DOI:
10.1039/D0RA04307A
(Paper)
RSC Adv., 2020,
10, 25780-25785
Preparative separation of mangiferin glycosides by high speed counter current chromatography and comparison of their antioxidant and antitumor activities†
Received
14th May 2020
, Accepted 1st July 2020
First published on 8th July 2020
Abstract
Mangiferin, a xanthonoid with various bioactivities. The low solubility of mangiferin limits the use in pharmacological fields. In this study, high-speed counter-current chromatography (HSCCC) was used to separate and purify mangiferin glycosides from the crude sample after enzymatic glycosylation of mangiferin. Two fructosyl mangiferin were successfully purified by HSCCC with a two-phase-solvent system composed of n-butanol–methanol–water (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:6, v/v). A total of 18 mg of mangiferin (I), 73 mg of β-D-fructofuranosyl-(2 → 6)-mangiferin (II), and 58 mg of β-D-difructofuranosyl-(2 → 6)-mangiferin (III) were obtained in one-step separation from 150 mg of the crude sample with purities of 99.2%, 98.7% and 98.9%, respectively. The chemical structures were identified by HRMS, 1H-NMR, 13C-NMR and 2D NMR. Mangiferin glycosides showed higher antioxidant and antitumor activities compared to that of mangiferin by employing DPPH scavenging effect, reducing power and cytotoxicity assay. Therefore, these novel fructosyl mangiferin exhibit a great potential to be developed into new medicines.
:1:6, v/v). A total of 18 mg of mangiferin (I), 73 mg of β-D-fructofuranosyl-(2 → 6)-mangiferin (II), and 58 mg of β-D-difructofuranosyl-(2 → 6)-mangiferin (III) were obtained in one-step separation from 150 mg of the crude sample with purities of 99.2%, 98.7% and 98.9%, respectively. The chemical structures were identified by HRMS, 1H-NMR, 13C-NMR and 2D NMR. Mangiferin glycosides showed higher antioxidant and antitumor activities compared to that of mangiferin by employing DPPH scavenging effect, reducing power and cytotoxicity assay. Therefore, these novel fructosyl mangiferin exhibit a great potential to be developed into new medicines.
1. Introduction
Mangiferin is a xanthone glucoside present in many plant species including Iridaceae, Gentianaceae and Anacardiacae, while it was originally isolated from Mangifera indica L.1 The functional groups of mangiferin have several aromatic and non-aromatic secondary hydroxyl groups, one primary glycosidic hydroxyl group and one lactonic carbonyl group.2 Owing to these functional groups, it shows multiple pharmacological activities, including the anti-oxidant,3,4 central nervous system-stimulating,5 analgesic,6 antitumor,7 antidiabetic8 and anti-inflammation.9 However, the solubility of mangiferin is a serious issue which limits its pharmacological uses. Glycosylation has been used to improve the solubility and bioactivity of many natural products.10 These glycosylation reactions in water are not efficient with lower yields due to the serious solubility issue of the acceptor.11 Enzymatic glycosylation in organic solvents supply numerous industrially attractive advantages, such as reversing of the thermodynamic equilibrium of hydrolysis reactions, increasing the solubility of hydrophobic substrates, and eliminating of microbial contamination. Screening and exploiting of organic solvent-stable glycosidases have been studied to improve the stability of enzymes while catalysing reactions in organic solvents.12 In our previous study, we developed an effective method to enzymatic synthesis of mangiferin glycosides in hydrophilic organic solvents, high concentrations and high yields of mangiferin glycosides were achieved in a low-cost process.13 However due to the structural similarity of the glycosylated mangiferin, it was difficult to directly separate and purify an enough amount of product from the crude sample by single chromatographic method.
High speed counter-current chromatography (HSCCC), a liquid–liquid separation chromatographic technology, has been widely applied for the large-scale separation of chemical compositions and biotransformation reactor.14,15 It provides unique advantages such as lager loading capacity and higher sample recovery compared to traditional techniques.16,17 For these reasons, HSCCC has been used to purify mangiferin from mango fruit peel, the ethyl acetate–n-butanol–water (4:1:5) solvent system showed ideal k value of 1.76 for mangiferin.18 Genistein and daidzein were separated for the first time from Hericium erinaceum mycelium using chloroform–dichloromethane–methanol–water (4:2:3:2) solvent system.19 Four glycosides from Gentianae radix have been efficiently purified using different solvent systems.20 To the best of our knowledge, no report has been published on the use of HSCCC for one-step purification of mangiferin glycosides. The aims of this study, therefore, were to isolate and purify two types of mangiferin glycosides with high purity using HSCCC, and evaluate their antioxidant and antitumor activities.
2. Materials and methods
2.1 Materials and regents
Mangiferin was purchased from Zelang (Jiangsu, China). 2,2-Diphenyl-1-picrylhydrazyl (DPPH) was obtained from Sigma (St. Louis, MO, USA). Methanol used for HPLC analysis was of chromatographic grade from Sinopharm (Shanghai, China). n-Butanol, ethyl acetate, hexane, methanol used for HSCCC separation were of analytical grade and purchased from Beijing Chemical Factory (Beijing, China). Water used for deionized by an osmosis Milli-Q system (Millipore, USA). All other chemicals were of analytical grade and purchased from Sunshine (Nanjing, China). β-Fructofuranosidase was purified from Arthrobacter nicotianae XM6 (CCTCC M2010164).
2.2 Apparatus
HSCCC was performed with a model TBE-300B high speed counter current chromatography (Tauto Biotech, Shanghai, China). The instrument was equipped with three multilayer coil separation columns connected in series (diameter of tube = 2.6 mm, total volume = 300 mL, the range of β values = 0.5–0.8) and a 20 mL sample loop. The revolution speed of the apparatus was adjustable, ranging from 0 to 1000 rpm. An HX 1050 constant-temperature circulator (Beijing Boyikang Lab Instrument Company, Beijing, China) was used to control separation temperature. Sepu 3000 chromatography workstation (Hangzhou Puhui Science Apparatus Co. Ltd., Hangzhou, China) was employed to record the chromatograms.
2.3 Enzymatic glycosylation of mangiferin
The glycosylation reaction (3.0 L) was conducted in 1/15 M Na2HPO4/KH2PO4 buffer (pH 6.47) containing 0.24 U mL−1 β-fructofuranosidase, 20% (w/v) sucrose and 60 mM mangiferin and 15% (v/v) dimethyl sulfoxide (DMSO). The reaction mixture was conducted at 35 °C with shaking at 180 rpm for 12 h, then the reaction was stopped at 100 °C for 5 min.13
2.4 Preparation of crude sample
After the reaction solution has been loaded on AB-8 macroporous resin column (80 cm × 3 cm), the column was eluted by 3 L of distilled water (pH 4.0) acidified with acetic acid to remove sucrose, and then was eluted by 1 L of 100% ethanol. The 100% ethanol fraction was collected and evaporated to dryness by rotary evaporation at 60 °C under reduced pressure.
2.5 Selection of the two-phase solvent systems
The selection of two-phase solvent system was based on the partition coefficient (k) of the target components, which can be determined by HPLC. 10 mg of sample was dissolved with 5 mL of the upper and lower phases of the solvent system, and the sample was mixed thoroughly. After partition equilibration, the upper phase and the lower phase solution was taken for HPLC analysis, respectively. The k value was defined as the peak area of component in the upper phase (AU) divided by that of in the lower phase (AL) (k = AU/AL).
2.6 Preparation of two-phase solvent system and sample solution
In the present study, n-butanol–methanol–water (6:1:6, v/v/v) was used as the two-phase solvent system for HSCCC separation. It was prepared by adding all the solvents to a separatory funnel according to the volume ratios and fully equilibrated. Then, the upper and lower phase were separated and degassed for 40 min shortly before using. The sample solution was prepared by dissolving 150 mg of crude sample into 10 mL of the lower phase of the selected solvent system.
2.7 HSCCC separation procedure
First, the multilayer coil column was entirely filled with the lower phase as the stationary phase. Then the upper phase as the mobile phase was pumped into the column at a flow rate of 1.0 mL min−1 during rotation. The apparatus was rotated at 850 rpm. After the hydrodynamic equilibrium was reached, 150 mg prepared sample solution was loaded into the column through the injection valve. The effluent from the outlet of the column was continuously monitored with a UV detector at 254 nm. Each peak fraction was manually collected according to the chromatographic peak profiles displayed on the recorder. After running, the solvents in the column were pushed out and the retention of stationary phase was measured.
2.8 Analysis and identification of HSCCC peak fractions
The crude sample, each fraction obtained by HSCCC were performed on Dionex P680A HPLC system. The analysis was performed in a Discovery ODS C18 column (250 mm × 4.6 mm, i.d., 5 μm) at 30 °C. Methanol/0.1% formic acid (v/v) was used as the mobile phase in gradient elution mode as follows: 0–20 min, 20–60% methanol.13 The effluent was monitored at 316 nm and the flow rate was 1 mL min−1. The molecular weight of each fraction was carried out on a TOF mass spectrometer (Micromass) equipped with an electrospray ion source was used in positive-ion mode. 1H, 13C and 2D NMR spectra of each fraction were obtained using a Bruker AV-500 spectrometer (Switzerland), operating at 500 MHz. Samples were dissolved in DMSO-d6 at room temperature with tetramethylsilane as the chemical shift reference.
2.9 Evaluation of antioxidant activity
The effects of reference control (ascorbic acid), mangiferin, β-D-fructofuranosyl-(2 → 6)-mangiferin (Mangiferin-F) and β-D-difructofuranosyl-(2 → 6)-mangiferin (Mangiferin-F2) on the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical were estimated according to the method of Singh.21 Briefly, 150 μL of various concentration samples (6.25, 12.5, 25, 50, 100 and 250 μM) were mixed with 150 μL of 150 μM DPPH solution. After being incubated at 37 °C for 30 min, the absorbance of the mixture was measured at 517 nm. The free radical scavenging activity of the reaction solution was calculated as a percentage of DPPH discolouration using the following equation: Radical scavenging activity (%) = (1 − Asample/Acontrol) × 100, where Asample is the absorbance of the solution when the sample has been added at a particular level, and Acontrol is the absorbance of the DPPH solution. The results were expressed as mean values ± standard deviations (n = 8).
The reducing power of mangiferin, mangiferin-F, mangiferin-F2 and ascorbic acid was determined according to the method of Sakariah22 with some modifications. Mixtures of 50 μL different concentrations of mangiferin, mangiferin-F, mangiferin-F2 and ascorbic acid, 50 μL of phosphate buffer (0.2 M, pH 6.6), and 50 μL of 1% potassium ferricyanide were prepared and incubated at 37 °C for 20 min. Next, 50 μL of 10% trichloroacetic acid was added to the mixtures, followed by centrifuging at 4000 rpm for 10 min. The supernatant (200 μL) was mixed with 50 μL of distilled water and 30 μL of 0.1% ferric chloride and the absorbance was measured at 700 nm after standing for 10 min. Higher absorbance of the reaction mixture indicated higher reducing power of the samples. The results were expressed as mean values ± standard deviations (n = 8).
2.10 Cell growth and maintenance
Human leukemia promyelocytic cells HL60, and human papillary thyroid cancer cells TPC1 were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). Two cells were cultured in RPMI-1640 with FBS (10%), 2 mM glutamine, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C in a humidified incubator with 5% CO2.
2.11 MTT assay
The cytotoxicity effects of mangiferin-F, mangiferin-F2 and mangiferin were analyzed using MTT assay.23 Approximately 2 × 104 cells suspended in 100 μL of growth medium were added to each well of a 96 well plates and were incubated. The cells were treated with mangiferin, mangiferin-F and mangiferin-F2 at final concentrations of 5, 10, 20, 40, 80, 160, 250 μM for 48 h. Cells treated with dimethyl sulfoxide (DMSO) alone (0.01%) were used as control. Each concentration was tested with 6 replicate wells. After 48 h, the medium containing the treatments was removed, and 100 μL of MTT solution (0.5 mg mL−1) was added to each well and incubated at 37 °C for 3 h. The formazan crystals were dissolved in 100 μL of DMSO, and the absorbance was measured at 490 nm with ELISA plate reader. The experiment was performed in triplicate.
3. Results and discussion
3.1 Selection of the two-phase solvent systems
The selection of a suitable two-phase solvent system is a crucial step in a HSCCC separation, which can provide the ideal partition coefficient (k) for each target compound. As previously reported, the k value should be in range of 0.5–2 to get a short elution time and a good resolution.24 The small k value usually results in poor separation resolution, while large k value tends to produce broader chromatographic peaks due to a longer elution time.
For separation of mangiferin and fructosyl mangiferins, a series of experiments were performed to optimize the two-phase solvent system for HSCCC separation. The k values of the target compounds were described in Table 1. The results shown that the solvent systems composed of ethyl acetate–water (1:1, v/v) and hexane–methanol–water (1:1:1, v/v/v) had too small k values for all compounds indicating poor resolution. The solvent system of n-butanol–water (1:1, v/v) gave large k value for compound I and small k value for compound III. So, the solvent system of n-butanol–water (1:1, v/v) was slightly modified. When n-butanol–ethanol–water (6:1:6, v/v/v) and n-butanol–ethyl acetate–water (5:1:6, v/v/v) were used as the two-phase solvent system, the k values of target compounds were suitable. Finally, the solvent system of n-butanol–methanol–water (6:1:6, v/v) was used to isolate the target compounds with best suitable k values (kI = 2.10, kII = 1.48 and kIII = 0.75). Based on previous researches, low flow rate and high revolution speed could both lead to increase the retention of the stationary phase.19,25 The flow rate of 1 mL min−1, and the retention speed of 850 rpm were selected as the suitable conditions to run the HSCCC separation. Under the optimized conditions, the retention of stationary phase was 56%, and the purified compounds I, II and III were obtained (Fig. 1). About 18 mg of compound I, 73 mg of compound II, and 58 mg of compound III were obtained from 150 mg of crude sample. As shown in Fig. 2, the purities of compounds I, II and III were 99.2%, 98.7%, and 98.9%, respectively.
Table 1 k values of the target compounds in two-phase solvent systems
| Solvent system |
Volume ratio (v/v) |
kI |
kII |
kIII |
| n-Butanol–water |
1:1 |
3.89 |
0.92 |
0.23 |
| Ethyl acetate–water |
1:1 |
0.15 |
0.05 |
0.01 |
| Hexane–methanol–water |
1:1:1 |
0.05 |
0.01 |
0.001 |
| n-Butanol–methanol–water |
6:1:6 |
2.10 |
1.48 |
0.75 |
| n-Butanol–ethyl acetate–water |
5:1:6 |
2.50 |
1.21 |
0.64 |
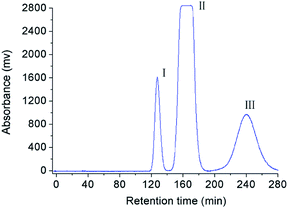 |
| | Fig. 1 HSCCC chromatogram of crude sample under the optimized condition. Solvent systems: n-butanol–methanol–water (6:1:6, v/v); stationary phase: lower phase; flow rate: 1 mL min−1; revolution speed: 850 rpm; sample amount: 150 mg; separation temperature: 25 °C; detection wavelength: 254 nm. | |
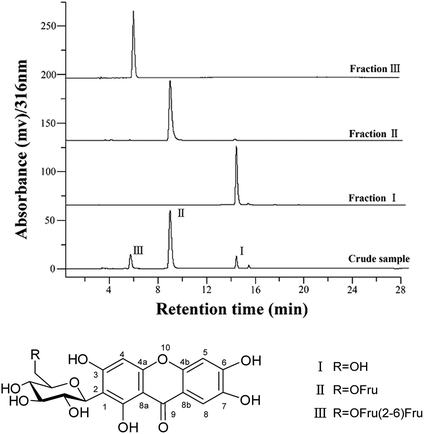 |
| | Fig. 2 HPLC analysis and chemical structures of compounds separated by HSCCC. | |
3.2 Identification of chemical structure
The chemical structure of compounds II and III was identified by HRMS and NMR spectra. The high-resolution ESI mass spectrum of compounds II showed an ion peak [M–H]− at m/z 583.1324 (calculated for C25H27O16, 583.1299) (Fig. S11-a†). Compared with mangiferin, the compound II has an additional formula C6H10O5, suggesting that compound II may be a mangiferin monosaccharide derivative. The 1H-NMR spectra in low field showed three ethylenic signals [δ 6.36 (1H, s), 6.86 (1H, s), 7.38 (1H, s)] (Fig. S1†). These were the characteristic mangiferin signals. The high field peak δ 4.59 (d, 9.8 Hz) was the core mangiferin glucose terminal hydrogen signal. The δ 73.1 ppm methylene may be β-sugar terminal carbon, which was connected with C–C bond. The δ 104.2 ppm quaternary carbon should be the other sugar terminal carbon (Fig. S2†). HSQC spectra can classify all the carbon signals except the quaternary carbon (Fig. S4†). The connections of each self-spinning segment can be inferred by HMBC spectra. From the 13C-NMR, 1H-NMR and 2D NMR spectra, the monosaccharide was identified as β-D-fructofuranose. The other diagnostic difference between compound II and mangiferin was the carbon signal at C-6′ of the glucose moiety, which was about 0.9 ppm downfield from those of mangiferin due to the known effects of O-glycosylation. Moreover, the carbon signal of C-5′ was about 6.3 ppm upfield from that of mangiferin. The HMBC spectrum showed long-range correlations between C-2′′ (δC 104.2) and H-6′ (δH 3.42, 3.52) of glucose moiety and H-1′′ (δH 3.30–3.40) of fructose moiety, thus proving the correlation between C-2′′ and H-6′ of compound II (Fig. S5†). Therefore, the chemical structure of compound II was elucidated as β-D-fructofuranosyl-(2 → 6)-mangiferin. The high-resolution ESI mass spectrum of compounds III showed an ion peak [M–H]− at m/z 745.1813 (calculated for C31H37O21, 745.1827) (Fig. S11-b†). Compared with mangiferin, the compound III has an additional formula C12H20O10, suggesting that compound III may be a mangiferin disaccharide derivative. The δ 73.2 ppm methylene should be β-sugar terminal carbon, which was connected with C–C bond. The δ 104.3 ppm and 104.2 ppm quaternary carbon should be fructose terminal carbon (Fig. S7†). The other diagnostic difference between compound III and mangiferin was the carbon signal at C-6′ (δC 62.9) of the glucose moiety, which was about 1.4 ppm downfield from those of mangiferin due to the known effects of O-glycosylation. And the carbon signal at C-6′′ (δC 62.5) was about 0.6 ppm downfield from those of compound II. The HMBC spectrum showed long-range correlations between C-2′′ (δC 104.3) and H-6′ (δH 3.51, 3.72) of glucose moiety and H-1′′ (δH 3.28–3.40) of fructose moiety, thus proving the correlation between C-2′′ and H-6′ of compound III (Fig. S10†). The HMBC spectrum also showed long-range correlations between C-2′′′ (δC 104.2) and H-6′′ (δH 3.42, 3.51) of fructose moiety and H-1′′′ (δH 3.26–3.37) of fructose moiety, thus proving the correlation between C-2′′′ and H-6′′ of compound III (Fig. S10†). Therefore, the chemical structure of compound III was elucidated as β-D-difructofuranosyl-(2 → 6)-mangiferin. The spectroscopic data for 1H and 13C-NMR (500 MHz, DMSO-d6) were summarized in Table 2.
Table 2 1H and 13C-NMR data for mangiferin and its glucosides (δ in ppm, J in Hz)
| No |
Mangiferin |
β-D-Fructofuranosyl-(2 → 6)-mangiferin |
β-D-Difructofuranosyl-(2 → 6)-mangiferin |
| 13C NMR (δC) |
1H NMRδH (J/Hz) |
13C NMR (δC) |
1H NMRδH (J/Hz) |
13C NMR (δC) |
1H NMRδH (J/Hz) |
| 1 |
161.7 |
13.75 (s, 1-OH) |
161.7 |
13.75 (s, 1-OH) |
161.7 |
13.75 (s, 1-OH) |
| 2 |
107.6 |
|
107.4 |
|
107.3 |
|
| 3 |
163.8 |
10.52 (s, 3-OH) |
163.8 |
|
163.7 |
|
| 4 |
93.3 |
6.37 (s) |
93.4 |
6.36 (s) |
93.4 |
6.37 (s) |
| 4a |
156.2 |
|
156.3 |
|
156.3 |
|
| 4b |
150.7 |
|
150.8 |
|
150.8 |
|
| 5 |
102.6 |
6.86 (s) |
102.6 |
6.86 (s) |
102.6 |
6.86 (s) |
| 6 |
153.9 |
10.63 (s, 6-OH) |
154.1 |
|
154.1 |
|
| 7 |
143.7 |
9.74 (s, 7-OH) |
143.7 |
|
143.7 |
|
| 8 |
108.1 |
7.38 (s) |
108.1 |
7.38 (s) |
108.1 |
7.38 (s) |
| 8a |
111.7 |
|
111.7 |
|
111.7 |
|
| 8b |
101.3 |
|
101.3 |
|
101.4 |
|
| 9 |
179.0 |
|
179.1 |
|
179.1 |
|
| 1′ |
73.1 |
4.60 (d, 9.8) |
73.1 |
4.59 (d, 9.8) |
73.2 |
4.60 (d, 9.8) |
| 2′ |
70.2 |
4.04 (t, 17.7) |
70.2 |
3.98 (t) |
70.3 |
4.00 (m) |
| 3′ |
78.9 |
3.12–3.23 (m) |
78.7 |
3.21 (m) |
78.3 |
3.22 (m) |
| 4′ |
70.6 |
|
70.7 |
3.21 (m) |
70.8 |
3.20 (m) |
| 5′ |
81.5 |
|
75.2 |
3.79 (t, 15.2) |
75.2 |
3.74–3.82 (m) |
| 6′ |
61.5 |
3.70 (d, 11.3), 3.40–3.43 (H, m) |
62.4 |
3.42, 3.52 (2H, m) |
62.9 |
3.51, 3.72 (2H, m) |
| 1′′ |
|
|
61.3 |
3.30–3.40 (2H, m) |
60.9 |
3.28, 3.40 (2H, m) |
| 2′′ |
|
|
104.2 |
|
104.3 |
|
| 3′′ |
|
|
79.6 |
3.30 (1H, m) |
80.3 |
3.59 (m) |
| 4′′ |
|
|
76.7 |
3.94 (t, 14.5) |
76.5 |
3.92–3.99 (m) |
| 5′′ |
|
|
82.1 |
3.52 (1H, m) |
75.7 |
3.74–3.82 (m) |
| 6′′ |
|
|
61.9 |
3.88, 3.58 (2H, m) |
62.5 |
3.42, 3.51 (2H, m) |
| 1′′′ |
|
|
|
|
61.3 |
3.26–3.37 (2H, m) |
| 2′′′ |
|
|
|
|
104.2 |
|
| 3′′′ |
|
|
|
|
79.6 |
3.30 (m) |
| 4′′′ |
|
|
|
|
76.3 |
3.92–3.99 (m) |
| 5′′′ |
|
|
|
|
82.0 |
3.53 (m) |
| 6′′′ |
|
|
|
|
62.2 |
3.51, 3.84 (2H, m) |
3.3 Antioxidant activity
The DPPH scavenging potential of mangiferin, mangiferin-F, mangiferin-F2 and ascorbic acid at different concentrations is shown in Fig. 3a. Significant DPPH radical scavenging activity was evident at 50 μM concentration of mangiferin (75.2%), mangiferin-F (77.1%), mangiferin-F2 (78.1%) and ascorbic acid (53.1%). The scavenging effect of mangiferin glycosides were higher than that of mangiferin and ascorbic acid. Fig. 3b shows the reducing powers of different concentrations of mangiferin, mangiferin-F, mangiferin-F2 and ascorbic acid using potassium ferricyanide reduction method. The reducing power of four samples correlated well with increasing concentrations. Moreover, the reducing power of mangiferin glycosides was relatively more pronounced than that of mangiferin and ascorbic acid. DPPH scavenging effect and reducing power of mangiferin glycosides suggests that glycosylation of mangiferin increases the antioxidant activities to a certain extent.
 |
| | Fig. 3 Antioxidant activity mangiferin and its glycosides. ((a) DPPH scavenging effect; (b) reducing power). | |
3.4 Cytotoxicity assay
MTT assay was performed to compare the cell cytotoxicity induced between mangiferin and its glycosides. As shown in Table 3, mangiferin showed effective cytotoxicity on TPC1 cells (IC50 4.2 μM) and HL60 cells (IC50 72.8 μM) after 48 h of incubation. The cytotoxicity of mangiferin glycosides were better than that of mangiferin, mangiferin-F presented IC50 values of 3.8 and 65.8 μM for TPC1 and HL60 tumor cell lines, respectively. However, there no obvious difference of antitumor activity between mangiferin-F and mangiferin-F2. This suggests that glycosylation of mangiferin can enhances the antitumor activity.
Table 3 The cytotoxicity activity of mangiferin and its glycosides
| Cells |
IC50 (Mean ± SD, μM) |
| Mangiferin |
Mangiferin-F |
Mangiferin-F2 |
| TPC1 |
4.2 ± 0.4 |
3.8 ± 0.3 |
3.7 ± 0.5 |
| HL60 |
72.8 ± 2.9 |
65.8 ± 1.7 |
65.2 ± 2.1 |
4. Conclusions
In summary, a HSCCC method was successfully established to separate and purify mangiferin and its glycosides. Two mangiferin glycosides were obtained in one-step isolation, and their purities were more than 98.7%. The DPPH scavenging activity and reducing power analysis demonstrated that β-D-fructofuranosyl-(2 → 6)-mangiferin and β-D-difructofuranosyl-(2 → 6)-mangiferin showed better antioxidant activities than that of mangiferin. The cytotoxicity of two mangiferin glycosides were also better than that of mangiferin, β-D-fructofuranosyl-(2 → 6)-mangiferin presented IC50 values of 3.8 and 65.8 μM for TPC1 and HL60 tumor cell lines, respectively.
Conflicts of interest
All authors declare no conflicts of interest.
Acknowledgements
This project was sponsored by National Key R&G Program of China (2018YFC1706201), Natural Science Foundation of Jiangsu (BK20161038), and PAPD.
References
- K. Sanugul, T. Akao, Y. Li, N. Kakiuchi, N. Nakamura and M. Hattori, Biol. Pharm. Bull., 2005, 28, 1672–1678 CrossRef CAS PubMed.
- S. Jaiswal, K. Ramesh, G. Kapusetti, A. K. Ray, B. Ray and N. Misra, Polym. Bull., 2015, 72, 1407–1416 CrossRef CAS.
- R. Boonnattakorn, V. Chonhenchob, M. Siddiq and S. P. Singh, Packag. Technol. Sci., 2015, 28, 241–252 CrossRef CAS.
- P. K. Jain, K. Kumar and A. Gajbhiye, Drug Dev. Ind. Pharm., 2013, 39, 1840–1850 CrossRef CAS PubMed.
- J. J. Jeong, S. E. Jang, S. R. Hyam, M. J. Han and D. H. Kim, Eur. J. Pharmacol., 2014, 740, 652–661 CrossRef CAS PubMed.
- B. B. Garrido-Suárez, G. Garrido, M. E. García and R. Delgado-Hernández, Phytother. Res., 2014, 28, 1646–1653 CrossRef PubMed.
- F. Gold-Smith, A. Fernandez and K. Bishop, Nutrients, 2016, 8, 396–421 CrossRef PubMed.
- J. Hou, D. L. Zheng, F. Gabriel, H. Y. Deng, L. Chen, J. L. Liang, Y. Jiang and Y. H. Hu, Can. J. Physiol. Pharmacol., 2015, 94, 332–340 CrossRef PubMed.
- Y. P. Zhao, W. H. Wang, X. H. Wu, X. Q. Ma, R. Z. Qu, X. M. Chen, C. H. Liu, Y. G. Liu, X. K. Wang, P. C. Yan, H. Zhang, J. R. Pan and W. W. Li, Int. Immunopharmacol., 2017, 45, 174–179 CrossRef CAS PubMed.
- I. Gill and R. Valivety, Angew. Chem., Int. Ed., 2000, 39, 3804–3808 CrossRef CAS PubMed.
- C. G. Yu, H. D. Xu, G. D. Huang, T. Chen, G. Y. Liu, N. Chai, Y. Ji, S. Y. Wang, Y. J. Dai and S. Yuan, Appl. Microbiol. Biotechnol., 2010, 86, 863–870 CrossRef CAS PubMed.
- X. M. Wu, J. L. Chu, B. Wu, S. Zhang and B. F. He, Bioresour. Technol., 2013, 129, 659–662 CrossRef CAS PubMed.
- X. M. Wu, J. L. Chu, J. Y. Liang and B. F. He, RSC Adv., 2013, 3, 19027–19032 RSC.
- D. J. Wang, X. Y. Song, H. J. Yan, M. M. Guo, R. M. Fu, H. L. Jiang, H. Zhu and X. Wang, RSC Adv., 2018, 8, 34321–34330 RSC.
- D. J. Wang, M. S. Khan, L. Cui, X. Y. Song, H. Zhu, T. Y. Ma and R. Sun, RSC Adv., 2019, 9, 4892–4899 RSC.
- J. Q. Yu, X. W. Sun, L. Zhao, X. Y. Wang and X. Wang, RSC Adv., 2020, 10, 11132–11138 RSC.
- D. Q. Li, J. Zhao, D. Wu and S. P. Li, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1021, 81–90 CrossRef CAS PubMed.
- F. Luo, Q. Lv, Y. Zhao, G. Hu, G. Huang, J. Zhang, C. Sun, X. Li and K. Chen, Int. J. Mol. Sci., 2012, 13, 11260–11274 CrossRef CAS PubMed.
- J. He, P. Fan, S. Feng, P. Shao and P. Sun, Molecules, 2018, 23, 560 CrossRef PubMed.
- B. Chen, Y. Peng, X. Wang, Z. Li and Y. Sun, Molecules, 2017, 22, 2002 CrossRef PubMed.
- N. Singh and P. S. Rajini, Food Chem., 2004, 85, 611–616 CrossRef CAS.
- G. K. Jayaprakasha, R. P. Singh and K. K. Sakariah, Food Chem., 2001, 73, 285–290 CrossRef CAS.
- L. Zhang and M. C. Wang, Biotechnol. Bioprocess., 2018, 23, 649–654 CrossRef CAS.
- Y. Ito, J. Chromatogr. A, 2005, 1065, 145–168 CrossRef CAS PubMed.
- X. M. Wu, J. L. Chu, T. T. Xu and B. F. He, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2013, 935, 70–74 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The HRMS and NMR spectroscopic data for compounds II and III. See DOI: 10.1039/d0ra04307a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b


