
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasticity control of poly(vinyl alcohol)–graphene oxide nanocomposites
Tatiana V. Panova *,
Anna A. Efimova,
Anna K. Berkovich and
Aleksander V. Efimov
*,
Anna A. Efimova,
Anna K. Berkovich and
Aleksander V. Efimov
Faculty of Chemistry, Lomonosov Moscow State University, Leninskiye Gory 1-3, 119991 Moscow, Russia. E-mail: tvk@genebee.msu.ru
First published on 23rd June 2020
Abstract
Composite films containing poly(vinyl alcohol) filled with different amounts of graphene oxide (2 and 4 wt%) were prepared by the solution casting technique, and the mechanical properties of the resulting materials were modified with different amounts of glycerol as a plasticizer. Two series of pure poly(vinyl alcohol) and graphene oxide-loaded films with fixed amounts of water were used for modification with glycerol, since water can also serve as a plasticizer for poly(vinyl alcohol). The morphology and physical properties of the plasticized and non-plasticized composites were studied; tensile tests were performed to investigate and compare their mechanical properties. Glycerol addition does not affect the excellent compatibility of the filler with the polymer matrix and uniform distribution of graphene oxide in poly(vinyl alcohol). For poly(vinyl alcohol)/graphene oxide films an increase of the Young's modulus and yield stress was found with an increase of the filler content; the Young's modulus for poly(vinyl alcohol) filled with 4 wt% of graphene oxide is almost two times higher than that of the pure polymer. Simultaneously, a sharp decrease of the elongation at break from 80% for pure PVA to about 5% for the PVA/GO composite with 4 wt% of GO is observed, and the film's brittleness dramatically increases. It was shown that the addition of glycerol to the composite films leads both to the Young's modulus decrease and tensile energy at break increase; here the Young's modulus decreases by 18 times after addition of 20 wt% of glycerol to the poly(vinyl alcohol) film filled with 4 wt% of graphene oxide. Thus, the use of plasticizer results in a significant increase of the ductile properties of graphene oxide filled poly(vinyl alcohol) composite films, and the higher the water content in the composite film, the more drastic the increase of the ductile properties observed.
Introduction
Polymer composites and nanocomposite materials are intensively investigated these days. The possibility to combine useful properties of several components in one material leads to the increasing amount of work in this field.1–11 Poly(vinyl alcohol) (PVA) is rather frequently used as a matrix for preparation of composite materials.12–20 PVA is a semi-crystalline polymer with excellent film-forming properties, and it is easily soluble in water which makes the processing of composite materials having PVA as a matrix from solutions eco-friendly. PVA also has one of the largest production outputs in the world of synthetic polymers, so it is easily available. Usually PVA is combined with nanofillers to modify the mechanical properties as well as its electroconductive, optical, and gas barrier properties, and several others.5,7,8 Graphene oxide (GO) is extremely popular material nowadays for using in nanocomposites as a filler.21–24 GO represents a two-dimensional material with a high aspect ratio having a single layer of sp2-hybridized carbon atoms, with planars and edges decorated with oxygen-containing functional groups, such as hydroxyl, epoxides, carbonyls, and carboxyls.25,26 GO is a commercially-available product, which can be obtained in large-scale amounts. Using GO one can modify the mechanical or thermal properties of the composite materials. Since GO contains multiply hydrophilic groups it can be easily dispersed in water, which makes it compatible with water-soluble polymers as a matrix and provides a uniform distribution of this filler in the resulting composite material. Thus, hydrogen bonding between the oxygen-containing groups situated on the graphene oxide layers and hydroxyl groups of the polymer matrix provides an excellent compatibility of GO with PVA matrix, which determines an interest to the PVA–GO nanocomposites.27–31 Pure PVA films are rather brittle with little or no thermoplasticity as a consequence of the polymer crystallinity; but PVA composite films containing GO are usually even more brittle. Plasticizers are usually added to modify the mechanical properties of PVA and to make the polymer more flexible. The choice of plasticizer is determined by the nature of a polymer, and glycerol is one of the common plasticizers used for PVA modification along with propylene glycol or other polyols.32–35 Plasticizer blended with polymer increases the interchain distances, and thus the free volume in a polymer material, enhancing the macromolecular mobility of polymer chains. Thus, all polymer structure became less dense, and chains flexibility improves. In the case of PVA–glycerol system, plasticizer can also destroy the hydrogen bonds between macromolecules, which additionally improve the chains mobility. It was shown that the addition of plasticizer could significantly change the plasticity of PVA films and increase its flexibility. With an increase of a plasticizer amount in the PVA the higher elongation at break can be achieved and the tensile strength became lower. Water also serves as a PVA plasticizing agent, but glycerol is much more preferable since it has significantly low vapor pressure compare to water, thus, the glycerol content in a polymer composite usually determines by the initial glycerol loading and does not change with the rises and falls of atmosphere relative humidity and temperature.Both pure and covalently modified with different polymers GO as well as GO based composites are also extensively investigated as candidate for potential biomedical applications, drug delivery systems, materials for transdermal drug delivery.36 The antibacterial activity of GO and GO containing polymer films was also found in ref. 37 and 38. From this point of view, the mechanical properties of the GO composite materials, and in particular the flexibility of composite films, are very important, suitable films should have a relative high tensile strength and elongation at break.
Thus, PVA forms very uniform composites with GO, however the plasticity of these composites decreases with the increase of GO content even at the addition of rather small amount of the filler. Up to now the amount of publications concerning the influence of glycerol as a plasticizer on the mechanical properties of PVA nanocomposites is very limited. The authors of ref. 39 studied the mechanical properties of PVA nanocomposite filled with graphene and found the changes of their mechanical properties as a result of PVA matrix plastification. In this work we tried to improve the mechanical properties, in particular, the ductility of PVA–GO composite films by adding the glycerol as a plasticizer.
Experimental
Materials
GO was supplied by PuntoQuantico (Italy), poly(vinyl alcohol) (PVA, 16/1, degree of hydrolysis 98.3–99.1%) was purchased from Reakhim (Russia), and glycerol was purchased from Panreac Applichem (Italy). All chemicals were used as received. Double distilled water was used for preparation of PVA aqueous solutions and GO aqueous dispersions.PVA solutions and GO dispersions
PVA solutions were prepared as follows: PVA powder was heated in water at 80 °C for 2 hours in an oven and then stirred for 12 h at 70 °C. Aqueous dispersion of GO was prepared by mixing of 44 mg of GO with 10 ml of water, succeeded by ultrasonication and vigorous stirring. Thus the stable dispersions containing 4.4 mg ml−1 GO were obtained.In Fig. 1 TEM images of air-dried GO dispersion are presented; separated GO sheets could be easily seen which evidences fine dispersibility of GO in the aqueous dispersion.
 | ||
| Fig. 1 TEM images of air-dried GO dispersion. | ||
Preparation of composite and reference films
Two series of PVA/GO composite films with 2 and 4 wt% of GO and different amount of glycerol as a plasticizer (0, 10, and 20 wt%) were prepared. Also the samples of pure PVA containing 0, 10, and 20 wt% of glycerol (Gly) were prepared for the reference.First, PVA/GO and PVA/GO/Gly aqueous dispersions were obtained. For the preparation of PVA/GO and PVA/GO/Gly aqueous dispersions, dry PVA, GO aqueous dispersion (4.4 mg ml−1), double distilled water, and Gly (for glycerol containing samples) were mixed. Final PVA concentration in dispersions was 5 wt%, GO concentrations were 1.1 mg ml−1 or 2.2 mg ml−1 (for 2 wt% and 4 wt% GO in the final dried film correspondingly), and the amount of glycerol added corresponds to 10 and 20 wt% in the final dried film. Dispersions were heated in an oven at 80 °C for 2 hours. Then the dispersions were stirred vigorously for 12 h at 70 °C.
PVA/GO and PVA/GO/Gly films, as well as pure PVA films with and without glycerol were prepared by solution casting technique from the PVA/GO or PVA/GO/Gly aqueous dispersions or PVA aqueous solution. Dispersions or solutions were cast onto a glass and air-dried on a horizontally aligned table for 48 h. The films obtained were uniform in thickness with smooth surface and were easily separated from the glass support. Using 10 ml of PVA/GO, PVA/GO/Gly dispersion or PVA, PVA/Gly solution, films with dimensions of about 7 × 5 cm and thickness ∼60–70 μm were obtained.
The resulting composite films were equilibrated for 5 days in a desiccator under saturated MgCl2 at 25 °C (relative humidity 33%). For the other series of the experiments, composite films were dried at 70 °C for 2 h. In all cases the amount of water in the samples was estimated using thermogravimetric analysis.
Transmission electron microscopy
The morphology of the initial GO aqueous dispersion as well as PVA/GO and PVA/GO/Gly composite films was studied via transmission electron microscopy using LEO 912 AB OMEGA microscope (Carl Zeiss, Germany). Aqueous GO dispersion was placed on a Formvar–carbon covered copper mesh and air dried. For the PVA/GO and PVA/GO/Gly composite films, ultrathin slices of composite films with a thickness of 100 nm were deposited on a copper mesh covered with a Formvar–carbon film.Synchronous thermal analysis
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed using a synchronous thermal analysis instrument STA 449 F3 Jupiter (Netzsch). Aluminum crucibles were used for the experiment, the sample weighed was 1–5 mg. The heating rate was 10 K min−1; prior to the analysis, the baseline TGA and DSC was recorded.The degree of crystallinity, χ, was calculated from the DSC data using the specific heat of fusion of pure PVA or PVA/GO composite and the specific heat of fusion of perfectly crystalline PVA:
| χ = ΔHm/ΔH0 × 100%, |
Mechanical analysis
The tensile tests of the composites were evaluated using an Instron 4300 multipurpose testing machine. The initial length of the specimen was 20 mm, and the width was 6 mm. Crosshead speed of 2 mm min−1 (10% min−1) was used. The toughness was defined as the area surrounded by the stress–strain curve.WAXS measurements
The structure of the PVA films and PVA/GO composites both pure and plasticized with glycerol was studied by the WAXS technique. X-rays scattering patterns were registered with Bruker D8 ADVANCE diffractometer equipped with germanium bent-crystal monochromator and LYNXEYE scintillation detector, using Ni-filtered CuKα radiation (λ = 1.5418 Å). Patterns were recorded in a diffraction angle range of 2θ = 10–50°. The incoherent and background scattering subtraction and X-rays patterns processing for the partition of semi crystalline and amorphous phases reflexes were performed in DIFFRAC.EVA and Origin 15Pro software as described in ref. 41 and 42.Thermomechanical analysis
The glass transition temperature was measured using thermomechanical analysis (TMA 402 F1 Hyperion, Netzsch) with penetration equipment. The heating rate of 3 K min−1 and constant force of 20 mN with the argon flow rate of 50 ml min−1 were used. The inflection point on a curve was used to compare data for different samples.Results and discussion
It is known that PVA films are characterized with rather poor plasticity and the addition of GO as a filler, even at the amounts of a few wt%, decreases their plasticity further and makes them more brittle. One of the possible ways to increase the plasticity of PVA/GO nanocomposites is the addition of a plasticizer, and here we tried to use glycerol as a ductility rising component. The structure and mechanical properties of PVA and its composites are also known to be very strongly dependent on the water content, since water serves as a very good plasticizer for PVA.43,44 The rises and falls of relative humidity could significantly change the water content in the polymer45 thus influence the physicomechanical properties. One can control the water content in the PVA and its composites by annealing it at the temperature, slightly lower than the glass transition temperature for the polymer, or by keeping the polymer samples at a constant humidity. So here we used two series of PVA and its composite films samples with different water contents.Pure PVA films or PVA/GO nanocomposite films containing 2 or 4 wt% of GO were obtained by solution casting technique and air-dried to a constant weight. Then two series of samples with different water content were prepared. The samples with minimal water content were obtained by drying of PVA or its composite films at 70 °C for 2 h, and for the second series, air-dried PVA or its composite films were equilibrated for 5 days at the 33% relative humidity.
The properties of PVA/GO composite films containing 2 wt% of GO were described earlier and it was shown that GO is uniformly distributed inside the polymer matrix, with stacks of several GO sheets or individual GO layers.29
In Fig. 2(a) the thermogravimetric curves of annealed for 2 h at 70 °C (further denoted as annealed) and equilibrated at 33% relative humidity (further denoted as dampened) samples are given.
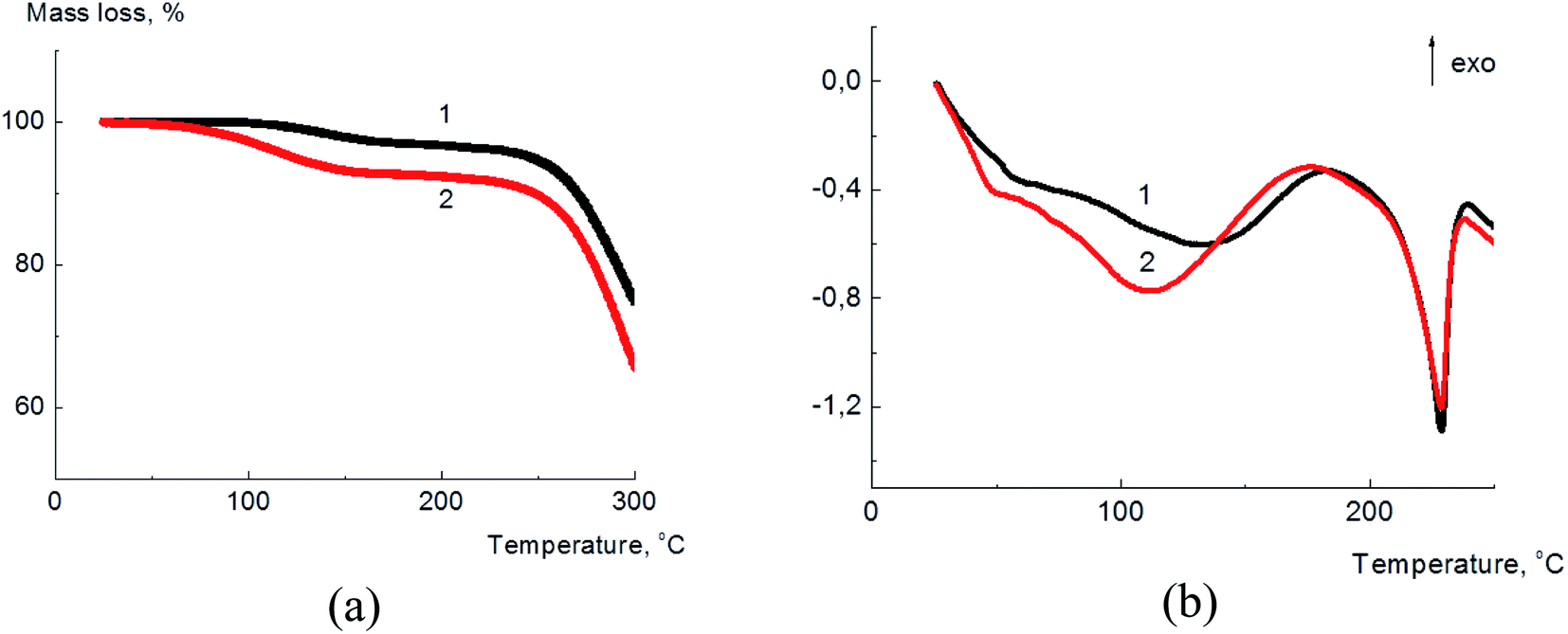 | ||
| Fig. 2 Thermogravimetric (a) and calorimetric (b) curves for annealed (1) dampened (2) PVA/GO composite films containing 2 wt% of GO. | ||
According to the TGA data, the water content in the annealed sample was about 2.8–3.1%, and for the dampened sample this value was about 5.3–5.5%. The increase of water content which serves as a plasticizer for PVA matrix, allows to suggest the rise of the composite ductility for the dampened sample and therefore the improvement of its mechanical properties.
The thermal transformations in the PVA/GO composite films were studied by DSC technique and the corresponding data for annealed (1) and dampened (2) films are shown in Fig. 2(b). The broad endothermic peak in the region of 80–120 °C corresponds to the water evaporation from the composite sample. The melting temperature for both samples almost coincides and was found to be ∼230 °C. The specific heat of fusion was found to be equal to 66.7 J g−1 and 63.2 J g−1 for the annealed and dampened samples, correspondingly. From these data the degree of PVA crystallinity (χ) in the composite films was calculated as follows:
| χ = ΔHm/ΔH0 × 100%, |
Let us now proceed to the consideration of plasticized with glycerol films. It is well known that glycerol represents an effective and well compatible plasticizer for PVA,32 therefore here we tried to modify the mechanical properties, in particular, the ductility of PVA/GO composite films by adding the glycerol into the PVA/GO composite. One of the key parameters determining the material ductile properties is the glass transition temperature therefore their values were measured for pure PVA films and PVA films containing glycerol. The glass transition temperature values (Tg) for PVA and PVA/Gly films annealed at 70 °C are presented in Table 1. One can see the significant decrease in the glass transition temperatures with the addition of glycerol: it drops from 94 °C for pure PVA film to 42 °C for PVA/Gly film containing 20 wt% of glycerol.
| Sample | % Gly | Tg, °C |
|---|---|---|
| PVA | 0 | 94 |
| PVA/Gly | 10 | 75 |
| PVA/Gly | 20 | 42 |
From the data on the decrease of the PVA glass transition temperature with the increase of glycerol content one can conclude that this plasticizer is very effective for PVA. These data are in a good agreement with the previous studies and theoretical values predicted with Kelley–Bueche equation which allow the calculation of the glass transition temperatures using thermal expansion coefficients and volume fractions of a polymer and plasticizer.46,47
As a next step we added glycerol into the PVA/GO composites during film preparation and first checked whether the addition of glycerol affects the distribution of GO in the polymer matrix. It should be noted that the addition of glycerol to the initial mixture for films casting does not change the processability of the composition, and visually significantly improves the flexibility of the films obtained. We used two PVA/GO compositions having 10 wt% and 20 wt% of glycerol, loaded with 2 and 4 wt% of GO. After casting and drying, smooth and uniform films were obtained in all cases. In Fig. 3 TEM images of the thin slices of PVA/GO composite film with 20 wt% of glycerol at different magnifications are shown, TEM image of the film without glycerol is also given for the comparison. One can see that the addition of glycerol to the film does not change the way of it distribution in the resulting film: the filler in both samples is distributed uniformly while slight stacking of GO sheets is observed. According to the TEM data, the number of stacked GO layers is up to 10, usually 3–5 GO layers.
 | ||
| Fig. 3 TEM images of PVA/GO film containing 2 wt% of GO (a) and PVA/GO/Gly film containing 2 wt% of GO and 20 wt% of glycerol (b–d) at different magnifications. | ||
Calorimetric curves for PVA/GO/Gly with 0, 10, and 20 wt% of glycerol are given in Fig. 4. The melting temperatures for the composites decreases with the increase of the amount of the plasticizer added, and its values are equal to 229, 221, and 215 °C for 0, 10, and 20 wt% of glycerol content, correspondingly.
 | ||
| Fig. 4 DSC curves for PVA/GO annealed sample (1), and PVA/GO/Gly annealed samples with 10 (2) and 20 (3) wt% of glycerol; all samples contain 2 wt% of GO. | ||
The calculation of the PVA crystallinity degree in the plasticized composite films using the DSC method applied earlier in our study will be not correct for the samples containing glycerol because of unavoidable glycerol evaporation during the sample heating, which begins at lower temperatures than PVA melting temperature. Therefore, we use X-ray diffraction technique to determine PVA degree of crystallinity in the composite sample with glycerol.
Fig. 5 shows typical radial X-rays diffraction curves for PVA/GO/Gly samples with different glycerol content and 2 wt% of GO in 2Θ range of 10°–50°.
 | ||
| Fig. 5 X-ray diffraction curves for (a) PVA/GO sample (1), and PVA/GO/Gly samples with 10 (2) and 20 (3) wt% of glycerol; all films contain 2 wt% of GO; (b) PVA (1), and PVA containing 10 (2) and 20 (3) wt% of glycerol. | ||
X-ray diffraction patterns for PVA and PVA/GO nanocomposites curves contain the main diffraction peak at 2Θ = 19.3° and the second one at 2Θ = 40.7°, both typical for the PVA crystalline phase. It should be noted that nanocomposite patterns does not show the pronounced peak typical for GO diffraction, which is observed at 2Θ = 10.6° (corresponds to the d = 0.83 nm).48 This is the evidence of the unformed distribution of GO inside the PVA matrix.
The degrees of crystallinity for PVA and its composites with GO in plasticized and unplasticized states were calculated from the X-ray diffraction spectra. The value of degree of crystallinity was calculated as a ratio of the integral scattering contribution for the crystalline phase to the total integral scattering intensity. The data handling for scattering peak and amorphous halo partition was performed using DIFFRAC.EVA and Origin 15Pro software. The example of the diffraction pattern treatment for pure PVA is shown in Fig. 6, here, the scattering curves are shown as a dependence of Is2 on s, where I is the scattering intensity, and s is a scattering vector calculated as
s = π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(Θ)/λ, sin(Θ)/λ, |
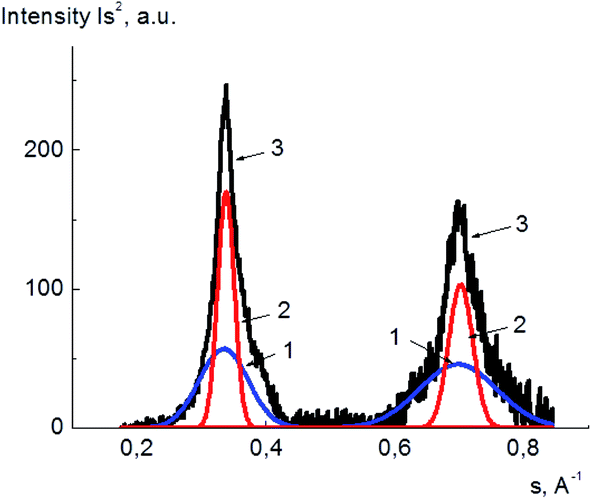 | ||
| Fig. 6 Example of PVA X-ray diffraction pattern treatment; amorphous phase (1), crystalline phase (2), the initial X-ray diffraction pattern (3). | ||
The crystallinity degrees calculated from X-ray scattering data along with the melting temperatures obtained from the DSC data for PVA and its composites are shown in Table 2.
| Sample | Melting temperature, °C | Crystallinity degree |
|---|---|---|
| PVA | 232 | 49 |
| PVA/10 wt% Gly | 230 | 48 |
| PVA/20 wt% Gly | 229 | 42 |
| PVA/2 wt% GO | 228 | 40 |
| PVA/2 wt% GO/10 wt% Gly | 226 | 37 |
| PVA/2 wt% GO/20 wt% Gly | 228 | 41 |
The crystallinity degree slightly decreases for the nanocomposite material compared to the pure PVA, which could be due to the restriction of the PVA chains movements near GO surface as a result of the formation of hydrogen bonds between oxygen-containing GO functional groups and hydroxyl groups of PVA. The variations of the crystallinity degree for PVA filled with GO were explained in ref. 45 with two opposite effects. GO can play a role of nucleation agent, providing sites for PVA crystallization, which results in crystallization degree increase. On the other hand, GO aggregates and single sheets could restrict PVA chains movement thus decreasing the crystallinity of the polymer in the nanocomposite. As a result, slight changes of crystallinity degree could be observed after addition of GO into PVA matrix depends, for example, on the way of a sample preparation.
The addition of glycerol to the PVA and GO filled nanocomposites leads to the decrease of the melting temperature and degree of crystallinity. Both effects are usually observed as a result of a plasticizer addition into a polymer, and it was as well shown for PVA plasticized with glycerol.49
However we can conclude that the addition of glycerol and GO into the PVA matrix does not lead to the significant changes of the polymer crystallinity degree, and in the PVA filled with GO still very high percent of the chains are included into the crystalline phase. One can suggest that with the plasticizer addition, along with the increase of the samples ductility, mechanical performance of the modified composite will remain at a good level, Young's modulus and yield stress will remain sufficiently high.
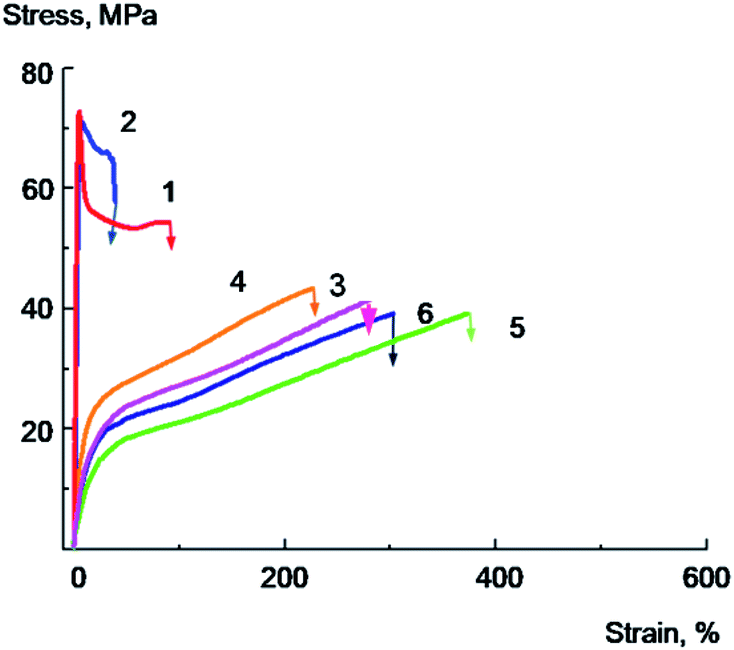
Mechanical performance of the composite films are the most important criteria for the evaluation of polymer films properties, and to study the mechanical properties of the PVA/GO composite films tensile tests were carried out. Typical stress–strain curves for annealed samples containing low amount of water for PVA and PVA/GO nanocomposites, both unmodified and modified with glycerol are shown in Fig. 7. The Young's modulus, yield point, stress at break, elongation at break were calculated from stress–strain curves; the tensile energy at break was determined as the area under the deformation curve. The data on mechanical properties are summarized in Table 3.
 | ||
| Fig. 7 Stress–strain curves for the composite annealed films: PVA (1), PVA/GO containing 2 wt% of GO (2), PVA/Gly containing 10 wt% of glycerol (3), PVA/GO/Gly containing 2 wt% of GO and 10 wt% of glycerol (4), PVA/Gly containing 20 wt% of glycerol (5), PVA/GO/Gly containing 2 wt% of GO and 20 wt% of glycerol (6). | ||
| Sample | % Gly | Young's modulus, MPa | Yield point, MPa | Stress at break (strength), MPa | Elongation at break, % | Tensile energy at break, J cm−3 |
|---|---|---|---|---|---|---|
| PVA | 0 | 4010 ± 240 | 103 ± 10 | 90 ± 5 | 35 ± 6 | 29.0 ± 3.0 |
| PVA/Gly | 10 | 2620 ± 120 | 75 ± 5 | 60 ± 4 | 25 ± 4 | 16.5 ± 2.0 |
| PVA/Gly | 20 | 1610 ± 130 | 45 ± −4 | 63 ± 4 | 250 ± 20 | 127.0 ± 12 |
| PVA/GO | 0 | 4750 ± 330 | 123 ± 8 | — | 5 ± 1 | 2.7 ± 0.3 |
| PVA/GO/Gly | 10 | 2820 ± 170 | 80 ± −5 | 65 ± 3 | 11 ± 1.5 | 5.2 ± 0.4 |
| PVA/GO/Gly | 20 | 1930 ± 90 | 51 ± 4 | 52 ± 4 | 55 ± 5 | 26.0 ± 3.0 |
Pure PVA film (Fig. 7, curve 1) deforms with a neck formation, which appears after the yield point achievement. The extensive area of a constant stress on a curve for PVA film corresponds to the neck propagation in the sample. The sample destroys at the deformation value of about 40–50% at the stage of the neck propagation.
The introduction of 2 wt% of GO leads to the 10–20% increase in the Young's modulus, strength rise, and a sharp decrease in tensile elongation of the composite film (Fig. 7, curve 2). As it is seen from the data, the plastic deformation is observed in the case of the initial PVA, whilst for the sample containing 2% of GO brittle destruction without reaching the yield strength is observed at a very low deformation values about 5–7%. The elongation at break for PVA/GO composite films in this case is about ten times less than that of the original polymer.
Let us enlarge on the PVA mechanical property changes as a result of GO nanofiller addition into the polymer matrix. Earlier it was shown that the mechanical parameters such as Young's modulus, yield point, and stress at break significantly increases after addition of rather small amounts of GO into PVA; 30–50% increase in Young's modulus and yield point were observed after addition of as small as tenth hundredths of one percent of GO. At the same time, the addition of GO into PVA results in the abrupt decrease of elongation at break and composite embrittlement.45,50 The possible reasons for such effects are extremely high rigidity and mechanical strength of GO (Young's modulus for GO is about 210–250 GPa, and its mechanical resistance is about 130 GPa), high degree of GO exfoliation in the composite material, uniform GO sheets distribution in a polymer matrix, and high adhesive power, which is determined by the formation of hydrogen bonds between GO nanosheets and PVA molecules.
At the same time, the strong interaction between GO surface and polymer matrix caused by the network of hydrogen bonds results in the restriction of polymer chains movement and as a result leads to the decrease of elongation at break for PVA–GO composite also making it brittle. The brittleness of the PVA as a result of GO addition into the composite film also can be caused by GO aggregates formation and single GO sheets hitching, especially at the high contents of the filler. GO and GO aggregates having high anisotropy coefficient appears to be the starting point of the crack under concentrated stress; this effect was described earlier in ref. 50.
The tensile properties changed significantly after addition of glycerol into the films. Typical stress–strain curves for PVA/Gly film containing 20 wt% of glycerol and PVA/GO/Gly film containing 2 wt% of GO and 20 wt% of glycerol are shown in Fig. 7 (curves 5 and 6, correspondingly). It can be seen that the introduction of glycerol leads to a significant decrease in the Young's modulus, the yield strength of both original PVA and PVA/GO composite (compare the curves 1 and 5 for PVA, and curves 2 and 6 for PVA/GO), the elongation at break for both materials dramatically increases. Thus, it has been found that for the PVA/GO composite a brittle to plastic transition is observed with the introduction of 20 wt% of glycerol. As it is follows from the Fig. 7, the unplasticized PVA/GO composite breaks brittle, not reaching the yield point (curve 2), in contrast to the PVA/GO/Gly composite containing 20 wt% glycerol, which deforms plastically (curve 6). The fracture work increases by an order of magnitude as it is seen from the Table 3. It should be noted that the samples with glycerol deforms without a neck formation, after reaching the yield point the stress increased monotonically with the deformation raising.
One can assume that the introduction of a plasticizer besides the effect on the PVA interchain interactions, leads to a decrease in local overstrains around the filler particles or particle aggregates that results in an increase of the crack growth resistance of the material and an increase of the films plasticity.
It should be noted that the introduction of 10 wt% of glycerol into a PVA/GO composite containing 2 wt% of GO is accompanied by a significant decrease in the glass transition temperature (the glass transition temperature decreases from 94 to 75 °C, Table 1), the Young's modulus and the yield point of the material (Table 3). However, this does not result in a significant increase in elongations and tensile energy at break. It can be seen from the Fig. 7 that the PVA/GO composite containing 10 wt% glycerol breaks in a quasi-brittle manner at the stage of neck formation, wherein the elongations at break do not exceed 10–15% (curve 4).
Apparently, the effect of increasing the composite film plasticity is observed when the glass transition temperature of the composite becomes more close to the temperature of the experiment (in our case, to the room temperature), when a sufficiently high level of molecular mobility in the plasticized polymer is achieved. These conditions are realized for PVA/GO/Gly composite containing 20% of glycerol, since the glass transition temperature of this material was found to be about 42 °C. It is important to note that plasticized PVA/GO composite films in spite of the significant glass transition temperature decrease demonstrate rather high yield point and stress at break values (see Fig. 7 and Table 3). This is the result of sufficiently high crystallinity degree (about 40%) of plasticized with glycerol composites.
The effect of increasing of the PVA/GO composite plasticity with the decrease of the glass transition temperature realized even more vividly for the dampened PVA samples equilibrated at 33% relative humidity which contain 5.3–5.5% of water as a plasticizer in addition to glycerol. Typical stress–strain curves for plasticized and unplasticized PVA and nanocomposite films equilibrated at 33% relative humidity with 2 wt% or 4 wt% of GO are presented in Fig. 8 and 9; the data on the mechanical properties are summarized in Table 4.
 | ||
| Fig. 8 Stress–strain curves for the dampened films: PVA (1), PVA/GO containing 2 wt% of GO (2), PVA/Gly containing 10 wt% of glycerol (3), PVA/GO/Gly containing 2 wt% of GO and 10 wt% of glycerol (4), PVA/Gly containing 20 wt% of glycerol (5), PVA/GO/Gly containing 2 wt% of GO and 20 wt% of glycerol (6). | ||
 | ||
| Fig. 9 Stress–strain curves for the dampened films: PVA/GO containing 4 wt% of GO (1), PVA/GO/Gly containing 4 wt% of GO and 10 wt% of glycerol (2) and PVA/GO/Gly containing 4 wt% of GO and 20 wt% of glycerol (3). | ||
| Sample | % GO | % Gly | Young's modulus, MPa | Yield point, MPa | Stress at break (strength), MPa | Elongation at break, % | Tensile energy at break, J cm−3 |
|---|---|---|---|---|---|---|---|
| PVA | 0 | 0 | 2875 ± 125 | 71 ± 4 | 54 ± 3 | 80.0 ± 9.0 | 35.0 ± 3.0 |
| PVA/Gly | 0 | 10 | 170 ± 25 | 24 ± 3 | 54 ± 5 | 270.0 ± 20.0 | 190 ± 25.0 |
| PVA/Gly | 0 | 20 | 90 ± 10 | 18 ± 3 | 39 ± 4 | 370.0 ± 20.0 | 198.5 ± 7.0 |
| PVA/GO | 2 | 0 | 2900 ± 150 | 71 ± 5 | 65 ± 3 | 38.0 ± 8.0 | 24.0 ± 4.0 |
| PVA/GO/Gly | 2 | 10 | 330 ± 40 | 26 ± 3 | 43 ± 3 | 230.0 ± 20.0 | 74.5 ± 8.0 |
| PVA/GO/Gly | 2 | 20 | 160 ± 30 | 22 ± 2 | 39 ± 3 | 305.0 ± 25.0 | 85.5 ± 6.0 |
| PVA/GO | 4 | 0 | 4500 ± 300 | — | 78 ± 4 | 4.5 ± 1.0 | 2.9 ± 0.2 |
| PVA/GO/Gly | 4 | 10 | 470 ± 50 | 18 ± 2 | 41 ± 4 | 155.0 ± 20.0 | 50.0 ± 6.0 |
| PVA/GO/Gly | 4 | 20 | 250 ± 25 | 27 ± 2 | 31 ± 3 | 175.0 ± 20.0 | 45.0 ± 4.5 |
The initial PVA sample is deformed with a neck formation; the elongation at break of the sample is equal to 80–100%; it characterized by lower Young's modulus and the yield point values, compared to annealed sample, which could be explained by the higher water content (Fig. 8, curve 1). The introduction of 2 wt% GO into the polymer leads to a decrease in the elongation at break up to 40%, the sample destroys at the stage of neck formation or at the initial stage of neck propagation (Fig. 8, curve 2). At the same time, a PVA/GO sample containing 4 wt% of GO is destroyed brittly without reaching the yield point, and the elongation at break value reaches only about 4–5% (Fig. 9, curve 1). It should be noted that PVA/GO nanocomposite films containing 4 wt% of glycerol and annealed at 70 °C for 2 h were extremely brittle, and it was hardly possible to conduct the reliable mechanic tests for these samples.
As it follows from the Fig. 8, 9 and Table 4, the introduction of glycerol into PVA/GO composites, along with a significant decrease in the Young's modulus and yield point, is accompanied by a remarkable increase in elongations at break and tensile energy at break. The failure mechanism changes from brittle to ductile already with the introduction of 10% glycerol. For PVA/GO sample containing 2 wt% of GO the introduction of 10% glycerol leads to an increase in elongation at break from 40 to 230%, and for the composite containing 4 wt% of GO, the addition of 20 wt% of glycerol gives the rise of elongation at break from 4 up to 150%, which is rather impressive. Plasticized with glycerol samples are deformed uniformly, without a neck formation, herewith the stress increases monotonically with deformation growth. It should be noted that PVA samples plasticized with glycerol equilibrated at 33% humidity are characterized by lower values of yield point and Young's modulus and higher tensile energy at break values compared to the PVA samples annealed at 70 °C.
The observed effects are, apparently related to the lower values of the glass transition temperatures of PVA samples plasticized with glycerol equilibrated at 33% relative humidity, compared with PVA samples, which were annealed at 70 °C. This is obviously due to the higher content of water that acts as an effective plasticizer for PVA samples.
As it was mentioned earlier, the transition from ductile to the brittle state after GO addition could be caused by the filler aggregates formation and the existence of the network of hydrogen bonds between PVA molecules and GO which restrict polymer chains movements. One can assume that the main reason of the plasticity increase after glycerol addition into the composite film is polymer matrix changes as a result of plasticization. The plasticizer reduces the restrictions of the polymer chains movements in the PVA/GO composite, which was caused by the presence of GO. Moreover, the plasticizer addition decreases local overstrains around GO and GO aggregates, which results in the increase of cracks growth resistance in the material and plasticity increase.
Thus the plasticizer allows the transformation of the PVA/GO nanocomposite films from brittle to plastic condition and provides a remarkable increase of elongation at break of the modified films. This effect could be additionally enhanced with an increase of water content in the PVA/GO/Gly nanocomposite, which should be taken into consideration when using PVA composite materials in the atmospheres with different humidity.
Conclusions
Glycerol was used as a plasticizer to modify the mechanical properties of PVA/GO composite films. The addition of glycerol does not disturb the uniform distribution of GO inside the PVA. The presence of up to 4 wt% of GO in the PVA matrix leads to the increase of Young's modulus and yield stress, however dramatically decreases the elongation at break with the increasing of film brittleness. Plasticized with glycerol PVA/GO films demonstrate both Young's modulus and yield stress decrease and significant elongation at break increase, the brittle to ductile transition is observed. The connection between the glass transition temperature of the composite material and mechanical properties of the films was shown. Glycerol does not crucially influence the PVA degree of crystallinity, and a significant part of PVA still exist in a crystalline state after the plasticization, which allow to maintain suitable mechanical properties along with the significant rise of the sample ductility. Water also serves as a plasticizer for PVA which was confirmed by the even more ductility improvement for the nanocomposite films equilibrated at 33% RH, compared to the annealed films with less water content.Funding
This work was supported by Russian Science Foundation (project no. 17-73-20266).Conflicts of interest
The authors declare no conflict of interest.References
- S. Kim, F. Fornasiero, H. G. Park, J. B. In, E. Meshot, G. Giraldo, M. Stadermann, M. Fireman, J. Shan, C. P. Grigoropoulos and O. Bakajin, J. Membr. Sci., 2014, 460, 91–98 CrossRef CAS.
- I. Pastoriza-Santos, C. Kinnear, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, Nat. Rev. Mater., 2018, 3, 375–391 CrossRef.
- U. Khan, P. May, A. O'Neill and J. N. Coleman, Carbon, 2010, 48, 4035–4041 CrossRef CAS.
- Z. Spitalsky, D. Tasis, K. Papagelis and C. Galiotis, Prog. Polym. Sci., 2010, 35, 357–401 CrossRef CAS.
- S. Morimune, M. Kotera, T. Nishino, K. Goto and K. Hata, Macromolecules, 2011, 44, 4415–4421 CrossRef CAS.
- L. Ge, Z. Zhu, F. Li, S. Liu, L. Wang, X. Tang and V. Rudolph, J. Phys. Chem. C, 2011, 115, 6661–6670 CrossRef CAS.
- T. Tyler, O. Shenderova, G. Cunningham, J. Walsh, J. Drobnik and G. McGuire, Diamond Relat. Mater., 2006, 15, 2078–2081 CrossRef CAS.
- D. Chen, X. Wang, T. Liu, X. Wang and J. Li, ACS Appl. Mater. Interfaces, 2010, 2, 2005–2011 CrossRef CAS.
- D. R. Paul and L. M. Robeson, Polymer, 2008, 49, 3187–3204 CrossRef CAS.
- K. Müller, E. Bugnicourt, M. Latorre, M. Jorda, Y. Echegoyen Sanz, J. M. Lagaron, O. Miesbauer, A. Bianchin, S. Hankin, U. Bölz, G. Pérez, M. Jesdinszki, M. Lindner, Z. Scheuerer, S. Castelló and M. Schmid, Nanomaterials, 2017, 7, 74 CrossRef PubMed.
- C. Harito, D. V. Bavykin, B. Yuliarto, H. K. Dipojonobc and F. C. Walsha, Nanoscale, 2019, 11, 4653–4682 RSC.
- A. Cesar, M. Isaza, J. M. Herrera Ramírez, J. E. Ledezma Sillas and J. M. Meza, J. Compos. Mater., 2018, 52, 1617–1626 CrossRef.
- D. Liu, X. Sun, H. Tian, S. S. Maiti and Z. S. Ma, Cellulose, 2013, 20, 2981–2989 CrossRef CAS.
- G. Chakraborty, K. Gupta, A. K. Meikap, R. Babu and W. P. J. Blau, J. Appl. Phys., 2011, 109, 033707 CrossRef.
- A. Pourjavadi, B. Pourbadiei, M. Doroudiana and S. Azari, RSC Adv., 2015, 5, 92428–92437 RSC.
- M. S. N. Salleh, M. Saadon, N. Razali, Z. Omar, A. S. Khalid, A. R. Mustaffa, M. M. Yashim and W. A. Rahman, IEEE Symposium on Humanities, Science and Engineering Research, 2012, vol. 4, pp. 523–526 Search PubMed.
- R. B. Siddaramaiah and R. Somashekar, J. Appl. Polym. Sci., 2004, 91, 630–635 CrossRef.
- W. Zhang, W. Li, J. Wang, C. Qin and L. Dai, Fibers Polym., 2010, 11, 1132–1136 CrossRef CAS.
- J. M. Gohil and P. Ray, J. Colloid Interface Sci., 2009, 338, 121–127 CrossRef CAS PubMed.
- S. A. Escobar, C. A. Merino and J. M. Meza, Rev. Mater., 2015, 20, 794–802 CAS.
- H. D. Huang, P. G. Ren, J. Chen, W. Q. Zhang, X. Ji and Z. M. Li, J. Membr. Sci., 2012, 409, 156–163 CrossRef.
- T. Ma, P. R. Chang, P. Zheng and X. Ma, Carbohydr. Polym., 2013, 94, 63–70 CrossRef CAS PubMed.
- P. P. Peregrino, M. J. Sales, M. F. P. da Silva, M. A. G. Soler, L. F. L. da Silva, S. G. C. Moreira and L. G. Paterno, Carbohydr. Polym., 2014, 106, 305–311 CrossRef CAS PubMed.
- S. L. Qiu, C. S. Wang, Y. T. Wang, C. G. Liu, X. Y. Chen, H. F. Xie, Y. A. Huang and R. S. Cheng, eXPRESS Polym. Lett., 2011, 5, 809–818 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- L. R. De Jesus, R. V. Dennis, S. W. Depner, C. Jaye, D. A. Fischer and S. Banerjee, J. Phys. Chem. Lett., 2013, 4, 3144–3151 CrossRef CAS PubMed.
- D. Liu, Q. Bian, Y. Li, Y. Wang, A. Xiang and H. Tian, Compos. Sci. Technol., 2016, 129, 146–152 CrossRef CAS.
- A. Sapalidis, Z. Sideratou, K. N. Panagiotaki, E. Sakellis, E. P. Kouvelos, S. Papageorgiou and F. Katsaros, Front. Mater., 2018, 5, 11 CrossRef.
- T. V. Panova, A. A. Efimova, A. V. Efimov and A. K. Berkovich, Colloid Polym. Sci., 2019, 297, 485–491 CrossRef CAS.
- J. Ma, Y. Li, X. X. Yin, Y. Xu, J. Yuea, J. Bao and T. Zhou, RSC Adv., 2016, 6, 49448–49458 RSC.
- X. Zhao, Q. Zhang and D. Chen, Macromolecules, 2010, 43, 2357–2363 CrossRef CAS.
- R. J. Fong, A. Robertson, P. E. Mallon and R. L. Thompson, Polymers, 2018, 10, 1036 CrossRef PubMed.
- Y. Han Cho, B. Chul Kim and K. Sik Dan, Macromol. Res., 2009, 17, 591–596 CrossRef.
- F. Lin, W. Wu, H. Sun and A. Xiang, J. Polym. Mater., 2011, 28, 577–586 CrossRef CAS.
- M. Mohsin, A. Hossin and Y. Haik, J. Appl. Polym. Sci., 2011, 122, 3102–3109 CrossRef CAS.
- J. Liu, L. Cui and D. Losic, Acta Biomater., 2013, 10, 9243–9257 CrossRef PubMed.
- S. Ruiz, J. A. Tamayo, J. D. Ospina, D. P. Porras, M. E. Zapata, J. H. Hernandez, C. H. Valencia, F. Zuluaga and C. D. Tovar, Biomolecules, 2019, 9(3), 109 CrossRef CAS PubMed.
- S. Ye, K. Shao, Z. Li, N. Guo, Y. Zuo, Q. Li, Z. Lu, L. Chen, Q. He and H. Han, ACS Appl. Mater. Interfaces, 2015, 7, 21571–21579 CrossRef CAS PubMed.
- M. Cobos, M. J. Fernandez and M. D. Fernandez, Nanomaterials, 2018, 8, 1013 CrossRef PubMed.
- S. V. Yurovskikh, S. N. Chvalun and W. S. Lyoo, Polym. Sci., Ser. A, 2001, 43, 278–284 Search PubMed.
- B. Wunderlich, Macromolecular physics, Academic Press, New York, 1976 Search PubMed.
- H. P. Klug and L. E. Alexander, X-Ray diffraction procedures: for polycrystalline and amorphous materials, Wiley-Interscience, 2nd edn, 1974 Search PubMed.
- X. Qi, X. Yao, S. Deng, T. Zhou and Q. Fu, J. Mater. Chem. A, 2014, 2, 2240–2249 RSC.
- M. V. Konidari, K. G. Papadokostaki and M. Sanopoulou, J. Appl. Polym. Sci., 2011, 120, 3381–3386 CrossRef CAS.
- J. Chen, Y. Gao, W. Liu, X. Shi, L. Li, Z. Wanga, Y. Zhang, X. Guo, G. Liu, W. Li and B. Beake, Carbon, 2015, 94, 845–855 CrossRef CAS.
- P. Sakellariou, A. Hassan and R. C. Rowe, Eur. Polym. J., 1993, 29, 937–943 CrossRef CAS.
- F. N. Kelley and F. Bueche, J. Polym. Sci., 1961, 50, 549–556 CrossRef CAS.
- M. Narayana and S. Jammalamadaka, Graphene, 2016, 5, 73–80 CrossRef CAS.
- J. Jang and D. K. Lee, Polymer, 2003, 44, 8139–8146 CrossRef CAS.
- T. Cheng-an, Z. Hao, W. Fang, Z. Hui, Z. Xiaorong and W. Jianfang, Polym. Polym. Compos., 2017, 25, 11–16 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |