63; H, 1.81; N, 5.02. Found: C, 41.88; H, 1.89; N, 5.09%. IR (ν(CO), CH2Cl2): 2091m, 2056vs, 2023m, 1996m, 1985m, 1951w, 1888w cm−1. 1H NMR (CD2Cl2): δ 8.58 (d, J 4.0 Hz, 1H), 8.49 (d, J 4.0 Hz, 1H), 8.12 (m, 2H), 7.85 (m, 1H), 7.77 (m, 2H), 7.68 (m, 2H), 7.61 (m, 1H), 7.23 (t, J 6.0 Hz, 1H), 7.14 (t, J 6.0 Hz, 1H), 6.95 (d, J 4.0 Hz, 1H), 6.75 (t, J 6.0 Hz, 1H), 4.57 (d, J 3.6, 1H).
2.5. Reaction of [Ru3(CO)10(NCMe)2] with 2-ethynylpyridine
To an MeCN solution (20 mL) of [Ru3(CO)10(NCMe)2] (0.15 g, 0.23 mmol) was added 2-ethynylpyridine (47 mg, 0.46 mmol) at −78 °C and the reaction mixture was allowed to warm to room temperature. The mixture was further stirred at room temperature for 30 min. The solvent was removed under reduced pressure and the residue chromatographed by TLC on silica gel. Elution with cyclohexane/CH2Cl2 (1:1, v/v) developed two bands. The faster-moving band was unreacted [Ru3(CO)10(NCMe)2] (trace), while the slower-moving band afforded 5 (50 mg, 30%).
2.6. Crystal structure determinations
Single crystals of 1–3, 5, and 6 suitable for X-ray diffraction analysis were grown by slow diffusion of n-hexane into a CH2Cl2 solution containing each product. Suitable crystals of 1 and 2 were mounted on an Agilent Super Nova dual diffractometer (Agilent Technologies Inc., Santa Clara, CA) using a Nylon loop and Paratone oil and the diffraction data were collected at 150(1) K using Mo-Kα radiation (λ = 0.71073). Unit cell determination, data reduction, and absorption corrections were carried out using CrysAlisPro.23 The structures were solved either with the Superflip24 structure solution program using Charge Flipping (for 1) or with the ShelXS25 structure solution program by direct methods (for 2) and refined by full-matrix least-squares based on F2 using ShelXL26 within the OLEX2 (ref. 27) graphical user interface. Suitable crystals of 3, 5, and 6 were mounted on a Bruker APEX-III CCD diffractometer using nylon loop and Paratone oil, and the diffraction data were collected at 193(1) K using Mo-Kα radiation (λ = 0.71073). Unit cell determination, data reduction, and absorption corrections were carried out using SAINT software.28 The structures were solved with the ShelXS25 structure solution program by direct methods and refined by full-matrix least-squares based on F2 using ShelXL26 within the OLEX2 (ref. 27) graphical user interface. All non-hydrogen atoms were anisotropically refined, while the hydrogen atoms (except those directly bonded to metals) were included using a riding model. The asymmetric unit of 3 also contains part of a CH2Cl2 molecule whose carbon atom is disordered and is refined over two sites using 50% occupancy. Pertinent crystallographic parameters are given in Table 1.| Compound | 1 | 2 | 3 | 5 | 6 |
| CCDC | 2009586 | 2009587 | 2009588 | 2009591 | 2009592 |
| Empirical formula | C17H6NO10Os3 | C16H6NO9Os3 | C17·5H5ClNO11Os3 | C22H10N2O8Ru3 | C29H15N3O8Ru3 |
| Formula weight | 954.83 | 926.82 | 1011.27 | 733.53 | 836.65 |
| Temperature (K) | 150(1) | 150(1) | 193(1) | 193(1) | 193(1) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | C2/c | P |
P |
|
|||||
| Unit cell dimensions | |||||
| a (Å) | 8.7108(4) | 8.1249(2) | 17.575(4) | 8.187(5) | 11.114(18) |
| b (Å) | 9.1324(4) | 15.2910(4) | 8.329(2) | 9.641(6) | 11.221(17) |
| c (Å) | 14.1005(6) | 15.3121(4) | 30.267(6) | 15.139(9) | 13.16(2) |
| α (°) | 100.068(4) | 100.068(4) | 90 | 97.65(3) | 75.91(5) |
| β (°) | 92.632(4) | 91.697(2) | 96.45(3) | 94.716(17) | 73.55(6) |
| γ (°) | 111.204(4) | 111.204(4) | 90 | 106.30(2) | 61.99(9) |
| Volume (Å3) | 1022.35(8) | 1901.52(8) | 4402.6(17) | 1127.7(12) | 1377(4) |
| Z | 2 | 4 | 8 | 2 | 2 |
| Density (calculated) (Mg m−3) | 3.102 | 3.237 | 3.051 | 2.160 | 2.017 |
| Absorption coefficient (mm−1) | 18.652 | 20.047 | 17.455 | 2.036 | 1.682 |
| F(000) | 846 | 1636 | 3600 | 704 | 812 |
| Crystal size (mm3) | 0.28 × 0.16 × 0.10 | 0.18 × 0.16 × 0.08 | 0.12 × 0.11 × 0.04 | 0.12 × 0.08 × 0.03 | 0.25 × 0.20 × 0.04 |
| 2θ range for data collection (°) | 6.854 to 51.998 | 6.336 to 53.996 | 4.664 to 54.482 | 4.462 to 54.31 | 4.472 to 54.436 |
| Reflections collected | 15181 |
30729 |
32329 |
32047 |
31001 |
| Independent reflections [Rint] | 4006 [Rint = 0.0553] | 4142 [Rint = 0.0623] | 4927 [Rint = 0.0428] | 5010 [Rint = 0.0468] | 6101 [Rint = 0.0720] |
| Data/restraints/parameters | 4006/0/281 | 4142/0/270 | 4927/0/312 | 5010/0/316 | 6101/0/388 |
| Goodness-of-fit on F2 | 1.017 | 1.103 | 1.135 | 1.039 | 1.052 |
| Final R indices [I > 2σ(I)] | R1 = 0.0269, wR2 = 0.0623 | R1 = 0.0211, wR2 = 0.0459 | R1 = 0.0261, wR2 = 0.0534 | R1 = 0.0323, wR2 = 0.0775 | R1 = 0.0687, wR2 = 0.1619 |
| R indices (all data) | R1 = 0.0294, wR2 = 0.0641 | R1 = 0.0239, wR2 = 0.0471 | R1 = 0.0318, wR2 = 0.0551 | R1 = 0.0497, wR2 = 0.0854 | R1 = 0.1010, wR2 = 0.1872 |
| Largest diff. peak and hole (e Å−3) | 2.26/−1.46 | 1.06/−1.50 | 1.22/−0.99 | 1.24/−0.83 | 3.27/−1.45 |
2.7. Computational methodology
All calculations were performed with the hybrid meta exchange-correlation functional M06,29 as implemented by the Gaussian 09 program package.30 The osmium and ruthenium atoms were described by Stuttgart–Dresden effective core potentials (ECP) and an SDD basis set,31 while a 6-31G(d′) basis set was employed for the remaining atoms.32 All optimizations were performed using an ultrafine grid and included Grimme's dispersion correction.33The reported geometries represent fully optimized ground states (positive eigenvalues) based on the Hessian matrix, and the natural charges (Q) and Wiberg bond indices were computed using Weinhold's natural bond orbital (NBO) program (version 3.1).34,35 The geometry-optimized structures presented here have been drawn with the JIMP2 molecular visualization and manipulation program.36
3. Results and discussion
3.1. Reaction of [H2Os3(CO)10] with 2-ethynylpyridine: formation of metalated alkenyl clusters
The reaction between [H2Os3(CO)10] and a slight excess of 2-ethynylpyridine proceeds rapidly at room temperature with the parent cluster fully consumed in ca. 10 min as confirmed by 1H NMR spectroscopy, which revealed the presence of new hydride resonances at δ −9.82, −15.79, −17.72, and −18.78. The four products were separated by preparative TLC, and, to our surprise, only three of the four products that were isolated exhibited 1H NMR data in agreement with the initial reaction mixture. The product hydride at δ −15.79 is unstable and transforms into a new product (band 3) during chromatographic separation. This premise was corroborated by 1H NMR control experiments that established the stability of the four initial products at room temperature over several days. The products unaffected by chromatography correspond to the first, second, and fourth bands on the TLC plate. Recrystallization attempts to isolate the hydride product associated with the resonance at δ −15.79 were not successful, and we employed chromatography to separate this product from the initial mixture. The products isolated by preparative TLC, in order of elution, were established as [HOs3(CO)10(μ-C5H4NCHCH)] (1), [HOs3(CO)9(μ3-C5H4NCCH2)] (2), [HOs3(CO)9(μ3-C5H4NCCCO2)] (3), and [HOs3(CO)10(μ-CHCHC5H4N)] (4) in yields of 25, 15, 30, and 20%, respectively (Scheme 2). We repeated the reaction depicted in Scheme 2 several times and confirmed that the distribution of the cluster products is unaffected when the cluster:alkyne ratio was changed from 1:2 to 1:10. Clusters 1 and 4 represent the two products previously described in the LHD report.11 Performing the reaction at 50 °C in n-hexane furnished the same four initial products, but the yield of 4 was greatly reduced. The lability of purified 4 was investigated under comparable conditions and was found to be unstable, transforming to 1 after 2 h at 60 °C. These data indicate that 4 is the product of kinetic substitution with 1 representing the thermodynamically preferred isomer, a feature that was confirmed by DFT calculations (vide infra).
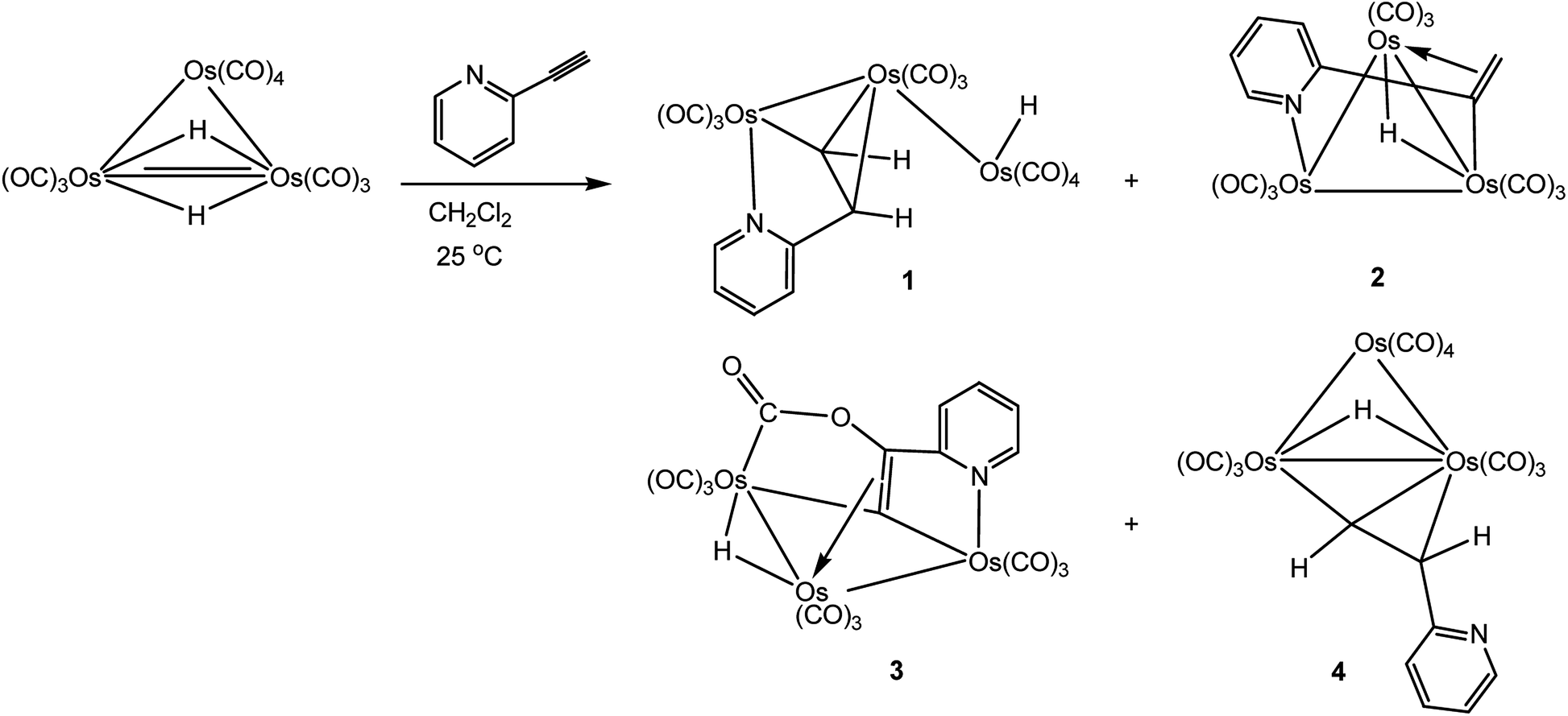 | ||
| Scheme 2 Products 1–4 isolated from the reaction of [H2Os3(CO)10] with 2-ethynylpyridine. | ||
The identity of clusters 1–4 was examined spectroscopically by IR and NMR, and the molecular structures for clusters 1–3 were established by X-ray crystallography. Our product 1 corresponds to the minor product in the LHD report, which incidentally is also labeled as 1 in the original report.11 Given the quality of earlier diffraction data collected for 1 (R = 0.1001), we collected a new data set for 1 and re-determined its solid-state structure (Fig. 1). Surprisingly, our structure is polymorphic to the original LHD structure. As with the original structure, we were unable to locate the terminal hydride ligand on the “spike” Os(CO)4 moiety. The location of the terminal hydride, while not established in the original report containing 1, is supported by the ligand distribution at the Os(CO)4 center in 1. Moreover, the solid-state structure of the related PMe2Ph-substituted derivative [HOs3(CO)9(PMe2Ph)(μ-C5H4NCHCH)], whose hydride shares a common site at the pendent osmium center as 1, was located during data reduction and verified at the “free” site at the Os(CO)3P center.11 Generally speaking, bridging hydrides are preferred at polynuclear clusters.37 The preference of a terminal versus an edge-bridging hydride in 1 was computationally verified, as discussed below.
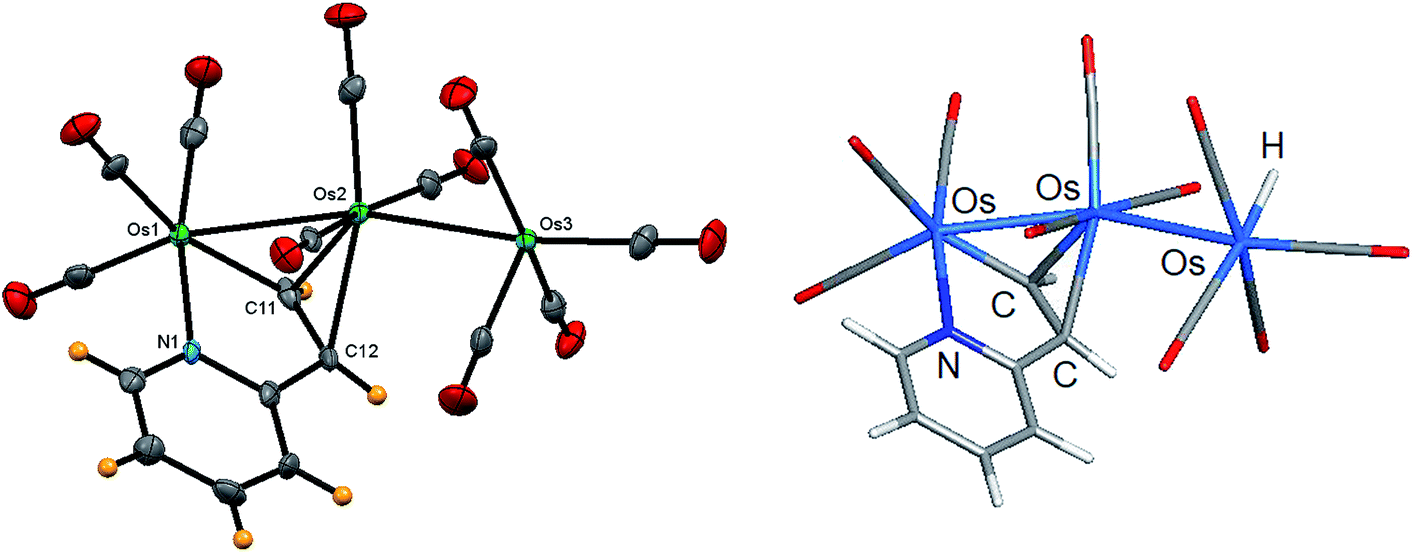 | ||
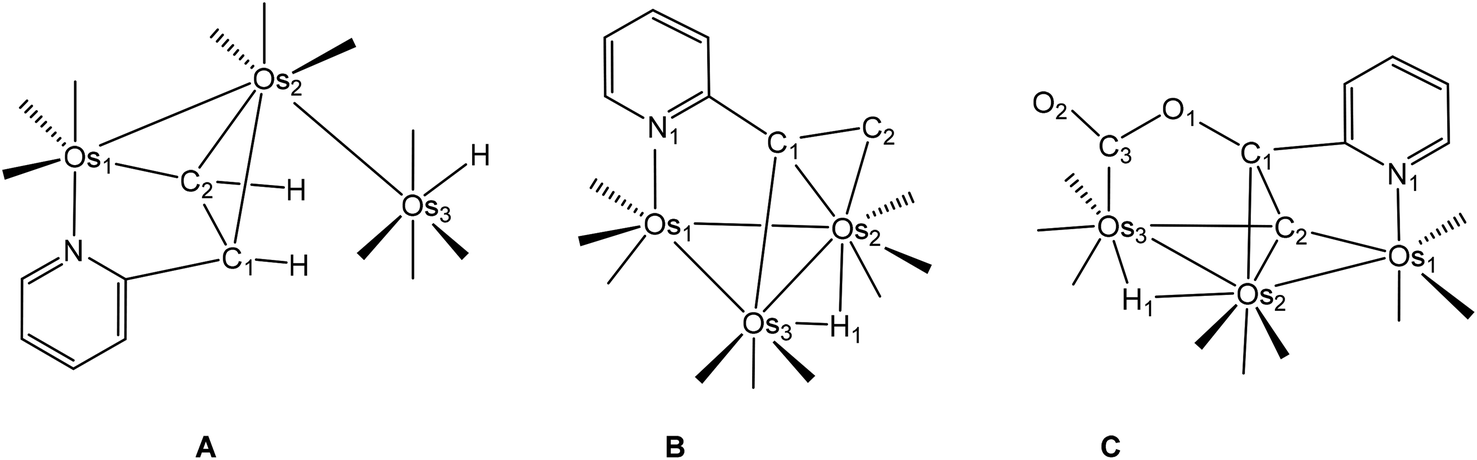
| Fig. 1 Solid-state molecular structure of [HOs3(CO)10(μ-C5H4NCHCH)] (1, left) showing 50% probability atomic displacement ellipsoids and DFT-optimized structure of A (right). Selected bond lengths (Å) and bond angles (°) for 1: Os(1)–Os(2) 2.8248(4), Os(2)–Os(3) 2.8945(4), Os(1)–N(1) 2.144(5), Os(1)–C(11) 2.048(6), Os(2)–C(11) 2.223(6), Os(2)–C(12) 2.346(6), C(11)–C(12) 1.437(9), Os(1)–Os(2)–Os(3) 159.593(12), N(1)–Os(1)–Os(2) 83.17(13), N(1)–Os(1)–C(11) 78.1(2), Os(1)–C(11)–Os(2) 82.7(2), C(11)–Os(2)–C(12) 36.5(2). | ||
Since the gross structural features of the two structures for 1 are similar, we will limit our structural discussion to a few highlights. The near-linear Os(1)–Os(2)–Os(3) linkage [159.593(12)°] confirms the polyhedral expansion of the metallic core and is a common phenomenon in trimetallic clusters with a 50e count.38 The C5H4NCHCH ligand bridges the Os(1)–Os(2) edge regiospecifically by μ-C,N coordination with the nitrogen occupying a terminal position on the metallic frame. The metalated C(11) atom and the N(1) donor are located syn to each other at the six-coordinate Os(1) center. The ethenyl moiety produced from the alkyne/OsH addition process functions as a π donor to Os(2) as defined by the C(11)–Os(2) and C(12)–Os(2) bonds, whose mean distance of 2.285 Å is >0.23 Å longer than the Os(1)–C(11) σ bond [2.084(6) Å]. These structural features are typical of clusters containing π,σ-donating ligands. The metalated ethenylpyridine ligand in 1 donates no electrons to the dangling Os(3) center, which also contains a terminal hydride ligand that resides at the “empty” coordination site trans to the O(8)–C(8)–Os(3) linkage. The two Os–Os vectors exhibit a mean distance of 2.8597 Å, with the bridged Os(1)–Os(2) bond [2.8248(4) Å] slightly shorter than the non-bridged Os(2)–Os(3) edge [2.8945(4) Å]. The terminal hydride was not located during data refinement, but its location at the Os(3) atom is supported by the arrangement of carbonyls about the Os(3) center.
The presence of the expected hydride at the Os(3) atom was verified by DFT calculations with the computed structure for A depicted in Fig. 1 alongside the experimental structure. Excellent agreement exists between the experimental and the computed structures. The preference for a terminal hydride in A versus a bridging hydride at the adjacent Os–Os vector was also examined by DFT through a series of step-scan calculations. Reducing the initial Os–Os–H angle of 83° in species A to 25°, well past the typical angle for an edge-bridging hydride of ca. 37°, led to an increase in total energy with no sign of a stable stationary point. All attempts to optimize a structure with a bridging hydride collapsed to species A, and we confidently estimate an energy difference of ≥13 kcal mol−1 in favor of the isomer with a terminal hydride. The natural charges (Q) and Wiberg bond indices were also examined, and these data are reported in Table 2. All three osmium atoms exhibit a negative charge that ranges from −0.96 [Os(1)] to −1.55 [Os(3)], as do the alkenyl carbons C(1) [–0.30] and C(2) [–0.24] and the pyridine N(1) [–0.42] atom. The charge on the terminal H(1) atom is 0.15 and is similar in magnitude to the edge-bridging hydride ligand in species B (cluster 2) and C (cluster 3). The two Os–Os vectors exhibit a mean Wiberg bond index of 0.35, consistent with their single-bond designation, and the metalated alkenyl moiety displays Wiberg indices of 0.46 and 0.47 for the Os(2)–C(1) and Os(2)–C(2) bonds that are a factor of two weaker than the σ-bonding component defined by the Os(1)–C(2) vector (0.87). Finally, the WBI for the Os(3)–H(1) bond is 0.45.
|
|||
|---|---|---|---|
| Natural charges (Q) | A | B | C |
| a Atom numbering for the participant atoms follows the structures depicted below the table caption. | |||
| Os1 | −0.96 | −1.01 | −0.96 |
| Os2 | −1.18 | −1.24 | −1.13 |
| Os3 | −1.55 | −1.21 | −1.29 |
| N1 | −0.42 | −0.40 | −0.41 |
| C1 | −0.30 | −0.14 | 0.23 |
| C2 | −0.24 | −0.39 | −0.15 |
| C3 | 0.75 | ||
| H1 | 0.15 | 0.14 | 0.13 |
| WBI | A | B | C |
|---|---|---|---|
| Os1–Os2 | 0.38 | 0.42 | 0.37 |
| Os1–Os3 | 0.48 | ||
| Os2–Os3 | 0.31 | 0.30 | 0.23 |
| Os1–N1 | 0.52 | 0.48 | 0.52 |
| Os1–C2 | 0.87 | 0.81 | |
| Os2–C1 | 0.46 | 0.43 | 0.42 |
| Os2–C2 | 0.47 | 0.44 | 0.50 |
| Os3–C1 | 0.67 | ||
| Os3–C2 | 0.74 | ||
| Os3–C3 | 0.77 | ||
| O1–C1 | 0.92 | ||
| O1–C3 | 0.89 | ||
| O2–C3 | 1.81 | ||
| Os1–H1 | |||
| Os2–H1 | 0.42 | 0.23 | |
| Os3–H1 | 0.45 | 0.35 | 0.41 |
Whereas the formation of 1 arises from an anti-Markovnikov Os–H/alkyne addition process, cluster 2 derives from a Markovnikov insertion product where the hydride adds to the terminal alkyne carbon to produce a methylene group. The molecular structure of 2 was established by X-ray crystallography, and the solid-state structure is shown in Fig. 2. The cluster core contains a closed osmium triangle where the Os–Os bond distances range from 2.7913(3) Å [Os(1)–Os(3)] to 2.8581(2) Å [Os(2)–Os(3)] with a mean distance of 2.8357 Å. Each osmium is bound to three carbonyls, and the 5e donor C5H4NCCH2 caps a metallic face through the N(1), C(15), and C(16) atoms. The Os(2) and Os(3) centers bind the latter two atoms of the ligand in a traditional σ,π-vinyl fashion, with the Os(3)–C(15) bond representing the σ component of the metalated alkenyl moiety. The longer Os(2)–C(15) [2.242(4) Å] and Os(2)–C(16) [2.306(5) Å] bonds represent the π component of this ligand, whose Os–C distances are comparable to those bond distances reported in related clusters.11,18,20 The hydride was not located during data reduction but was assumed to span the Os(2)–Os(3) edge based on the disposition of CO ligands about the Os–Os bond. The locus for the hydride was subsequently verified by DFT calculations, and the optimized structure is depicted alongside the experimental structure in Fig. 2. The optimized structure of B closely mirrors the experimental structure with the bridging hydride sharing the Os(2)–Os(3) vector bridged by the alkenyl ligand. The hydride is tipped slightly below the metallic plane opposite the polyhedral face capped by the C5H4NCCH2 ligand. The osmium atoms and the Os-bound N(1), C(1), and C(2) atoms of the activated ligand all display a negative charge, while the edge-bridging hydride exhibits a positive charge of 0.14. The computed WBI for the Os–Os bonds in B range from 0.30 [Os(2)–Os(3)] to 0.48 [Os(1)–Os(2)] with a mean index of 0.40. The Os–N and Os–C WBIs in B associated with the capping C5H4NCCH2 ligand parallel those data reported for A. Finally, the Os–H bond indices for the edge-bridging hydride are comparable in magnitude to the WBIs in related hydride clusters investigated by us.39
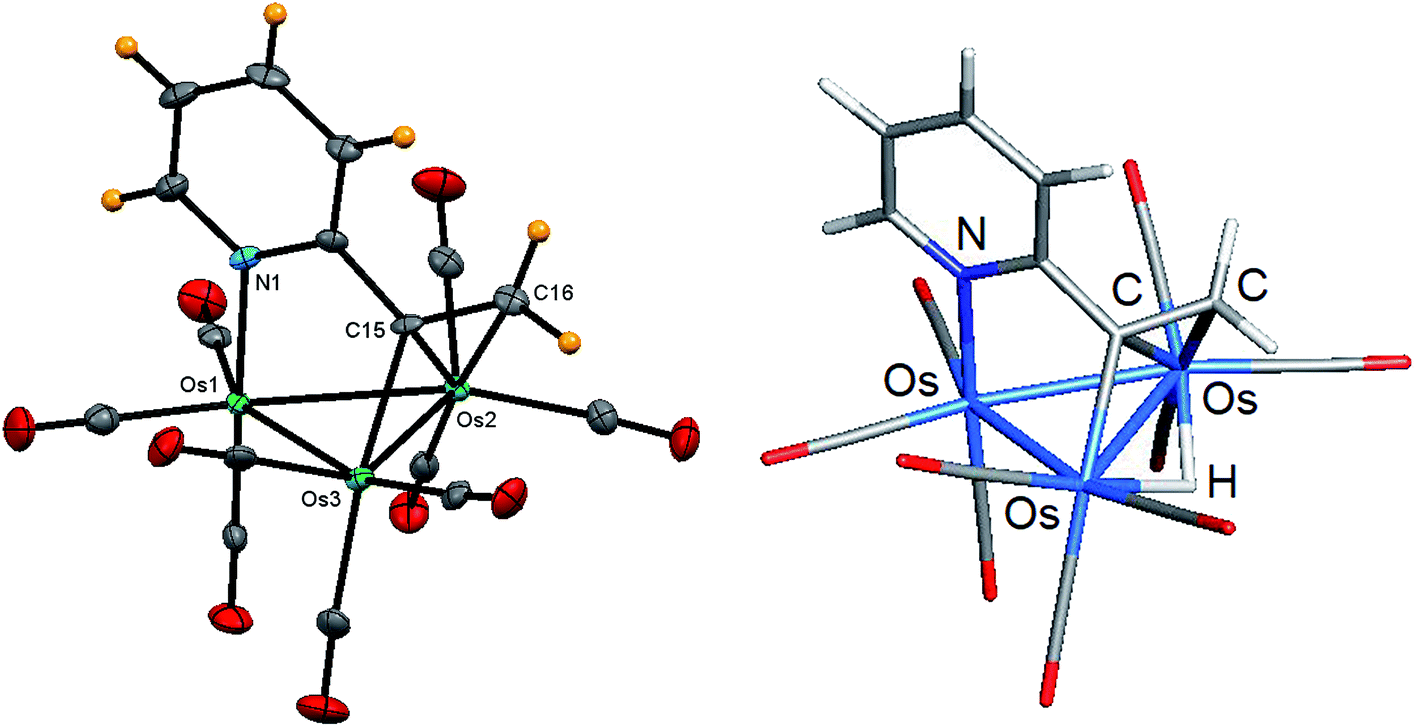 | ||
| Fig. 2 Solid-state molecular structure of [HOs3(CO)9(μ3-C5H4NCCH2)] (2, left) showing 50% probability atomic displacement ellipsoids and DFT-optimized structure of B (right). Selected bond lengths (Å) and bond angles (°) for 2: Os(1)–Os(2) 2.8576(3), Os(2)–Os(3) 2.8581(2), Os(1)–Os(3) 2.7913(3), Os(1)–N(1) 2.162(4), Os(2)–C(15) 2.242(4), Os(2)–C(16) 2.306(5), Os(3)–C(15) 2.122(4), C(15)–C(16) 1.393(7), N(1)–Os(1)–Os(2) 86.03(10), N(1)–Os(1)–Os(3) 85.64(10), C(15)–Os(3)–Os(2) 50.93(12), Os(2)–C(15)–Os(3) 81.78(15), C(15)–Os(2)–C(16) 35.63(17). | ||
The spectroscopic properties of 2 are consistent with the solid-state structure. The 1H NMR spectrum reveals a pair of doublets at δ 8.56 (J 6.0 Hz) and δ 6.92 (J 8.0 Hz) and a pair of multiplets at δ 7.64 and δ 6.98 that are readily ascribed to the ABCD spin system of the pyridyl ring. The two singlets are δ 4.30 and 2.63 are assigned to the hydrogens of the vinyl moiety, with the lower-field singlet confidently assigned to the hydrogen situated syn to the hydride ligand (δ −17.70) based on NOESY 1H NMR experiments.
Recall, one of the four initial products produced in the reaction between [H2Os3(CO)10] and 2-ethynylpyridine is not stable over silica gel, transforming to cluster 3 during chromatographic separation. The hydride signal for the intermediate appears at δ −15.79 and it shifts 0.97 ppm upfield after chromatography. We had to settle on the characterization of this unknown intermediate using isolated 3. NMR analysis of the crude reaction mixture suggests that the silica gel-unstable intermediate does not contain any vinyl hydrogens, leading us to propose the transformation depicted in Scheme 3 as an explanation of the precursor to 3. Here the dimetalated alkenyl moiety of the intermediate undergoes ring expansion through an oxygen capture pathway that is assisted by an ancillary CO ligand at the Os(CO)4 center. No change in the initial product mixture was observed over several hours when exposed to oxygen, suggesting that the stationary support serves as the source of oxygen in this reaction.
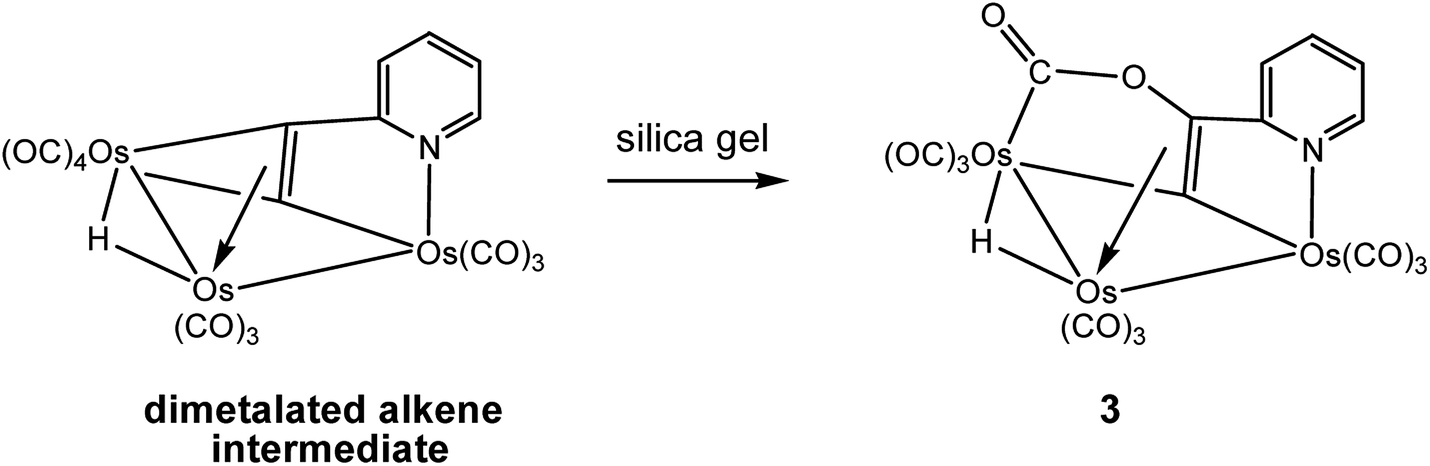 | ||
| Scheme 3 SiO2-promoted conversion of the proposed alkenyl-substituted intermediate to 3. | ||
Fig. 3 shows the solid-state molecular structure of 3 with selected bond distances and angles reported in the caption. The molecule contains 50e, assuming the face-capping C5H4NCCCO2 ligand functions as a 7e donor, and the opened triosmium core is in concert with the overall electron count.38 The Os(1)–Os(2) [2.8464(9) Å] and Os(2)–Os(3) [2.9561(10) Å] bond distances are consistent with their single-bond designation and the Os–Os bond distances in clusters 1 and 2. Each metal center contains three terminal CO ligands that are situated at mutually cis sites to furnish Os(CO)3 units possessing one axial and two equatorial CO groups. The hydride, which was located crystallographically, spans the Os(2)–Os(3) edge, which is significantly longer (ca. 0.1 Å) than the non-hydride bridged Os(1)–Os(2) vector. The C5H4NCCCO2 ligand may be viewed as a doubly metalated σ,σ,π-alkenyl ligand with the Os(1)–C(11) and Os(3)–C(11) bonds corresponding to the σ components and the π bond represented by the Os(2)–C(11) and Os(2)–C(12) bonds. The mean distance of 2.078 Å for the former two Os–C bonds is ca. 0.16 Å shorter than the mean distance displayed by the latter two Os–C(π) bonds of the alkenyl linkage. The carboxylate ligand, which is defined by the C(10), O(10), and O(11) atoms, exhibits bond distances and angles that are unremarkable and require no comment. The presence of the carboxylate group in 3 was supported by IR spectroscopy based on a low-energy ν(CO) band at 1693 cm−1. The optimized structure of C is depicted alongside the experimental structure, and excellent agreement between the two structures is noted. Apart from the positive charge of 0.23 computed for the C(1) atom of alkenyl ligand, whose adjacent O(1) atom likely serves to withdraw electron density for the metalated carbon center, the charges and WBIs for C mirror the data reported for species A and B.
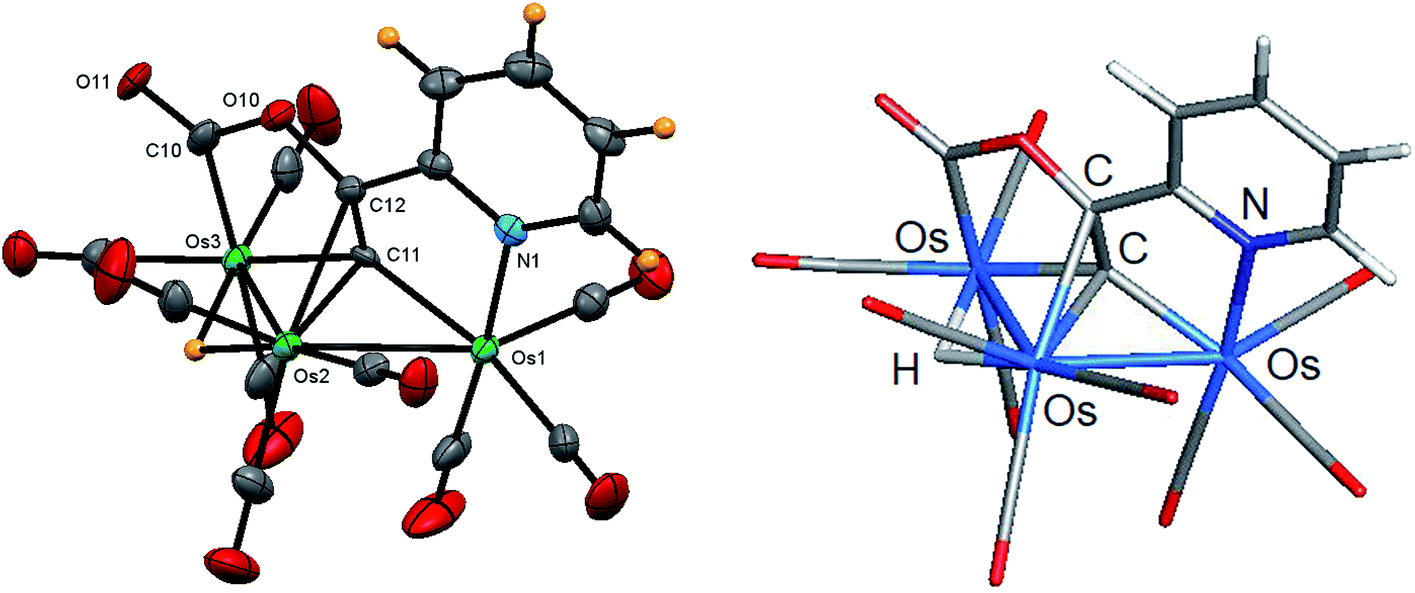 | ||
| Fig. 3 Solid-state molecular structure of [HOs3(CO)9(μ3-C5H4NCCCO2)] (3, left) showing 50% probability atomic displacement ellipsoids and DFT-optimized structure of C (right). Selected bond lengths (Å) and bond angles (°) for 3: Os(1)–Os(2) 2.8464(9), Os(2)–Os(3) 2.9561(10), Os(1)–N(1) 2.137(5), Os(1)–C(11) 2.076(5), Os(2)–C(11) 2.171(5), Os(2)–C(12) 2.301(6), Os(3)–C(10) 2.101(6), Os(3)–C(11) 2.079(5), C(11)–C(12) 1.410(8), Os(1)–Os(2)–Os(3) 83.83(2), N(1)–Os(1)–Os(2) 84.82(13), C(10)–Os(3)–Os(2) 80.57(15), C(11)–Os(2)–C(12) 36.6(2). | ||
The slowest moving band isolated from the TLC plate was confirmed as cluster 4, and this material corresponds to the major product reported in the LHD study.11 The initial identity of this product was formulated based on the solution spectroscopic data. The 1H NMR spectrum supported a product with a single hydride and trans alkenyl moiety while the IR spectrum was consistent with a triangular cluster having ten terminal CO ligands. These data supported a product involving the addition of the Os–H bond to the alkyne triple bond. The ancillary pyridyl ligand remains free and functions as a non-coordinated spectator ligand. Accordingly, the product was formulated as possessing a structure similar to the known vinyl cluster [HOs3(CO)10(μ,η2-CHCH2)],40 as depicted by the major product in Scheme 1.
As with the earlier LHD study, we were unable to grow X-ray quality crystals of this product and could not unequivocally establish its molecular structure. Accordingly, we examined the LHD-proposed structure of 4 (species A_alt) by electronic structure calculations [Fig. 4]. Both the hydride and alkenyl moiety share the same edge of the Os3 polyhedron. Species A and A_alt are isomers, and the former is computed to be 8.9 kcal mol−1 (ΔG) more stable. Independent control experiments confirmed 4 (kinetic isomer) as the precursor to 1 (thermodynamic isomer). Repeating the reaction between [H2Os3(CO)10] and 2-ethynylpyridine in refluxing hexane furnished cluster 1 at the expense of cluster 4. The yields of 2 and 3 remained unchanged. Heating pure 4 in CDCl3 in a sealed NMR tube at 60 °C led to the conversion to 1 after 2 h, confirming cluster 1 as the thermodynamically preferred isomer. We also examined the transformation of 4 → 1 at 100 °C in toluene-d8, but the reaction was accompanied by visible cluster decomposition. Subsequent studies confirmed that cluster 1 is unstable at 100 °C.
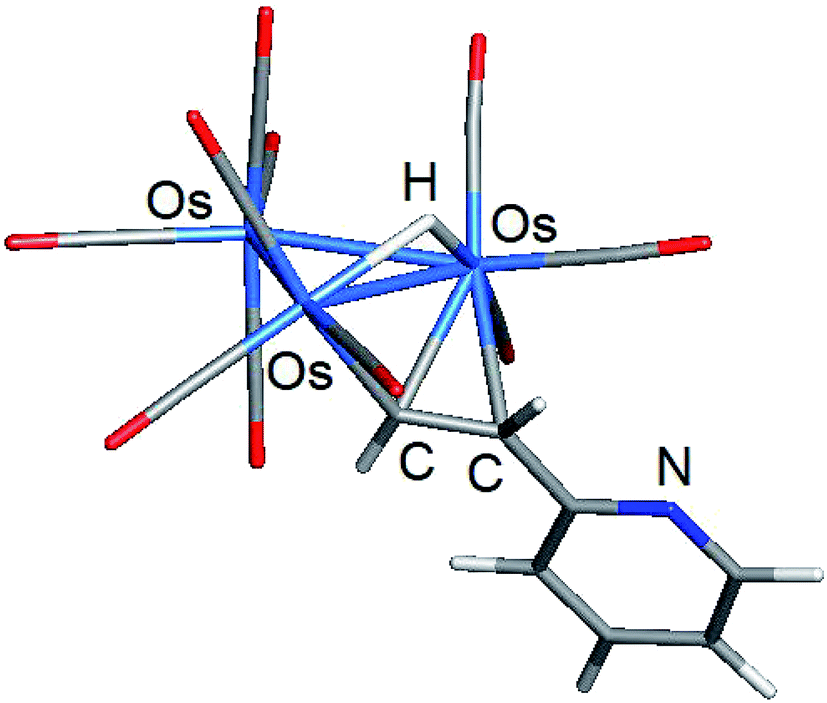 | ||
| Fig. 4 DFT-optimized structure of major product A_alt from the room temperature reaction of [H2Os3(CO)10] and 2-ethynylpyridine. | ||
3.2. Reactions of [Ru3(CO)12−n(NCMe)n] (n = 0, 2) with 2-ethynylpyridine: C–C bond formation
The reaction between [Ru3(CO)12] and 2-ethynylpyridine follows a different pathway compared to the reactivity described for [H2Os3(CO)10]. The dominant manifolds in the reaction of [Os3(CO)10(μ-H)2] with 2-ethynylpyridine involve hydride transfer to the alkyne moiety and C–H bond activation of the alkyne functionality. The reaction of [Ru3(CO)12] with excess 2-ethynylpyridine is dominated by C–C bond coupling of the alkyne moiety of 2-ethynylpyridine to furnish [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CH}] (5) and [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CHCHC(C5H4N)}] (6) in 21 and 15% yield, respectively (Scheme 4). Cluster 5 does not serve as a precursor to 6 since control experiments in refluxing thf confirmed that 5 was inert to further 2-ethynylpyridine insertion. In an attempt to isolate a ruthenium cluster with a single metalated 2-ethynylpyridine ligand, we also treated [Ru3(CO)10(NCMe)2] with two equivalents of 2-ethynylpyridine at ambient temperature, but this reaction afforded only 5 in 30% yield. The two products were separated by chromatography and characterized by solution methods (IR and NMR) and each molecular structure established by X-ray crystallography.
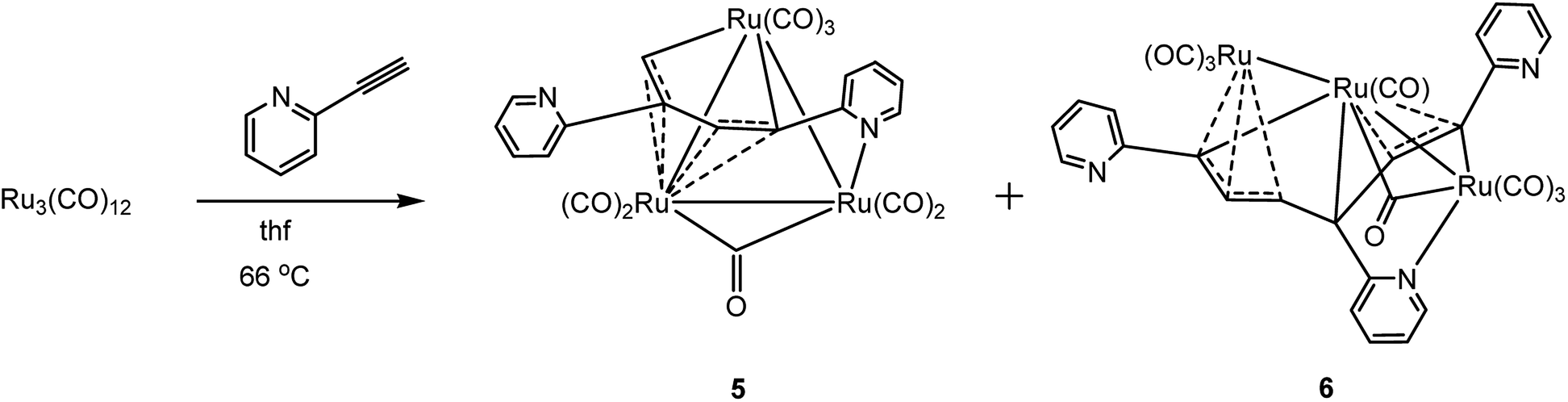 | ||
| Scheme 4 Reaction of [Ru3(CO)12] with 2-ethynylpyridine. | ||
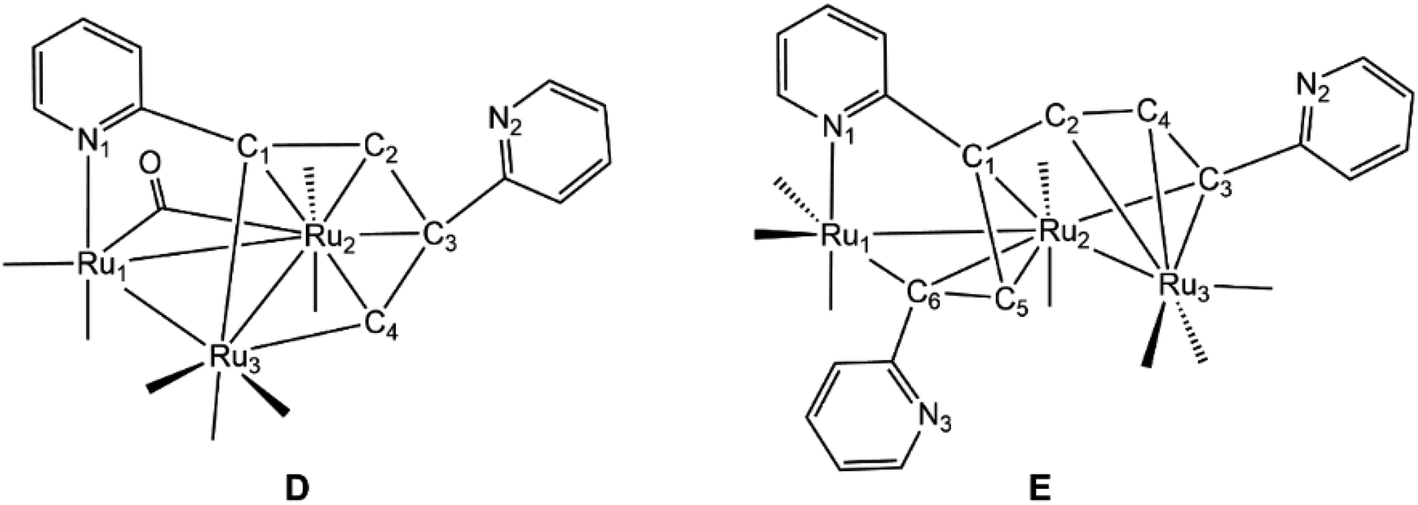
The solid-state molecular structure of 5 is depicted in Fig. 5 along with selected bond distances and angles contained in the figure caption. The triangular Ru3 cluster is ligated by seven terminal carbonyls, an edge-bridging carbonyl, and a μ3-C5H4NCCHC(C5H4N)CH ligand. The Ru–Ru distances range between 2.7868(15) and 2.8747(15) Å with the Ru(2)–Ru(3) edge, which accommodates the bridging carbonyl ligand, being the longest of the three Ru–Ru bonds. The C5H4NCCHC(C5H4N)CH ligand, which is formed via head-to-tail C–C coupling of the ethynyl moieties, functions as an 8e donor ligand that is best viewed as a dimetalated 2-pyridyl-substituted butadiene ligand. The Ru(1)–C(9) and Ru(1)–C(12) vectors, which represent the two metalated bonds associated with the ruthenacyclopentadiene ring defined by the Ru(1)–C(9)–C(10)–C(11)–C(12) atoms, display a mean distance of 2.074 Å. This ligand donates an additional four electrons to the Ru(2) center via the butadiene component of the ruthenacyclopentadiene ring and two electrons through coordination of the pyridyl N(1) moiety to the Ru(3) center. The Ru(3)–N(1) [2.150(4) Å] bond distance and the Ru–C bond distances for the coordinated butadiene moiety [Ru(1)–C(9) 2.081(4), Ru(1)–C(12) 2.066(4), Ru(2)–C(9) 2.279(4), Ru(2)–C(10) 2.241(4), Ru(2)–C(11) 2.250(4), Ru(2)–C(12) 2.233(4)Å] are similar to those distances found in related clusters.17,20 The cluster is electronically saturated based on an electron count of 48e, and the optimized structure of D (Fig. 5) mirrors the experimental structure. Table 3 reports the natural charges and Wiberg bond indices for D. The three rutheniums, the two nitrogens, and the coordinated carbon atoms of the butadiene moiety of the μ3-C5H4NCCHC(C5H4N)CH ligand all exhibit a negative charge. The metalated Ru(3)–C(1) and Ru(3)–C(4) bonds of the ligand display a mean WBI of 0.72 that is twofold stronger than the mean WBI index of 0.36 for the Ru(2)–C(π) bonds involving C(1)–C(4) atoms of the butadiene moiety. Finally, the solution spectroscopic data for 5 also support the solid-state structure. The IR spectrum exhibits seven carbonyl stretching bands between 2068–1937 cm−1 for the terminal carbonyls along with a low-energy absorption at 1792 cm−1 assigned to the bridging carbonyl ligand. The 1H NMR spectrum displays a series of pyridyl multiplets and alkenyl protons consistent with the solid-state structure.
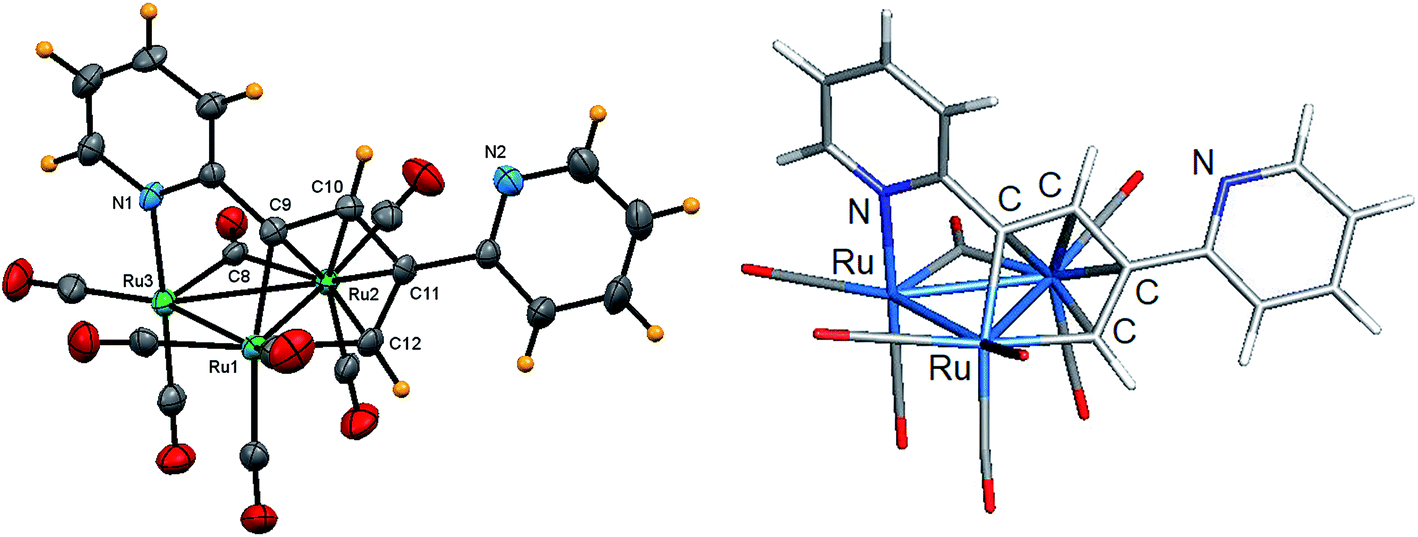 | ||
| Fig. 5 Solid-state molecular structure of [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CH}] (5, left) showing 50% probability atomic displacement ellipsoids and DFT-optimized structure of D (right). Selected bond lengths (Å) and bond angles (°) for 5: Ru(1)–Ru(2) 2.7868(15), Ru(2)–Ru(3) 2.8747(15), Ru(1)–Ru(3) 2.8426(16), Ru(3)–N(1) 2.150(4), Ru(1)–C(9) 2.081(4), Ru(1)–C(12) 2.066(4), Ru(2)–C(9) 2.279(4), Ru(2)–C(10) 2.241(4), Ru(2)–C(11) 2.250(4), Ru(2)–C(12) 2.233(4), Ru(2)–C(8) 2.137(4), Ru(3)–C(8) 1.980(4), C(9)–C(10) 1.391(5), C(10)–C(11) 1.450(5), C(11)–C(12) 1.398(6), N(1)–Ru(3)–Ru(1) 84.58(9), N(1)–Ru(3)–Ru(2) 89.28(9), Ru(2)–C(8)–Ru(3) 88.49(16). | ||
|
||
|---|---|---|
| Natural charges (Q) | D | E |
| a Atom numbering for the participant atoms follows the structures depicted below the table caption. | ||
| Ru1 | −0.85 | −1.09 |
| Ru2 | −0.98 | −0.84 |
| Ru3 | −1.27 | −1.11 |
| N1 | −0.41 | −0.41 |
| N2 | −0.49 | −0.48 |
| N3 | −0.48 | |
| C1 | −0.07 | −0.16 |
| C2 | −0.16 | −0.27 |
| C3 | −0.04 | −0.06 |
| C4 | −0.18 | −0.12 |
| C5 | −0.12 | |
| C6 | −0.09 | |
| WBI | D | E |
|---|---|---|
| Ru1–Ru2 | 0.29 | 0.32 |
| Ru1–Ru3 | 0.24 | |
| Ru2–Ru3 | 0.30 | 0.40 |
| Ru1–N1 | 0.43 | 0.47 |
| Ru1–C6 | 0.71 | |
| Ru2–C1 | 0.38 | 0.48 |
| Ru2–C2 | 0.31 | |
| Ru2–C3 | 0.28 | 0.65 |
| Ru2–C4 | 0.41 | |
| Ru2–C5 | 0.22 | |
| Ru2–C6 | 0.37 | |
| Ru3–C1 | 0.68 | |
| Ru3–C2 | 0.50 | |
| Ru3–C3 | 0.53 | |
| Ru3–C4 | 0.75 | 0.30 |
Fig. 6 shows the solid-state molecular structure of 6 and the caption contains selected bond distances and angles. The cluster contains 50-valence electrons and exhibits an expanded triruthenium core that possesses two formal Ru–Ru bonds. The coordination sphere of 6 contains eight carbonyl groups and a C5H4NCCHC(C5H4N)CHCHC(C5H4N) ligand, the latter which is formed via alkyne C–C bond coupling of three 2-ethynylpyridine ligands. The alkyne-based ligand functions as a 10e donor and may be considered as a dimetalated tris(2-pyridyl-substituted) conjugated triene ligand. The two Ru–Ru bonds defined by the Ru(1)–Ru(2) and Ru(1)–Ru(3) vectors exhibit a mean distance of 2.769 Å that is similar in magnitude to the shortest Ru–Ru bond distance observed in 5. Each terminal ruthenium is bound to three carbonyls, while the central ruthenium is bound to two carbonyls, one of which is nominally semibridging in nature based on a bond angle of 159.5(9)° for the Ru(1)–C(1)–O(1) linkage. The dimetalated tris(pyridyl-substituted) triene ligand coordinates all three ruthenium atoms using the alkenyl carbons (σ and π fashion) and one of the pyridyl nitrogens [Ru(3)–N(2)]. The triene moiety displays an altered C–C backbone [C(9)–C(10) 1.417(13), C(10)–C(11) 1.428(13), C(11)–C(12) 1.465(13), C(12)–C(13) 1.480(12), C(13)–C(14) 1.405(14) Å] that effectively transforms the three CC π units into a discrete pair of 3e donating allyl ligands that bind the Ru(1) and Ru(2) atoms. The metalated components of the ligand are represented by the Ru(1)–C(9) and Ru(3)–C(14) vectors, which display a mean bond distance of 2.086 Å. The Ru(3)–N(2) [2.138(9) Å] and Ru–C(triene) [Ru(1)–C(9) 2.093(10), Ru(1)–C(12) 2.146(9), Ru(1)–C(13) 2.270(9), Ru(1)–C(14) 2.291(9), Ru(2)–C(9) 2.154(9), Ru(2)–C(10) 2.189(9), Ru(2)–C(11) 2.249(10), Ru(3)–C(14) 2.079(10) Å] bond distances involving this ligand are similar to the bond distances in 5 and related ruthenium clusters.17,20 The optimized structure of E is shown alongside the crystallographic structure in Fig. 6. The computed charges and bond indices for E parallel the data reported for D.
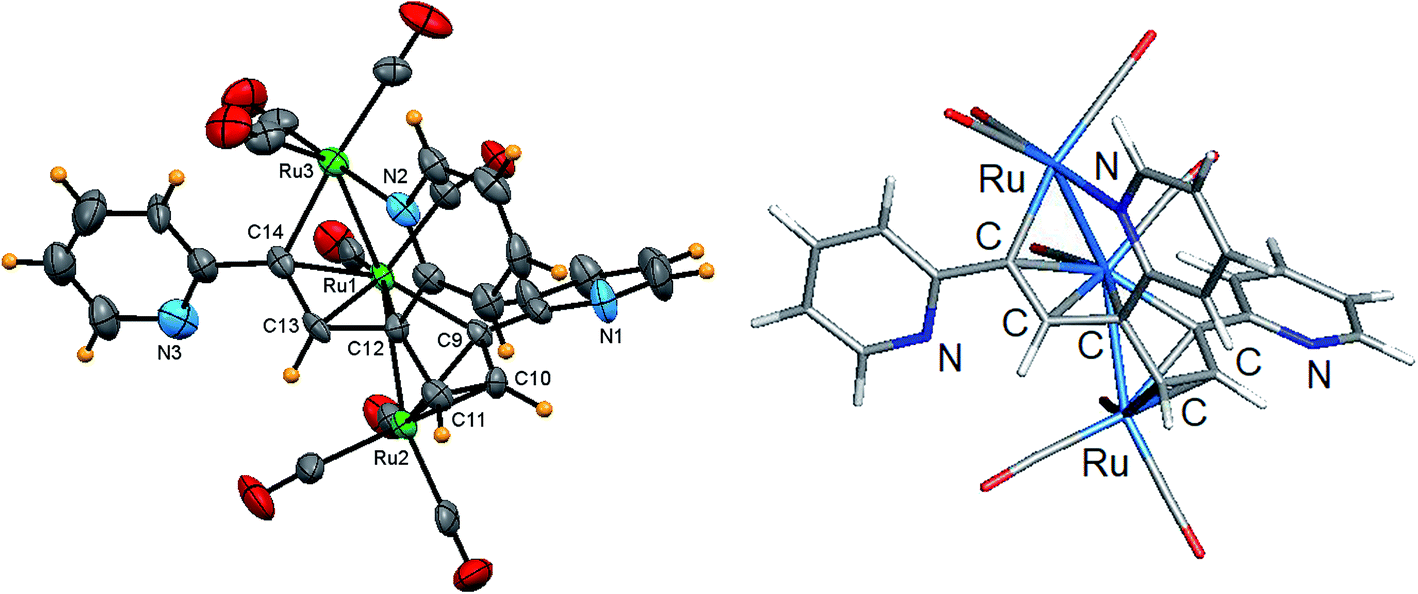 | ||
| Fig. 6 Solid-state molecular structure of [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CHCHC(C5H4N)}] (6, left) showing 50% probability atomic displacement ellipsoids and DFT-optimized structure of E (right). Selected bond lengths (Å) and bond angles (°) for 6: Ru(1)–Ru(2) 2.764(4), Ru(1)–Ru(3) 2.774(3), Ru(3)–N(2) 2.138(9), Ru(1)–C(9) 2.093(10), Ru(1)–C(12) 2.146(9), Ru(1)–C(13) 2.270(9), Ru(1)–C(14) 2.291(9), Ru(2)–C(9) 2.154(9), Ru(2)–C(10) 2.189(9), Ru(2)–C(11) 2.249(10), Ru(3)–C(14) 2.079(10), Ru(1)–C(1) 1.905(10), Ru(3)–C(1) 2.541(10), C(9)–C(10) 1.417(13), C(10)–C(11) 1.428(13), C(11)–C(12) 1.465(13), C(12)–C(13) 1.480(12), C(13)–C(14) 1.405(14), Ru(2)–Ru(1)–Ru(3) 152.05(6), N(2)–Ru(3)–Ru(1) 84.4(2), N(2)–Ru(3)–C(14) 90.1(4), Ru(1)–C(1)–O(1) 159.5(9), Ru(3)–C(1)–O(1) 123.7(7). | ||
4. Conclusions
The reactivity of 2-ethynylpyridine at triosmium and triruthenium centers has been investigated, and we have isolated and characterized four new trimetallic clusters. The reaction of [H2Os3(CO)10] with excess 2-ethynylpyridine furnished the new triosmium clusters [HOs3(CO)9(μ3-C5H4NCCH2)] (2) and [HOs3(CO)9(μ3-C5H4NCCCO2)] (3) along with the previously reported clusters [HOs3(CO)10(μ-C5H4NCHCH)] (1) and [HOs3(CO)10(μ-CHCHC5H4N)] (4).11 Cluster 4 transforms into 1 at elevated temperature, confirming the former as the kinetic product of substitution. Clusters 1, 2, and 4 are apparently formed by the addition of an Os–H bond across CC bond of 2-ethynylpyridine, whereas 3 is formed via C–H bond activation of the alkyne functionality of 2-ethynylpyridine through an unstable intermediate that affords 3 on chromatographic work-up. In contrast, the reaction between [Ru3(CO)12] and 2-ethynylpyridine affords [Ru3(CO)7(μ-CO)(μ3-C5H4NCCHC(C5H4N)CH)] (5) and [Ru3(CO)7(μ-CO){μ3-C5H4NCCHC(C5H4N)CHCHC(C5H4N)}] (6) via C–C coupling of the alkyne moiety of 2-ethynylpyridine at ruthenium centers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Ministry of Science and Technology, the Government of the People's Republic of Bangladesh (SG and SEK) and the Robert A. Welch Foundation (Grant B-1093-MGR) are acknowledged. The DFT calculations were performed at UNT through CASCaM, which is an NSF-supported facility (CHE-1531468).References
- L. Zhang, L. Dang, T. B. Wen, H. H.-Y. Sung, I. D. Williams, Z. Lin and G. Jia, Organometallics, 2007, 26, 2849–2860 CrossRef CAS.
- A. G. Wong-Foy, L. M. Henling, M. Day, J. A. Labinger and J. E. Bercaw, J. Mol. Catal. A: Chem., 2002, 189, 3–16 CrossRef CAS.
- J. Navarro, E. Sola, M. Martín, I. T. Dobrinovitch, F. J. Lahoz and L. A. Oro, Organometallics, 2004, 23, 1908–1917 CrossRef CAS.
- H.-F. Klein, S. Camadanli, R. Beck, D. Leukel and U. Flörke, Angew. Chem., Int. Ed., 2005, 44, 975–977 CrossRef CAS PubMed.
- O. V. Ozerov, M. Pink, L. A. Watson and K. G. Caulton, J. Am. Chem. Soc., 2004, 126, 2105–2113 CrossRef CAS PubMed.
- M. L. Buil, M. A. Esteruelas, E. Goni, M. Oliván and E. Oñate, Organometallics, 2006, 25, 3076–3083 CrossRef CAS.
- J. N. Coalter III, W. E. Streib and K. G. Caulton, Inorg. Chem., 2000, 39, 3749–3756 CrossRef PubMed.
- P. Barrio, M. A. Esteruelas and E. Oñate, Organometallics, 2004, 23, 3627–3639 CrossRef CAS.
- M. A. Esteruelas, F. J. Fernández-Alvarez, M. Oliván and E. Oñate, J. Am. Chem. Soc., 2006, 128, 4596–4597 CrossRef CAS PubMed.
- B. Eguillor, M. A. Esteruelas, M. Oliván and E. Oñate, Organometallics, 2005, 24, 1428–1438 CrossRef CAS.
- K. Burgess, H. D. Holden, B. F. G. Johnson, J. Lewis, M. B. Hursthouse, N. P. C. Walker, A. J. Deeming, P. J. Manning and R. Peters, J. Chem. Soc., Dalton Trans., 1985, 85–90 RSC.
- W.-Y. Wong and W.-T. Wong, J. Organomet. Chem., 1996, 513, 27–29 CrossRef CAS.
- J. P.-K. Lau and W.-T. Wong, Inorg. Chem. Commun., 2003, 6, 174–177 CrossRef CAS.
- S. Chan, W.-Y. Wong and W.-T. Wong, J. Organomet. Chem., 1994, 474, C30–C33 CrossRef CAS.
- W.-Y. Wong, S. Chan and W.-T. Wong, J. Organomet. Chem., 1995, 493, 229–237 CrossRef CAS.
- S. E. Kabir, F. Ahmed, A. Das, M. R. Hassan, D. T. Haworth, S. V. Lindeman, G. M. G. Hossain, T. A. Siddiquee and D. W. Bennett, J. Organomet. Chem., 2008, 693, 1696–1702 CrossRef CAS.
- K. A. Azam, D. W. Bennett, M. R. Hassan, D. T. Haworth, G. Hogarth, S. E. Kabir, S. V. Lindeman, L. Salassa, S. R. Simi and T. A. Siddiquee, Organometallics, 2008, 27, 5163–5166 CrossRef CAS.
- M. R. Al-Mamun, S. Ghosh, S. E. Kabir and G. Hogarth, J. Organomet. Chem., 2017, 849–850, 80–87 CrossRef CAS.
- R. H. Naulty, M. P. Cifuentes, M. G. Humphrey, S. Houbrechts, C. Boutton, A. Persoons, G. A. Heath, D. C. R. Hockless, B. Luther-Davies and M. Samoc, J. Chem. Soc., Dalton Trans., 1997, 4167–4174 RSC.
- M. M. Hossain, N. Akter, S. Ghosh, V. N. Nesterov, M. G. Richmond, G. Hogarth and S. E. Kabir, RSC Adv., 2019, 9, 21025–21030 RSC.
- E. Sappa and M. Valle, Inorg. Synth., 1977, 26, 365–369 Search PubMed.
- G. A. Foulds, B. F. G. Johnson and J. Lewis, J. Organomet. Chem., 1985, 296, 147–153 CrossRef CAS.
- CrysAlisPro; Oxford Diffraction, Yarnton, England, 2015 Search PubMed.
- (a) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; (b) L. Palatinus and A. van der Lee, J. Appl. Crystallogr., 2008, 41, 975–984 CrossRef CAS; (c) L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- SAINT Version 8.37, Bruker AXS, Inc., Madison, WI, 2015 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. J. Frisch et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- (a) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS; (b) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- (a) JIMP2, version 0.091, a free program for the visualization and manipulation of molecules: M. B. Hall and R. F. Fenske, Inorg. Chem., 1972, 11, 768–775 CrossRef CAS; (b) J. Manson, C. E. Webster and M. B. Hall, Texas A&M University, College Station, TX, 2006, http://www.chem.tamu.edu/jimp2/index.html.
- D. M. P. Mingos and D. J. Wales, Introduction to Cluster Chemistry, Prentice-Hall, Englewood Cliffs, NJ, 1990 Search PubMed.
- D. M. P. Mingos, Acc. Chem. Res., 1984, 17, 311–319 CrossRef CAS.
- N. C. Bhoumik, M. T. R. Joy, S. Ghosh, M. G. Richmond and S. E. Kabir, Inorg. Chim. Acta, 2020, 510, 119733 CrossRef CAS and references therein.
- (a) A. J. Deeming, S. Hasso and M. Underhill, Dalton Trans., 1975, 1614–1620 RSC; (b) J. B. Keister and J. R. Shapley, J. Organomet. Chem., 1975, 85, C29–C31 CrossRef CAS; (c) E. C. Bryan, B. F. G. Johnson and J. Lewis, Dalton Trans., 1977, 1328–1330 RSC; (d) A. G. Orpen, A. V. Rivera, E. G. Bryan, D. Pippard, G. M. Sheldrick and K. D. Rouse, Chem. Commun., 1978, 723–724 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: IR and 1H NMR spectral data on clusters 2, 3, 5, and 6. Atomic coordinates and energies for all DFT-optimized structures are available upon request (MGR). CCDC 2009586 (1), 2009587 (2), 2009588 (3), 2009591 (5), and 2009592 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05393g |
| This journal is © The Royal Society of Chemistry 2020 |