
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation and characterization of crosslinks, including Tyr–Trp species, on one electron oxidation of free Tyr and Trp residues by carbonate radical anion†
Juan David Figueroa‡
a,
Ana María Zárate‡a,
Eduardo Fuentes-Lemus a,
Michael J. Daviesb and
Camilo López-Alarcón*a
a,
Michael J. Daviesb and
Camilo López-Alarcón*a
aPontificia Universidad Católica de Chile, Facultad de Química y de Farmacia, Departamento de Química Física, Santiago, Chile. E-mail: clopezr@uc.cl
bUniversity of Copenhagen, Department of Biomedical Sciences, Copenhagen, Denmark
First published on 8th July 2020
Abstract
Dityrosine and ditryptophan bonds have been implied in protein crosslinking. This is associated with oxidative stress conditions including those involved in neurodegenerative pathologies and age-related processes. Formation of dityrosine and ditryptophan derives from radical–radical reactions involving Tyr˙ and Trp˙ radicals. However, cross reactions of Tyr˙ and Trp˙ leading to Tyr–Trp crosslinks and their biological consequences have been less explored. In the present work we hypothesized that exposure of free Tyr and Trp to a high concentration of carbonate anion radicals (CO3˙−), under anaerobic conditions, would result in the formation of Tyr–Trp species, as well as dityrosine and ditryptophan crosslinks. Here we report a simple experimental procedure, employing CO3˙− generated photochemically by illumination of a Co(III) complex at 254 nm, that produces micromolar concentrations of Tyr–Trp crosslinks. Analysis by mass spectrometry of solutions containing only the individual amino acids, and the Co(III) complex, provided evidence for the formation of o,o′-dityrosine and isodityrosine from Tyr, and three ditryptophan dimers from Trp. When mixtures of Tyr and Trp were illuminated in an identical manner, Tyr–Trp crosslinks were detected together with dityrosine and ditryptophan dimers. These results indicate that there is a balance between the formation of these three classes of crosslinks, which is dependent on the Tyr and Trp concentrations. The methods reported here allow the generation of significant yields of isolated Tyr–Trp adducts and their characterization. This technology should facilitate the detection, and examination of the biological consequences of Tyr–Trp crosslink formation in complex systems in future investigations.
1. Introduction
The formation and accumulation of crosslinked proteins is associated with the etiology of multiple human diseases, as well as ageing-related processes.1–4 Consequently, there is a considerable interest from biochemical and health researchers in the detection, characterization, and quantification of such species. Crosslinking of proteins can be triggered by oxidative stress conditions, including those initiated by free radicals (FR˙ in Scheme 1), with this resulting in the formation of secondary free radicals on the side chains of susceptible amino acids (protein˙ in Scheme 1).5 Multiple reaction pathways are known for protein˙, including, under aerobic environments, those involving O2 (pathway A in Scheme 1) that yield protein peroxyl radicals (protein–OO˙) and subsequently hydroperoxides. The latter can extend the original damage through further metal-ion catalyzed reactions.1,6,7 Other oxygenated products, such as alcohols and carbonyl groups are commonly produced, with hydroperoxides being intermediates in the formation of some of these species.1,6,7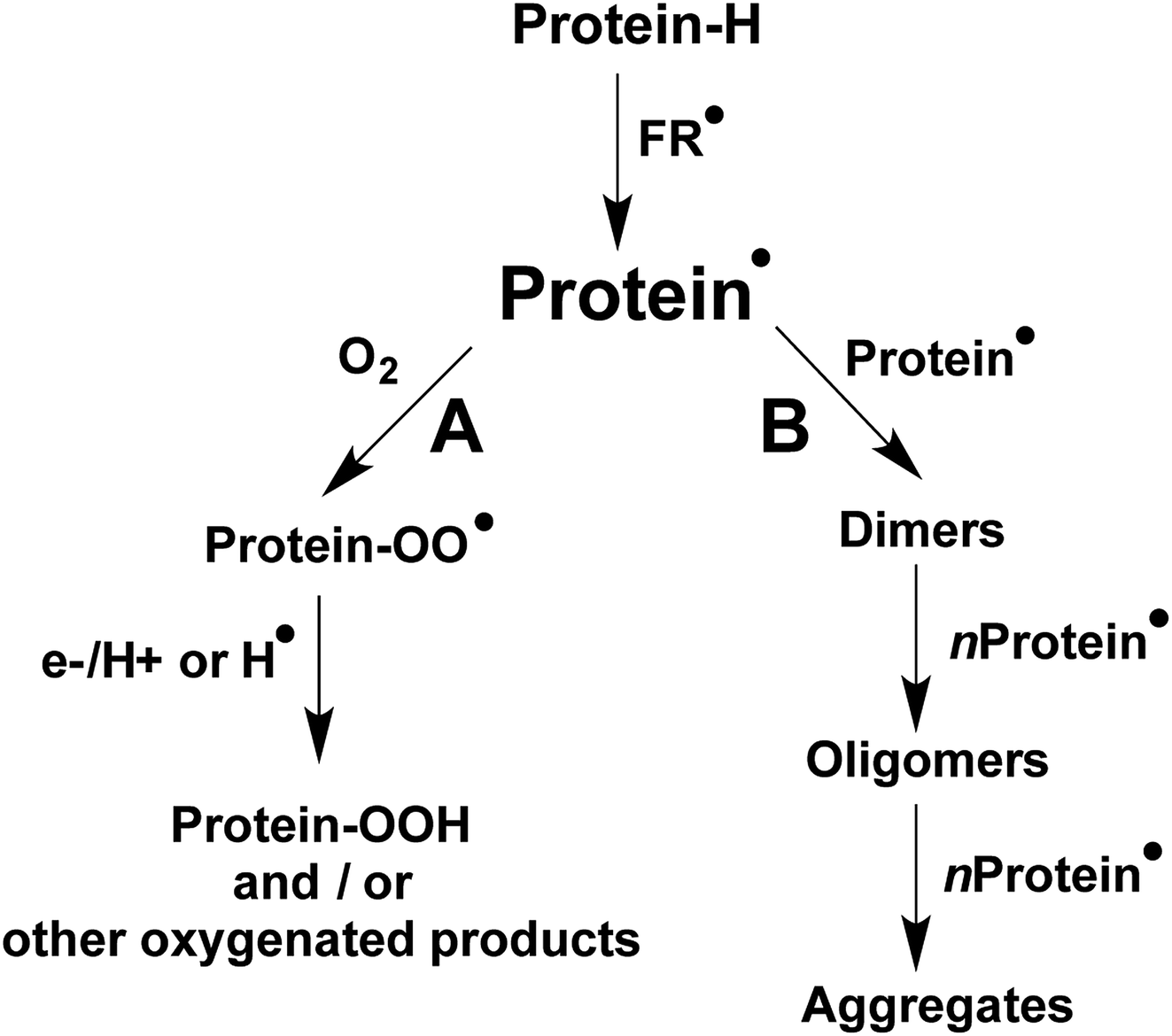 | ||
| Scheme 1 Reactions pathways derived from protein secondary free radicals (protein˙). Pathway A represents the main reactions mediated by O2, while pathway B shows protein crosslinking (both inter- and intra-molecular processes) resulting from radical–radical reactions. | ||
Depending on the structural characteristics of the proteins, their environment, and the O2 concentration, new covalent bonds can also be formed by protein˙, as a consequence of radical–radical reactions. As presented in pathway B of Scheme 1, such processes can result in inter-molecular crosslinks, mediating protein dimerization, oligomerization and aggregation, as well intra-molecular crosslinks. The formation of both inter- and intra-molecular cross-linked species can affect the biological functionality of proteins.
Disulfide and dityrosine bonds are produced physiologically during posttranslational processing of proteins, with this giving stabilized tertiary and quaternary structures.5,8–10 This allows proteins to generate the conformation and structure required for their biological function.5,8–11 In contrast to this physiological role, the formation of other disulfides and dityrosine has been associated with the etiology and development of oxidative stress-related pathological conditions and diseases.12 These species can be generated from self-reactions of thiyl (Cys˙) and tyrosyl (Tyr˙) radicals (with dimerization rate constants, k, ≈ 108 and 109 M−1 s−1).9,10 Other reactions can also contribute to the formation of disulfides (e.g. reactions of sulfenic acids and RS-NO species).13–15 The formation of such non-intended crosslinks can alter the native structure of proteins, and give rise to dysfunctional species. For example, internal disulfide bonds promote the stabilization of an unfolded domain of γ-crystallins, resulting in inter-molecular interactions and protein aggregation, a process involved in the development of cataracts.12,16 Dityrosine bonds have been implicated in the pathogenesis of neurodegenerative pathologies,17,18 including the crosslinking of α-synuclein in Parkinson's disease, with the formation of oligomers favoring intracellular amyloid fibril deposition in Lewy bodies,17 and the dimerization of amyloid-beta (Aβ) in Alzheimer's disease, which promotes aggregation and deposition in amyloid plaques.18 The role of dityrosine in these pathologies has been strongly supported by analytical investigations (e.g. by fluorescence, electrochemical, and mass spectroscopy, MS) of post-mortem brain sections.17,19 Such studies, as well as others that have reported elevated levels of dityrosine in atherosclerotic plaques, human blood fluids, and bovine milk, have resulted in the widespread use of dityrosine as a marker of protein damage.20–25 Dietary intake of dityrosine has also been linked with toxicity, on the basis of studies that have reported metabolic alterations in mice after intragastric administration of pure dityrosine.26
Trp oxidation has also been associated with both positive and negative biological effects. Recent studies have shown that some Trp oxidation products, such as specific hydroperoxide isomers, can act as important signaling molecules and mediators of vascular tone,27 but other reports have linked the formation of ditryptophan resulting from radical–radical reactions of Trp˙ (with k ≈ 2–6 × 108 M−1 s−1)28 with multiple pathological conditions.29,30 Thus, the formation of ditryptophan from peroxidase reactions has been implicated in the dimerization of human superoxide dismutase 1 (hSOD1),31 and the formation of non-amyloid hSOD1 aggregates in amyotrophic lateral sclerosis (ALS).32–34 Ditryptophan bonds have also been detected in crosslinked lysozyme, caseins, crystallin proteins, and mixed lysozyme-hSOD1 species, as a result of protein modification by multiple oxidants and photochemical reactions.35–40
Despite the increasing number of reports on ditryptophan formation, the significance of this species in human disease, and the factors that modulate its formation are not fully understood. Recent data indicate that ditryptophan yields are diminished under aerobic conditions due to the occurrence of competing reactions involving O2, and that multiple ditryptophan isomers, including both C–C and C–N linked species can be formed.41,42 Reaction with O2 may limit the participation of Tyr˙ and Trp˙ in protein crosslinking, though the rate constants for reaction of both radicals with O2 are modest (k 4 ≤ 106 M−1 s−1 for Trp˙ (ref. 43) and < 103 M−1 s−1 for Tyr˙ (ref. 44)). The properties and structure of a protein can also modulate the steady state concentration of these species,43 and their reaction with other species including O2−˙ (ref. 45) and reducing agents such as GSH and ascorbic acid.44,46,47 In contrast to these relatively slow reactions, the rate constants for dimerization of Tyr˙ and Trp˙ are rapid (k > 108 M−1 s−1 (ref. 28 and 44)), and this might result in cross reaction between Tyr˙ and Trp˙ leading to Tyr–Trp adducts; these species are poorly characterized. Tyr–Trp crosslinks have been reported in a cytochrome c peroxidase mutant, with these generated via a peroxide-dependent process,48 in the vicinity of the active site of a catalase-peroxidase from Haloarcula marismortui,49 and in oxidized samples of lysozyme and glucose 6-phosphate dehydrogenase.36,37,50 Studies on these species have however been limited by a lack of information on the structure of these species, methods that allow their detection and characterization, and authentic standards that would facilitate their quantification.
In the present work we hypothesized that exposure of free Tyr and Trp to a high concentration of oxidants under anaerobic conditions, would result in the formation of significant yields of Tyr–Trp. We report a novel and simple experimental procedure, employing photochemically-generated carbonate anion radicals (CO3˙−), to produce Tyr–Trp crosslinks at micromolar concentrations allowing their characterization. This can be achieved with minimal formation of ditryptophan and dityrosine, suggesting that the formation of these crossed dimers is a favorable reaction. These data should be useful for future investigations aimed at understanding the role that Tyr–Trp crosslinks play in biology, pathology, pharmacology and food technology.
2. Materials and methods
2.1 Reagents
Tryptophan (Trp), tyrosine (Tyr), and diethylenetriaminepentaacetic acid (DTPA) were purchased from Sigma Aldrich. The ortho,ortho isomer of dityrosine (o,o′-dityrosine) was supplied by Toronto Research Chemicals (Toronto, CA). Methanol, disodium hydrogen phosphate and sodium dihydrogen phosphate were obtained from Merck (Darmstadt, Germany). All the solvents employed were HPLC or LC-MS grade. [Co(NH3)5(CO3)]NO3 was synthesized according to the procedure reported by Basolo et al.51 Thus, 30 g of Co(NO3)2 × 6H2O in 15 mL of water was added to 45 g of ammonium carbonate dissolved in 45 mL of water, followed by the addition of 75 mL of concentrated ammonia. The solution was bubbled with air for 24 h, and cooled in an ice bath, and the solid product recrystallized by dissolving it in 55 mL of water at 90 °C and then slowly cooling in an ice bath. Pure crystals were isolated, and used in the experiments.2.2 Illumination set-up
Solutions (1.9 mL) were placed into a quartz cell (internal dimensions: 4.2 × 1.0 × 0.5 cm) sealed with a septum and illuminated inside a photoreactor containing eight Xe lamps (λmax 254 nm) with four lamps each side of the cell (optical path 0.5 cm). Solutions were stirred constantly, and the incident light absorbed (Iabs = 2.3 × 10−5 Einstein per min, in 1.9 mL of illuminated solution) was assessed using as reference the decomposition quantum of Tyr (Φ = 9 ×10−3 mol per Einstein) at 254 nm.52 Considering a quantum yield of CO3˙− formation from the photodecomposition of [Co(NH3)5(CO3)]NO3 of Φ = 9 × 10−2 mol per Einstein,53 a rate of CO3˙− production of 367 μM min−1 was determined. This implies that after 3 min illumination a total dose of CO3˙− of 1.1 mM was produced.2.3 Consumption of Trp and Tyr elicited by CO3˙−
Solutions (1.9 mL) containing either Trp, Tyr or their mixtures (50–1000 μM final concentration) and DTPA (0.1 mM) were prepared in phosphate buffer (75 mM, pH 7.4) and illuminated for 3 min as indicated in Section 2.2, in the absence and presence of [Co(NH3)5(CO3)]NO3 (4 mM). Prior to illumination, solutions were bubbled with N2 for 50 min. Subsequently, 262 μL of a fresh [Co(NH3)5(CO3)]NO3 stock solution (29 mM in phosphate buffer 75 mM, pH 7.4) were added (to give a final concentration of 4 mM) and N2 bubbled for a further 10 min. After illumination (3 min), the samples were centrifuged at 2800g for 5 min and stored at −80 °C until analysis.Consumption of Trp and Tyr was assessed by transferring 300 μL of the samples to HPLC vials and analysis using an Agilent 1200 system equipped with an Agilent 1260 series auto sampler (with the samples kept at 4 °C) and Agilent 1260 series diode array (DAD) and fluorescence (FL) detectors. Samples (20 μL) were injected on to a reversed phase column (Hibar® 250 × 4.3 mm RP-18 endcapped (particle size 5 μm) Phurospher®STAR, Millipore, maintained at 40 °C), and eluted using a gradient of buffer A (0.1% formic acid in water) and buffer B (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 methanol: water containing 1% formic acid) at a flow rate of 0.8 mL min−1. Buffer A was kept at 100% for the first 2 min, before decreasing to 20% over 30 min. Subsequently, buffer A was returned to 100% over 35 min with a further equilibration phase of 5 min. The DAD detector was set at 280 nm, while FL detection was carried out using λex 280 and λem 360 nm. Data analysis was carried out using OpenLab Software (Santa Clara, CA). Quantification of Trp and Tyr was determined from the area under the curve of the corresponding chromatographic peaks employing calibration curves constructed using commercial standards.
:50 methanol: water containing 1% formic acid) at a flow rate of 0.8 mL min−1. Buffer A was kept at 100% for the first 2 min, before decreasing to 20% over 30 min. Subsequently, buffer A was returned to 100% over 35 min with a further equilibration phase of 5 min. The DAD detector was set at 280 nm, while FL detection was carried out using λex 280 and λem 360 nm. Data analysis was carried out using OpenLab Software (Santa Clara, CA). Quantification of Trp and Tyr was determined from the area under the curve of the corresponding chromatographic peaks employing calibration curves constructed using commercial standards.
2.4 Detection of dityrosine, ditryptophan and Tyr–Trp by mass spectrometry
Samples were analyzed using an Ekspert Ultra-High Performance Liquid Chromatography (UPLC) ABSciex 4500 system coupled to a triple quadrupole detector (Triple Quad™). The chromatographic conditions, stationary and elution buffers, were the same as those described in Section 2.3. Mass detection was carried out under a positive polarity mode employing an Information Dependent Acquisition (IDA) scanning method with the first criteria being the search for intense peaks from 1 to 1 exceeding 1000.0 counts. The acquisition period was 40.45 min with a mass tolerance of 250.0 mDa, and an enhanced resolution scan was employed to confirm the charge state and/or isotope pattern. No dynamic background subtraction nor rolling collision energy were employed. In addition, selected reaction monitoring (SRM) MS was used with positive polarity, and employing a Turbo Spray™ system as ion source (electrospray ionization), to collect Q1 and Q3 of Trp, Tyr, and their oxidation products. MS2 spectra were collected using a step size of 0.1 Da of selected parent ions, with a MS2 scan range of 50–1000 Da. The collision energy and declustering potentials were 15 and 56 V, respectively. N2 was used as nebulizing gas, the temperature inside the nebulization chamber was set at 600 °C.The SRM mode was also set to detect ions arising from specific oxidation products corresponding to Trp+16, Trp+32 (hydroperoxides and N-formylkynurenine), and Tyr+16 (3,4-dihydroxyphenylalanine, DOPA). For crosslinked species, the following ions were examined: dityrosine (Q1 361/Q3 315), ditryptophan (Q1 407/Q3 203) and Tyr–Trp (Q1 384/Q3 203, or Q1 384/Q3 367).
2.5 Quantification of dityrosine by HPLC-FL and UPLC-MS
Production of dityrosine was analyzed and quantified by fluorescence (FL), as described previously,54 and by MS. Samples (10 μL) were injected on to a reversed phase column (Hibar® 250 × 4.3 mm RP-18 endcapped (particle size 5 μm) Phurospher®STAR, Millipore) maintained at 40 °C, and separated by gradient elution using buffer A (100 mM sodium perchlorate, 10 mM, H3PO4) and buffer B (80% aqueous methanol), at flow rate of 0.8 mL min−1. Fluorescence detection was carried out using λex 280 and λem 410 nm. Mass detection was achieved employing SRM mode to follow the fragmentation of the base peak 361 → 315 employing 86 V for declustering potential (DP), a collision energy (CE) of 85 V and a collision cell exit potential (CXP) of 18 V. Quantification of dityrosine was determined from the area under the curve (AUC) of the chromatographic peaks employing calibration curves (1–100 μM for FL detection, 1–50 μM for mass analysis) generated using the commercial o,o′-dityrosine standard (ESI Fig. 1 and 2†). Analysis of FL data was carried out using OpenLab Software (Santa Clara, CA), while for MS analysis, calibration curves were generated, and data analysis carried out using the software Analyst (AB Sciex, Framingham, MA, USA).2.6 Data and statistical analysis
The data reported are from measurements carried out in triplicate on at least two or three independent experiments. Analysis employed Graphpad Prisma 7.0a software. With the exception of ESI Fig. 1 and 2,† data are presented as means ± SD. Statistical analysis of data presented in Fig. 5A for χTyr = 0.95 and 1, was carried out by Student's t-test, using Graphpad Prisma 7.0a software.3. Results
UV illumination of [Co(NH3)5(CO3)]NO3 at 254 nm results in the release of CO3˙−.39,53 Under the experimental conditions employed the complex was illuminated at a rate of 9 ×10−3 Einstein per min, giving a CO3˙− flux of 367 μM min−1.53 Reaction of this species with Tyr and Trp, results in one-electron oxidation of the aromatic rings, and formation of the Tyr-derived phenoxyl radical, Tyr˙, and the Trp-derived indolyl radical, Trp˙, as a result of rapid deprotonation of the initial radical–cations. In the absence of O2, subsequent rapid radical–radical reactions generate the respective dimers.3.1 Formation dityrosine and ditryptophan elicited by CO3˙− from the individual parent amino acids
Illumination of individual solutions of Tyr and Trp (each 500 μM) with [Co(NH3)5(CO3)]NO3 (4 mM), resulted in the consumption of both residues, with 332 and 298 μM of Tyr and Trp, remaining after 3 min (Fig. 1A). In control illumination systems, in the absence of the Co(III) complex, no Tyr, and approximately 50 μM Trp consumption was detected (Fig. 1A). As shown in Fig. 1B, the consumption of both amino acids mediated by CO3˙−, increased in a manner that was dependent on the initial concentration of the amino acid. Both amino acids showed a similar slight down curvature in the consumption versus initial concentration plots, probably as a result of increasing light absorption by the amino acid instead of the [Co(NH3)5(CO3)]NO3 complex. The similarity of the curves for both residues suggests a similar stoichiometry of reaction. For both amino acids, a stoichiometric factor (n), defined as the moles of CO3˙− trapped per mole of amino acid, of about 5.5, and 3.7 was determined for initial concentrations of 0.5 and 1 mM, respectively.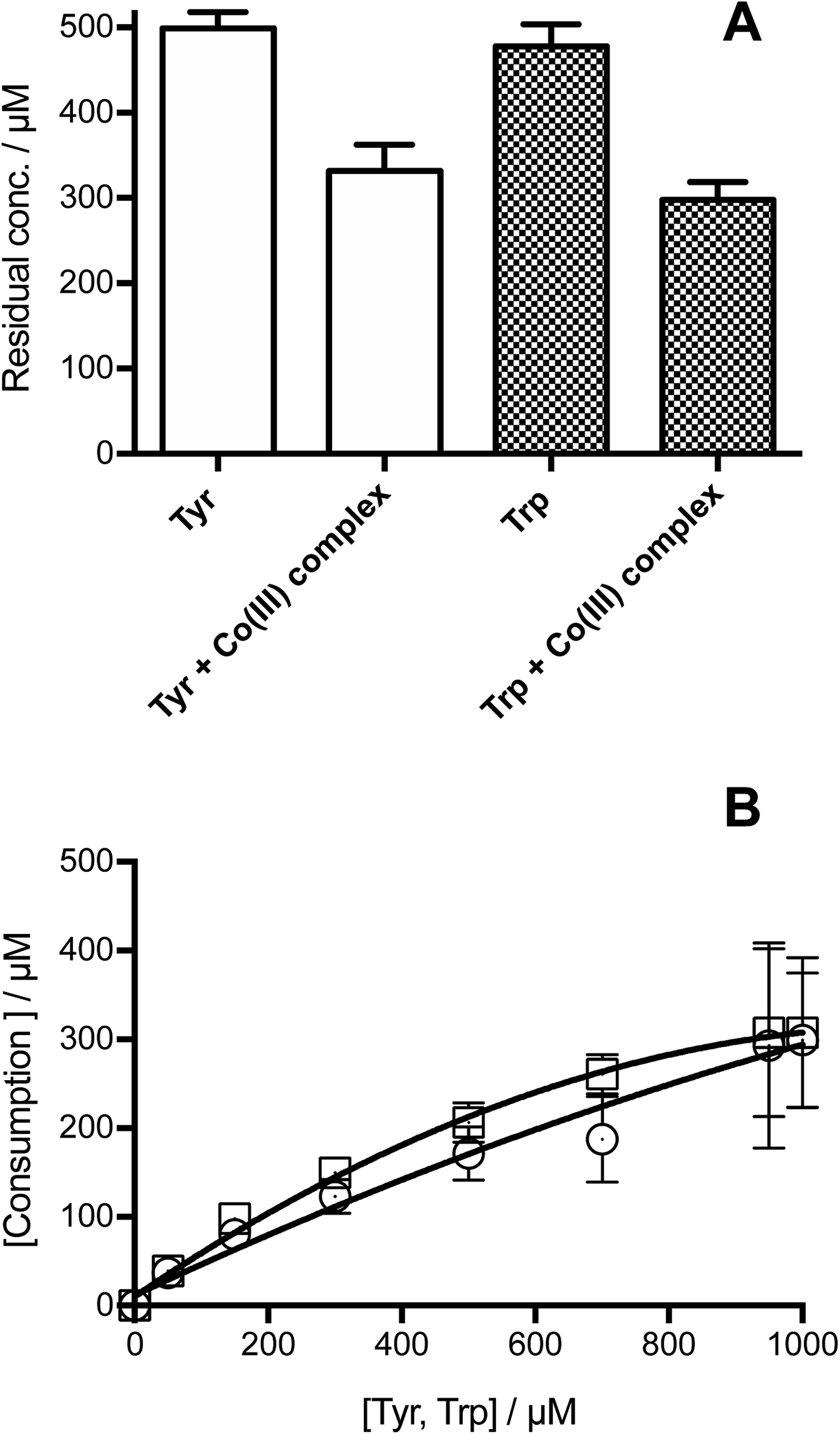 | ||
| Fig. 1 Illumination of free Tyr, or free Trp, as individual amino acids in the presence of [Co(NH3)5(CO3)]NO3 results in consumption of Tyr and Trp. Panel A: consumption of Tyr and Trp determined for solutions containing each amino acid at 0.5 mM illuminated at 254 nm for 3 min in the absence or presence of [Co(NH3)5(CO3)]NO3 (4 mM), under a N2 atmosphere. Panel B: dependence of the consumed Tyr (○) and Trp (□) with their initial concentration. All solutions were prepared in phosphate buffer (75 mM, pH 7.4, containing 0.1 mM DTPA) and bubbled with N2 for 1 h prior to illumination. Consumption of amino acids was assessed by HPLC with fluorescence detection as described in Material and methods. | ||
The formation of specific Tyr and Trp oxidation products, both dimers and oxygenated (m/z +16 and +32) species, was analyzed by MS. In all cases, no oxygenated products were detected consistent with effective deoxygenation of the solutions. ESI Fig. 3† depicts a representative chromatogram for m/z +32 ions, obtained for Trp under aerobic and anaerobic atmospheres. UPLC-MS analysis of illuminated Tyr-containing solutions and the Co(III) complex, showed the presence of two peaks with m/z 361, with these eluting at 11.7 and 14.2 min (Fig. 2A, peaks a and b, respectively). These signals showed similar MS/MS spectra with fragmentations 361 → 315, and 361 → 344 (Fig. 2B and C). This is in agreement with dimerization of Tyr˙ and the formation of two isomers linked by C–C and C–O bonds (o,o′-dityrosine and isodityrosine), respectively. Peak a (at 11.7 min, Fig. 2A) co-eluted with the commercial standard of o,o′-dityrosine (ESI Fig. 2A†), allowing its identification as this isomer. Analysis by HPLC-FL showed the presence of a single peak that eluted at the same time as the commercial standard (ESI Fig. 4†), consistent with the known fluorescent properties of o,o′-dityrosine. Peak b (detected by UPLC-MS, Fig. 2A) was not observed by HPLC-FL, indicating that this is (non-flurorescent)55 isodityrosine. Based on the ion intensities of the obtained mass spectra, apparently a higher yield of the o,o′-dityrosine isomer than isodityrosine, was produced.
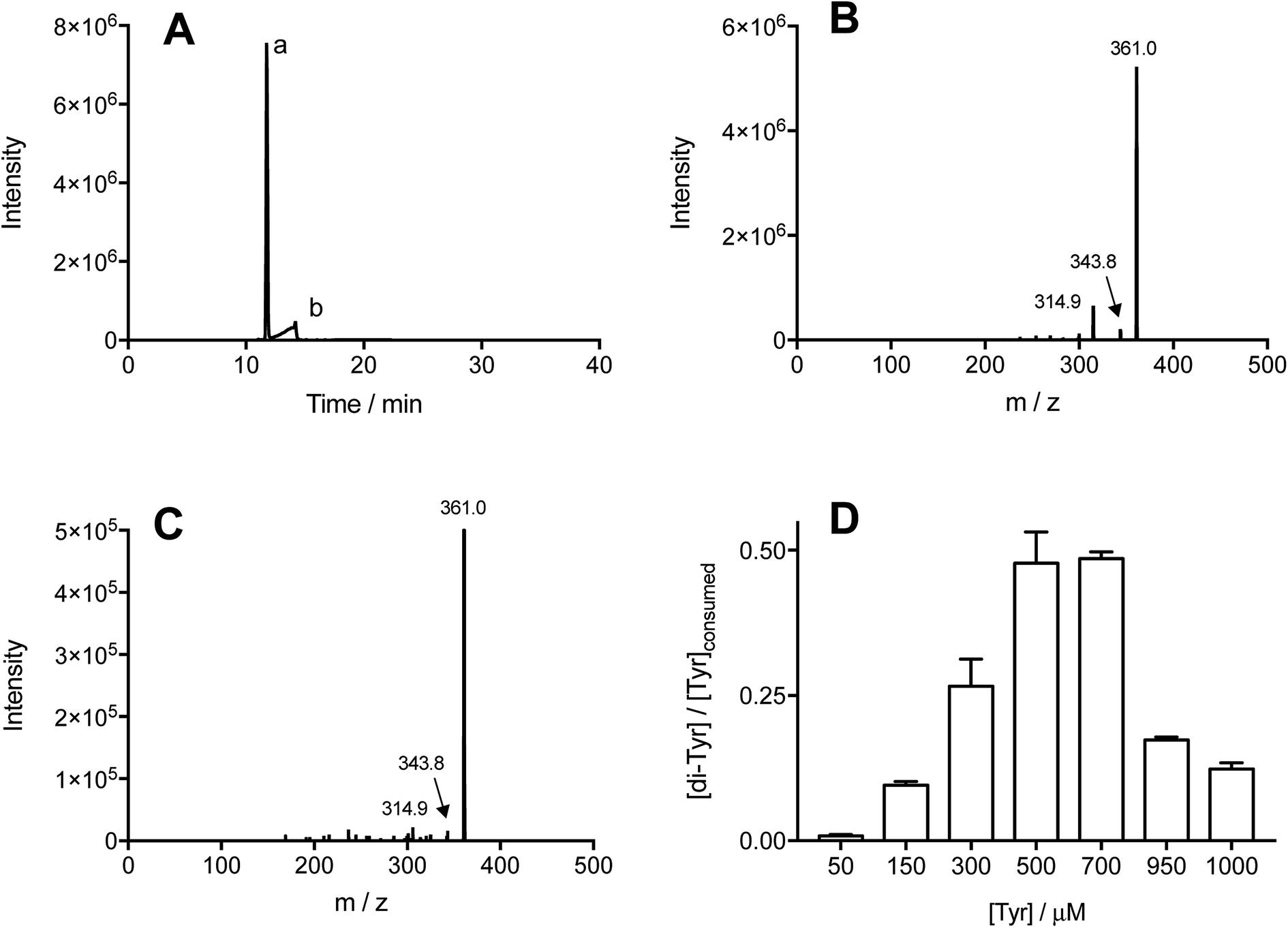 | ||
| Fig. 2 Illumination of free Tyr in the presence of [Co(NH3)5(CO3)]NO3 induces formation of di-Tyr. Tyr solutions (1 mM) in phosphate buffer (75 mM, pH 7.4, containing 0.1 mM DTPA) were illuminated for 3 min at 254 nm in the presence 4 mM of the Co(III) complex, under a N2 atmosphere. Formation of di-Tyr was detected by mass spectrometry (MS, selected reaction monitoring, SRM) as described in the Material and methods section. Panel A: MS/MS chromatogram obtained for ions that undergo the transition m/z 361 → 315. Panels B and C present representative MS/MS spectra obtained for peak a (11.7 min) and peak b (14.2 min) presented in A, respectively. Panel D shows the ratio between di-Tyr concentration and consumed Tyr, for experiments carried out at Tyr concentrations between 50 and 1000 μM. | ||
Commercial o,o′-dityrosine standard was used to construct calibration curves for both the UPLC-MS and HPLC-FL experiments (ESI Fig. 1 and 2†). As UPLC-MS allowed the detection of both isomers, this technique was used to quantify these materials in working solutions, with the assumption that isodityrosine responded in a similar manner to o,o′-dityrosine. To assess the total production yield of dityrosine (the di-Tyr acronym representing both isomers), solutions containing Tyr (50–1000 μM) and [Co(NH3)5(CO3)]NO3 (4 mM) were illuminated (as above), and the sum of the area under the curve (AUC) of the peaks corresponding to o,o′-dityrosine, and isodityrosine isomers interpolated in the calibration curve ([AUC] = 507781 + 931044 [o,o′-dityrosine, μM], r2 = 0.9990). The data obtained for di-Tyr formation (expressed as μM) were employed to determine the mass balance for Tyr oxidation. If the consumed Tyr leads exclusively to di-Tyr, the expected ratio [di-Tyr]/[Tyr]consumed should be 0.5; as two moles of Tyr are necessary to produce one mole of di-Tyr. Fig. 2D presents the experimentally determined ratio at different initial Tyr concentrations. At both low (50–300 μM) and also at high (950–1000 μM) Tyr concentrations, values <0.5 were determined, whereas at 500 and 700 μM Tyr, the ratio was close to the 0.5 expected for complete transformation of Tyr to di-Tyr.
In the case of Trp, MS analysis of solutions illuminated in the presence of [Co(NH3)5(CO3)]NO3, provided evidenced the exclusive formation of ditryptophan dimers (di-Trp, ions with m/z 407). Three peaks were detected at retention times >20 min (Fig. 3A). Two of these (20.5 and 21.2 min) showed low intensities (<150000 counts for [Trp] = 1 mM, Fig. 3A), while the third peak (31.2 min) was of significantly higher intensity (2 × 106 counts for Trp 1 mM, Fig. 3A). This implies that under these conditions (N2 atmosphere) formation of one di-Trp isomer is favored (Fig. 3A). The MS/MS spectrum of this peak, and also the lower intensity species, showed characteristic m/z 407 → 203 transitions (Fig. 3B). Analysis by HPLC with DAD and FL detection showed the presence of a peak at 32.7 min with a low intensity (mUA = 1.16 at 280 nm, ESI Fig. 5†). This species displayed a UV-vis spectrum with a band at 290 nm, and an emission fluorescence band at 346 nm on excitation at 280 nm (Fig. 3C). This signal is attributed to the di-Trp isomer detected at 31.2 min by UPLC-MS.
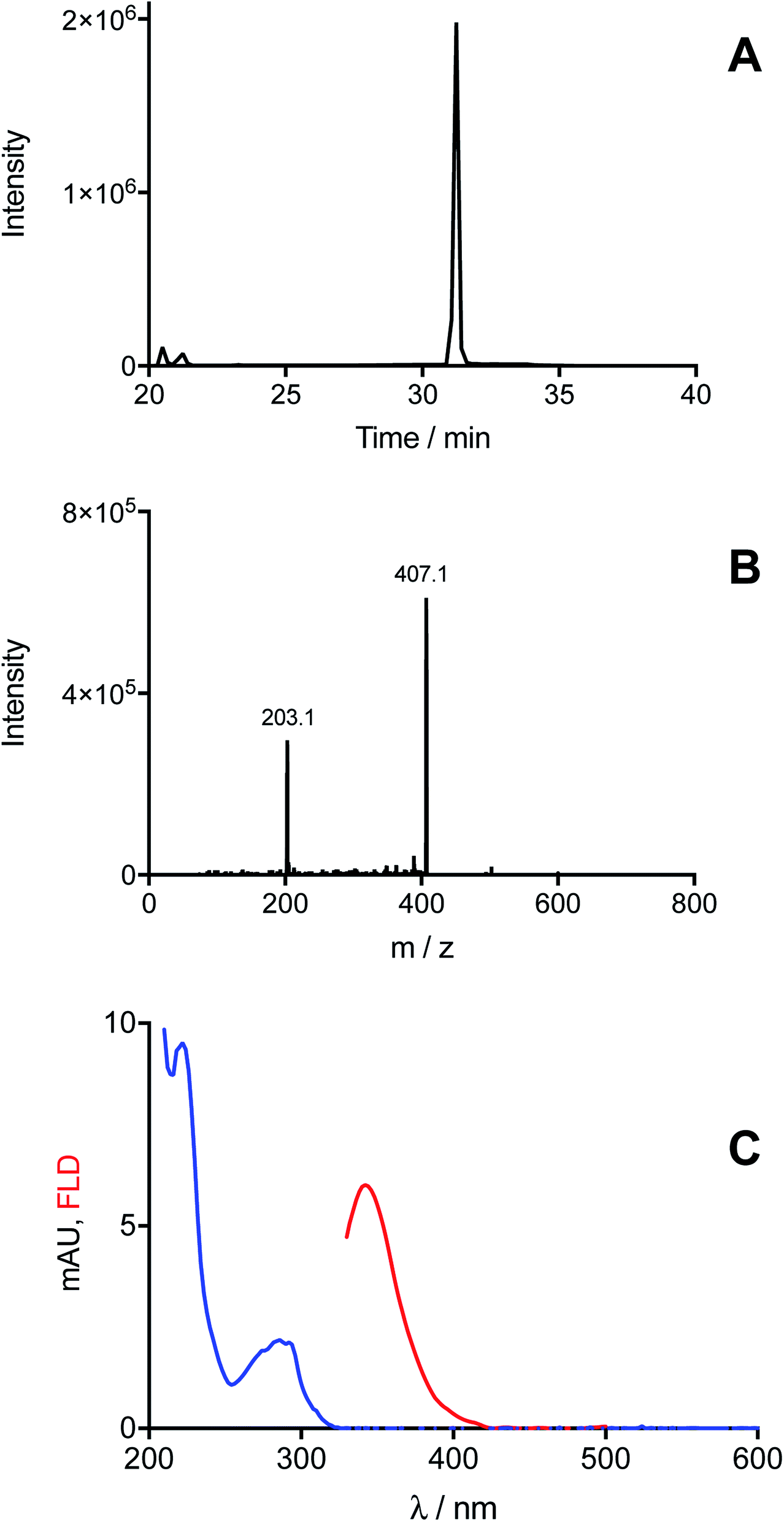 | ||
| Fig. 3 Illumination of free Trp in the presence of [Co(NH3)5(CO3)]NO3 induces formation of di-Trp. Trp solutions (1 mM) in phosphate buffer (75 mM, pH 7.4, containing 0.1 mM DTPA) were illuminated for 3 min at 254 nm in the presence 4 mM of the Co(III) complex, under a N2 atmosphere. Production of di-Trp was detected by MS (selected reaction monitoring, SRM). Panel A: MS/MS chromatogram obtained for ions that undergo the transition m/z 407 → 203. Panel B shows a representative MS/MS spectrum obtained for the peak detected at 31.2 min. Panel C represents the UV-visible (blue line) and fluorescence emission spectrum (red line) of di-Trp separated by HPLC and detected by DAD and fluorescence detectors. | ||
3.2 Formation of Tyr–Trp induced by CO3˙− in solutions containing Tyr and Trp
Illumination, under an atmosphere of N2, of solutions containing Tyr (500 μM), Trp (500 μM), and [Co(NH3)5(CO3)]NO3 (4 mM), resulted in the exclusive production of di-Tyr, di-Trp and Tyr–Trp crosslinks, as determined by UPLC-MS. The chromatograms showed the presence of three peaks (at 19.2, 21.4 and 23.6 min) with m/z 384, with MS/MS spectra showing fragmentations to give ions at 367 and/or 203 (Fig. 4A). Each of the peaks (a, b and c in Fig. 4A) showed 384 → 367 fragmentations, while only two (peaks b and c in Fig. 4A) showed 384 → 203 transitions. These data indicate that at least three different Tyr–Trp isomers are formed with slightly different MS/MS fragmentation patterns. Peak a showed fragmentation to 367, without significant formation of a m/z 203 ion (Fig. 4B), whilst peaks b and c generated both the 367 and 203 ions, but with different intensity patterns (Fig. 4C and D). In addition to these Tyr–Trp species, di-Tyr (361 → 315) and di-Trp (407 → 203) were also detected at variable levels (ESI Fig. 6†).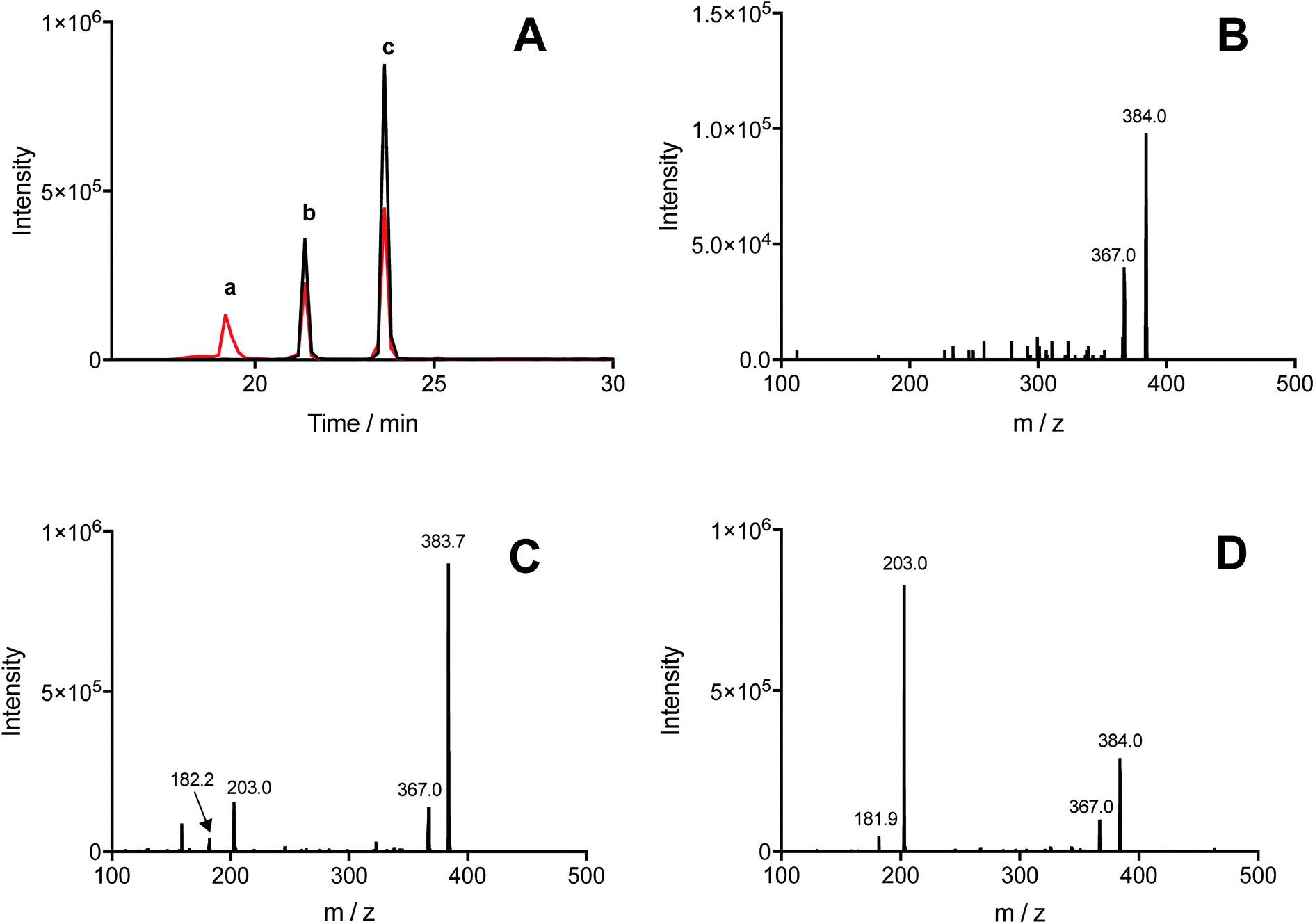 | ||
| Fig. 4 Illumination of solutions containing a mixture of free Tyr and Trp in the presence of the [Co(NH3)5(CO3)]NO3 results in the formation of Tyr–Trp adducts. Solutions containing 500 μM of Tyr and 500 μM of Trp (1 mM total concentration) in phosphate buffer (75 mM, pH 7.4, containing 0.1 mM DTPA) were illuminated for 3 min at 254 nm in the presence of 4 mM of the Co(III) complex, under a N2 atmosphere. Tyr–Trp adducts were detected by MS (selected reaction monitoring, SRM) in samples exposed to light, but not in controls (non-illuminated solutions). Panel A: MS/MS chromatogram obtained for ions undergoing a m/z 384 → 203 transition (black line) and m/z 384 → 367 transitions (red line). Panels B, C, and D show representative MS/MS spectra of peaks a, b and c, indicated in panel A (19.2, 21.4, and 23.6 min), respectively. | ||
The dependence of each signal (di-Tyr, di-Trp and Tyr–Trp) on the concentration of the parent amino acids (Tyr and Trp) were analyzed. Data obtained with a total amino acid concentration of 500 μM are presented in Fig. 5, where χTyr = 0 indicates a solution containing only Trp, and χTyr = 1 corresponds to a solution containing only Tyr. As indicated in Fig. 5A, between χTyr values of 0.05 and 0.95, the production of di-Tyr increased in a χTyr dependent manner. Interestingly, at χTyr = 1.0 (i.e. only 500 μM Tyr) the AUC for the di-Tyr peaks showed no significant differences that those obtained for χTyr = 0.95 (p* < 0.05) (Fig. 5A). In the case of di-Trp, higher AUC values were observed at χTyr = 0 and 0.05 (Fig. 5B). At χTyr > 0.5, low intensity di-Trp peaks were detected by UPLC. As expected, Tyr–Trp adducts (their UPLC-MS peaks) were only detected between χTyr = 0.05 and 0.95 (Fig. 5C). The AUC of such peaks (i.e. those with transitions 384 → 367) showed a bell-shaped curve with a maximum at χTyr = 0.5 (Fig. 5C). At a total amino acid concentration of 1 mM, the relationship between di-Tyr, di-Trp and Tyr–Trp crosslinks with the value of χTyr was similar, however a more marked decrease in di-Trp was observed at 0.3, while the maximum yield of Tyr–Trp was reached at 0.7 (ESI Fig. 7†).
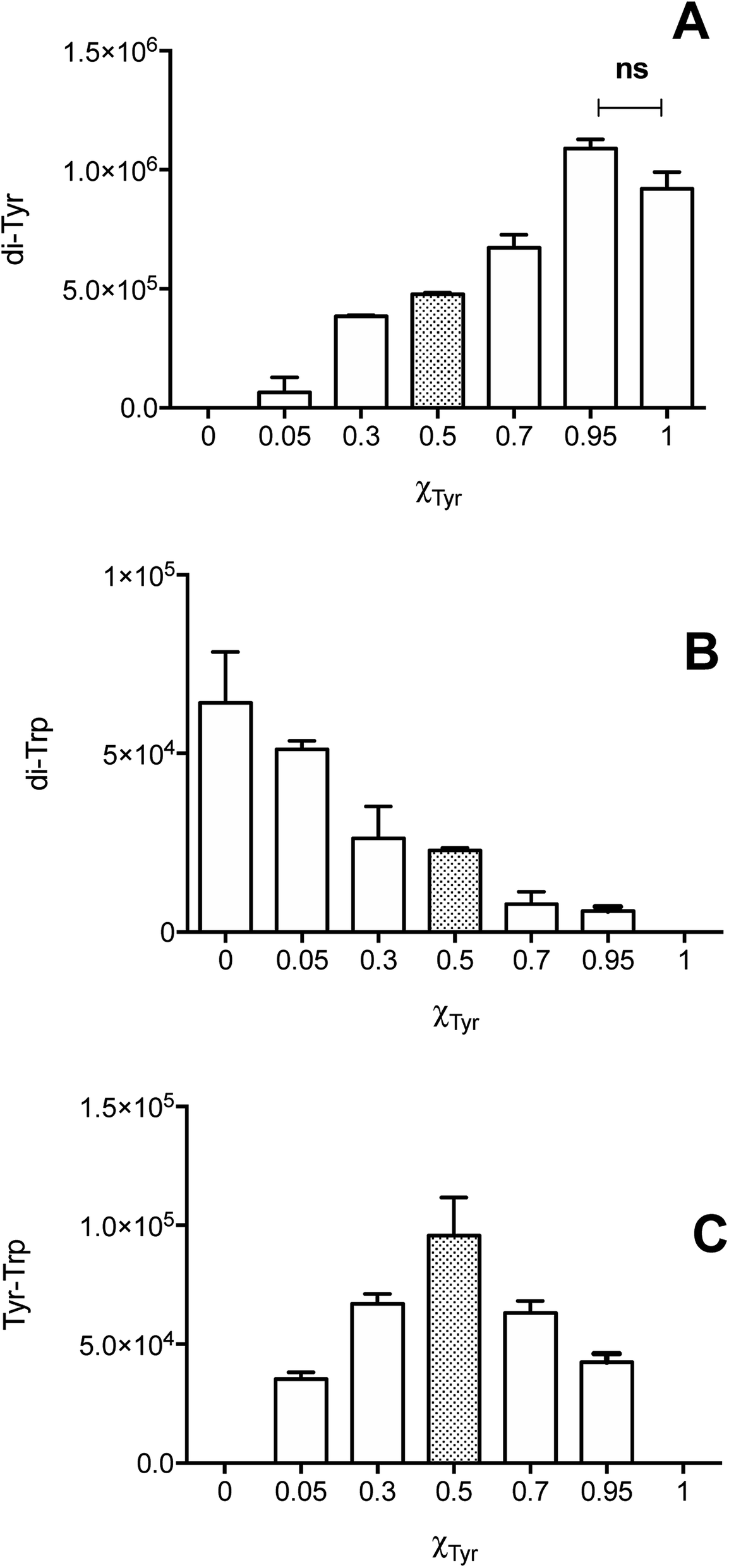 | ||
| Fig. 5 Formation of di-Tyr, di-Trp and Tyr–Trp depends on the concentration of Tyr and Trp. Solutions containing Tyr and Trp (500 μM total concentration) in phosphate buffer (75 mM, pH 7.4, containing 0.1 mM DTPA) were illuminated for 3 min at 254 nm in the presence of 4 mM [Co(NH3)5(CO3)]NO3, under a N2 atmosphere. The area under the curve of the chromatographic peaks for di-Tyr (m/z 361 → 315 transition, panel A), di-Trp (m/z 407 → 203 transition, panel B), and Tyr–Trp (m/z 384 → 367 transition, panel C), were determined by MS as described in the Material and methods section. The concentrations of Tyr and Trp were expressed as molar fraction of Tyr (χTyr), with χTyr = 0 indicating a solution containing 500 μM Trp with no Tyr, and χTyr = 1, a solution containing 500 μM Tyr and no Trp. In panel A, ns indicates no significant difference (p > 0.05) as determined by Student's t-test. | ||
The consumption of both amino acids in these mixed amino acid systems illuminated in the presence of [Co(NH3)5(CO3)]NO3 was assessed by HPLC-FL. As shown in Fig. 6A, with a total amino acid concentration of 1 mM, Tyr consumption increased as its initial concentration increased (i.e. increasing χTyr values), though with a slight down curvature. Trp consumption showed the inverse relationship with χTyr, also showing a curvature. These results are similar to those obtained detected for the individual amino acid solutions (Fig. 1B).
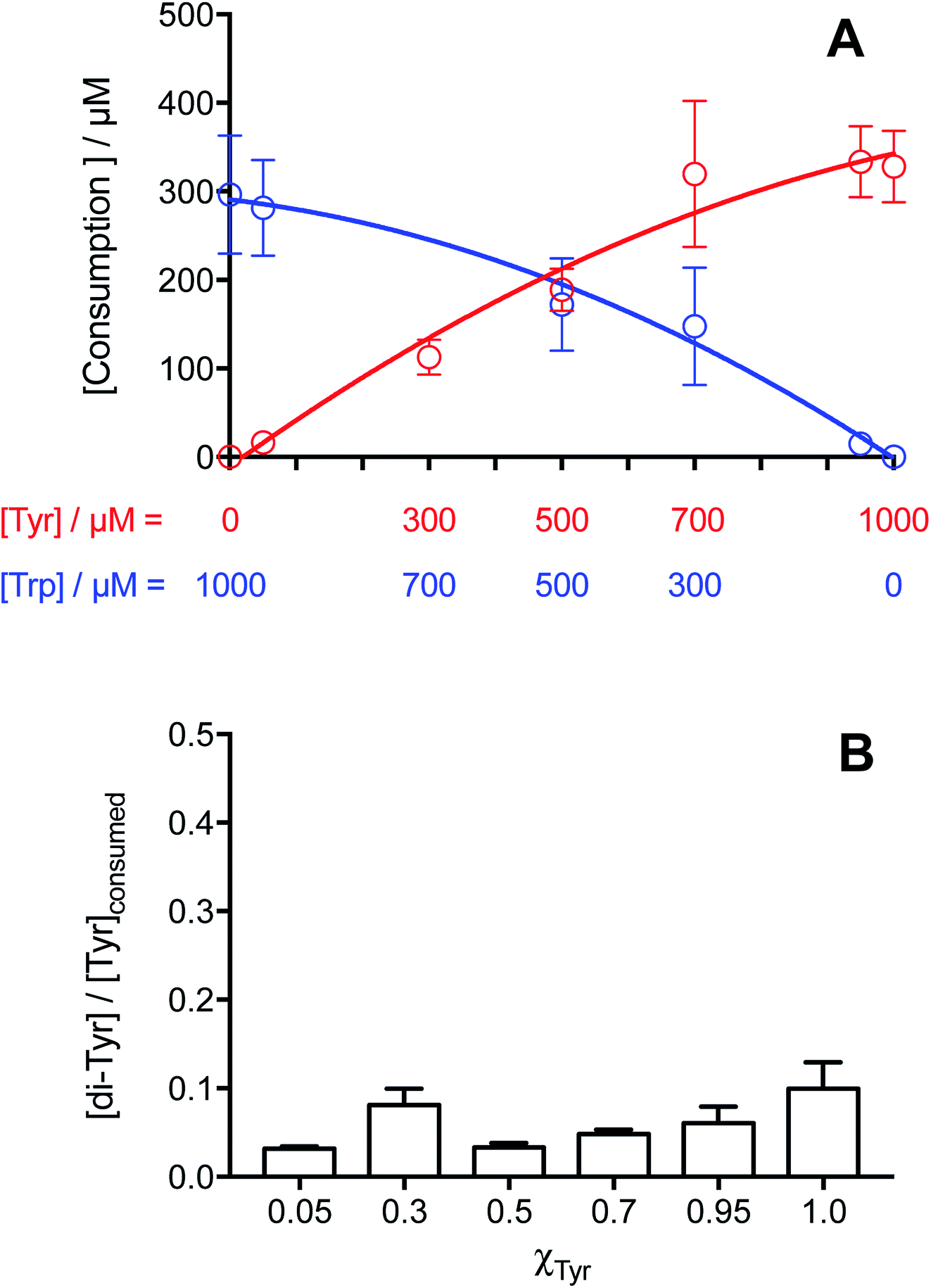 | ||
| Fig. 6 Consumption of Tyr and Trp in solutions containing both amino acids and [Co(NH3)5(CO3)]NO3 depends on the concentration of each amino acid residue. Panel A depicts the consumption (in μM) of Tyr (red symbols) and Trp (blue symbols) at the different molar fractions (expressed as amino acid concentration). Panel B shows the ratio between [di-Tyr] and consumed Tyr, for experiments carried out employing various Tyr and Trp mixtures. | ||
For the χTyr values where the mass balance implies near complete conversion of Tyr to di-Tyr, Tyr consumption should equate solely to the formation of di-Tyr and Tyr–Trp crosslinks. In this zone, low values of the [di-Tyr]/[Tyr]consumed ratio were determined (Fig. 6B), together with a similar level of Tyr and Trp consumption. In such conditions, the difference between the concentration of Tyr consumed and di-Tyr formed should equate to the extent of formation of Tyr–Trp. At χTyr = 0.5, the consumption of Tyr was ∼189 μM, and the yield of di-Tyr formed was 6 μM (and hence a [di-Tyr]/[Tyr]consumed ratio of ∼0.03), giving a yield of Tyr–Trp formation of ∼177 μM. This value in similar to that for Trp consumption, and consistent with the very low levels of di-Trp detected (ESI Fig. 7B†).
4. Discussion
Oxidation of proteins, leading to crosslinking, oligomerization and aggregation, is closely associated with a number of human pathologies, and particularly neurodegenerative diseases.1–4 It has been reported that dityrosine formation, arising from self-reaction of two Tyr˙, contributes to the observed inter-molecular crosslinks. Furthermore, the detection of this species is increasing being used as a marker of protein damage and oxidative stress.20–23 It is therefore increasingly accepted that Tyr˙ are critical intermediates in both protein oxidation in general, and especially protein crosslinking. The reactions of other protein-derived radicals (protein˙ in Scheme 1) are less well explored, even though it is well established that Trp residues are one of the most susceptible residues to oxidation.56 Thus formation of Trp˙, and cross-reaction with Tyr˙ to produce Tyr–Trp crosslinks, was hypothesized to be an alternative pathway to the formation of protein dimers and aggregation. This tenet is supported by the detection of both intra- and inter-molecular Tyr–Trp bonds in a number of systems.36,37,48–50,57 In spite of this, little effort has been expended on understanding the factors that control the formation, accumulation and biological consequences of the generation of this type of species. This is probably, at least in part, due to the absence of robust methods to detect and quantify these adducts. In the present work we have shown that exposure of mixtures of free Tyr and Trp to high concentrations of CO3˙−, generates, under anaerobic conditions, high concentrations of Tyr–Trp crosslinks. The system involves photochemical decomposition of [Co(NH3)5(CO3)]NO3 (reaction (1)) to produce CO3˙−,39,53 a strong oxidant (E0 = 1.78 V, pH 7.0) that induces one-electron oxidation of targets. Little data support the occurrence of addition reactions with this species.58CO3˙− is generated in biological environments by secondary reactions of peroxynitrite in the presence of CO2, during turnover of the enzyme xanthine oxidase, by the bicarbonate-dependent peroxidase activity of hSOD1,58 and via reaction of hydroxyl radicals (from the Fenton reaction) with the ubiquitous biological species bicarbonate.59 In the case of hSOD1 peroxidase activity, CO3˙− have been implicated in protein dimerization and the formation of non-amyloid hSOD1 aggregates.31–34
For Tyr and Trp, reaction with CO3˙− (reactions (2) and (3)) occurs with rate constants of 4.5 × 107 and 7 × 108 M−1 s−1, respectively.60
| [Co(NH3)5(CO3)]NO3 + hν → CO3˙− + Co2+ + other products | (1) |
| Tyr + CO3˙− → Tyr˙ + HCO3− | (2) |
| Trp + CO3˙− → Trp˙ + HCO3− | (3) |
In the absence of O2, the major likely reactions are self-reactions of Tyr˙ and Trp˙, to give dityrosine and ditryptophan, respectively, and cross-reaction between these species to generate Tyr–Trp dimers (reactions (4) to (6)).
| 2Tyr˙ → di-Tyr | (4) |
| 2Trp˙ → di-Trp | (5) |
| Tyr˙ + Trp˙ → Tyr–Trp | (6) |
As shown in Fig. 1A, similar extents of Tyr and Trp consumption were detected after illumination of solutions containing the Co(III) complex and 500 μM of either Tyr or Trp. This indicates that, independent of the differences in the kinetics of oxidation (with k for Trp being significantly higher, see above), both amino acids are consumed with similar stoichiometries. The relationship between the consumption of Tyr and Trp with their initial concentration, also showed a similar behavior, highlighting a downward curvature at initial concentrations >500 μM (Fig. 1B). The increase in Tyr and Trp consumption with higher initial concentrations implies that excess CO3˙− are generated in the system, with the alternative fate of excess CO3˙− being reaction (7), which has k 1.5 × 107 M−1 s−1.61
| CO3˙− + CO3˙− → CO2 + CO42− | (7) |
Therefore, under our experimental conditions, one-electron oxidation of Tyr and Trp (reactions (4) and (5), respectively) occurs in parallel with self-reaction of CO3˙− (reaction (7)). These data are consistent with the observed high stoichiometric factor (n) detected for both amino acids, and imply that not all the generated CO3˙− react with Tyr or Trp.
Analysis of dityrosine formation in solutions of free Tyr exposed to CO3˙− showed the formation of two isomers by UPLC-MS, with a single species detected by HPLC-FL (Fig. 2A and ESI Fig. 4†). This behavior indicates that both the thermodynamically-favored (C–C bonded) species o,o′-dityrosine (ΔG = −195.4 kJ mol−1), and the alternative C–O bonded isomer isodityrosine (ΔG = −119.2 28.5 kJ mol−1) are produced.62 Interestingly, lower concentrations of Tyr gave rise to a lower yield of the isodityrosine isomer relative to o,o′-dityrosine (data not shown) suggesting that its production is favored by a high steady state concentration of Tyr˙. It should be noted that the yield of dityrosine is often quantified by fluorescence (e.g. after HPLC separation), an approach that does not detect isodityrosine. This is unfortunate as the latter appears to be important in some biological contexts, including the crosslinking of plant cell glycoproteins,63–65 and reactions mediated by human phagocytes.55 In order to determine whether (under the conditions employed) the extent of Tyr consumption could be explained solely by the formation of dityrosine isomers (di-Tyr), the ratio of [di-Tyr]/[Tyr]consumed was determined at different initial Tyr concentrations. As depicted in Fig. 2D, at Tyr concentrations lower than 500 μM (i.e. 50, 150, and 300 μM), as well as at high concentrations (950 and 1000 μM), values significantly <0.5 were determined. This may be due to the occurrence of further reactions of di-Tyr isomers, including the formation of oligomers. In the low concentration regime, the high fluxes of CO3˙− might be expected to facilitate such processes, whilst with high Tyr concentrations, a decreased yield of CO3˙− production due to a higher absorption of light of the parent Tyr could explain the low values of the [di-Tyr]/[Tyr]consumed ratio. With moderate concentrations of Tyr (e.g. 500 and 700 μM) (Fig. 2D) (ESI Fig. 7A†), values close to the expected value of 0.5 were obtained, indicating that all the oxidized Tyr are converted to di-Tyr (reaction (4)). Thus, there appears to be a delicate balance between the concentrations of CO3˙− and Tyr required to produce a maximum (quantitative) yield of di-Tyr.
Dimerization of Trp residues and consequent protein crosslinking has been less well studied than with di-Tyr, though a number of examples have been reported in the literature.31,36,37,40 Pulse radiolysis studies indicate that dimerization of Trp˙ occurs with k ∼1 × 108 M−1 s−1 for both free and peptide Trp residues.28 Exposure of Trp-containing peptides and lysozyme to Co60 γ-radiation, peroxynitrous acid (ONOOH), or CO3˙− derived from the bovine SOD1/hydrogen peroxide system, has been reported to yield at least four ditryptophan isomers (detected as ions with m/z 407 by MS), and with different retention times; this consistent with the delocalization of the unpaired electron across the indole ring of Trp˙ with significant spin density present at N1 and C3 of the pyrrole ring, as well as carbon atoms of the benzene ring.28 Interestingly, this study did not detected Trp dimers with CO3˙−,28 a result that is inconsistent with the current study; this may be due to the presence of O2 and or the low flux of CO3˙− employed in this previous work.28,66 Photo-oxidation of free Trp and its N-acetyl derivative employing the photosensitizers kynurenic acid41 and riboflavin42 has also resulted in the detection of ditryptophan isomers, with these processes being influenced by the presence of O2. Of particular interest is the detection of five ditryptophan dimers, and a trimer on exposure of free Trp to high intensity 365 nm light in the presence of riboflavin, a system that generates a high steady state concentration of Trp˙.42 In the present work, we took advantage of the photolabile [Co(NH3)5(CO3)]NO3 complex to produce CO3˙−, and high yields of Trp dimers. This resulted in the detection of four ditryptophan species (di-Trp, detected as species with m/z 407 ions, and fragmentation ions with m/z 203), however one of these species was considerably favored (Fig. 3A).
UPLC-MS/MS studies of Trp and Tyr mixtures (250 μM each amino acid) exposed to CO3˙−, showed formation of di-Tyr, di-Trp and Tyr–Trp crosslinks. The latter were evidenced by the presence of two peaks with m/z 384 (consistent with the expected mass of a single-charged ion from a Tyr–Trp crosslink) and fragmentation ions with m/z 367, 203 and 182 (ESI Fig. 6†). Interestingly, with solutions containing 500 μM of each amino acid, the same chromatographic species were detected, together with an additional species that eluted at 19.2 min, and only gave a fragment ion with m/z 367, and not m/z 203 and 182 ions (Fig. 4). The transition m/z 384 → 203 is consistent with the formation of Tyr and Trp ions, while the ion with m/z 367 results from deamination at the α-carbon of either the Tyr or Trp moiety (Fig. 7). We speculate that the m/z 384 → 203 fragmentation arises from the presence of species with relatively weak C–O or C–N bonds (structures 1 and 4 in Fig. 7), whereas the species that only undergoes deamination (i.e. m/z 384 → 367) has a much stronger C–C bond (i.e. structure 3 in Fig. 7). In a number of these cases, the species are likely to be stereoisomer pairs, as addition of the Tyr phenoxyl radical, or the C3 radical on the phenol ring, could occur at C3 on the indole ring of Trp via either face. Another alternative crosslinked species where the radical centered on the indole nitrogen of Trp, links to the phenol oxygen of Tyr (i.e. structure 2 in Fig. 7), is likely to be very labile due to the weak nature of the N–O bond, and is unlikely to be detected under the MS conditions employed here.
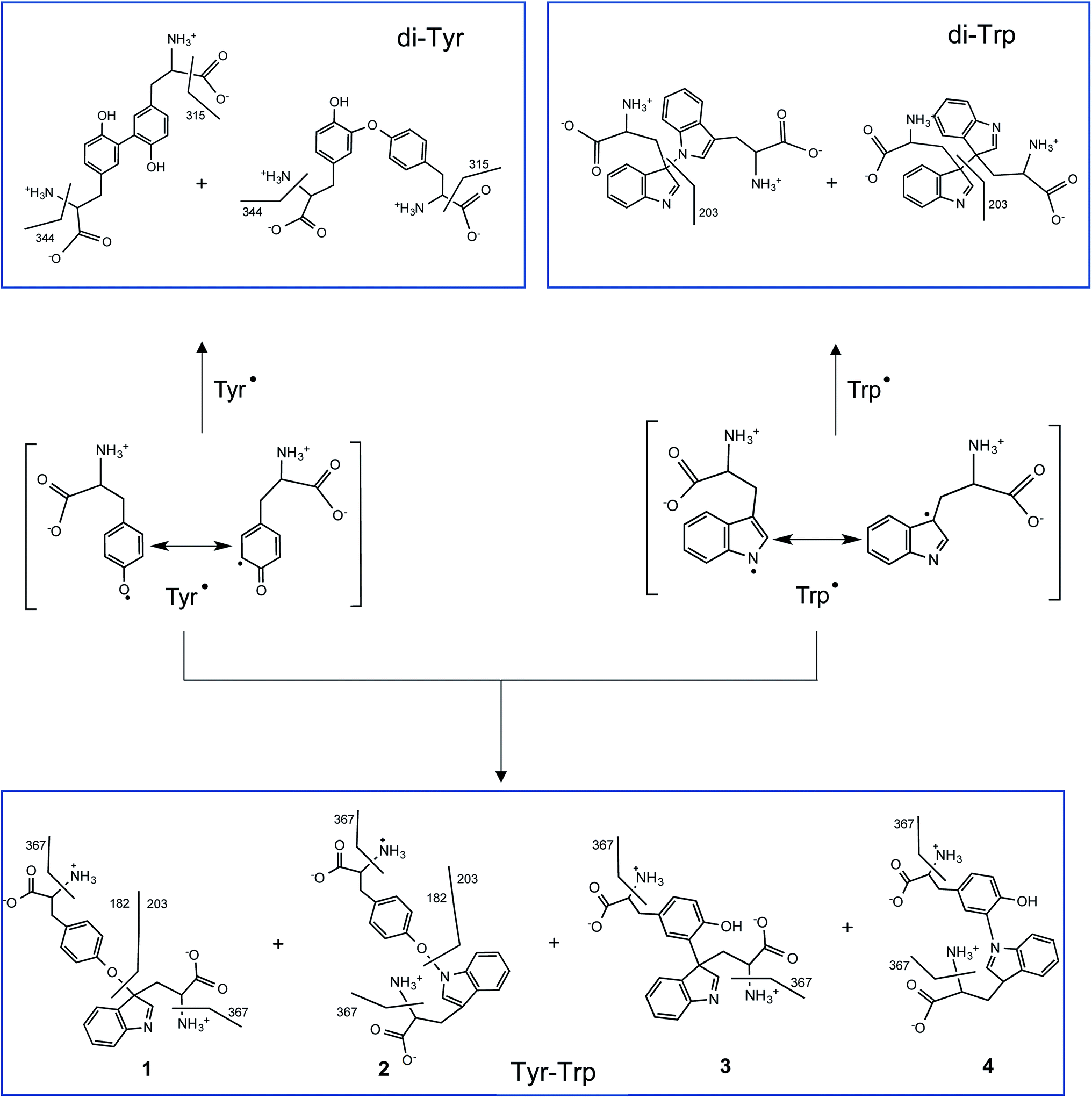 | ||
| Fig. 7 Proposed radical–radical reactions giving rise to di-Tyr, di-Trp and Tyr–Trp formation. It should be noted that only some of the possible resonance structures are depicted for the delocalization of the unpaired electron in Tyr˙ and Trp˙. Crosslinks involving C–O, C–C, O–N and C–N bonds, and their proposed fragmentation pathways are presented. Some of the di-Trp and Tyr–Trp dimers are likely to be stereoisomeric pairs (those formed at C3 of Trp due to reaction at either face of the indole ring, see Carroll et al.,28) these are not indicated for reasons of clarity. | ||
Crosslinking between Tyr˙ and Trp˙ (reaction (6)) to give Tyr–Trp, appears to compete with self-reactions of these radicals to give di-Tyr and di-Trp (reactions (4) and (5)). This competition will depend on the kinetic rate constants of each process, as well as the concentrations of Tyr˙ and Trp˙. If the rate constant for reaction (6) is of the same order of magnitude as those for reactions (4) and (5), as would seem likely, it would be expected that the efficiency of Tyr–Trp formation would depend on the relative concentrations of Tyr˙ and Trp˙ and hence the concentrations of the parent amino acids. On this basis equimolar concentrations of Tyr and Trp might be expected to favor the formation of similar yields of Tyr˙ and Trp˙ and hence higher levels of Tyr–Trp than di-Tyr and di-Trp. This is consistent with the observed data that show that the maximum Tyr–Trp AUC is observed at around χTyr = 0.5 (Fig. 5C and ESI Fig. 7C†). Overall these results suggest that cross-termination reactions of Tyr˙ and Trp˙ is a facile and competitive process, especially when compared to self-reaction of Tyr˙ (to give di-Tyr) and Trp˙ (to give di-Trp), with the pattern of Tyr and Trp consumption assessed in mixtures of both amino acids resembling the behavior observed when each amino acid was studied separately (Fig. 1B versus Fig. 6A). However, the absolute quantification of each of isomer species and potential remains to be accomplished, with only some of the potential complement of di-Trp and Tyr–Trp isomers detected by MS both in this study and previously.28 Whether the ‘missing’ isomers (cf. Fig. 7) are not formed in significant yields, or are formed but just not detected, remains to be established.
5. Conclusions
Photochemical decomposition of [Co(NH3)5(CO3)]NO3 has been shown to produce high yields of CO3˙−, and hence dimers from Tyr (di-Tyr) and Trp (di-Trp), together with high (micromolar) concentrations of Tyr–Trp crosslinks under an atmosphere of N2. The relative yield of each species depends on the initial concentrations of Tyr and Trp, as well as the yields of Tyr˙ and Trp˙. The experimental methods presented here allow ready formation of Tyr–Trp adducts, and these should be easily adapted to allow generation of authentic standards, and isotope-labelled materials for use in more complex systems where these species might be detected and characterized by the specific parent and fragment ions, and the corresponding transitions, by MS. These data will hopefully guide future investigations into the presence, and biological consequences of Tyr–Trp crosslink formation.Conflicts of interest
There are no conflicts to declare.Abbreviations
| di-Tyr | o,o′-Dityrosine (3,3′-dityrosine) and isodityrosine |
| di-Trp | Ditryptophan dimers |
| HPLC | High performance liquid chromatography |
| UPLC | Ultra performance liquid chromatography |
| MS | Mass spectrometry |
| FL | Fluorescence spectrophotometry |
Acknowledgements
This work was supported by Fondecyt grant no 1180642 (to CLA) and the Novo Nordisk Foundation (Laureate grant: NNF13OC0004294 to MJD). CLA also thanks FONDEQUIP (EQM130032) for an equipment grant.References
- M. J. Davies, Biochem. J., 2016, 473, 805–825 CrossRef CAS PubMed.
- A. Krisko and M. Radman, Open Biol., 2019, 9, 180249 CrossRef PubMed.
- E. H. Sharman, Reactive oxygen species and protein oxidation in neurodegenerative disease, Springer, Cham, 2016, pp. 199–212 Search PubMed.
- E. R. Stadtman, Ann. N. Y. Acad. Sci., 2006, 928, 22–38 CrossRef PubMed.
- C. L. Hawkins and M. J. Davies, Biochim. Biophys. Acta, 2001, 1504, 196–219 CrossRef CAS.
- C. L. Hawkins and M. J. Davies, J. Biol. Chem., 2019, 294, 19683–19708 CrossRef CAS PubMed.
- C. López-Alarcón, A. Arenas, E. Lissi and E. Silva, Biomol. Concepts, 2014, 5, 119–130 Search PubMed.
- M. J. Davies, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1703, 93–109 CrossRef CAS PubMed.
- B. Halliwell and J. M. C. Gutteridge, Free radicals in biology and medicine, 2015 Search PubMed.
- C. Houée-Lévin, K. Bobrowski, L. Horakova, B. Karademir, C. Schöneich, M. J. Davies and C. M. Spickett, Free Radical Res., 2015, 49, 347–373 CrossRef PubMed.
- B. P. Partlow, M. B. Applegate, F. G. Omenetto and D. L. Kaplan, ACS Biomater. Sci. Eng., 2016, 2, 2108–2121 CrossRef CAS.
- E. Lévy, N. El Banna, D. Baïlle, A. Heneman-Masurel, S. Truchet, H. Rezaei, M.-E. Huang, V. Béringue, D. Martin and L. Vernis, Int. J. Mol. Sci., 2019, 20, 3896 CrossRef PubMed.
- K. Wolhuter, H. J. Whitwell, C. H. Switzer, J. R. Burgoyne, J. F. Timms and P. Eaton, Mol. Cell, 2018, 69, 438–450.e5 CrossRef CAS PubMed.
- K. A. Broniowska and N. Hogg, Antioxid. Redox Signaling, 2012, 17, 969–980 CrossRef CAS PubMed.
- A. C. Krämer, A. Torreggiani and M. J. Davies, J. Agric. Food Chem., 2017, 65, 10258–10269 CrossRef PubMed.
- E. Serebryany, J. C. Woodard, B. V. Adkar, M. Shabab, J. A. King and E. I. Shakhnovich, J. Biol. Chem., 2016, 291, 19172–19183 CrossRef CAS PubMed.
- Y. K. Al-Hilaly, L. Biasetti, B. J. F. Blakeman, S. J. Pollack, S. Zibaee, A. Abdul-Sada, J. R. Thorpe, W.-F. Xue and L. C. Serpell, Sci. Rep., 2016, 6, 39171 CrossRef CAS PubMed.
- Y. K. Al-Hilaly, T. L. Williams, M. Stewart-Parker, L. Ford, E. Skaria, M. Cole, W. Bucher, K. L. Morris, A. Sada, J. R. Thorpe and L. C. Serpell, Acta Neuropathol. Commun., 2013, 1, 83 CrossRef PubMed.
- K. Hensley, M. L. Maidt, Z. Yu, H. Sang, W. R. Markesbery and R. A. Floyd, J. Neurosci., 1998, 18, 8126–8132 CrossRef CAS PubMed.
- C. Giulivi, N. J. Traaseth and K. J. A. Davies, Amino Acids, 2003, 25, 227–232 CrossRef CAS PubMed.
- G.-R. Wu, M. Cheserek, Y.-H. Shi, L.-Y. Shen, J. Yu and G.-W. Le, Ann. Nutr. Metab., 2015, 66, 44–50 CrossRef CAS PubMed.
- T. DiMarco and C. Giulivi, Mass Spectrom. Rev., 2007, 26, 108–120 CrossRef CAS PubMed.
- A. Annibal, G. Colombo, A. Milzani, I. Dalle-Donne, M. Fedorova and R. Hoffmann, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1019, 147–155 CrossRef CAS PubMed.
- E. Fuentes-Lemus, E. Silva, F. Leinisch, E. Dorta, L. G. Lorentzen, M. J. Davies and C. López-Alarcón, Food Chem., 2018, 256, 119–128 CrossRef CAS PubMed.
- T. K. Dalsgaard, J. H. Nielsen, B. E. Brown, N. Stadler and M. J. Davies, J. Agric. Food Chem., 2011, 59, 7939–7947 CrossRef CAS PubMed.
- Y. Yang, H. Zhang, B. Yan, T. Zhang, Y. Gao, Y. Shi and G. Le, J. Agric. Food Chem., 2017, 65, 6957–6971 CrossRef CAS PubMed.
- C. P. Stanley, G. J. Maghzal, A. Ayer, J. Talib, A. M. Giltrap, S. Shengule, K. Wolhuter, Y. Wang, P. Chadha, C. Suarna, O. Prysyazhna, J. Scotcher, L. L. Dunn, F. M. Prado, N. Nguyen, J. O. Odiba, J. B. Baell, J.-P. Stasch, Y. Yamamoto, P. Di Mascio, P. Eaton, R. J. Payne and R. Stocker, Nature, 2019, 566, 548–552 CrossRef CAS PubMed.
- L. Carroll, R. F. Anderson, C. Lopez-Alarcon and M. J. Davies, Free Radic. Biol. Med., 2017, 113, 132–142 CrossRef CAS PubMed.
- M. Ehrenshaft, L. J. Deterding and R. P. Mason, Free Radic. Biol. Med., 2015, 89, 220–228 CrossRef CAS PubMed.
- J. Mizdrak, R. J. W. Truscott, J. F. Jamie and M. J. Davies, Free Radic. Biol. Med., 2008, 44, 1108–1119 CrossRef CAS PubMed.
- D. B. Medinas, A. H. Iglesias and O. Augusto, Free Radic. Biol. Med., 2010, 49, 1046–1053 CrossRef CAS PubMed.
- F. R. Coelho, A. Iqbal, E. Linares, D. F. Silva, F. S. Lima, I. M. Cuccovia and O. Augusto, J. Biol. Chem., 2014, 289, 30690–30701 CrossRef CAS PubMed.
- E. Pokrishevsky, L. McAlary, N. E. Farrawell, B. Zhao, M. Sher, J. J. Yerbury and N. R. Cashman, Sci. Rep., 2018, 8, 15590 CrossRef PubMed.
- S. Grimm, A. Hoehn, K. J. Davies and T. Grune, Free Radical Res., 2011, 45, 73–88 CrossRef CAS PubMed.
- P. S. Sherin, E. A. Zelentsova, E. D. Sormacheva, V. V. Yanshole, T. G. Duzhak and Y. P. Tsentalovich, Phys. Chem. Chem. Phys., 2016, 18, 8827–8839 RSC.
- E. Fuentes-Lemus, M. Mariotti, J. Reyes, F. Leinisch, P. Hägglund, E. Silva, M. J. Davies and C. López-Alarcón, Free Radic. Biol. Med., 2020, 152, 61–73 CrossRef CAS PubMed.
- E. Fuentes-Lemus, M. Mariotti, P. Hägglund, F. Leinisch, A. Fierro, E. Silva, C. López-Alarcón and M. J. Davies, Free Radic. Biol. Med., 2019, 143, 375–386 CrossRef CAS PubMed.
- E. Fuentes-Lemus, E. Silva, P. Barrias, A. Aspee, E. Escobar, L. G. Lorentzen, L. Carroll, F. Leinisch, M. J. Davies and C. López-Alarcón, Free Radic. Biol. Med., 2018, 124, 176–188 CrossRef CAS PubMed.
- V. Paviani, R. F. Queiroz, E. F. Marques, P. Di Mascio and O. Augusto, Free Radic. Biol. Med., 2015, 89, 72–82 CrossRef CAS PubMed.
- E. D. Savina, Y. P. Tsentalovich and P. S. Sherin, Free Radic. Biol. Med., 2020, 152, 482–493 CrossRef CAS PubMed.
- E. D. Sormacheva, P. S. Sherin and Y. P. Tsentalovich, Free Radic. Biol. Med., 2017, 113, 372–384 CrossRef CAS PubMed.
- E. Silva, P. Barrias, E. Fuentes-Lemus, C. Tirapegui, A. Aspee, L. Carroll, M. J. Davies and C. López-Alarcón, Free Radic.. Biol. Med., 2019, 131, 133–143 CrossRef CAS PubMed.
- L. P. Candeias, P. Wardman and R. P. Mason, Biophys. Chem., 1997, 67, 229–237 CrossRef CAS PubMed.
- E. P. L. Hunter, M. F. Desrosiers and M. G. Simic, Free Radic. Biol. Med., 1989, 6, 581–585 CrossRef CAS PubMed.
- L. Carroll, D. I. Pattison, J. B. Davies, R. F. Anderson, C. Lopez-Alarcon and M. J. Davies, Free Radic. Biol. Med., 2018, 118, 126–136 CrossRef CAS PubMed.
- J. M. Gebicki, T. Nauser, A. Domazou, D. Steinmann, P. L. Bounds and W. H. Koppenol, Amino Acids, 2010, 39, 1131–1137 CrossRef CAS PubMed.
- L. K. Folkes, M. Trujillo, S. Bartesaghi, R. Radi and P. Wardman, Arch. Biochem. Biophys., 2011, 506, 242–249 CrossRef CAS PubMed.
- B. Bhaskar, C. E. Immoos, H. Shimizu, F. Sulc, P. J. Farmer and T. L. Poulos, J. Mol. Biol., 2003, 328, 157–166 CrossRef CAS PubMed.
- Y. Yamada, T. Fujiwara, T. Sato, N. Igarashi and N. Tanaka, Nat. Struct. Biol., 2002, 9, 691–695 CrossRef CAS PubMed.
- F. Leinisch, M. Mariotti, M. Rykaer, C. Lopez-Alarcon, P. Hägglund and M. J. Davies, Free Radic. Biol. Med., 2017, 112, 240–252 CrossRef CAS PubMed.
- F. Basolo, R. K. Murmann, J. E. Whitney and C. L. Rollinson, Inorganic Syntheses., 1953, 4, 171–176 CAS.
- F. Jin, J. Leitich and C. von Sonntag, J. Photochem. Photobiol., A, 1995, 92, 147–153 CrossRef CAS.
- V. W. Cope, S.-N. Chen and M. Z. Hoffman, J. Am. Chem. Soc., 1973, 95, 3116–3121 CrossRef CAS.
- C. L. Hawkins, P. E. Morgan and M. J. Davies, Free Radic. Biol. Med., 2009, 46, 965–988 CrossRef CAS PubMed.
- J. S. Jacob, D. P. Cistola, F. F. Hsu, S. Muzaffar, D. M. Mueller, S. L. Hazen and J. W. Heinecke, J. Biol. Chem., 1996, 271, 19950–19956 CrossRef CAS PubMed.
- J. Butler, E. J. Land, W. A. Prütz and A. J. Swallow, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1982, 705, 150–162 CrossRef CAS.
- P. Hägglund, M. Mariotti and M. J. Davies, Expert Rev. Proteomics, 2018, 15, 665–681 CrossRef PubMed.
- D. B. Medinas, G. Cerchiaro, D. F. Trindade and O. Augusto, IUBMB Life, 2007, 59, 255–262 CrossRef CAS PubMed.
- E. Illés, A. Mizrahi, V. Marks and D. Meyerstein, Free Radic. Biol. Med., 2019, 131, 1–6 CrossRef PubMed.
- S. Chen and M. Z. Hoffman, Radiat. Res., 1973, 56, 40–47 CrossRef CAS PubMed.
- O. Augusto, M. G. Bonini, A. M. Amanso, E. Linares, C. C. X. Santos and S. L. De Menezes, Free Radic. Biol. Med., 2002, 32, 841–859 CrossRef CAS PubMed.
- S. Karmakar and A. Datta, J. Phys. Chem. B, 2017, 121, 7621–7632 CrossRef CAS PubMed.
- S. Waffenschmidt, J. P. Woessner, K. Beer and U. W. Goodenough, Plant Cell, 1993, 5, 809–820 CAS.
- M. A. Held, L. Tan, A. Kamyab, M. Hare, E. Shpak and M. J. Kieliszewski, J. Biol. Chem., 2004, 279, 55474–55482 CrossRef CAS PubMed.
- S. C. Fry, Biochem. J., 1982, 204, 449 CrossRef CAS PubMed.
- J. D. Figueroa, E. Fuentes-Lemus, E. Dorta, V. Melin, J. Cortés-Ríos, M. Faúndez, D. Contreras, A. Denicola, B. Álvarez, M. J. Davies and C. López-Alarcón, Redox Biol., 2019, 24, 101207 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04051g |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |