
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis of 2-hydroxy-4H-quinolizin-4-one scaffolds by integrating coenzyme a ligases and a type III PKS from Huperzia serrata†
Juan Wang‡
,
Ning Ding‡,
Yun Wu,
Xiaoping Shi,
Bowen Qi,
Xiao Liu ,
Xiaohui Wang,
Jun Li,
Pengfei Tu and
Shepo Shi*
,
Xiaohui Wang,
Jun Li,
Pengfei Tu and
Shepo Shi*
Modern Research Center for Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing 100029, China. E-mail: shishepo@163.com
First published on 22nd June 2020
Abstract
2-Hydroxy-4H-quinolizin-4-one scaffolds were enzymatically synthesized by integrating three enzymes including phenylacetate-CoA ligase (PcPCL) from an endophytic fungus Penicillium chrysogenum MT-12, malonyl-CoA synthase (AtMatB) from Arabidopsis thaliana, and a type III polyketide synthase (HsPKS3) from Chinese club moss Huperzia serrata. The findings paved the way to produce these kinds of structurally interesting alkaloids by engineered microorganisms.
Introduction
Quinolizinones represent an important group of heterocyclic molecules with a bridgehead nitrogen atom, displaying unique physicochemical attributes, such as polar zwitterionic character and low log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value, and a broad spectrum of bioactivities, including selective activation of M1 muscarinic receptor, inhibition of HIV integrase and phosphoinositide-3-kinase (PI3K), and anti-ulcerative and anti-allergic activities.1 Noteworthily, a number of quinolizinones, being the bioisosteres of quinolone-type antibiotics, also indicate promising potential to be new antibacterial agents (Fig. 1).2 In addition, quinolizinones tend to be readily reduced to partially- or fully-saturated heterocyclic systems,3 generally termed as quinolizidines which widely occur in the plant kingdom, particularly in the families of Leguminosae, Lycopodiaceae, Huperziaceae and so on, exhibiting great bioactivity diversity (Fig. 1).4 Encouraged by the significant importance of quinolizinones in new drug research and development, strategies to construct quinolizinone scaffolds have been extensively reported.3c,5 However, their chemical syntheses always involved multistep and environmentally harmful reactions. Accordingly, investigations on the enzymatic syntheses of quinolizinone scaffolds are definitely meaningful and interesting, which would also pave the way to produce pharmaceutically important quinolizinones by engineered microorganisms.
P value, and a broad spectrum of bioactivities, including selective activation of M1 muscarinic receptor, inhibition of HIV integrase and phosphoinositide-3-kinase (PI3K), and anti-ulcerative and anti-allergic activities.1 Noteworthily, a number of quinolizinones, being the bioisosteres of quinolone-type antibiotics, also indicate promising potential to be new antibacterial agents (Fig. 1).2 In addition, quinolizinones tend to be readily reduced to partially- or fully-saturated heterocyclic systems,3 generally termed as quinolizidines which widely occur in the plant kingdom, particularly in the families of Leguminosae, Lycopodiaceae, Huperziaceae and so on, exhibiting great bioactivity diversity (Fig. 1).4 Encouraged by the significant importance of quinolizinones in new drug research and development, strategies to construct quinolizinone scaffolds have been extensively reported.3c,5 However, their chemical syntheses always involved multistep and environmentally harmful reactions. Accordingly, investigations on the enzymatic syntheses of quinolizinone scaffolds are definitely meaningful and interesting, which would also pave the way to produce pharmaceutically important quinolizinones by engineered microorganisms.
 | ||
| Fig. 1 Examples of synthetic quinolizinones (1–3) and natural products containing quinolizidine ring system (4–6). | ||
It has been well reported that type III polyketide synthases (PKSs), a class of architecturally simple homodimeric proteins belonging to chalcone synthase superfamily, could utilize coenzyme A (CoA) thioesters as starters to perform iteratively decarboxylative condensation reactions with malonyl-CoAs to produce a vast array of molecules with diverse structures and biological activities.6 Remarkably, some plant specific type III PKSs represented as acridone synthase (ACS) and quinolone synthase (QNS) could efficiently employ bulky N-methylanthraniloyl-CoA as a starter to synthesize acridone and quinolone alkaloids, where the formation mechanism involved a canonical C–N bond forming reaction (Scheme 1A and 1B).7 Accordingly, non-physiological substrates containing nitrogen atoms have been applied to mimetically generate complex polyketide-alkaloid hybrids with important bioactivities.8 However, restricted by the limited knowledge on the promiscuous substrate tolerance and the uncontrollable cyclization modes of the polyketide intermediates, synthesis of designated molecules using type III PKSs is currently a great challenge. On the other hand, reactions catalyzed by type III PKSs require commercially unavailable or extremely expensive CoA thioesters as substrates, which also greatly confines the application of type III PKSs as ideal biocatalysts.
 | ||
| Scheme 1 Enzymatic synthesis of alkaloids by plant specific type III PKSs. (A) Acridone scaffold by ACS; (B) quinolone scaffold by QNS, and (C) quinolizinone scaffolds by integrating PcPCL, AtMatB, and HsPKS3. | ||
In the present study, enzymatic synthesis of 2-hydroxy-4H-quinolizin-4-one scaffolds by integrating three enzymes including phenylacetate-CoA ligase from an endophytic fungus Penicillium chrysogenum MT-12 (PcPCL),9 malonyl-CoA synthase from Arabidopsis thaliana (AtMatB),10 and a type III PKS from Chinese club moss Huperzia serrata (HsPKS3) was described (Scheme 1C).11
Results and discussion
Retrosynthetic analysis suggested that enzymatic synthesis of 2-hydroxy-4H-quinolizin-4-one scaffold might be achieved via intramolecular cyclization of the diketide intermediate produced by a certain type III PKS from the condensation of 2-pyridylacetyl-CoA and one molecule of malonyl-CoA (Scheme 2). The great challenge is to screen out a suitable type III PKS which could readily accept 2-pyridylacetyl-CoA as a starter to perform the aforementioned condensation and intramolecular cyclization reactions. Accordingly, 2-pyridylacetyl-CoA was chemically synthesized and used as a probe for the screening of a type III PKSs library comprising ten proteins (Fig. S1†). Only two type III PKSs (HsPKS1 and HsPKS3) from H. serrata,11,12 as a result, could expectedly catalyze the generation of a product with same molecular weight m/z 162.0552 ([M + H]+), consistent with an empirical molecular formula of C9H7NO2 (calc for C9H8NO2, 162.0555). Evaluated by the peak area showed on the HPLC charts, the crude yield of the product generated by HsPKS3 is about 2.5 times that of HsPKS1, the temperature and pH optima were 30–40 °C and 7.0–8.0, respectively. In order to unambiguously determine the structure of the enzymatic product, large-scale co-incubation of 2-pyridylacetyl-CoA, malonyl-CoA, and the recombinant enzyme HsPKS3 was performed, and the enzymatic product was purified by prep-HPLC (2.3 mg, yield ∼72%). The NMR spectroscopic data showed nine carbon resonances at δC 168.9, 161.8, 144.8, 131.2, 127.4, 125.6, 114.6, 96.5, and 95.2, six proton resonances at δH 6.01 (1H, s), 6.39 (1H, s), 6.92 (1H, t, J = 6.0 Hz), 7.34 (1H, t, J = 8.0 Hz), 7.44 (1H, d, J = 9.0 Hz), and 8.81 (1H, d, J = 7.0 Hz). Comprehensive analysis of the 1D- and 2D-NMR spectra (Table S1 and Fig. S2–S5†) allowed unambiguous assignments of all the proton and carbon resonances. Accordingly, the structure of the product was unequivocally determined as 2-hydroxy-4H-quinolizinone-4-one (P1) (Scheme 1C), a very useful scaffold for facile access to quinolizione and quinolizine skeletons with great structural complexity and biological diversity.13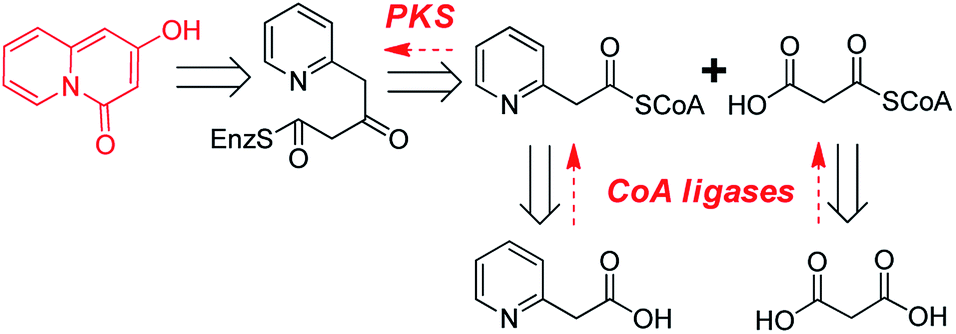 | ||
| Scheme 2 Retrosynthetic analysis of the enzymatic synthesis of 2-hydroxy-4H-quinolizin-4-one scaffold. | ||
Next, we keep going forward to seek for the right CoA ligases which could efficiently convert commercially available 2-(pyridine-2-yl) acetic acid and malonic acid to 2-pyridylacetyl-CoA and malonyl-CoA. At4CL, an A. thaliana originated 4-coumarate coenzyme A ligase (4CL) with the versatile capabilities of employing diverse phenylpropanoic acid analogues to form their corresponding CoA thioesters,9b,14 was firstly selected as a candidate. Unfortunately, the purified At4CL could not convert 2-(pyridine-2-yl)acetic acid to its CoA thioester. It has been reported that phenylacetate-CoA ligase from P. chrysogenum (PcPCL) could produce phenylacetate-CoA from phenylacetic acid.9 Considering the high similarity of 2-(pyridine-2-yl)acetic acid and phenylacetic acid, we assumed that PcPCL might accept 2-(pyridine-2-yl)acetic acid to produce its CoA thioester. Thus, PcPCL was cloned from P. chrysogenum MT-12, an endophytic fungus we previously isolated from H. serrata.15 Expectedly, the recombinant PcPCL could also efficiently accept 2-(pyridine-2-yl)acetic acid, besides phenylpropanoic acid analogues such as p-coumaric acid, ferulic acid, and 4-phenylpropanoic acid, to generate 2-pyridylacetyl-CoA (52 mg, yield ∼58%) (Fig. S6–S8†). Notably, PcPCL could also accept 2-(5-F-pyridine-2yl) acetic acid and 2-(6-F-pyridine-2yl) acetic acid to produce their corresponding CoA thioesters (Fig. S9 and S10†). For the formation of the 2-pyridylacetyl-CoA derivatives, kinetic analysis indicated that the values of KM and kcat are of the same magnitude as those reported for p-coumaric acid (Table S2†).9 Regarding to the enzymatic synthesis of malonyl-CoA, a gene annotated as malonyl-CoA synthase (AtMatB) was amplified from A. thaliana and heterologously expressed in E. coli according to our previous report (Fig. S11†).10
To avoid the tedious and time-consuming purification of CoA thioester intermediates, one-pot synthesis of quinolizinone scaffolds was developed. Under the optimized reaction condition (pH 7.5 and temperature 37 °C), the 2-hydroxy-4H-quinolizinone-4-one scaffold (P1) was successfully synthesized from 2-(pyridine-2-yl) acetic acid and malonic acid by the one-pot incubation of PcPCL, AtMatB, and HsPKS3 (Fig. 2A). When malonic acid was replaced by methylmalonic acid, the one-pot reaction generated P2 with molecular weight m/z 176.0703 ([M + H]+) which is 14 Da larger than that of P1 (Scheme 1C and Fig. 2B), suggesting the additional presence of a methyl group in P2. Large-scale reactions combined with HPLC purification allowed the accumulation of 8 mg (yield ∼45%) sample for measuring the NMR spectra of P2. Comparison of the NMR spectroscopic data of P2 with those of P1 revealed that the olefinic proton resonated at δH 6.01 (1H, s) was replaced by a methyl group [δH 2.01 (3H, s), δC 10.1], demonstrating the successful synthesis of 2-hydroxy-3-methyl-4H-quinolizinone-4-one (P2) (Table S1 and Fig. S12–S13†). When malonic acid was replaced by the bulky ethylmalonic acid or allylmalonic acid, 2-hydroxy-3-ethyl-4H-quinolizinone-4-one (P3) and 2-hydroxy-3-allyl-4H-quinolizinone-4-one (P4) could also be produced but with poor yields (Fig. S14 and S15†), which should be caused by the facts of that the bulky malonic acid derivatives could not be efficiently accepted by AtMatB.10 Notably, when 2-(pyridine-2-yl)acetic acid was replaced by 2-(5-fluoro-pyridine-2-yl)acetic acid, 7-fluoro-2-hydroxy-4H-quinolizin-4-one (P5) could be detected from the one-pot reaction mixture by LC-MS (Fig. S16†). In contrast, (6-fluoro-pyridine-2-yl)acetyl-CoA could not be accepted by HsPKS3 to generate the expected quinolizinone scaffold. Large-to-small substitutions of the amino residues lining the active-site cavity of HsPKS3 revealed that a mutant, HsPKS3 N221G, could obviously increase the yields of P3 and P4 (Fig. S17†). In contrast, the other single-site mutants, F225L, F275L, and S348G did not obviously change the yields of the end products. Noteworthily, the only one residue mutation (HsPKS3 N221G) also made the production of 2-hydroxypyrido[2,1-a]isoindole-4,6-dione, a known pyridoisoindole scaffold produced by HsPKS1 (Fig. S18†).8a
 | ||
| Fig. 2 LC-MS charts for the one-pot formation of (A) 2-hydroxy-4H-quinolizinone-4-one (P1) and (B) 2-hydroxy-3-methyl-4H-quinolizinone-4-one (P2) by PcPCL, AtMatB, and HsPKS3. The HPLC chromatograms were measured at 230 nm. | ||
With the merits of environmental friendly conditions, low processing cost, and easy separation of the end products, enzymes catalyzed one-pot multistep reactions have recently attracted significant attention.16 The method reported here circumvented the separation of CoA thioester intermediates, which is tedious, time-consuming, and challenging due to the high hydrophilicity and the easy degradation of CoA thioesters. Additionally, the one-pot reaction could also provide a recycling system that coenzyme A released from the CoA thioesters by HsPKS3 could be reused by the CoA ligases (PcPCL and AtMatB). Predictably, this method would be more practical and economic using an ATP regeneration system since ATP is the critical and costly cofactor in the cascade reactions.17 Nevertheless, this work sets the stage for the construction of an engineered microorganism to produce 2-hydroxy-4H-quinolizin-4-one scaffolds.
Experimental section
Chemicals
Chemicals and reagents were purchased from Sigma Aldrich (St. Louis, MO, USA), J&K Scientific Ltd. (Beijing, China), BioDee Biotechnology Co. Ltd. (Beijing, China), unless noted otherwise. Restriction enzymes, DNA polymerase and DNA ligase were purchased from Takara Biotechnology Co. Ltd. (Dalian, China). Primer synthesis and DNA sequencing were performed at Invitrogen (Shanghai, China). Malonyl-CoA was purchased from Sigma Aldrich (St. Louis, MO, USA). Chemical shifts (δ) were recorded with reference to solvent signals (1H NMR: CD3OD 3.31 ppm; DMSO-d6 2.50 ppm; 13C NMR: CD3OD 49.0 ppm; DMSO-d6 39.5 ppm). HRESIMS analyses were performed with LCMS-IT-TOF system (Shimadzu, Kyoto, Japan) equipped with an electrospray ionization source.Expression and protein purification of type III PKSs
We have previously reported three type III PKSs (AsCHS1, AsPKS1, and AsPKS2) from Aquilaria sinensis,18 two type III PKSs (HsPKS1 and HsPKS3) from Huperzia serrata,11,14 two quinolone synthases (ErQNS1 and ErQNS2) from Euodia rutaecarpa,19 and two type III PKSs (ClDCS and ZoCURS) from Curcuma longa and Zingiber officinale.20 The preparation of the nine recombinant proteins were performed according to our previous reports. For the preparation of the recombinant protein HsPKS2, the plasmid encoding the known HsPKS2 from H. serrata was constructed according to the previous report, and the expression and protein purification were performed according to the same procedure reported in the literatures.21Cloning, expression, and protein purification of coenzyme A ligases
For the preparation of phenylacetate-CoA ligase (PCL), the gene encoding PCL (AJ001540) was amplified via polymerase chain reaction (PCR) from genomic DNA isolated from Penicillium chrysogenum MT-12 using Pyrobest™ DNA Polymerase. The PCR mixture (50 μL) contained 25 ng of plasmid DNA, 5 μL of 10× Pyrobest buffer, 4 μL dNTPmix (2.5 mM each), 50 pmol of each primer (forward primer 5′-CC G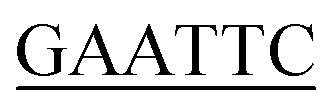 ATGGTTTTTTTACCTCC-3′ and reverse primer 5′-CCC
ATGGTTTTTTTACCTCC-3′ and reverse primer 5′-CCC GATCTTGCTACCAG CC-3′), 1.25 units of Pyrobest™ DNA Polymerase, and ddH2O 30.25 μL. The gene was amplified using a Mastercycler nexus (Eppendorf, Hamburg, Germany), with the following parameters: 94 °C for 1 min followed by 35 cycles of 98 °C for 5 s, 54 °C for 45 s, 72 °C for 1 min and 30 s. The amplified gene was cloned into a pMD19-T vector and sequenced. After removal of the introns by overlapping PCR method, a full length of pcl gene was finally obtained using N- and C-terminal specific primers 5′-CGC
GATCTTGCTACCAG CC-3′), 1.25 units of Pyrobest™ DNA Polymerase, and ddH2O 30.25 μL. The gene was amplified using a Mastercycler nexus (Eppendorf, Hamburg, Germany), with the following parameters: 94 °C for 1 min followed by 35 cycles of 98 °C for 5 s, 54 °C for 45 s, 72 °C for 1 min and 30 s. The amplified gene was cloned into a pMD19-T vector and sequenced. After removal of the introns by overlapping PCR method, a full length of pcl gene was finally obtained using N- and C-terminal specific primers 5′-CGC ATGGTTTT TTTACCTCCAAAGG-3′ (the BamHI site is underlined) and 5′-CCC
ATGGTTTT TTTACCTCCAAAGG-3′ (the BamHI site is underlined) and 5′-CCC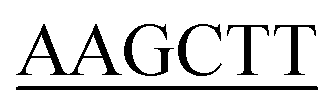 TTAATGATGGTGATGGTGATGGATCTTGCTACCGCCTTTC-3' (the Hind III site is underlined). The amplified full-length pcl gene was digested with BamHI/Hind III and cloned into the BamHI/Hind III sites of pMAL-C2X vector. After confirmation of the sequence, the plasmid containing the full-length cDNA encoding PcPCL was transformed into E. coli BL21(DE3). The cells harboring the plasmid were cultured to an OD600 of 0.6 in LB medium containing 100 μg mL−1 of Ampicillin at 37 °C, and 0.5 mM isopropyl-1-thio-β-D-galactopyranoside was then added to induce protein expression. The culture was incubated further at 17 °C for 16 h. The E. coli cells were harvested by centrifugation at 7500g for 10 min using Ependorf 5810R centrifuge (Ependorf, Germany), and resuspended in TANG buffer [50 mM Tris/HCl, pH 7.5, 200 mM NaCl, 0.002% sodium azide and 10% (w/v) glycerol] containing 0.1% (v/v) PMSF. Cells were disrupted by sonication, and centrifuged at 8000g for 40 min. The supernatant was loaded on a Ni2+ Sepharose 6 Fast Flow column (Biohao). After washing the column with Tang buffer, the recombinant protein was subsequently eluted with TANG buffer containing 500 mM imidazole. The protein solution was then concentrated by millipore column and diluted five-fold with 20 mM KPB buffer (pH 7.8), containing 10% (v/v) glycerol and 100 mM NaCl. The purified protein was analyzed by SDS-PAGE, and protein concentration was determined by the BCA method (Protein Assay, BIOMIGA) with the bovine serum albumin as standard.
TTAATGATGGTGATGGTGATGGATCTTGCTACCGCCTTTC-3' (the Hind III site is underlined). The amplified full-length pcl gene was digested with BamHI/Hind III and cloned into the BamHI/Hind III sites of pMAL-C2X vector. After confirmation of the sequence, the plasmid containing the full-length cDNA encoding PcPCL was transformed into E. coli BL21(DE3). The cells harboring the plasmid were cultured to an OD600 of 0.6 in LB medium containing 100 μg mL−1 of Ampicillin at 37 °C, and 0.5 mM isopropyl-1-thio-β-D-galactopyranoside was then added to induce protein expression. The culture was incubated further at 17 °C for 16 h. The E. coli cells were harvested by centrifugation at 7500g for 10 min using Ependorf 5810R centrifuge (Ependorf, Germany), and resuspended in TANG buffer [50 mM Tris/HCl, pH 7.5, 200 mM NaCl, 0.002% sodium azide and 10% (w/v) glycerol] containing 0.1% (v/v) PMSF. Cells were disrupted by sonication, and centrifuged at 8000g for 40 min. The supernatant was loaded on a Ni2+ Sepharose 6 Fast Flow column (Biohao). After washing the column with Tang buffer, the recombinant protein was subsequently eluted with TANG buffer containing 500 mM imidazole. The protein solution was then concentrated by millipore column and diluted five-fold with 20 mM KPB buffer (pH 7.8), containing 10% (v/v) glycerol and 100 mM NaCl. The purified protein was analyzed by SDS-PAGE, and protein concentration was determined by the BCA method (Protein Assay, BIOMIGA) with the bovine serum albumin as standard.
For the preparation of malonyl-CoA synthase (AtMatB), the plasmid encoding the known MatB from A. thaliana was constructed according to our previous report, and the expression and protein purification were performed according to the same procedure reported in the literature.22
Chemical synthesis of 2-pyridylacetyl-CoA
2-Pyridylacetic acid (5 mmol) prepared from 2-pyridylacetic acid hydrochloride via alkalization using sodium hydroxide and N-hydroxysuccinimide (5 mmol) were dissolved in a flask containing 90 mL anhydrous tetrahydrofuran (THF). Then, the solution of 8 mmol dicyclohexyl carbodimide (DCC) in 10 mL THF was dropped slowly into the flask. After being stirred for 5 h under room temperature, the mixture was filtered to remove the white precipitates. The filtrate was evaporated under reduced pressure and crystallized in absolute ethanol. Then, the solution of 0.12 mmol crystals in 5 mL acetone was dropped into the flask containing 10 mL mixture of CoA (60 μmol) and NaHCO3 (0.3 mmol) solutions. After being stirred for 5 h at 0 °C, the acetone was completely removed by vacuum evaporation. Then, ammonium acetate was added to the water phase to a final concentration of 4%, and the mixture was loaded onto Sep-Pak cartridge preconditioned with 4% ammonium acetate. The column was eluted with 4% ammonium acetate solution to completely remove the free CoA, and then 2-pyridylacetyl-CoA was obtained by elution with ddH2O. The structure was elucidated by HRESIMS and 1H NMR techniques.2-Pyridylacetyl-CoA was obtained as white powder. 1H NMR (500 MHz, in CD3OD): δH 8.47 (1H, d, J = 5.5 Hz), 7.81 (1H, td, J = 7.5, 2.0 Hz), 7.43 (1H, d, J = 7.5 Hz), 7.34 (1H, td, J = 7.5, 5.5 Hz), 4.27 (2H, s), 8.57 (1H, s), 8.20 (1H, s), 6.13 (1H, d, J = 6.0 Hz), 3.45 (2H, overlapped), 4.50 (1H, s), 4.08 (1H, s), 4.82 (1H, s), 4.27 (1H, s), 4.02 (1H, m), 3.58 (1H, m), 3.36 (4H, s), 3.02 (2H, m), 2.39 (2H, m), 1.08 (3H, s), 0.83 (3H, s). HRESIMS: m/z [M + H]+ 887.1551, calc for C28H42N8O17P3S, 887.1596.
Enzymatic synthesis of 2-hydroxy-4H-quinolizin-4-one by HsPKS3
The reaction mixture contained 50 nmol of 2-pyridylacetyl-CoA, 50 nmol of malonyl-CoA, 0.4 nmol of the purified recombinant protein HsPKS3, in a final volume of 500 μL of 100 mM potassium phosphate buffer (pH 7.0). The reaction was incubated at 37 °C for 12 h. The products were then extracted three times with 800 μL of ethyl acetate. After removal of solvent by evaporation, the residue was redissolved by MeOH and analyzed by LC-MS on an Agilent ZORBAX SB C18 Column (4.6 mm I.D. × 250 mm, 5 μm) with a flow rate of 1.0 mL min−1. The gradient elution for analyzing was performed with aqueous acetonitrile (MeCN). 0–5 min, 5–30% MeCN; 5–10 min, 30–40% MeCN; 10–15 min, 40% MeCN; 15–22 min, 40–60% MeCN; 22–25 min, 60–95% MeCN.For large scale reaction: 20 μmol of 2-pyridylacetyl-CoA, 20 μmol of malonyl-CoA and 0.2 μmol of recombinant HsPKS3 was dissolved in 100 mL 100 mM potassium phosphate buffer (pH 7.0) and incubated at 37 °C for 12 h. The reaction mixture was extracted by 100 mL ethyl acetate for three times. After removal of solvent under reduced pressure, the residue was dissolved in 0.5 mL of MeOH and purified by semi-preparative HPLC using Agilent ZORBAX Eclipse XDB C18 Column (4.6 mm I.D. × 250 mm, 5 μm). The structure of the purified product was determined by HRESIMS and NMR techniques.
Enzymatic synthesis of 2-pyridylacetyl-CoA derivatives by PcPCL and kinetic analysis
The reaction mixture contained 0.10 mmol acid, 62.5 μmol MgCl2, 1.6 mmol NaCl, 0.15 mmol CoA, 32 μmol ATP, and 10 nmol of the purified PcPCL, in a final volume of 10 mL of 100 mM Tris–HCl buffer (pH 7.5). The reaction was incubated at 30 °C for 6 h. The products were then purified by Sep-Pak cartridge using the method mentioned in the section of synthesizing 2-pyridylacetyl-CoA. The enzymatic product was analyzed by LC-MS on an Shimadzu shim-pack XR-ODS II (2.0 mm I.D. × 100 mm, 2.2 μm) column with a flow rate of 0.2 mL min−1. The gradient elution for analyzing was performed with aqueous acetonitrile containing 5 mM ammonium formate: 0–10 min, 5% MeCN; 10–20 min, 5–100% MeCN.Kinetic parameters were determined according to the method previously reported. By varying the concentration of the acid at the fixed concentration of ATP, CoA, and Mg2+, the apparent kinetic parameters for 2-pyridylacetic acid derivatives were determined by fitting the initial rates of CoA thioesters formation with the Michaelis–Menten equation.23
Enzymatic synthesis of malonyl-CoA by AtMatB
The reaction mixture contained 0.10 mmol malonic acid, 62.5 μmol MgCl2, 1.6 mmol NaCl, 0.15 mmol CoA, 32 μmol ATP, and 10 nmol of the purified AtMatB, in a final volume of 10 mL of 100 mM potassium phosphate buffer (pH 7.0). The reaction was incubated at 30 °C for 6 h. The products were then purified by Sep-Pak cartridge using the method mentioned in the section of synthesizing 2-pyridylacetyl-CoA. The enzymatic product was analyzed by LC-MS on an Shimadzu shim-pack XR-ODS II (2.0 mm I.D. ×100 mm, 2.2 μm) column with a flow rate of 0.2 mL min−1. The gradient elution for analyzing was performed with aqueous acetonitrile containing 5 mM ammonium formate: 0–10 min, 5% MeCN; 10–20 min, 5–100% MeCN.One-pot synthesis of quinolizinone scaffolds
The reaction mixture contained 0.1 mmol 2-pyridylacetic acids, 0.1 mmol malonic acids, 62.5 μmol MgCl2, 60 μmol ATP, 0.45 mmol CoA, 10 nmol PcPCL, and 10 nmol AtMatB, in a final volume of 10 mL of 100 mM potassium phosphate buffer (pH 7.5). The reaction was incubated at 30 °C for 1 h, then 10 nmol HsPKS3 was added into the reaction mixture, and the reaction was incubated at 33 °C for another 6 h. The products were then extracted three times with ethyl acetate. After removal of solvent by evaporation, the residue was redissolved by MeOH and analyzed by HPLC and LC-MS on an Agilent ZORBAX SB C18 Column (4.6 mm I.D. × 250 mm, 5 μm) with a flow rate of 1.0 mL min−1. The gradient elution for analyzing was performed with aqueous acetonitrile (MeCN): 0–5 min, 5–30% MeCN; 5–10 min, 30–40% MeCN; 10–15 min, 40% MeCN; 15–22 min, 40–60% MeCN; 22–25 min, 60–95% MeCN, 25–35 min.Construction of HsPKS3 mutants
HsPKS3 mutants (N221G, F225L, F275L, S348G) were constructed using the Mutagenesis with In-Fusion HD Cloning Plus Kit (Takara) following the standard protocol and a pair of complementary mutagenic primers as follows (mutated codons are underlined); N221G, forward primer: 5′-CCCAAATCTCATA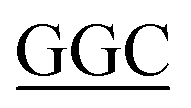 TCTGCTT-3′, reverse primer: 5′-AAGCAGA
TCTGCTT-3′, reverse primer: 5′-AAGCAGA TATGAGATTTGGG-3′; F225L, forward primer: 5′-TTCTGCTTTA
TATGAGATTTGGG-3′; F225L, forward primer: 5′-TTCTGCTTTA GGCGATGGTGC-3′, reverse primer: 5′-GCACCATCGCC
GGCGATGGTGC-3′, reverse primer: 5′-GCACCATCGCC TAAAGCAGAA-3′; F275L, forward primer: 5′-AG CTGGGATGGTA
TAAAGCAGAA-3′; F275L, forward primer: 5′-AG CTGGGATGGTA TTACTGAT-3′, reverse primer: 5′-ATCAGTAA
TTACTGAT-3′, reverse primer: 5′-ATCAGTAA TACCATCCCAGCT-3′; S348G, forward primer: 5′-GGAAACATG
TACCATCCCAGCT-3′; S348G, forward primer: 5′-GGAAACATG A GCCCTTCTGTG-3′, reverse primer: 5′-CACAGAAGGGCT
A GCCCTTCTGTG-3′, reverse primer: 5′-CACAGAAGGGCT CATGTTTCC-3′.
CATGTTTCC-3′.
Conclusions
In summary, the present work describes the one-pot formation of quinolizinone scaffolds by integrating phenylacetate-CoA ligase (PcPCL), malonyl-CoA ligase (AtMatB), and a type III polyketid synthase (HsPKS3). Here, both the formation of 2-pyridylacetyl-CoA by PcPCL and the synthesis of quinolizinone scaffold from 2-pyridylacetyl-CoA by HsPKS3 have never been reported. The findings not only expanded the biosynthetic repertoire of type III PKSs but also paved the way to produce pharmaceutically important quinolizinones by engineered microorganisms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 81573312, 81773842), the Drug Innovation Major Project (No. 2018ZX09711001-006).Notes and references
- (a) Y. Kitaura, T. Oku, H. Hirai, T. Yamamoto and M. Hashimoto(Fujisawa Pharmaceutical Co., Ltd.), US Pat. 4698349, 1987; (b) M. Satoh, H. Aramaki, H. Nakamura, M. Inoue, H. Kawakami, H. Shinkai, Y. Matsuzaki and K. Yamataka(Japan Tobacco Inc.) US Pat. 7745459B2, 2010; (c) S. D. Kuduk, R. K. Chang, C. N. D. Marco, W. J. Ray, L. Ma, M. Wittmann, M. A. Seager, K. A. Koeplinger, C. D. Thompson, G. D. Hartman and M. T. Bilodeau, ACS Med. Chem. Lett., 2010, 1, 263 CrossRef CAS PubMed; (d) S. D. Kuduk, R. K. Chang, T. J. Greshock, W. J. Ray, L. Ma, M. Wittmann, M. A. Seager, K. A. Koeplinger, C. D. Thompson, G. D. Hartman and M. T. Bilodeau, ACS Med. Chem. Lett., 2012, 3, 1070 CrossRef CAS PubMed; (e) M. R. Shukla, N. K. Jana, S. J. Mahangare, P. P. Vidhirte, D. R. Lagad, J. A. Tarage, S. A. Kulkarni, V. P. Palle and R. K. Kamboj(Lupin Limited), US Pat. 9944639B2, 2018.
- (a) Q. Li, L. A. Mitscher and L. L. Shen, Med. Res. Rev., 2000, 20, 231 CrossRef CAS PubMed; (b) J. A. Wiles, A. Hashimoto, J. A. Thanassi, J. J. Cheng, C. D. Incarvito, M. Deshpande, M. J. Pucci and B. J. Bradbury, J. Med. Chem., 2006, 49, 39 CrossRef CAS PubMed.
- (a) V. Boekelheide and J. P. Lodge, J. Am. Chem. Soc., 1951, 73, 3681 CrossRef CAS; (b) Y. Sato, Chem. Pharm. Bull., 1959, 7, 247 CrossRef CAS; (c) H. Yu, G. Y. Zhang and H. M. Huang, Angew. Chem., Int. Ed., 2015, 54, 10912 CrossRef CAS PubMed.
- (a) W. A. Ayer, L. S. Trifonov, in The Alkaloids: Chemistry and Pharmacology, ed. G. A. Cordell, A. Brossi, Academic Press, New York, 1994, vol. 45, pp. 233–266 Search PubMed; (b) S. Ohmiya, K. Saito, I. Murakoshi, in The Alkaloids: Chemistry and Pharmacology, ed. G. A Cordell, Academic Press, New York, 1995, vol. 47, pp. 1–114 Search PubMed; (c) X. Q. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752 RSC; (d) S. K. Mandal, R. Biswas, S. S. Bhattacharyya, S. Paul, S. Dutta, S. Pathak and A. R. Khuda-Bukhsh, Eur. J. Pharmacol., 2010, 626, 115 CrossRef CAS PubMed; (e) N. Walker, C. Howe, M. Glover, H. McRobbie, J. Barnes, V. Nosa, V. Parag, B. Bassett and C. Bullen, N. Engl. J. Med., 2014, 371, 2353 CrossRef PubMed; (f) Y. Tang, J. Xiong, J. J. Zhang, W. Wang, H. Y. Zhang and J. F. Hu, Org. Lett., 2016, 18, 4376 CrossRef CAS PubMed; (g) Y. B. Zhang, X. L. Zhang, N. H. Chen, Z. N. Wu, W. C. Ye, Y. L. Li and G. C. Wang, Org. Lett., 2017, 19, 424 CrossRef CAS PubMed; (h) W. J. Ni, C. J. Li, Y. X. Liu, H. J. Song, L. Z. Wang, H. B. Song and Q. M. Wang, J. Agric. Food Chem., 2017, 65, 2039 CrossRef CAS PubMed; (i) Y. B. Zhang, L. Yang, D. Luo, N. H. Chen, Z. N. Wu, W. C. Ye, Y. L. Li and G. C. Wang, Org. Lett., 2018, 20, 594 CrossRef PubMed; (j) W. W. Li, T. Y. Wang, B. Cao, B. Liu, Y. M. Rong, J. J. Wang, F. Wei, L. Q. Wei, H. Chen and Y. X. Liu, Mol. Med. Rep., 2019, 20, 455 CAS.
- (a) J. E. Douglass and D. A. Hunt, J. Org. Chem., 1977, 42, 3974 CrossRef CAS; (b) T. Kappe and Y. Linnau, Monatsh. Chem., 1983, 114, 349 CrossRef CAS; (c) W. Eberbach and W. Maier, Tetrahedron Lett., 1989, 30, 5591 CrossRef CAS; (d) A. G. Birchler, F. Q. Liu and L. S. Liebeskind, J. Org. Chem., 1994, 59, 7737 CrossRef CAS; (e) S. R. Natarajan, M. H. Chen, S. T. Heller, R. M. Tynebor, E. M. Crawford, M. X. Cui, K. Z. Han, J. C. Dong, B. Hu, H. Wu and S. H. Chen, Tetrahedron Lett., 2006, 47, 5063 CrossRef CAS; (f) Y. S. Xu, C. C. Zeng, Z. G. Jiao, L. M. Hu and R. G. Zhong, Molecules, 2009, 14, 868 CrossRef CAS PubMed; (g) C. W. Muir, A. R. Kennedy, J. M. Redmond and A. J. B. Watson, Org. Biomol. Chem., 2013, 11, 3337 RSC; (h) T. A. Alanine, W. R. J. D. Galloway, T. M. McGuire and D. R. Spring, Eur. J. Org. Chem., 2014, 2014, 5767 CrossRef CAS; (i) H. T. B. Bui, D. D. Vo, Y. N. T. Chau, C. T. K. Tu, H. V. Mai and K. V. Truong, Synth. Commun., 2015, 45, 2861 CrossRef CAS; (j) A. Rosas-Sánchez, R. A. Toscano, J. G. López-Cortés and M. C. Ortega-Alfaro, Dalton Trans., 2015, 44, 578 RSC; (k) Z. Q. Xie, S. Luo and Q. Zhu, Chem. Commun., 2016, 52, 12873 RSC; (l) J. Li, Y. D. Yang, Z. G. Wang, B. Y. Feng and J. S. You, Org. Lett., 2017, 19, 3083 CrossRef CAS PubMed; (m) J. Diesel, A. M. Finogenova and N. Cramer, J. Am. Chem. Soc., 2018, 140, 4489 CrossRef CAS PubMed; (n) C. C. Dong, J. F. Xiang, L. J. Xu and H. Y. Gong, J. Org. Chem., 2018, 83, 9561 CrossRef CAS PubMed; (o) Z. W. Chen, T. G. Liu, X. Y. Ma, P. Liang, L. P. Long and M. Ye, Synlett, 2019, 30, 863 CrossRef CAS; (p) X. B. Zhou, A. R. Chen, W. Du, Y. W. Wang, Y. Peng and H. M. Huang, Org. Lett., 2019, 21, 9114 CrossRef CAS PubMed.
- (a) J. Schroder, in Comprehensive Natural Products Chemistry, ed. U. Sankawa, Elsevier, Oxford, 1999, vol. 1, pp. 749–771 Search PubMed; (b) M. B. Austin and J. P. Noel, Nat. Prod. Rep., 2003, 20, 79 RSC; (c) I. Abe and H. Morita, Nat. Prod. Rep., 2010, 27, 809 RSC; (d) H. Morita, I. Abe and H. Noguchi, in Comprehensive Natural Products II: Chemistry and Biology, ed. H. W. Liu and L. Mander, Elsevier, Oxford, 2010, vol. 1, pp. 171–225 Search PubMed; (e) H. Morita, C. P. Wong and I. Abe, J. Biol. Chem., 2019, 294, 15121 CrossRef CAS PubMed.
- (a) K. T. Junghanns, R. E. Kneusel, D. Gröger and U. Matern, Phytochemistry, 1998, 49, 403 CrossRef CAS PubMed; (b) R. Lukačin, K. Springob, C. Urbanke, C. Ernwein, G. Schröder, J. Schröder and U. Matern, FEBS Lett., 1999, 448, 135 CrossRef; (c) M. S. Resmi, P. Verma, R. S. Gokhale and E. V. Soniya, J. Biol. Chem., 2013, 288, 7271 CrossRef CAS PubMed; (d) T. Mori, Y. Shimokawa, T. Matsui, K. Kinjo, R. Kato, H. Noguchi, S. Sugio, H. Morita and I. Abe, J. Biol. Chem., 2013, 288, 28845 CrossRef CAS PubMed; (e) T. Matsui, T. Kodama, T. Mori, T. Tadakoshi, H. Noguchi, I. Abe and H. Morita, J. Biol. Chem., 2017, 292, 9117 CrossRef CAS PubMed.
- (a) H. Morita, M. Yamashita, S. P. Shi, T. Wakimoto, S. Kondo, R. Kato, S. Sugio, T. Kohno and I. Abe, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13504 CrossRef CAS PubMed; (b) T. Wakimoto, T. Mori, H. Morita and I. Abe, J. Am. Chem. Soc., 2011, 133, 4746 CrossRef CAS PubMed.
- (a) M. J. Koetsier, P. A. Jekel, M. A. Van den Berg, R. A. L. Bovenberg and D. B. Janssen, Biochem. J., 2009, 417, 467 CrossRef CAS PubMed; (b) M. K. Go, J. Y. Chow, V. W. N. Cheung, Y. P. Lim and W. S. Yew, Biochemistry, 2012, 51, 4568 CrossRef CAS PubMed.
- Y. Y. Liu, X. H. Wang, T. Mo, Y. R. Yan, Y. L. Song, Y. F. Zhao, J. Li, S. P. Shi, X. Liu and P. F. Tu, RSC Adv., 2017, 7, 21028 RSC.
- J. Wang, X. H. Wang, X. Liu, J. Li, X. P. Shi, Y. L. Song, K. W. Zeng, L. Zhang, P. F. Tu and S. P. Shi, Org. Lett., 2016, 18, 3550 CrossRef CAS PubMed.
- K. Wanibuchi, P. Zhang, T. Abe, H. Morita, T. Kohno, G. S. Chen, H. Noguchi and I. Abe, FEBS J., 2007, 274, 1073 CrossRef CAS PubMed.
- (a) B. S. Thyagarajan and P. V. Gopalakrishnan, Tetrahedron, 1965, 21, 945 CrossRef CAS; (b) Y. Quevedo-Acosta, I. D. Jurberg and D. Gamba-Sánchez, Org. Lett., 2020, 22, 239 CrossRef CAS PubMed.
- T. Mo, J. Wang, B. W. Gao, L. Zhang, X. Liu, X. H. Wang, P. F. Tu and S. P. Shi, Chin. J. Org. Chem., 2015, 35, 1052 CrossRef CAS.
- B. W. Qi, X. Liu, T. Mo, Z. X. Zhu, J. Li, J. Wang, X. P. Shi, K. W. Zeng, X. H. Wang, P. F. Tu, I. Abe and S. P. Shi, J. Nat. Prod., 2017, 80, 2699 CrossRef CAS PubMed.
- J. Muschiol, C. Peters, N. Oberleitner, N. Oberleitner, M. D. Mihovilovic, U. T. Bornscheuer and F. Rudroff, Chem. Commun., 2015, 51, 5798 RSC.
- (a) J. N. Andexer and M. Richter, ChemBioChem, 2015, 16, 380 CrossRef CAS PubMed; (b) X. Zhang, H. Wu, B. Huang, Z. Li and Q. Ye, J. Biotechnol., 2017, 241, 163 CrossRef CAS PubMed.
- (a) B. W. Gao, X. H. Wang, X. Liu, S. P. Shi and P. F. Tu, Bioorg. Med. Chem. Lett., 2015, 25, 1279 CrossRef CAS PubMed; (b) X. H. Wang, Z. X. Zhang, X. J. Dong, Y. Y. Feng, X. Liu, B. W. Gao, J. L. Wang, L. Zhang, J. Wang, S. P. Shi and P. F. Tu, Biochem. Biophys. Res. Commun., 2017, 486, 1040 CrossRef CAS PubMed.
- L. Zhang, J. Wang, X. J. Dong, Y. Y. Liu, B. W. Qi, S. P. Shi and P. F. Tu, Proc. 11th Natl. Nat. Org. Chem. Conf. Chin. Chem. Soc., 2016, p. 442 Search PubMed.
- L. Zhang, B. W. Gao, X. H. Wang, Z. X. Zhang, X. Liu, J. Wang, T. Mo, Y. Y. Liu, S. P. Shi and P. F. Tu, RSC Adv., 2016, 6, 12519 RSC.
- J. C. Ye, P. Zhang, J. Y. Sun, C. T. Guo, G. K. Chen, I. Abe and H. Noguchi, Acta Pharm. Sin., 2011, 46, 1273 CAS.
- Y. Y. Liu, X. H. Wang, T. Mo, Y. R. Yan, Y. L. Song, Y. F. Zhao, J. Li, S. P. Shi, X. Liu and P. F. Tu, RSC Adv., 2017, 7, 21028 RSC.
- M. J. Koetsier, P. A. Jekel, M. A. Van den Berg, R. A. Bovenberg and D. B. Janssen, Biochem. J., 2009, 417, 467 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04133e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |