DOI:
10.1039/D0RA04104A
(Paper)
RSC Adv., 2020,
10, 24037-24044
Efficient and stereoselective one-pot synthesis of benzo[b]oxazolo[3,4-d][1,4]oxazin-1-ones†
Received
7th May 2020
, Accepted 5th June 2020
First published on 23rd June 2020
Abstract
An efficient and mild one-pot convergent synthesis protocol has been developed for benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one derivatives through the Mitsunobu reaction and sequential cyclization. Various tricyclic fused benzoxazinyl-oxazolidinones (20 examples) were obtained in good to excellent yields and high enantioselectivities with facile operation. Furthermore, four stereoisomers were afforded respectively in high ee values (>97.8%) via using different chiral 2,3-epoxy-4-trityloxybutanol. This methodology has been applied to the synthesis of key intermediates of drug candidates.
Introduction
Oxazolidinone was a key scaffold for some pharmaceuticals. As shown in Fig. 1, Linezolid (1)1 was the first approved (2000) oxazolidinone antibacterial drug for the treatment of Gram-positive bacterial infection with the off-label use to treat complicated MDR-TB and XDR-TB. Another oxazolidinone drug Tedizolid phosphate was approved by FDA in June 2014 for the treatment of MRSA skin infections.2 Furthermore, Rivaroxaban launched in 2011 is a FXa inhibitor as an anticoagulant drug containing the oxazolidinone moiety.3 Benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one, a [6,6,5] tricyclic fused benzoxazinyl-oxazolidinone, was developed based on the oxazolidinone scaffold and is the essential structural framework of several bioactive compounds with antibacterial,4 anticoagulant,5 and antituberculosis6 activities (Fig. 2).
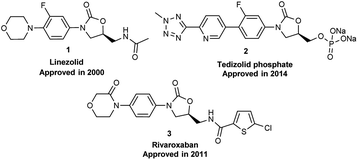 |
| | Fig. 1 Chemical structure of oxazolidinone drugs. | |
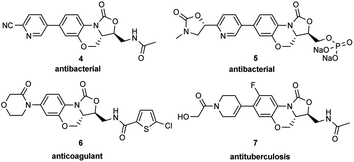 |
| | Fig. 2 Bioactive compounds containing benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one scaffold. | |
To the best of our knowledge, four synthetic routes for the synthesis of benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one derivatives have been reported (Scheme 1). Nicolaou's group reported the synthesis of tricyclic fused benzoxazinyl-oxazolidinone via Dess–Martin periodinane-mediated cascade cyclization of γ,δ-unsaturated urethanes in moderate yields (36–50%).7 Yang's group disclosed the synthesis of tricyclic fused oxazolidinone via n-BuLi mediated intramolecular cyclization reaction in linear synthetic routes (6–7 steps).4b,8 Chen et al. reported the synthesis of FXa inhibitors exemplified by 6 from substituted benzyl(2-hydroxyphenyl)carbamate and methyl 3-(bromomethyl)oxirane-2-carboxylate in the presence of Cs2CO3, which gave a moderate cyclization yield (54%).9 Malhotra et al. provided an approach for the synthesis of chiral benzoxazinyl-oxazolidinone from benzo[b][1,4]-oxazine-3-carboxylic acid via 3–5 steps of derivatization including acylation, reduction, Grignard reaction and cyclization reaction.10 Although these methods provided access to benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one analogues, the reported routes suffered from limitations to some extent such as ineffective stereoselectivity, limited substrates scope, unsatisfactory yields, harsh reaction conditions or tedious synthesis steps. Therefore, the development of a practical and efficient approach to benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one is still required.
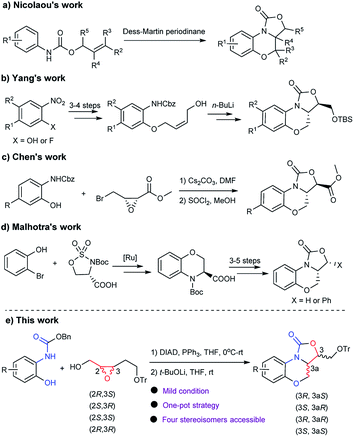 |
| | Scheme 1 Synthetic approaches to benzo[b]oxazolo[3,4-d][1,4]oxazin-1-ones. | |
In recent years, our group have developed a series of benzoxazinyl-oxazolidinones as anti-TB agents.6,11 To improve the efficiency of synthesis and avoid oxidation of the sulfide in the thiomorpholine, a convergent protocol has been developed,11 however, tedious column purification and harsh cyclization conditions posed a hurdle for further derivatization and large-scale preparation. As our continuing efforts toward the development of novel benzoxazinyl-oxazolidinones as anti-TB agents, herein, we report the first efficient protocol for one-pot and convergent synthesis of tricyclic fused benzoxazinyl-oxazolidinone compounds by the reaction of benzyl(2-hydroxyphenyl)carbamate with 2,3-epoxy-4-trityloxybutanol through the Mitsunobu reaction and sequential cyclization in mild conditions (Scheme 1).
Results and discussion
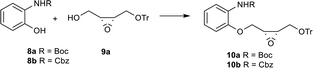
As the key substrate in the Mitsunobu reaction, ((2R,3S)-3-((trityloxy)methyl)oxiran-2-yl)methanol 9a could be synthesized easily from (Z)-4-(trityloxy)but-2-en-1-ol by Sharpless epoxidation in excellent yield with high ee value according to the reported procedure,5a and the trityl protection group could be removed smoothly under acid condition as well. Thus, at the outset of our attempt, N-Boc or N-Cbz substituted 2-aminophenol derivatives 8a and 8b, compound 9a were chosen as model substrates to optimize the Mitsunobu reaction conditions (Table 1). When R is Boc of 8a, the reaction furnished 10a in good yield (81%) in the presence of DEAD and PPh3 in anhydrous THF (entry 1). Employment of DIAD resulted in a similar yield (entry 2).
Table 1 Optimization of Mitsunobu reaction conditionsa
|
| Entry |
R |
Azo agents |
Phosphine |
Solvent |
Yieldb (%) |
| Reaction conditions: 8a or 8b (1 mmol), 9a (1.35 mmol), azo agent (1.5 mmol), PPh3 (1.5 mmol), solvent (8 mL), 0 °C–rt, 4 h. Isolated yields. |
| 1 |
Boc |
DEAD |
PPh3 |
THF |
81 |
| 2 |
Boc |
DIAD |
PPh3 |
THF |
80 |
| 3 |
Boc |
TMAD |
PPh3 |
THF |
Trace |
| 4 |
Boc |
ADDP |
PPh3 |
THF |
Trace |
| 5 |
Cbz |
ADDP |
PPh3 |
THF |
Trace |
| 6 |
Cbz |
DEAD |
PPh3 |
THF |
94 |
| 7 |
Cbz |
DIAD |
PPh3 |
THF |
90 |
| 8 |
Cbz |
DEAD |
PPh3 |
MeCN |
82 |
| 9 |
Cbz |
DIAD |
PPh3 |
MeCN |
91 |
However, this reaction did not work when TMAD or ADDP was used (entries 3 and 4). When R is Cbz of 8b, only a trace product was observed in the presence of ADDP (entry 5). DEAD and DIAD could promote the reaction of 8b and 9a efficiently to afford the desired product 10b in 94% and 90% yields (entries 6 and 7), respectively, which was superior to the corresponding N-Boc substrate 8a. Another polar aprotic solvent, MeCN, was also tested and the yields for 10b using DEAD or DIAD as azo agent were found to be similar with THF (entries 8 and 9). Based on the above results and the safety consideration of dialkyl azodicarboxylates,12 the combination of DIAD and PPh3 was selected to be the optimized conditions and N-Cbz-2-aminophenol was a privilege substrate.
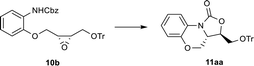
Next, we examined the optimization of the cyclization reaction conditions (Table 2). 11aa was obtained in 69% yield, when 10b was treated with n-BuLi (1.3 equiv.) from −78 °C to room temperature (entry 1). Consideration of danger of n-BuLi and harsh reaction condition, this reaction was not suitable for scale-up preparation. We noted that the lithium ion plays a very important role in region-chemical control and cyclization in the synthesis of Linezolid.13 It prompted us to replace dangerous n-BuLi by other lithium salts such as LiOH·H2O, Li2CO3 and t-BuOLi. As depicted in Table 2, LiOH·H2O and Li2CO3 failed to produce the expected product (entries 2 and 3). To our delight, t-BuOLi was found very effective for this reaction (entry 4), generating 11aa in an excellent yield (86%) at room temperature. However, the annulation process did not occur in the presence of Cs2CO3 and K2CO3 (entries 5 and 6). Although t-BuOLi could also promote the cyclization in the solvent of MeCN with the desired product 11aa obtained in 85% yield after 12 h at room temperature, it was difficult to stir the reaction mixture due to the solid precipitation formed (entry 7). The results displayed that t-BuOLi was an excellent base for the annulation process given the stability and mild condition compared with n-BuLi. Thus, 1.3 equiv. of t-BuOLi in THF at room temperature was identified as the optimal cyclization condition.
Table 2 Optimization of cyclization reaction conditionsa
|
| Entry |
Base |
Solvent |
Temp. |
Yieldb (%) |
| Reaction conditions: 10b (1 mmol), base (1.3 mmol), solvent (8 mL), rt, 6 h. Isolated yields. Reaction time was 12 h. |
| 1 |
n-BuLi |
THF |
−78 °C–rt |
69 |
| 2 |
LiOH·H2O |
THF |
rt |
— |
| 3 |
Li2CO3 |
THF |
rt |
— |
| 4 |
t-BuOLi |
THF |
rt |
86 |
| 5 |
Cs2CO3 |
THF |
rt |
— |
| 6 |
K2CO3 |
THF |
rt |
— |
| 7c |
t-BuOLi |
MeCN |
rt |
85 |
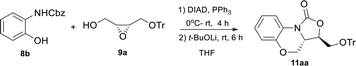
Considering the tedious column chromatography purification after the Mitsunobu reaction, we wonder if one-pot strategy could be applied to simplify the process. Since the reaction solvent (THF) was not only suitable for the Mitsunobu reaction (step 1), but also for the cyclization reaction (step 2), we explored the possibility of cyclization reaction by adding t-BuOLi directly into the mixture when the Mitsunobu reaction was completed without any purified treatment (Table 3). When 1.3 or 1.5 equiv. of t-BuOLi was used, no product was detected (entries 1 and 2). We were pleased to find that the target compound 11aa was obtained in 60% yield using 2.0 equiv. of t-BuOLi. The yield of 11aa was further improved (83%) when 2.5 equiv. of t-BuOLi was employed. It indicated that diisopropyl hydrazine-1,2-dicarboxylate, the byproduct of the Mitsunobu reaction, may form the complex with Li+ to cause the consumption of the base. The result demonstrated that the desired product 11aa can be synthesized by simple one-pot operation with higher isolated yield (83%) compared to the stepwise process (77%).
Table 3 Optimization of one-pot reaction conditionsa
|
| Entry |
Equiv. of t-BuOLi |
Yieldb (%) |
| Reaction conditions: 8b (1 mmol), 9a (1.35 mmol), DIAD (1.5 mmol), PPh3 (1.5 mmol), THF (8 mL), 0 °C–rt, 4 h; then t-BuOLi, THF, rt, 6 h. Isolated yields. |
| 1 |
1.3 |
— |
| 2 |
1.5 |
— |
| 3 |
2.0 |
60 |
| 4 |
2.5 |
83 |
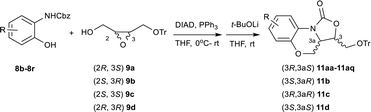
Encouraged by the success of one-pot protocol, subsequently, the generality of this one-pot process leading to the benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one skeleton using various combinations of substrates was investigated. As summarized in Table 4, first, the reaction of benzyl(2-hydroxyphenyl)carbamate 8b with different chiral 2,3-epoxy-4-trityloxybutanol was explored. Delightfully, four isomers were afforded smoothly in good to excellent yields and high ee values ((3R,3aS)-11aa: 83% yield, 98.9% ee; (3S,3aR)-11b: 67% yield, 97.8% ee; (3R,3aR)-11c: 73% yield, >99% ee; (3S,3aS)-11d: 80% yield, >99% ee).
Table 4 Scope of one-pot synthesis of benzo[b]oxazolo[3,4-d][1,4]oxazin-1-onesb
|
| Reaction conditions: 8b–8r (1 mmol), 9a–9d (1.35 mmol), DIAD (1.5 mmol), PPh3 (1.5 mmol), THF (8 mL), 0 °C–rt, 8 h for 11ab and 11ac, 6 h for 11ad and 11ae, 4 h for the others; then t-BuOLi (2.5 mmol), THF, rt, 1–3 h. Isolated yields via column chromatography. Isolated yields via trituration with MeOH and filtration. |
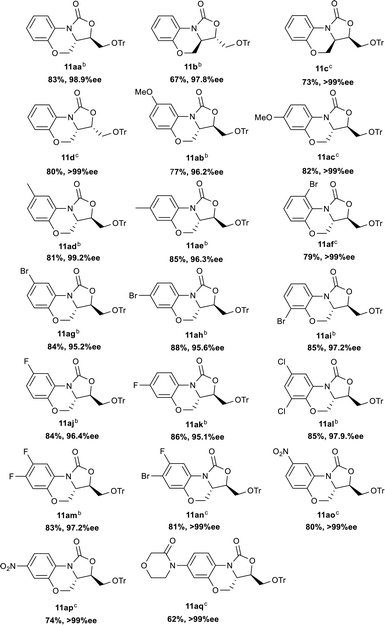 |
Benzyl(2-hydroxyphenyl)carbamate substituted with electron-donating groups such as methyl and methoxy delivered the corresponding target compounds 11ab–11ae in 77–85% yields with excellent enantioselectivities (96.2 to >99% ee). Halogenated benzyl(2-hydroxyphenyl)carbamates furnished the halogenated target compounds 11af–11an in 79–88% yields with high enantiomeric purity (95.1 to >99% ee). As anticipated, the electron-deficient benzyl(2-hydroxyphenyl)carbamates bearing nitro group performed efficiently to deliver the corresponding target compounds 11ao and 11ap in 80% yield, >99% ee and 74% yield, >99% ee respectively. In addition, the reaction was also efficient when benzyl(2-hydroxyphenyl)carbamate bearing morpholin-3-one group, leading to the desired product 11aq in 62% yield and >99% ee, a key intermediate for the synthesis of anticoagulant agent 6 shown in Fig. 1. It was worth noting that the reaction time of the Mitsunobu reaction was different depending on different substrate. When the substrate 8 containing electron-withdrawing substituent on the phenyl ring or non-substituted, 4 hours was sufficient for the Mitsunobu reaction (11aa, 11b–11d and 11af–11ap), while benzyl(2-hydroxyphenyl)carbamate containing electron-donating groups, the reaction took 6–8 hours (11ab–11ae). It is apparently that the electron-withdrawing group is beneficial to speed up the reaction due to increasing the acidity of the phenol hydroxyl, exemplified by 4 h for 11ap (R is 4-NO2) vs. 8 h for 11ac (R is 4-MeO). Taking the advantage of some rigid tricyclic fused oxazolidinones with low solubility in MeOH, we simplified the purification only via trituration with MeOH and then by filtration to afford the pure products. Notably, the excellent enantioselectivity (>99% ee) was observed after the simple operation, represented by 11c, 11d, 11ac, 11af, and 11an–11aq.
To further explore the scalability and potential synthetic application of the present method, the gram-scale reaction and application were carried out. As displayed in Schemes 2 and 3, 11ah was obtained in good yield (1.7 g, 74%, >99% ee) after triturating in MeOH. Next, the deprotection of the triphenylmethyl group of 11ah was performed well under mild condition. After aqueous workup and trituration with DCM/PE (1/1), compound 12 was obtained in 89% yield, and then the Suzuki–Miyaura reaction of 12 and bis(pinacolato)diboron provided the corresponding borate ester 13 (80%) which was an important intermediate to synthesize the clinical candidate 5 (Fig. 1) as an antibacterial agent.4c,14
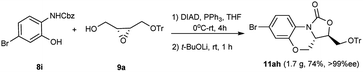 |
| | Scheme 2 Gram-scale synthesis of compound 11ah. | |
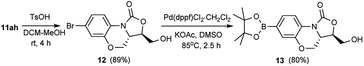 |
| | Scheme 3 Application of the methodology. | |
Conclusion
In summary, we have developed a practical, efficient and operationally simple one-pot convergent protocol to afford the tricyclic fused benzoxazinyl-oxazolidinones through the Mitsunobu reaction and sequential cyclization. The present methodology is a stereoselective process which was controlled by the chiral 2,3-epoxy-4-trityloxybutanol, delivering a series of benzo[b]oxazolo[3,4-d][1,4]oxazin-1-ones in good to excellent yields (62–88%) with high ee values (95.1 to >99% ee). In addition, this one-pot strategy was suitable for large-scale preparation, which has been successfully employed to afford the important intermediates of some promising candidate compounds such as 5 and 6.
Experimental
General
Unless otherwise stated, all the solvents and reagents were purchased from commercial suppliers and were used without further purification. All melting points were measured with a micro melting point apparatus (MP-J3, Yanaco) and were uncorrected. 1H NMR or 13C NMR spectra were recorded on Varian 400 MHz or 500 MHz spectrometer using CDCl3, DMSO-d6 or acetone-d6 as solvents and tetramethylsilane (TMS) as an internal standard. HR-MS spectra were obtained on a ThermoFisher Exactive Plus mass spectrometer (ESI, ThermoFisher Scientific, Bremen, Germany). Optical rotations were measured with a Rudolph Research Analytical (Autopol IV-T).
General procedure for the synthesis of target compounds
To a mixture of 8 (1.0 equiv.), 9 (1.35 equiv.), PPh3 (1.5 equiv.) in anhydrous THF (8 mL) at 0 °C was added dropwise a solution of DIAD (1.5 equiv.) in anhydrous THF (0.3 mL). The reaction mixture was stirred at 0 °C for 30 min. Then, the reaction was allowed to warm to room temperature and monitored by TLC. Upon the completion of reaction, t-BuOLi (2.5 equiv.) was added, and the reaction mixture was stirred at room temperature for 1–3 h. After the reaction completed, water (20 mL) was added to quench the reaction, the reaction mixture was extracted with EtOAc (15 mL × 3). The combined organic phase was washed with brine (15 mL), dried over anhydrous Na2SO4, and evaporated in vacuum. The residue was purified by column chromatography or triturating with MeOH to give the products 11.
(3R,3aS)-3-((Trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11aa). White solid (385 mg, 83% yield); mp: 175–177 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 80/20; flow rate: 1 mL min−1; λ = 220 nm; T = 20 °C): tR = 12.06 min (99.46%), tR = 14.30 min (0.54%). The enantiomeric purity of 11aa was determined to be 98.9% ee. [α]21D = −33.9 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.98–7.94 (m, 1H), 7.53–7.48 (m, 6H), 7.39–7.32 (m, 6H), 7.31–7.26 (m, 3H), 7.07–6.96 (m, 2H), 6.95–6.90 (m, 1H), 4.66–4.59 (m, 1H), 4.54 (dd, J = 2.8, 10.4 Hz, 1H), 4.14–4.08 (m, 1H), 4.02 (t, J = 10.0 Hz, 1H), 3.61 (dd, J = 4.0, 10.4 Hz, 1H), 3.46 (dd, J = 4.4, 10.4 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.7, 145.7, 144.7, 129.5, 128.9, 128.2, 125.2, 124.9, 122.1, 120.4, 117.7, 87.8, 75.5, 67.2, 64.9, 53.7. HR-MS (ESI): m/z [M + Na]+ calcd for C30H25NNaO4: 486.1676; found: 486.1670.
(3S,3aR)-3-((Trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11b). White solid (310 mg, 67% yield); mp: 184–186 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 80/20; flow rate: 1 mL min−1; λ = 220 nm; T = 20 °C): tR = 12.05 min (1.10%), tR = 14.23 min (98.90%). The enantiomeric purity of 11b was determined to be 97.8% ee. [α]21D = +33.1 (c = 1, CHCl3). 1H NMR (500 MHz, acetone-d6) δ: 7.96 (dd, J = 1.0, 7.5 Hz, 1H), 7.56–7.44 (m, 6H), 7.40–7.32 (m, 6H), 7.32–7.24 (m, 3H), 7.08–6.96 (m, 2H), 6.94 (dd, J = 1.5, 8.0 Hz, 1H), 4.66–4.58 (m, 1H), 4.53 (dd, J = 3.0, 10.5 Hz, 1H), 4.16–4.06 (m, 1H), 4.02 (t, J = 10.5 Hz, 1H), 3.61 (dd, J = 4.0, 10.5 Hz, 1H), 3.46 (dd, J = 4.0, 10.5 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.7, 145.8, 144.7, 129.5, 128.9, 128.2, 125.2, 124.9, 122.1, 120.4, 117.7, 87.8, 75.5, 67.2, 64.9, 53.7. HR-MS (ESI): m/z [M + Na]+ calcd for C30H25NNaO4: 486.1676; found: 486.1663.
(3R,3aR)-3-((Trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11c). White solid (340 mg, 73% yield); mp: 249–251 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 85/15; flow rate: 0.7 mL min−1; λ = 220 nm; T = 20 °C): tR = 20.17 min (100.00%). The enantiomeric purity of 11c was determined to be >99% ee. [α]25D = −32.6 (c = 1, DMSO). 1H NMR (400 MHz, acetone-d6) δ: 8.28–8.24 (m, 1H), 7.48–7.42 (m, 6H), 7.33–7.22 (m, 9H), 7.09–6.98 (m, 2H), 6.95–6.90 (m, 1H), 5.06–5.00 (m, 1H), 4.56–4.49 (m, 1H), 4.40 (dd, J = 3.2, 10.4 Hz, 1H), 4.11 (t, J = 10.0 Hz, 1H), 3.60 (dd, J = 4.4, 11.2 Hz, 1H), 3.09 (dd, J = 2.8, 10.8 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.3, 145.2, 144.4, 129.4, 128.9, 128.2, 125.6, 124.5, 122.5, 118.5, 117.9, 88.1, 74.6, 64.3, 63.0, 54.2. HR-MS (ESI): m/z [M + Na]+ calcd for C30H25NNaO4: 486.1676; found: 486.1666.
(3S,3aS)-3-((Trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11d). White solid (371 mg, 80% yield); mp: 250–252 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 85/15; flow rate: 0.7 mL min−1; λ = 220 nm; T = 20 °C): tR = 21.88 min (100.00%). The enantiomeric purity of 11d was determined to be >99% ee. [α]25D = +35.8 (c = 1, DMSO). 1H NMR (400 MHz, acetone-d6) δ: 8.28–8.24 (m, 1H), 7.48–7.42 (m, 6H), 7.33–7.22 (m, 9H), 7.09–6.98 (m, 2H), 6.95–6.90 (m, 1H), 5.06–5.00 (m, 1H), 4.56–4.49 (m, 1H), 4.40 (dd, J = 3.2, 10.4 Hz, 1H), 4.11 (t, J = 10.0 Hz, 1H), 3.60 (dd, J = 4.4, 11.2 Hz, 1H), 3.09 (dd, J = 2.8, 10.8 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.4, 145.2, 144.4, 129.4, 128.9, 128.2, 125.6, 124.5, 122.5, 118.5, 117.9, 88.1, 74.6, 64.3, 63.0, 54.2. HR-MS (ESI): m/z [M + Na]+ calcd for C30H25NNaO4: 486.1676; found: 486.1672.
(3R,3aS)-8-Methoxy-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ab). White solid (379 mg, 77% yield); mp: 145–147 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 70/30; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 12.82 min (98.09%), tR = 15.68 min (1.91%). The enantiomeric purity of 11ab was determined to be 96.2% ee. [α]21D = −17.8 (c = 1, CHCl3). 1H NMR (500 MHz, acetone-d6) δ: 7.61 (d, J = 2.5 Hz, 1H), 7.56–7.46 (m, 6H), 7.42–7.32 (m, 6H), 7.32–7.25 (m, 3H), 6.85 (d, J = 9.0 Hz, 1H), 6.62 (dd, J = 2.5, 9.0 Hz, 1H), 4.65–4.57 (m, 1H), 4.47 (dd, J = 3.0, 10.5 Hz, 1H), 4.14–4.04 (m, 1H), 3.95 (t, J = 10.5 Hz, 1H), 3.77 (s, 3H), 3.60 (dd, J = 3.5, 10.5 Hz, 1H), 3.46 (dd, J = 4.5, 10.5 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 155.1, 154.6, 144.7, 139.6, 129.5, 128.9, 128.2, 125.1, 118.1, 111.0, 105.3, 87.9, 75.5, 67.0, 64.8, 56.0, 54.0. HR-MS (ESI): m/z [M + Na]+ calcd for C31H27NNaO5: 516.1781; found: 516.1762.
(3R,3aS)-7-Methoxy-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ac). White solid (403 mg, 82% yield); mp: 220–222 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 70/30; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 13.88 min (100.00%). The enantiomeric purity of 11ac was determined to be >99% ee. [α]21D = −27.1 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.78 (d, J = 9.2 Hz, 1H), 7.51–7.44 (m, 6H), 7.36–7.30 (m, 6H), 7.28–7.22 (m, 3H), 6.58 (dd, J = 2.8, 8.8 Hz, 1H), 6.48 (d, J = 2.8 Hz, 1H), 4.58–4.53 (m, 1H), 4.49 (dd, J = 1.6, 9.2 Hz, 1H), 4.06–3.93 (m, 2H), 3.73 (s, 3H), 3.56 (dd, J = 3.6, 10.4 Hz, 1H), 3.40 (dd, J = 4.4, 10.4 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 157.8, 154.9, 146.6, 144.7, 129.5, 128.9, 128.2, 121.4, 118.1, 108.3, 103.0, 87.8, 75.4, 67.0, 65.0, 55.9, 53.6. HR-MS (ESI): m/z [M + Na]+ calcd for C31H27NNaO5: 516.1781; found: 516.1780.
(3R,3aS)-8-Methyl-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ad). White solid (388 mg, 81% yield); mp: 155–157 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 90/10; flow rate: 1 mL min−1; λ = 220 nm; T = 25 °C): tR = 14.86 min (99.62%), tR = 20.00 min (0.38%). The enantiomeric purity of 11ad was determined to be 99.2% ee. [α]21D = −26.9 (c = 1, CHCl3). 1H NMR (500 MHz, acetone-d6) δ: 7.80 (s, 1H), 7.56–7.45 (m, 6H), 7.41–7.32 (m, 6H), 7.32–7.24 (m, 3H), 6.85 (d, J = 9.0 Hz, 1H), 6.81 (d, J = 9.0 Hz, 1H), 4.64–4.56 (m, 1H), 4.47 (d, J = 10.5 Hz, 1H), 4.13–4.04 (m, 1H), 3.97 (t, J = 10.5 Hz, 1H), 3.60 (d, J = 10.0 Hz, 1H), 3.46 (dd, J = 3.5, 10.5 Hz, 1H), 2.30 (s, 3H). 13C NMR (100 MHz, acetone-d6) δ: 154.7, 144.7, 143.6, 131.5, 129.5, 128.9, 128.2, 125.8, 124.5, 120.5, 117.4, 87.8, 75.4, 67.2, 64.9, 53.9, 21.0. HR-MS (ESI): m/z [M + H]+ calcd for C31H28NO4: 478.2013; found: 478.2005.
(3R,3aS)-7-Methyl-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ae). White solid (407 mg, 85% yield); mp: 237–239 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 80/20; flow rate: 1 mL min−1; λ = 220 nm; T = 25 °C): tR = 11.75 min (98.15%), tR = 14.16 min (1.85%). The enantiomeric purity of 11ae was determined to be 96.3% ee. [α]21D = −27.0 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.81 (d, J = 8.0 Hz, 1H), 7.52–7.46 (m, 6H), 7.39–7.31 (m, 6H), 7.31–7.25 (m, 3H), 6.84–6.78 (m, 1H), 6.76 (d, J = 0.8 Hz, 1H), 4.62–4.56 (m, 1H), 4.50 (dd, J = 2.8, 10.0 Hz, 1H), 4.10–4.03 (m, 1H), 3.99 (t, J = 10.0 Hz, 1H), 3.60 (dd, J = 4.0, 10.4 Hz, 1H), 3.44 (dd, J = 4.4, 10.4 Hz, 1H), 2.26 (s, 3H). 13C NMR (100 MHz, acetone-d6) δ: 154.8, 145.6, 144.7, 135.1, 129.5, 128.9, 128.2, 122.8, 122.3, 120.2, 118.0, 87.8, 75.4, 67.3, 64.9, 53.8, 21.0. HR-MS (ESI): m/z [M + Na]+ calcd for C31H27NNaO4: 500.1832; found: 500.1808.
(3R,3aS)-9-Bromo-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11af). Off-white solid (427 mg, 79% yield); mp: 234–236 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 80/20; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 14.85 min (99.51%), tR = 19.54 min (0.49%). The enantiomeric purity of 11af was determined to be 99.0% ee. [α]21D = −85.4 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.59–7.51 (m, 6H), 7.39–7.31 (m, 6H), 7.31–7.24 (m, 4H), 7.11 (t, J = 8.4 Hz, 1H), 6.94 (dd, J = 1.6, 8.4 Hz, 1H), 4.64–4.54 (m, 2H), 4.04 (t, J = 10.8 Hz, 1H), 3.99–3.93 (m, 1H), 3.62 (dd, J = 3.6, 10.8 Hz, 1H), 3.38 (dd, J = 4.0, 10.8 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.5, 149.6, 144.7, 129.6, 128.9, 128.3, 128.1, 126.7, 123.8, 119.4, 117.0, 87.8, 75.5, 67.5, 64.8, 52.0. HR-MS (ESI): m/z [M + Na]+ calcd for C30H24BrNNaO4: 564.0781; found: 564.0778.
(3R,3aS)-8-Bromo-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ag). Pink solid (454 mg, 84% yield); mp: 207–209 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 90/10; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 16.33 min (97.61%), tR = 21.17 min (2.39%). The enantiomeric purity of 11ag was determined to be 95.2% ee. [α]21D = −9.0 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 8.14 (d, J = 2.4 Hz, 1H), 7.53–7.46 (m, 6H), 7.40–7.32 (m, 6H), 7.32–7.25 (m, 3H), 7.18 (dd, J = 2.8, 8.8 Hz, 1H), 6.91 (d, J = 8.8 Hz, 1H), 4.69–4.63 (m, 1H), 4.57 (dd, J = 3.2, 10.4 Hz, 1H), 4.18–4.11 (m, 1H), 4.06 (t, J = 10.4 Hz, 1H), 3.62 (dd, J = 4.0, 10.8 Hz, 1H), 3.48 (dd, J = 4.4, 10.4 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.6, 145.0, 144.6, 129.5, 128.9, 128.2, 127.8, 126.3, 122.5, 119.6, 113.5, 87.9, 75.8, 67.1, 64.7, 53.4. HR-MS (ESI): m/z [M + Na]+ calcd for C30H24BrNNaO4: 564.0781; found: 564.0756.
(3R,3aS)-7-Bromo-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ah). White solid (478 mg, 88% yield); mp: 208–210 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 90/10; flow rate: 0.6 mL min−1; λ = 210 nm; T = 25 °C): tR = 27.91 min (2.21%), tR = 29.96 min (97.79%). The enantiomeric purity of 11ah was determined to be 95.6% ee. [α]21D = −12.9 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.90 (d, J = 8.8 Hz, 1H), 7.52–7.47 (m, 6H), 7.39–7.32 (m, 6H), 7.32–7.25 (m, 3H), 7.18 (dd, J = 2.0, 8.4 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 4.68–4.63 (m, 1H), 4.57 (dd, J = 2.4, 9.6 Hz, 1H), 4.16–4.10 (m, 1H), 4.07 (t, J = 9.6 Hz, 1H), 3.62 (dd, J = 3.6, 10.4 Hz, 1H), 3.46 (dd, J = 4.4, 10.8 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.6, 146.6, 144.6, 129.5, 128.9, 128.2, 125.1, 124.4, 121.7, 120.6, 116.7, 87.9, 75.7, 67.3, 64.8, 53.4. HR-MS (ESI): m/z [M + Na]+ calcd for C30H24BrNNaO4: 564.0781; found: 564.0772.
(3R,3aS)-6-Bromo-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ai). White solid (460 mg, 85% yield); mp: 213–215 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 95/5; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 29.35 min (1.38%), tR = 31.89 min (98.62%). The enantiomeric purity of 11ai was determined to be 97.2% ee. [α]21D = −34.8 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.98 (dd, J = 1.6, 8.0 Hz, 1H), 7.55–7.46 (m, 6H), 7.40–7.32 (m, 6H), 7.32–7.25 (m, 4H), 6.96 (t, J = 8.0 Hz, 1H), 4.74–4.62 (m, 2H), 4.24–4.09 (m, 2H), 3.63 (dd, J = 3.6, 10.4 Hz, 1H), 3.46 (dd, J = 4.0, 10.4 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.6, 144.6, 142.9, 129.5, 128.9, 128.7, 128.2, 126.3, 122.8, 119.8, 111.1, 87.8, 75.4, 67.8, 64.8, 53.5. HR-MS (ESI): m/z [M + Na]+ calcd for C30H24BrNNaO4: 564.0781; found: 564.0757.
(3R,3aS)-8-Fluoro-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11aj). White solid (405 mg, 84% yield); mp: 180–182 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 90/10; flow rate: 1 mL min−1; λ = 220 nm; T = 25 °C): tR = 16.99 min (98.18%), tR = 19.75 min (1.82%). The enantiomeric purity of 11aj was determined to be 96.4% ee. [α]21D = −26.9 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.76 (dd, J = 3.2, 10.4 Hz, 1H), 7.54–7.47 (m, 6H), 7.40–7.32 (m, 6H), 7.32–7.26 (m, 3H), 6.95 (dd, J = 5.2, 8.8 Hz, 1H), 6.84–6.78 (m, 1H), 4.68–4.62 (m, 1H), 4.54 (dd, J = 3.2, 10.4 Hz, 1H), 4.18–4.10 (m, 1H), 4.02 (t, J = 10.4 Hz, 1H), 3.62 (dd, J = 4.0, 10.8 Hz, 1H), 3.48 (dd, J = 4.4, 10.4 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 157.8 (d, J = 235 Hz), 154.6, 144.6, 141.9 (d, J = 3 Hz), 129.5, 128.9, 128.2, 125.4 (d, J = 12 Hz), 118.6 (d, J = 9 Hz), 111.4 (d, J = 24 Hz), 106.6 (d, J = 28 Hz), 87.9, 75.8, 67.0, 64.7, 53.7. HR-MS (ESI): m/z [M + Na]+ calcd for C30H24FNNaO4: 504.1582; found: 504.1570.
(3R,3aS)-7-Fluoro-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ak). White solid (412 mg, 86% yield); mp: 178–180 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 90/10; flow rate: 0.7 mL min−1; λ = 210 nm; T = 25 °C): tR = 23.65 min (97.54%), tR = 25.39 min (2.46%). The enantiomeric purity of 11ak was determined to be 95.1% ee. [α]21D = −29.5 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.94 (dd, J = 5.6, 8.8 Hz, 1H), 7.54–7.45 (m, 6H), 7.41–7.32 (m, 6H), 7.32–7.24 (m, 3H), 6.87–6.78 (m, 1H), 6.74 (dd, J = 2.8, 10.0 Hz, 1H), 4.67–4.60 (m, 1H), 4.60–4.53 (m, 1H), 4.16–4.00 (m, 2H), 3.61 (dd, J = 4.0, 10.8 Hz, 1H), 3.45 (dd, J = 4.4, 10.8 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 160.1 (d, J = 240 Hz), 154.9, 146.7 (d, J = 12 Hz), 144.6, 129.5, 128.9, 128.2, 121.6 (d, J = 10 Hz), 121.4 (d, J = 2 Hz), 108.8 (d, J = 22 Hz), 104.9 (d, J = 26 Hz), 87.8, 75.5, 67.5, 64.9, 53.3. HR-MS (ESI): m/z [M + Na]+ calcd for C30H24FNNaO4: 504.1582; found: 504.1578.
(3R,3aS)-6,8-Dichloro-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11al). Off-white solid (449 mg, 85% yield); mp: 205–207 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 90/10; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 12.24 min (98.95%), tR = 14.46 min (1.05%). The enantiomeric purity of 11al was determined to be 97.9% ee. [α]21D = −15.1 (c = 1, CHCl3). 1H NMR (500 MHz, acetone-d6) δ: 7.99 (s, 1H), 7.55–7.45 (m, 6H), 7.42–7.32 (m, 6H), 7.32–7.24 (m, 3H), 7.21 (s, 1H), 4.77–4.65 (m, 2H), 4.29–4.10 (m, 2H), 3.64 (dd, J = 3.5, 10.5 Hz, 1H), 3.50 (dd, J = 4.0, 10.5 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.4, 144.6, 141.0, 129.5, 128.9, 128.2, 127.1, 126.2, 124.9, 123.1, 118.4, 88.0, 75.8, 67.6, 64.6, 53.3. HR-MS (ESI): m/z [M + Na]+ calcd for C30H23Cl2NNaO4: 554.0896; found: 554.0880.
(3R,3aS)-7,8-Difluoro-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11am). White solid (413 mg, 83% yield); mp: 222–224 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 90/10; flow rate: 0.7 mL min−1; λ = 210 nm; T = 25 °C): tR = 20.21 min (98.59%), tR = 22.95 min (1.40%). The enantiomeric purity of 11am was determined to be 97.2% ee. [α]21D = −24.6 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 7.88 (dd, J = 8.4, 12.0 Hz, 1H), 7.52–7.47 (m, 6H), 7.39–7.32 (m, 6H), 7.32–7.25 (m, 3H), 6.96 (dd, J = 7.6, 11.6 Hz, 1H), 4.70–4.63 (m, 1H), 4.58 (dd, J = 2.8, 10.4 Hz, 1H), 4.17–4.10 (m, 1H), 4.06 (t, J = 10.4 Hz, 1H), 3.62 (dd, J = 3.6, 10.4 Hz, 1H), 3.47 (dd, J = 4.4, 10.4 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 153.7, 145.5 (dd, J = 13, 240 Hz), 143.5 (dd, J = 13, 237 Hz), 143.2, 140.7 (dd, J = 2, 10 Hz), 128.2, 128.0, 127.2, 119.6 (dd, J = 3, 9 Hz), 107.2 (d, J = 23 Hz), 106.0 (d, J = 21 Hz), 86.2, 74.6, 65.8, 63.6, 51.5. HR-MS (ESI): m/z [M + Na]+ calcd for C30H23F2NNaO4: 522.1487; found: 522.1474.
(3R,3aS)-7-Bromo-8-fluoro-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11an). Off-white solid (185 mg, 81% yield); mp: 221–223 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 80/20; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 9.87 min (100.00%). The enantiomeric purity of 11an was determined to be >99% ee. [α]21D = −9.7 (c = 1, CHCl3). 1H NMR (500 MHz, acetone-d6) δ: 7.88 (d, J = 10.0 Hz, 1H), 7.55–7.41 (m, 6H), 7.41–7.32 (m, 6H), 7.32–7.25 (m, 3H), 7.22 (d, J = 6.5 Hz, 1H), 4.74–4.63 (m, 1H), 4.57 (dd, J = 3.0, 10.5 Hz, 1H), 4.22–4.11 (m, 1H), 4.06 (t, J = 10.5 Hz, 1H), 3.63 (dd, J = 4.0, 11.0 Hz, 1H), 3.49 (dd, J = 4.0, 10.5 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.4, 154.2 (d, J = 236 Hz), 144.6, 142.6 (d, J = 2 Hz), 129.5, 128.9, 128.2, 125.2 (d, J = 10 Hz), 121.6, 107.4 (d, J = 30 Hz), 102.6 (d, J = 23 Hz), 88.0, 76.0, 67.1, 64.6, 53.4. HR-MS (ESI): m/z [M + Na]+ calcd for C30H23BrFNNaO4: 582.0687; found: 582.0682.
(3R,3aS)-8-Nitro-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ao). Yellow solid (405 mg, 80% yield); mp: 235–237 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 60/40; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 20.31 min (100.00%). The enantiomeric purity of 11ao was determined to be >99% ee. [α]21D = +5.3 (c = 1, CHCl3). 1H NMR (500 MHz, acetone-d6) δ: 8.88 (d, J = 2.0 Hz, 1H), 7.97 (dd, J = 2.5, 9.0 Hz, 1H), 7.55–7.46 (m, 6H), 7.40–7.32 (m, 6H), 7.32–7.24 (m, 3H), 7.17 (d, J = 9.0 Hz, 1H), 4.78–4.67 (m, 2H), 4.30–4.19 (m, 2H), 3.66 (dd, J = 3.5, 10.5 Hz, 1H), 3.55 (dd, J = 4.5, 10.5 Hz, 1H). 13C NMR (100 MHz, acetone-d6) δ: 154.6, 151.0, 144.6, 142.7, 129.5, 128.9, 128.3, 125.2, 120.8, 118.3, 115.6, 88.0, 76.0, 67.7, 64.7, 53.0. HR-MS (ESI): m/z [M + Na]+ calcd for C30H24N2NaO6: 531.1527; found: 531.1525.
(3R,3aS)-7-Nitro-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ap). Yellow solid (374 mg, 74% yield); mp: 220–222 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 60/40; flow rate: 1 mL min−1; λ = 210 nm; T = 25 °C): tR = 21.46 min (100.00%). The enantiomeric purity of 11ap was determined to be >99% ee. [α]21D = +34.0 (c = 1, CHCl3). 1H NMR (400 MHz, acetone-d6) δ: 8.23 (d, J = 8.8 Hz, 1H), 7.94 (dd, J = 2.8, 10.2 Hz, 1H), 7.78 (d, J = 2.8 Hz, 1H), 7.53–7.48 (m, 6H), 7.39–7.33 (m, 6H), 7.32–7.26 (m, 3H), 4.78–4.72 (m, 1H), 4.68 (dd, J = 2.8, 10.4 Hz, 1H), 4.31–4.24 (m, 1H), 4.19 (t, J = 10.4 Hz, 1H), 3.67 (dd, J = 3.6, 10.4 Hz, 1H), 3.52 (dd, J = 4.4, 10.8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 153.3, 144.1, 143.2, 142.9, 129.8, 128.2, 128.0, 127.2, 118.6, 116.9, 112.1, 86.3, 74.9, 65.7, 63.4, 51.8. HR-MS (ESI): m/z [M + Na]+ calcd for C30H24N2NaO6: 531.1527; found: 531.1524.
(3R,3aS)-7-(3-Oxomorpholino)-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11aq). Off-white solid (1.6 g, 62% yield). Mp: 211–213 °C. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/ethyl acetate, 50/50; flow rate: 0.5 mL min−1; λ = 254 nm; T = 25 °C): tR = 26.30 min (100.00%). The enantiomeric purity of 11aq was determined to be >99% ee. [α]25D = −11.6 (c = 1, CHCl3). 1H NMR (400 MHz, DMSO-d6) δ: 7.85 (d, J = 9.2 Hz, 1H), 7.54–7.19 (m, 15H), 7.16–6.91 (m, 2H), 4.66–4.57 (m, 1H), 4.53–4.42 (m, 1H), 4.18 (s, 2H), 4.05–3.90 (m, 4H), 3.70 (t, J = 5.2 Hz, 2H), 3.48 (dd, J = 3.2, 10.8 Hz, 1H), 3.37–3.25 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ: 165.9, 153.8, 144.3, 143.3, 137.8, 128.2, 128.0, 127.2, 121.6, 119.2, 118.4, 114.2, 86.2, 74.4, 67.7, 65.9, 63.6, 63.4, 51.9, 49.0. HR-MS (ESI): m/z [M + H]+ calcd for C34H31N2O6: 563.2177; found: 563.2201.
Gram-scale synthesis and application
(3R,3aS)-7-Bromo-3-((trityloxy)methyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (11ah). Compound 11ah was prepared from 8i (1.4 g, 4.36 mmol) and compound 9a (2.0 g, 5.89 mmol) following the general procedure. After workup, MeOH was added to the residue. The mixture was stirred for 1 h, and then the slurry was filtered, the filter cake was washed with MeOH to give the product (1.7 g, 74%) as an off-white solid. The product was analyzed by chiral HPLC (IC, 5 μm, 4.6 mm × 250 mm; eluent: hexane/isopropanol, 90/10; flow rate: 0.6 mL min−1; λ = 210 nm; T = 25 °C): tR = 29.71 min (100.00%). The enantiomeric purity of 11ah was determined to be >99% ee.
(3R,3aS)-7-Bromo-3-(hydroxymethyl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (12). To a solution of compound 11ah (1.6 g, 3 mmol) in MeOH (1.6 mL) and DCM (16 mL) was added TsOH·H2O (1.14 g, 6 mmol). The reaction mixture was stirred at room temperature for 4 h. Saturated NaHCO3 (6 mL) was added and stirred vigorously for 10 min. The mixture was extracted with DCM. The combined organic layer was dried over anhydrous Na2SO4 and concentrated in vacuum. The residue was stirred in a mixture of DCM (10 mL) and PE (10 mL) for 10 min. After filtration, the filter cake was dried to give compound 12 (0.8 g, 89%) as an off-white solid. Mp: 148–150 °C; [α]25D = −25.4 (c = 0.22, DMSO). 1H NMR (400 MHz, CDCl3) δ: 7.89 (dd, J = 1.6, 8.0 Hz, 1H), 7.15–7.09 (m, 2H), 4.50 (dd, J = 3.2, 10.4 Hz, 1H), 4.41–4.33 (m, 1H), 4.22–4.12 (m, 1H), 4.03 (dd, J = 4.0, 12.4 Hz, 1H), 3.96–3.82 (m, 2H), 2.18 (brs, 1H). 13C NMR (100 MHz, CDCl3) δ: 153.4, 144.9, 125.0, 122.2, 120.4, 120.2, 117.0, 75.9, 66.6, 62.1, 52.1. HR-MS (ESI): m/z [M + H]+ calcd for C11H11BrNO4: 299.9866; found: 299.9876.
(3R,3aS)-3-(Hydroxymethyl)-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one (13). To a mixture of compound 12 (100 mg, 0.33 mmol), bis(pinacolato)diboron (168 mg, 0.66 mmol), KOAc (98 mg, 1 mmol) and Pd(dppf)Cl2·CH2Cl2 (14 mg) was added DMSO (3 mL) under argon. The reaction mixture was heated at 85 °C for 2.5 h. The mixture was cooled to room temperature and water was added. The resulting mixture was extracted with ethyl acetate and the organic phase was washed with water and brine, dried over anhydrous Na2SO4, filtered and the filtrate was concentrated in vacuo. The residue was purified by chromatography on silica gel eluted with EtOAc/PE (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give 13 (91 mg, 80%) as a white solid. Mp: 184–186 °C. 1H NMR (400 MHz, CDCl3) δ: 8.02 (d, J = 8.0 Hz, 1H), 7.43 (dd, J = 1.2, 8.0 Hz, 1H), 7.40 (d, J = 1.2 Hz, 1H), 4.48 (dd, J = 3.2, 10.4 Hz, 1H), 4.39–4.32 (m, 1H), 4.23–4.15 (m, 1H), 4.02 (dd, J = 3.6, 12.4 Hz, 1H), 3.95–3.81 (m, 2H), 1.34 (s, 12H). 13C NMR (100 MHz, CDCl3) δ: 153.3, 143.6, 128.4, 125.6, 123.1, 118.3, 83.9, 76.0, 66.4, 62.1, 52.5, 24.9, 24.8. HR-MS (ESI): m/z [M + H]+ calcd for C17H23BNO6: 348.1613; found: 348.1630.
:1) to give 13 (91 mg, 80%) as a white solid. Mp: 184–186 °C. 1H NMR (400 MHz, CDCl3) δ: 8.02 (d, J = 8.0 Hz, 1H), 7.43 (dd, J = 1.2, 8.0 Hz, 1H), 7.40 (d, J = 1.2 Hz, 1H), 4.48 (dd, J = 3.2, 10.4 Hz, 1H), 4.39–4.32 (m, 1H), 4.23–4.15 (m, 1H), 4.02 (dd, J = 3.6, 12.4 Hz, 1H), 3.95–3.81 (m, 2H), 1.34 (s, 12H). 13C NMR (100 MHz, CDCl3) δ: 153.3, 143.6, 128.4, 125.6, 123.1, 118.3, 83.9, 76.0, 66.4, 62.1, 52.5, 24.9, 24.8. HR-MS (ESI): m/z [M + H]+ calcd for C17H23BNO6: 348.1613; found: 348.1630.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial supports from the National Natural Science Foundation of China (no. 81502917) and the National Science & Technology Major Project of China (no. 2015ZX09102007-013) are greatly appreciated.
Notes and references
- K. J. Shaw and M. R. Barbachyn, Ann. N. Y. Acad. Sci, 2011, 1241, 48 CrossRef CAS PubMed.
- W. B. Im, S. H. Choi, J.-Y. Park, S. H. Choi, J. Finn and S.-H. Yoon, Eur. J. Med. Chem., 2011, 46, 1027 CrossRef CAS PubMed.
- S. Roehrig, A. Straub, J. Pohlmann, T. Lampe, J. Pernerstorfer, K.-H. Schlemmer, P. Reinemer and E. Perzborn, J. Med. Chem., 2005, 48, 5900 CrossRef CAS PubMed.
-
(a) Q.-S. Xin, H.-X. Fan, B. Guo, H.-L. He, S. Gao, H. Wang, Y. Q. Huang and Y.-S. Yang, J. Med. Chem., 2011, 54, 7493 CrossRef CAS PubMed;
(b) B. Guo, H.-X. Fan, Q.-S. Xin, W.-J. Chu, H. Wang and Y.-S. Yang, Bioorg. Med. Chem. Lett., 2013, 23, 3697 CrossRef CAS PubMed;
(c) B. Guo, H.-X. Fan, Q.-S. Xin, W.-J. Chu, H. Wang, Y.-Q. Huang, X.-Y. Chen and Y.-S. Yang, J. Med. Chem., 2013, 56, 2642 CrossRef CAS PubMed.
-
(a) T. Xue, S. Ding, B. Guo, Y.-R. Zhou, P. Sun, H.-Y. Wang, W.-J. Chu, G.-Q. Gong, Y.-Y. Wang, X.-Y. Chen and Y.-S. Yang, J. Med. Chem., 2014, 57, 7770 CrossRef CAS PubMed;
(b) C. Xu and Y.-J. Ren, Bioorg. Med. Chem. Lett., 2015, 25, 4522 CrossRef CAS PubMed.
- H.-Y. Zhao, Y. Lu, L. Sheng, Z.-S. Yuan, B. Wang, W.-P. Wang, Y. Li, C. Ma, X.-L. Wang, D.-F. Zhang and H.-H. Huang, ACS Med. Chem. Lett., 2017, 8, 533 CrossRef CAS PubMed.
- K. C. Nicolaou, P. S. Baran, Y.-L. Zhong and K. Sugita, J. Am. Chem. Soc., 2002, 124, 2212 CrossRef CAS PubMed.
- Y.-J. Cui, Y.-X. Dang, Y.-S. Yang and R.-Y. Ji, J. Heterocycl. Chem., 2006, 43, 1071 CrossRef CAS.
- S.-H. Chen, Z.-Z. Ding, G.-L. Gong, X.-B. Yan, W. Huang, F. Guo, B.-L. Duan, R.-L. Gao, P.-F. Zhou, X.-H. Lu and G.-M. Dong, CN Pat., 105503903B, New Drug Research and Development Co., Ltd. of North China Pharmaceutical Corporation, 2019.
- R. Malhotra, T. K. Dey, S. Basu and S. Hajra, Org. Biomol. Chem., 2015, 13, 3211 RSC.
- H.-H. Huang, D.-F. Zhang and H.-Y. Zhao, WO Pat., 2018177302A1, Institute of Materia Medica, Chinese Academy of Medical Sciences, 2018.
- A. Berger and K. D. Wehrstedt, J. Loss Prev. Process Ind., 2010, 23, 734 CrossRef CAS.
- M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed., 2003, 42, 2010 CrossRef CAS PubMed.
- P. Chen, Y.-Y. Zhang, C.-H. Shi, X. Meng, Y.-S. Yang, X.-L. Zhou and B. Guo, J. Chem. Res., 2019, 43, 380 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures for the substrates and their spectroscopic characterization, 1H and 13C NMR spectra for compounds, chiral HPLC chromatograms of compounds 9 and 11. See DOI: 10.1039/d0ra04104a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab and
Haihong Huang*ab
*ab and
Haihong Huang*ab









