DOI:
10.1039/D0RA00639D
(Paper)
RSC Adv., 2020,
10, 23573-23581
Essential geometric and electronic properties in stage-n graphite alkali-metal-intercalation compounds
Received
21st January 2020
, Accepted 22nd April 2020
First published on 30th June 2020
Abstract
The rich and unique properties of the stage-n graphite alkali-metal-intercalation compounds are fully investigated by first-principles calculations. According to the main features, the lithium and non-lithium (Na, K, Rb, Cs) systems are quite different from each other in stacking configurations, intercalant alkali-metal-atom concentrations, free conduction electron densities, atom-dominated and (carbon, alkali metal)-co-dominated energy bands, and interlayer charge density distributions. The close relations between the alkali-metal-doped metallic behaviors and the geometric symmetries are clarified through the interlayer atomic interactions. The stage-1 graphite alkali-metal-intercalation compounds possess the highest charge distribution for all stage-n types; moreover, those of the lithium systems are greater than those of the non-lithium systems. The lithium systems also have the largest blue shift of the Fermi level among all alkali metal systems.
Introduction
Graphene is a single layer with pure carbon honeycomb lattice.1 Bulk graphite,2 which could be regarded as a layered graphene system,3–6 has attracted a lot of theoretical and experimental research in basic science,4,6–8 engineering7 and applications.8 3D graphite systems might present AA, AB, ABC9 and turbostratic stackings.10 All of them belong to semimetals under the interlayer hopping integrals of C-2pz orbitals. In general, such condensed-matter systems become n- or p-type metals, depending on the kinds of intercalated atoms or molecules.11 For example, the intercalation of alkali metal atoms12 or FeCl3 (ref. 13) into graphite creates many free conduction electrons and valence holes, as observed in a pure metal. Also, graphite could serve as the best anode material14 in commercial Li+-based batteries,15 mainly owing to the lowest cost, the most stable structure for intercalation, and the outstanding ion transport under charging and discharging processes. When the Li+ ions are released from the cathode, they will transport through the electrolyte, and then intercalate into the graphitic system. Most importantly, the flexible interlayer spacings between graphene layers are capable of providing sufficient positions for the various intercalant concentrations.16 Any intermediate states and meta-stable configurations, which are created during the ion/atom intercalation, could survive through the very strong σ bondings in graphitic sheets. As a result, graphite is rather suitable for studying structural transformations in chemical reactions. For example, the close relations between graphene and intercalant layers in lattice symmetries are expected to present a dramatic transformation before and after the intercalation/de-intercalation processes.
Up to now, there have been a lot of theoretical and experimental studies on the fundamental properties in graphite-related systems. The former covers the phenomenological models and number simulation methods. For example, the tight-binding model, the random-phase approximation, and the Kubo formula are, respectively, utilized to investigate the electronic properties and magnetic quantization behaviors,17 Coulomb excitations18 and impurity screenings,19 and optical absorption spectra.20 Furthermore, first-principles calculations21 are available for understanding the optimal stacking configurations, intercalant lattices, π, σ and intercalation-induced energy bands, free conduction electrons/valence holes, and density of states (DOS).
Previous studies of alkali-metal-intercalation graphite compounds have primarily focused on Li, Na and K.22–24 However, certain critical physical quantities and pictures are absent from the previous studies, such as the atom-dominated band structures, the spatial charge densities between intercalant and graphene layers, the interlayer orbital hybridization of intercalant and carbon atom, and the atom- and orbital-projected van Hove singularities.
The main focuses of our work are the features of geometric and electronic properties in stage-n graphite alkali-metal-intercalation compounds (Li, Na, K, Rb, Cs) using the first-principles method.21 We aim to use first-principles calculation to understand the relation between the kinds of intercalants, concentrations and the essential properties, e.g., lattice symmetries of intercalant layers, the dominances of valence and conduction states by carbon and/or alkali metal atoms, the blue shift of the Fermi level, the interlayer charge density variations after the alkali-metal intercalations, and the atom- and orbital-decomposed DOS.
Theoretical calculations
First-principles density functional theory (DFT)26–29 calculations are used to investigate the optimal geometric structures, charge distribution, and electronic, magnetic and optical characteristics of intercalation compounds. In this paper, we performed the calculations with the Vienna ab initio simulation package (VASP), which evaluates an approximate solution within DFT by solving the Kohn–Sham equations. The Perdew–Burke–Ernzerhof formula used in VASP, which depends on the local electron density, is utilized to deal with many-particle Coulomb effects. As for the frequent electron-crystal scatterings, they are characterized by the projector-augmented wave pseudopotentials. The electron Bloch wave functions are solved from the linear superposition of plane waves, with a maximum kinetic energy of 500 eV. The current study on stage-n graphite compounds shows that the first Brillouin zone is sampled by 9 × 9 × 9 and 100 × 100 × 100 k-point meshes within the Monkhorst–Pack scheme, respectively, for the optimal geometry and band structure. Moreover, the convergence condition of the ground state energy is set to be ∼10−5 eV between two consecutive evaluation steps, where the maximum Hellmann–Feynman force for each ion is below 0.01 eV Å−1 during the atom relaxations.25
Results and discussion
(1) Unique stacking configurations and intercalant distributions
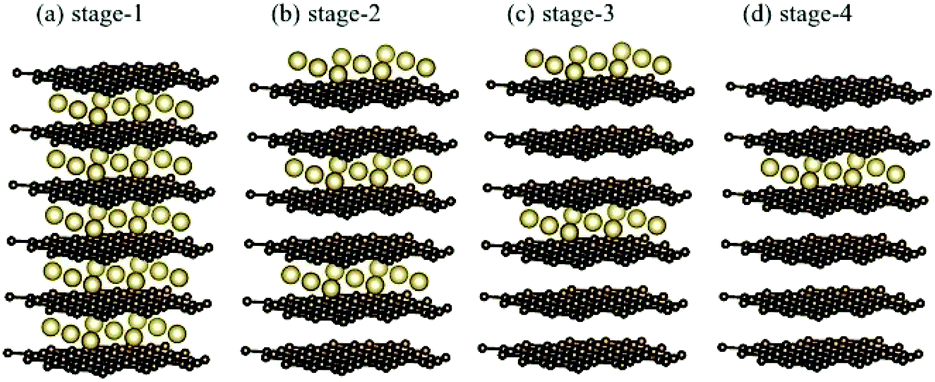
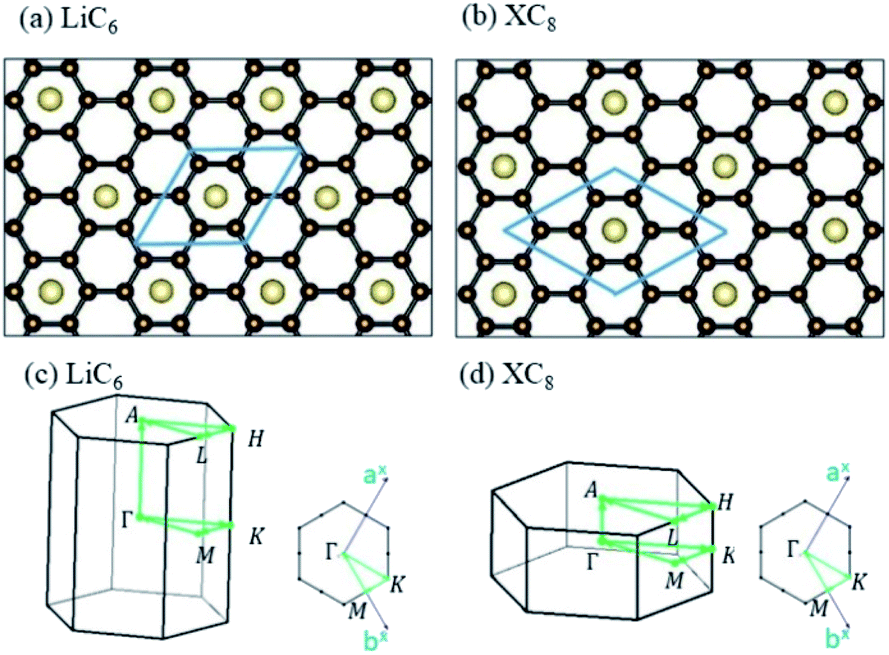
The normal stacking configurations, which are formed by layered graphene systems, include AAA, ABA, ABC and AAB. The interlayer spacings of graphitic layers are able to provide very suitable chemical environments for alkali metal intercalations and de-intercalations, i.e., they are very useful during the charging and discharging processes. Chemical modifications hardly affect the planar honeycomb lattices and thus do not change the orthogonal feature of π and σ bondings. Very interesting, the optimal geometric properties are rather sensitive to the (x, y)-plane distribution and concentration of alkali metal atoms, e.g., the interlayer distances of neighboring planes and an enlarged primitive unit cell. According to the highest concentration, the graphite alkali-metal-intercalation materials could be classified into two categories: lithium and non-lithium ones. There exist LiC6n or AC8n for the stage-n materials [Fig. 1(a)–(d)], as examined from experimental measurements and the theoretical predictions. The specific distance between carbon and intercalant layer is shortest/longest for Li/Cs [Table 1], only directly reflecting the effective atomic radius. Although all the alkali metal atoms present hollow-site optimal positions, their distribution symmetries are dominated by Li atoms or non-Li ones. The layered LiC6n and AC8n systems, respectively, possess a three- and four-times enhancement in the unit cell [Fig. 2(a) and (b)]; therefore, their reduced first Brillouin zones exhibit different high-symmetry points [Fig. 3(a)]. This is expected to have a strong effect on the initial π-electronic states of the Dirac-cone structure. More chemical bonds, which arise from the alkali metal and carbon atoms, are produced in graphite intercalation compounds compared with pristine systems. Such bondings need to be taken into account for the diverse essential properties. In addition to the intercalant distribution, the stacking configuration of two neighboring graphitic sheets also affects the ground state energy. The current study clearly shows that only the stage-1 LiC6 presents AA stacking, and the other compounds exhibit AB stacking. That is to say, all the graphite alkali-metal-intercalation materials exhibit the AB-stacked configurations except for the former system.
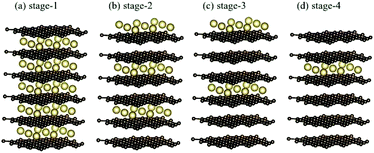 |
| | Fig. 1 The stage-n graphite alkali-metal-intercalation compounds: (a) n = 1, (b) n = 2, (c) n = 3, and (d) n = 4. | |
Table 1 The stage-dependent ground state energies, the optimal geometric properties, the blue shifts of the Fermi levels, and the red shifts of the initial σ-electronic states for the stage-n graphite alkali-metal-intercalation compounds
| |
Stage-1, alkali-metal-doped graphite |
Stage-2, alkali-metal-doped graphite |
| LiC6 |
NaC8 |
KC8 |
RbC8 |
CsC8 |
LiC12 |
NaC16 |
KC16 |
RbC16 |
CsC16 |
| Interlayer distance with intercalation (Å) |
3.815, 3.74,32 3.76 (ref. 30) (exp.) |
4.601, 4.52 (ref. 32) |
5.324, 5.40,32 5.35 (ref. 31) (exp.) |
5.824, 5.65 (ref. 32) |
6.032, 5.94 (ref. 32) |
|
|
|
|
|
| Ground state energy (eV) |
−57.29 |
−74.96 |
−75.13 |
−75.04 |
−75.12 |
−112.52 |
−148.78 |
−148.93 |
−148.84 |
−148.94 |
| Blue shift of EF in DOS |
1.80 |
1.54 |
1.48 |
1.41 |
1.58 |
1.37 |
1.23 |
1.17 |
1.12 |
1.10 |
| Blue shift of EF |
|
|
|
|
|
|
|
|
|
|
| In band structure (eV) |
1.28–2.20 |
1.47–1.66 |
1.47–1.51 |
1.60–1.64 |
1.59–1.65 |
1.34–1.38 |
1.19–1.20 |
1.15–1.16 |
1.08–1.09 |
1.06–1.06 |
| Red shifts of initial σ-electronic (eV) |
1.25 |
1.24 |
1.14 |
1.13 |
1.0 |
1.22 |
0.99 |
0.93 |
0.86 |
0.82 |
| |
Stage-3, alkali-metal-doped graphite |
Stage-4, alkali-metal-doped graphite |
| LiC18 |
NaC24 |
KC24 |
RbC24 |
CsC24 |
LiC24 |
NaC32 |
KC32 |
RbC32 |
CsC32 |
| Interlayer distance with intercalation (Å) |
3.864 |
4.878 |
5.360 |
6.056 |
6.058 |
|
|
|
|
|
| Ground state energy (eV) |
−166.99 |
−222.64 |
−222.81 |
−222.69 |
−222.79 |
−223.15 |
−289.80 |
−296.55 |
−296.42 |
−296.48 |
| Blue shift of EF in DOS |
1.07 |
1.01 |
0.98 |
0.94 |
0.90 |
0.89 |
0.83 |
0.80 |
0.77 |
0.78 |
| Blue shift of EF |
|
|
|
|
|
|
|
|
|
|
| In band structure (eV) |
1.22–1.23 |
1.02–1.03 |
0.57–0.58 |
0.84–0.84 |
0.76–0.77 |
0.31–0.33 |
0.63–0.64 |
0.63–0.63 |
0.51–0.52 |
0.41–0.41 |
| Red shifts of initial σ-electronic (eV) |
1.21 |
1.02 |
0.96 |
0.66 |
0.60 |
1.23 |
0.90 |
0.88 |
0.70 |
0.58 |
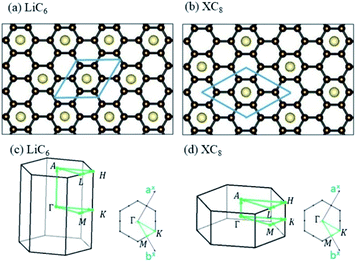 |
| | Fig. 2 The planar structures of (a) LiC6 and (b) XC8 [X = Na, K, Rb and Cs]. The first Brillouin zones of (c) LiC6 and (d) XC8 [X = Na, K, Rb and Cs]. | |
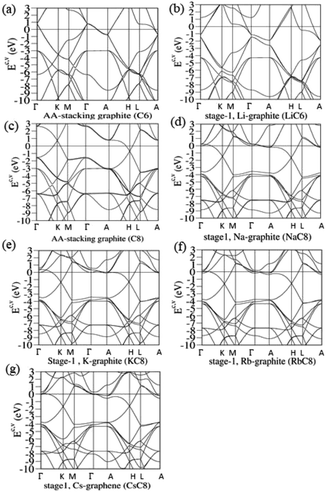 |
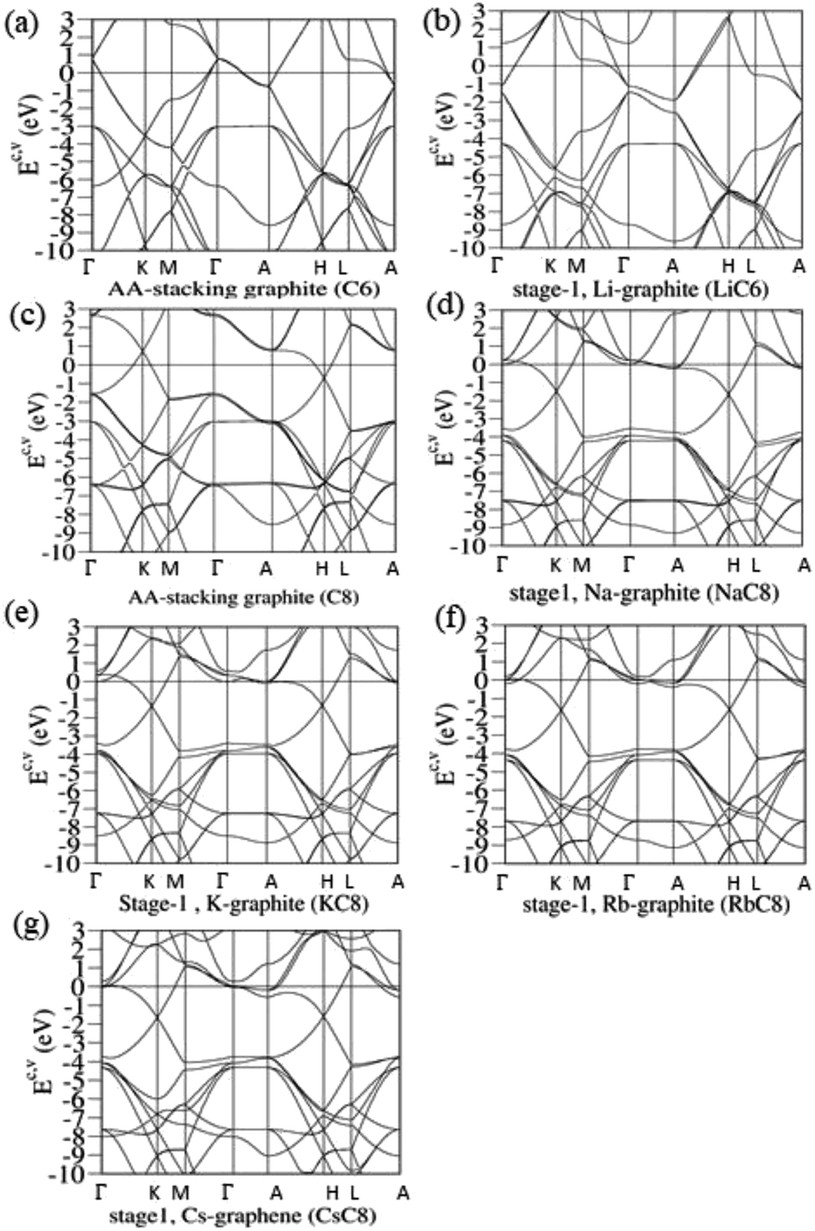
| | Fig. 3 The rich band structures of the pristine graphite and stage-1 graphite alkali-metal-intercalation compounds: (a) AA, (b) LiC6, (c) AB stackings without intercalations, (d) NaC8, (e) KC8, (f) RbC8 and (g) CsC8. | |
(2) Metallic and semi-metallic behaviors
Without alkali metal atom intercalations, pristine graphite exhibits unique semi-metallic behavior through weak, but significant van der Waals interactions. A semiconducting zero-gap graphene is dramatically changed into a semi-metallic system for any few-layer stackings, or infinite-layer graphites. For example, there exist the unusual overlaps of valence and conduction bands along the KH path [Fig. 3(c); or the ΓK path under the reduced Brillouin zone in Fig. 3(b)] in AA-, AB- and ABC-stacked graphites through single-orbital interlayer atomic interactions. According to the VASP, the simple hexagonal/rhombohedral graphite [Fig. 3(b)], with the highest/lowest stacking symmetry, presents the largest/lowest free carrier density of conduction electrons and valence holes. In general, the π valence bands, which are purely related to C-2pz, show the initial states at the stable K valley [Fig. 3(c)], the saddle-point structure near the M point [Ev ∼ −3 eV], and its termination in the Γ valley [the whole π-band width more than 7 eV].
The band structures of lithium- and non-lithium graphite intercalation compounds sharply contrast with each other, such as those of stage-1 LiC6 and XC8 compounds in Fig. 3(d) and (e)–(h), respectively. For the former, the Fermi level is transferred from the middle of Dirac-cone structures into the conduction ones near the Γ valley after the intercalation of lithium atoms. That is to say, EF presents a blue shift. Apparently, the free carriers are due to the outmost 2s orbitals of lithium atoms. The occupied and unoccupied states are highly asymmetric to each other about the Fermi level. The initial π/π* valence/conduction states are mainly determined by the stacking configurations in the chemical intercalations [the distribution symmetry of Li-intercalants in Fig. 2(a)], or the corresponding relation between the original and reduced first Brillouin zone. There exists an observable energy spacing of Ec ∼ 0.31–0.63 eV along the Γ–A path in the modified valence and conduction Dirac-cone structures. The creation of discontinuous states in honeycomb lattices might arise from the different ionization energies of Li-2s and C-2pz orbitals [the distinct on-site Coulomb potential energies]. The whole π valence-band width of LiC6, being created by the C-2pz orbitals, could be identified to about 7.32 eV/7.40 eV from the electronic energy spectrum along the Γ–M–K–Γ/A–H–L–A or Γ–K–M–Γ/A–L–H–A paths. This result clearly illustrates the well-behaved π bondings in graphite alkali-metal-intercalation compounds. Other σ bondings behave so, as indicated from the initial states at the Γ point of Ev ∼ −4.20 eV. Their orthogonal relation remains the same after chemical modification, being very useful in establishing the tight-binding model for graphite alkali-metal-intercalation compounds.
The main features of the band structure, as clearly indicated in Fig. 3(e)–(h), are dramatically changed for the other alkali-metal-atom intercalations. Both stage-1 AC8 and LiC6 have the totally different distribution configurations [Fig. 2(a) and (b)], and so do the reduced first Brillouin zones [Fig. 3(a)]. For the former, the Dirac-cone structures of the π and π* bands are initiated from the stable K/K′ valleys, but not the Γ ones. The energy spacing of separated Dirac points is small or almost vanishing; furthermore, the Fermi level is situated above the conduction point about ∼1.35–1.50 eV. In addition to EF, whether or not the second conduction energy subband is partially occupied depends on the kinds of alkali metal atoms, such as the alkali-metal-induced free electrons in the first and second subbands for KC8/RbC8/CsC8 [Fig. 3(f)/(g)/(h)]. The low-energy bands belong to the single states, without the split double degeneracy [Fig. 3(d) for LiC6]. The whole π-band energy spectra are identified from the K–Γ–K–M–Γ and H–A–H–L–A paths, leading to a width of ∼7.31–7.41 eV. Very interesting, one pair of π-valence subbands come to exist near Ev ∼ −4.0 eV, being accompanied by the initial pair of σ valence bands. This further illustrates the zone-folding effects on band structure and the well separation of π and σ bondings.
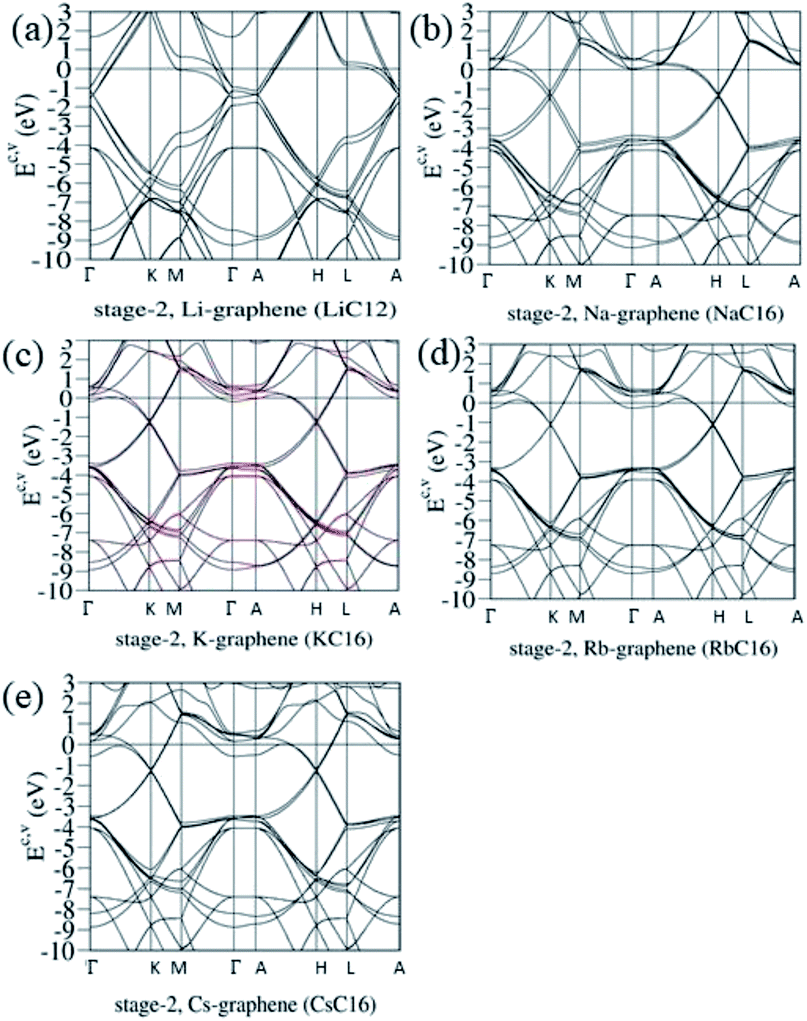
Electronic energy spectra strongly depend on the n stage of graphite intercalation compounds, as clearly illustrated in Fig. 4(a)–(e). Stage-2 LiC12 and AC16 have different first Brillouin zones on the (kx, ky) plane, and their kz-ranges are associated with the distances between two intercalant planes [Fig. 1]. Compared with stage-1 band structures [Fig. 3(d)–(g)], the number of Dirac cones becomes double in the stage-2 cases, in which the further modifications include the reduced blue shift of the Fermi level, the enhanced anisotropy, the induced energy spacings between valence and conduction Dirac points, and the diverse energy relations near the band-edge states. For example, four/two valence and conduction pairs arise from the Γ/K valley [or the A/H valley] for LiC12/AC16 [Fig. 4(a) and (b)–(e)]. Furthermore, the whole π-band widths could be roughly estimated from the ΓKMΓ/KMKΓ path [or the ALHA/HLHA path]. The low-energy essential properties are dominated by the π bondings of C-2pz orbitals. The above-mentioned obvious changes directly reflect the great enhancement of the interlayer atomic interactions due to the C–C bonds in two neighboring graphitic sheets/graphene-intercalant layers. Very interesting, all the stage-2 compounds exhibit a pair of σ bands at the Γ and A valleys, and the energy dispersions along ΓA are negligible. These results indicate the mutual orthogonality of the planar σ and perpendicular π bondings. Such a phenomenon is expected to survive in any stage-n graphite alkali-metal-intercalation compounds.
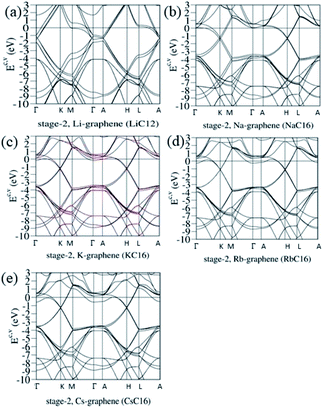 |
| | Fig. 4 The unusual electronic energy spectra of the AB-stacked stage-2 graphite alkali-metal-intercalation compounds: (a) LiC12, (b) NaC16, (c) KC16, (d) RbC16, and (e) CsC16. | |
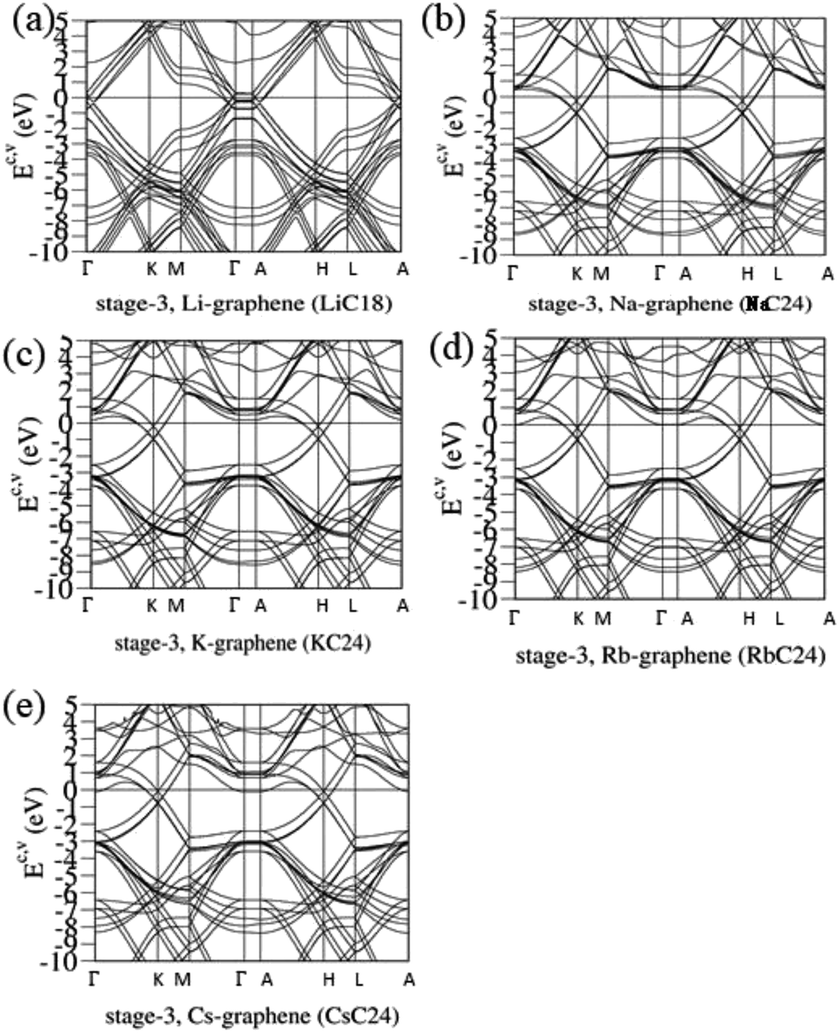
The blue shifts about the Fermi level and the free conduction electrons decline quickly with an increase of the n number. The concentration of alkali metal atoms is greatly reduced, and so is the charge transfer from their outmost s-orbitals to carbon 2pz-orbitals. Such results are clearly revealed in the stage-3 systems. Fig. 5 shows the slight modifications of the Dirac-cone structures and the intersecting of the Fermi level with most of the conduction bands. LiC18 [Fig. 5(a)] and AC24 [Fig. 5(b)–(e)], respectively, possess six and three pairs of linear valence and conduction bands near the Γ/A and K/H valleys. Apparently, there are significant changes in the observable energy spacing between valence and conduction Dirac points, the anisotropic Fermi velocities, and the distinct Fermi momenta slopes. The blue shifts of EF are estimated to be 0.45, 0.58, 0.55, 0.53, and 0.5 eVs. Very interesting, the electronic energy spectra become more complicated under the stronger zone-folding effects. The main features of electronic properties are closely related to the weak, but important Li–C bonds, e.g., the minor contributions of alkali metal atoms on each electronic state. That is, the diversified n-type dopings are created by the critical Li–C and C–C bondings.
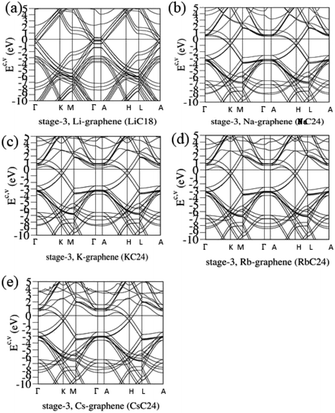 |
| | Fig. 5 The rich band structures for the AB-stacked stage-3 graphite alkali-metal-intercalation compounds: (a) LiC18, (b) NaC24, (c) KC24, (d) RbC24, and (e) CsC24. | |
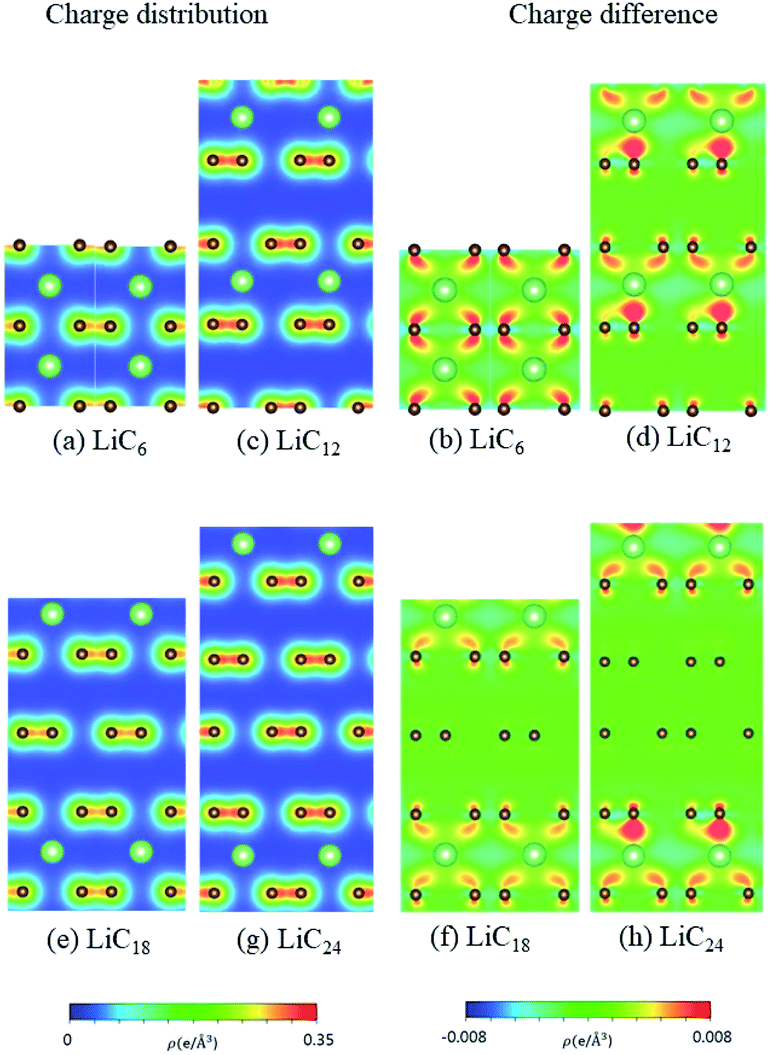
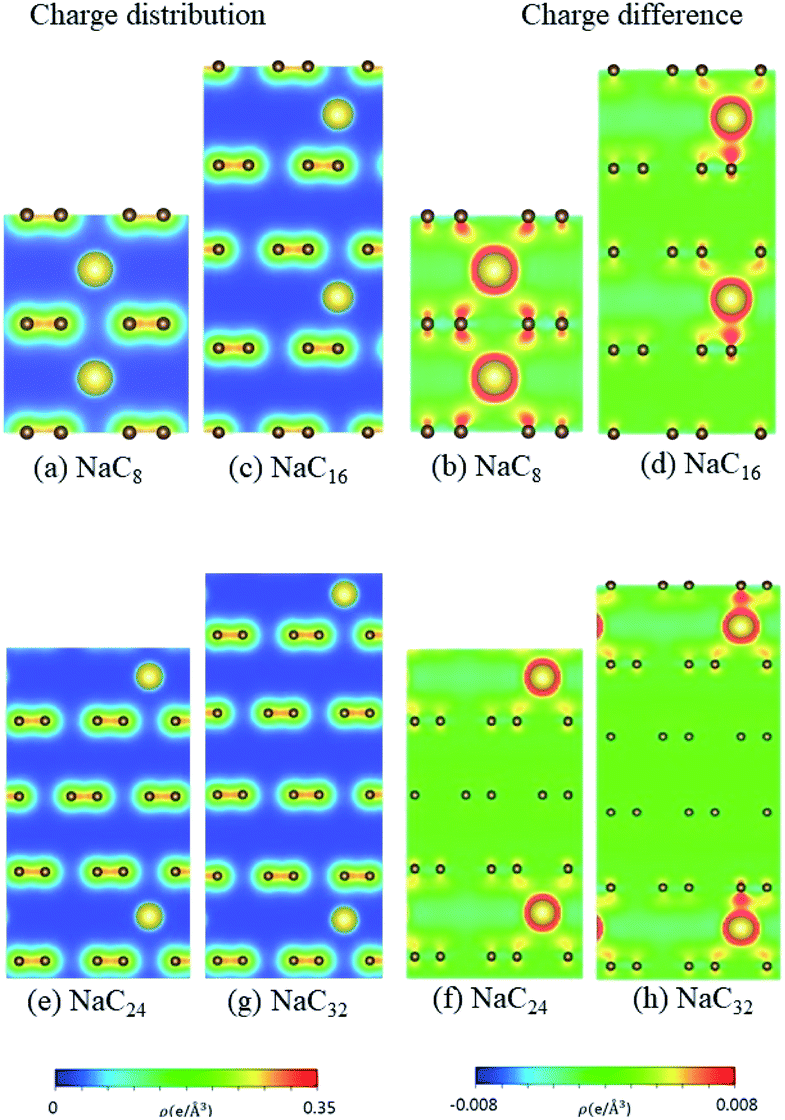
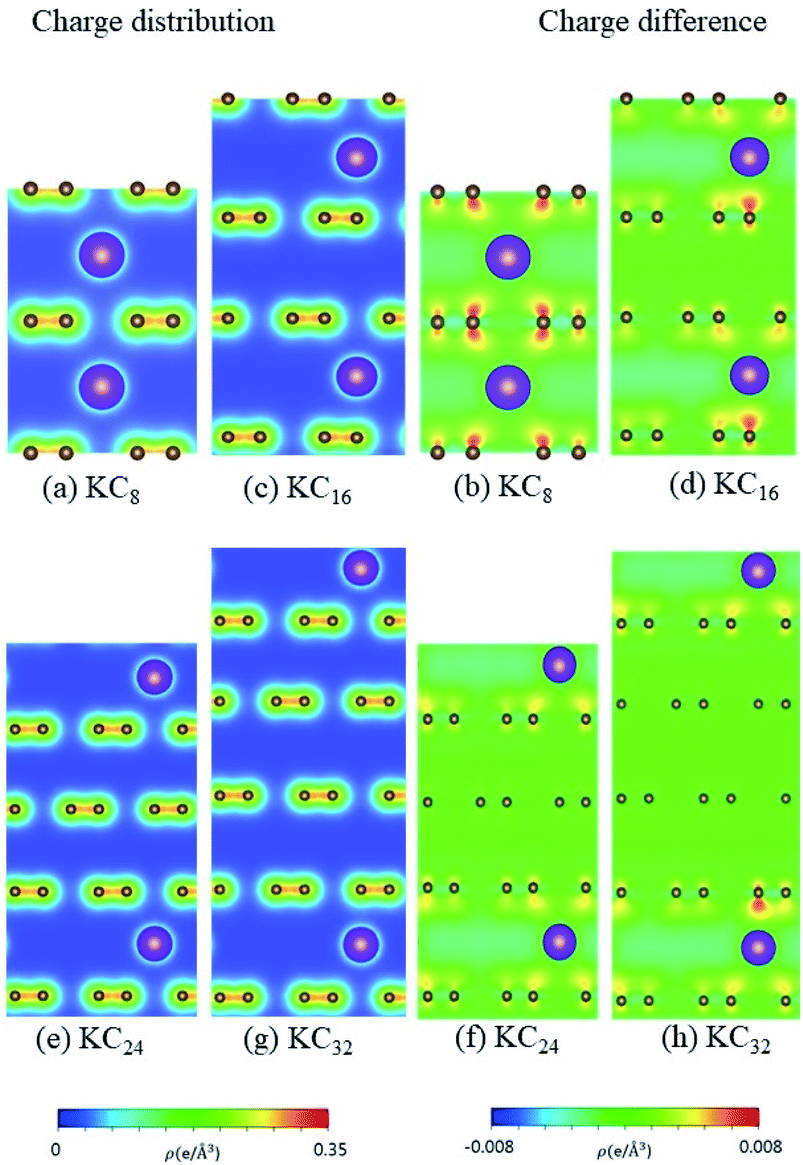
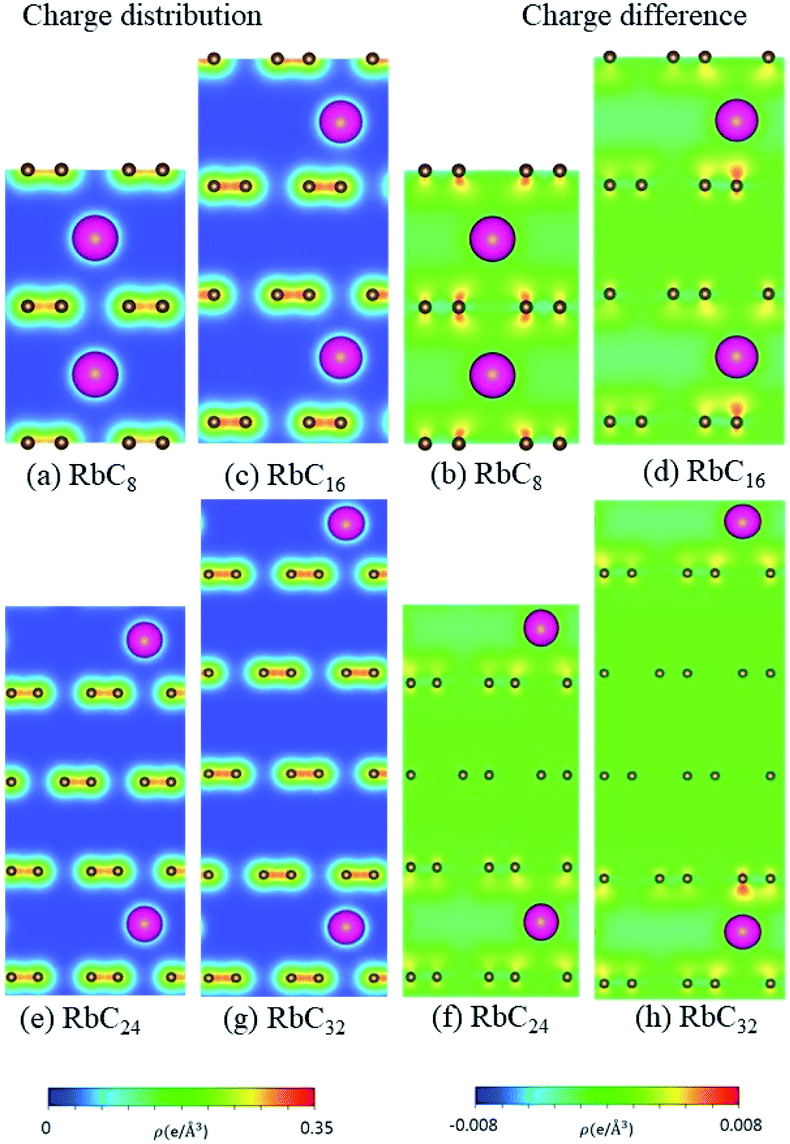
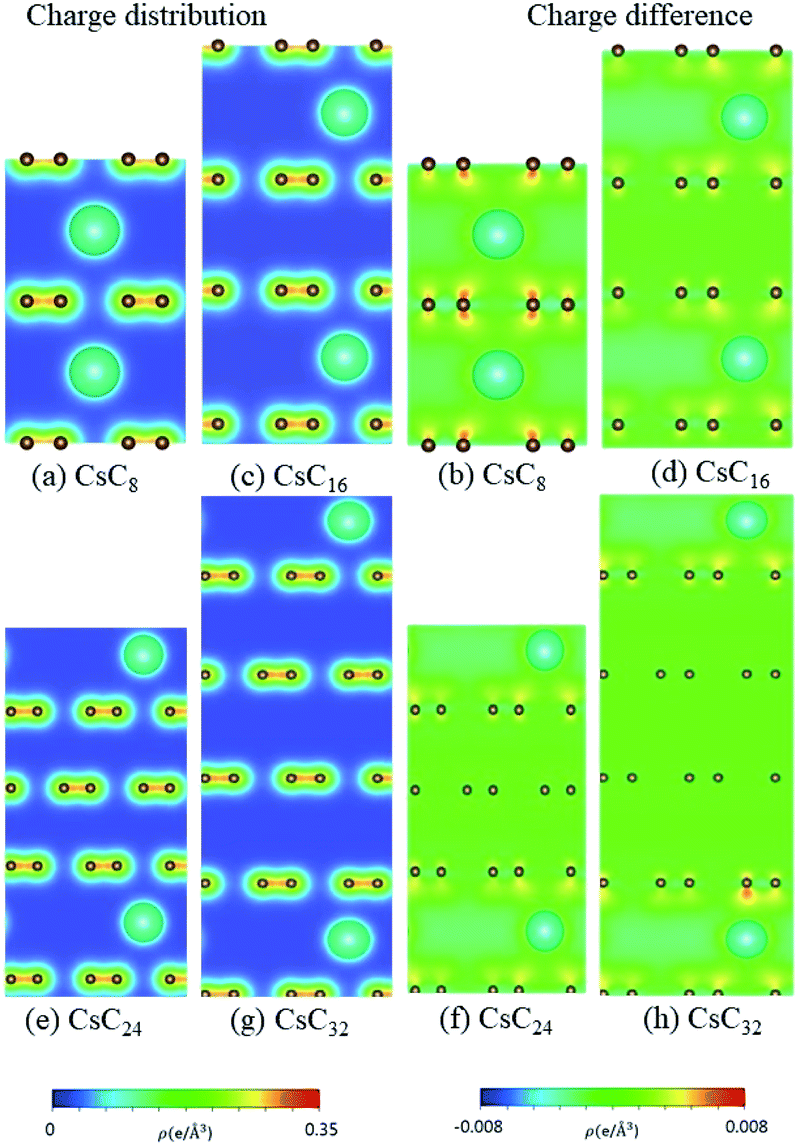
According to the spatial charge distributions and their variations after chemical modifications [Fig. 8–12], the chemical bondings hardly depend on the distributions and concentrations of alkali metal adatoms. After the alkali-metal-atom intercalations, we can observe that the changes depend on the concentrations and the kinds of alkali metal atoms. Comparing the charge distributions ρ with the charge variation Δρ, we can find the orbital hybridization in A-C bonds (A = Li, Na, K, Rb, Cs), and the charge density almost remains the same in the graphitic layers without direct intercalation with the alkali metal atoms, e.g., the middle one layer/two layers in stage-3/stage-4 compounds. Moreover, with increasing n number, Δρ becomes small [Δρ: stage-1 > stage-2 > stage-3 > stage-4]. We also see the evidence of 2s–2pz hybridization in A–C bonds in the charge variation. Obviously, the stage-1 compounds exhibit the strongest electron transfer from alkali metal atoms to carbons, as shown by the red region in Δρ [Fig. 8–12]. The variation reduces when the radius of alkali metal atoms gets larger, that is to say the electron transfer from A to carbon atoms is strongest in Li but weakest in Cs [radius: Li < Na < K < Rb < Cs; variation: Li > Na > K > Rb > Cs].
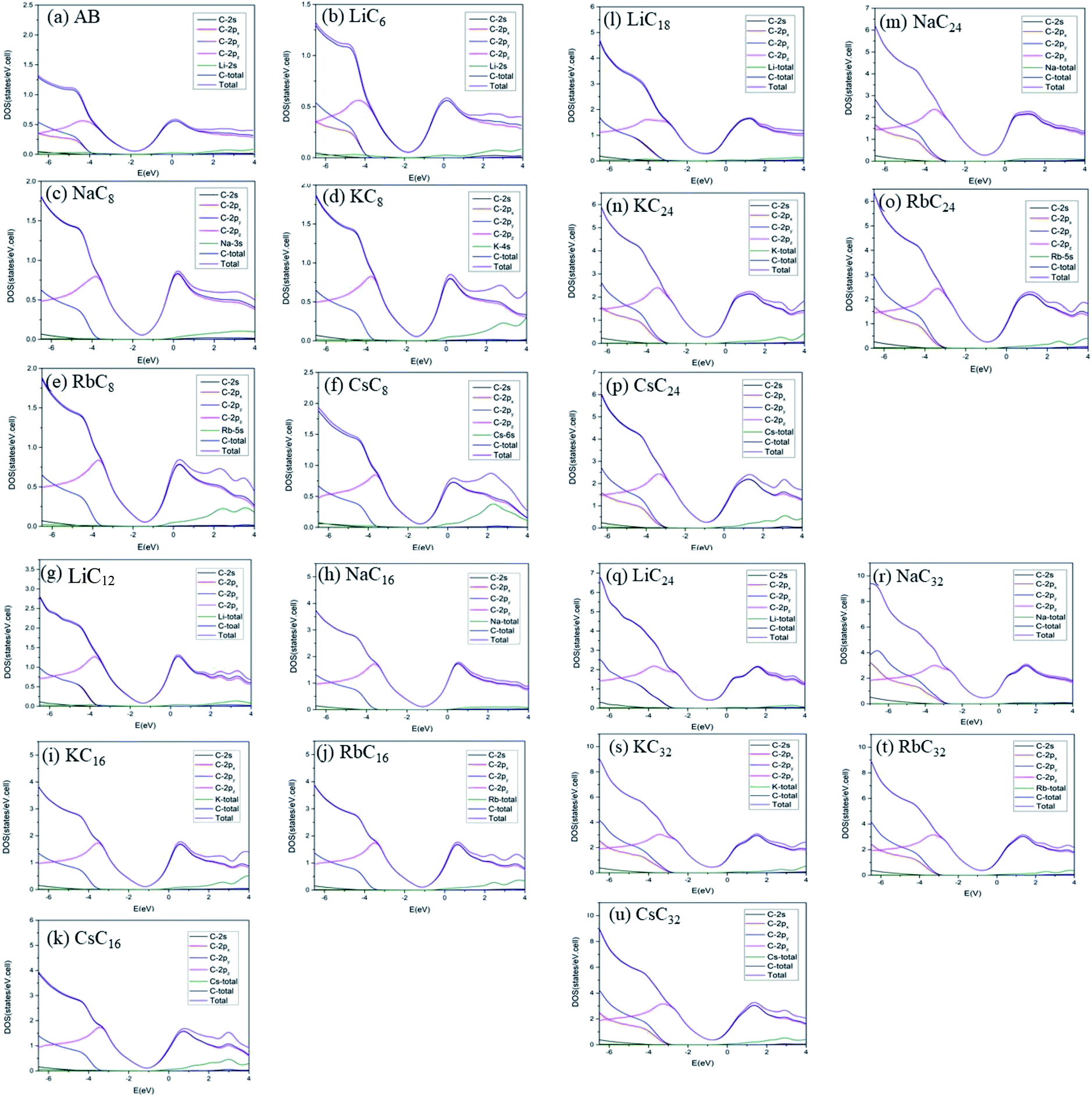
The atom- and orbital-dependent DOSs [Fig. 6] allow a full understanding of the metallic behavior and the close relations among the different chemical bondings. Although band structures become very complicated under the alkali-metal-atom intercalations, the main features of van Hove singularities are sufficiently clear for the identifications of diverse phenomena through a suitable broadening factor. The low-energy DOSs in stage-1 and stage-2 graphite alkali-metal-intercalation compounds present a prominent peak just at the Fermi level [EF = 0], regardless of the kind of alkali metal atoms. Furthermore, there exists a valley structure, with a minimum value, at its left-hand neighbor. When such a characteristic is combined with the similar ones at EF, the Fermi level is deduced to exhibit a blue shift. That is to say, EF is situated at the conduction energy subbands [EF roughly lies in the center of valence and conduction bands] roughly after [before] the alkali-metal-atom intercalations. It should be noticed that the contributions due to the alkali metal atoms are weak, but rather important. The blue shifts of stage-1 LiC6, NaC8, KC8, RbC8 and CsC8, and stage-2 LiC12, NaC16, KC16, RbC16 and CsC16 are, respectively, estimated to be 1.80, 1.54, 1.48, 1.41, 1.58, 1.37, 1.23, 1.17, 1.12, 1.10 eV.
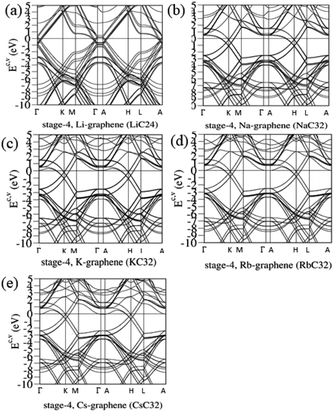 |
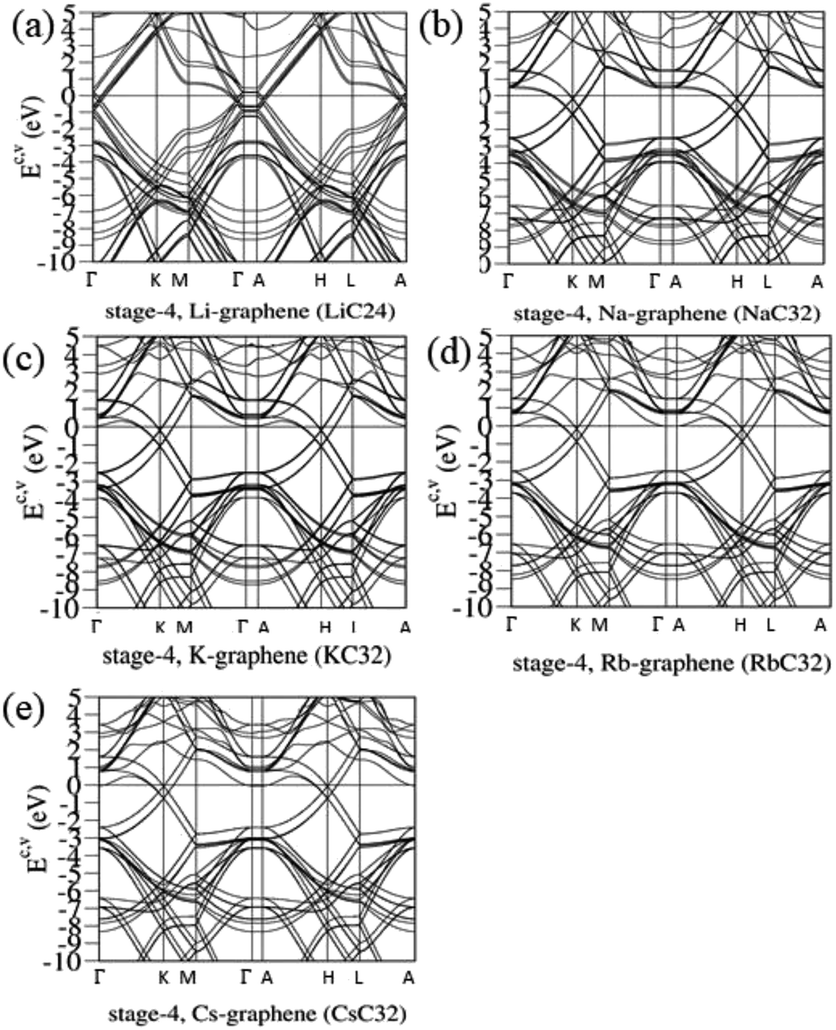
| | Fig. 6 The rich band structures for the AB-stacked stage-4 graphite alkali-metal-intercalation compounds: (a) LiC24, (b) NaC32, (c) KC32, (d) RbC32, and (e) CsC32. | |
The van Hove singularities in graphite intercalation compounds [Fig. 7(b)–(u)], which survive in the specific energy ranges, are mainly determined by the carbon or alkali metal atoms and their orbitals. Most important, DOSs in the critical energy range of −5.0 eV < E < 3.0 eV, being relatively easily examined from experimental STS measurements, are dominated by the C-2pz orbitals [the pink curves]. Furthermore, the minor contributions related to the outmost s-orbital of alkali metal atoms [the green curves], especially in the conduction energy spectrum, play a critical role in determining the blue shift of the Fermi level. As for the C-[2px, 2py] orbitals [the red and light blue curves], their contributions are initiated from ∼E < −4.0 eV, while they are absent in the opposite energy range. The red shift of the initial σ valence bands is about 1.0 eV, compared with those of the pristine simple hexagonal and Bernal graphites [Fig. 7(a)]. Only the LiC6 case [Fig. 7(b)] exhibits the split contributions of 2px and 2py orbitals. This result directly reflects the anisotropic distribution configuration. Also, the C-2s orbitals come to exist at the deeper energies of ∼E < −5.0 eV. Apparently, the above-mentioned features indicate the good separation of C-2pz and C-[2s, 2px, 2py] orbital contributions, and thus the normal perpendicular orbital hybridizations of π and σ chemical bonds. That such bonding behavior is strongly linked with the significant interlayer 2pz-orbitals due to the carbon-alkali metal bonds can account for the featured electronic properties, e.g., the main features of band structures and DOSs in Fig. 3–7. The clear identifications of stage-1 and stage-2 of graphite alkali-metal-intercalation compounds could be achieved from the low-energy features of van Hove singularities, as indicated in Fig. 7(b)–(f) and (g)–(k), respectively. Compared with those of the former, the blue shifts of the Fermi levels are relatively small, in which they are, respectively, ∼1.20 eV, 1.15 eV, 1.10 eV, 1.06 eV and 1.03 eV for LiC12, NaC16, KC16, RbC16, and CsC16. Most important, their DOSs at EF do not belong to the local maxima. Such a result directly reflects whether or not the band-edge states of conduction bands are somewhat away from the Fermi level [Fig. 4(b)–(e)]. This significant difference between stage-2 and stage-1 systems further illustrates the intersecting of EF and conduction bands, thus leading to the n-type doping cases after the alkali-metal-atom intercalations. Previous researches mainly show the structures, total energy, enthalpy of formations and the DOS.22–24,31–33 The geometric structures, especially the interlayer distances, are slightly different between these papers and experiment; for instance the LiC6 distance is 3.73/3.76 Å in ref. 24/ref. 23, 3.81 Å in this paper and 3.76 Å in experiment30 [other previously reported details in Table 1]. The results of our theoretical calculations are close to those of previous studies; furthermore, we give the details of geometric structures, ground state energies, blue shift (both in band structures and DOS) and spatial charge distributions.
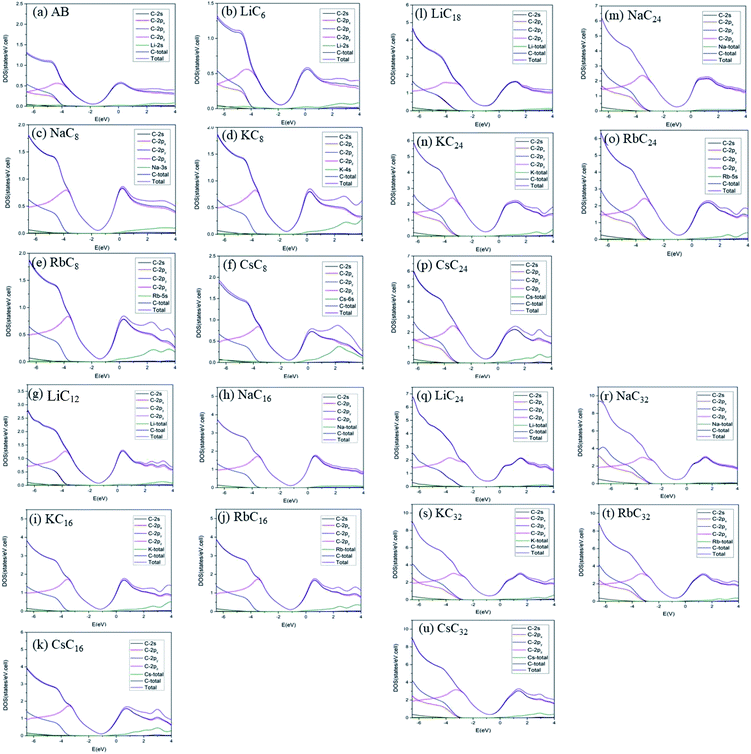 |
| | Fig. 7 The atom- and orbital-decomposed density of states for (a) AA, (b) LiC6, (c) NaC8, (d) KC8, (e) RbC8, (f) CsC8, (g) LiC12, (h) NaC16, (i) KC16, (j) RbC16, and (k) CsC16, (l) LiC18, (m) NaC24, (n) KC24, (o) RbC24, (p) CsC24, (q) LiC24, (r) NaC32, (s) KC32, (t) RbC32, (u) CsC32. The black, red, light blue, pink, green, deep blue, and purple curves represent, respectively, C-2s, C-2px, C-2py, C-2pz, alkali metal, carbon, and compound. | |
Conclusion
The fundamental properties of (Li, Na, K, Rb, Cs)-intercalated graphite compounds under the distinct stage configurations have been investigated by means of first-principles calculations. Due to the band structures, DOS and spatial charge distributions [Fig. 3–12], the weak but significant van der Waals interactions, which arise from the interlayer 2pz–2pz and 2pz–s orbital hybridizations in C–C and C–A bonds, respectively, make the most important contribution to the low-lying π-electronic structure and thus dominate the essential physical properties. The dramatic changes include the blue shift of the Fermi level/the red shift of the σ bands [the n-type doping behaviors], the greatly enhanced asymmetric electron and hole energy spectra, and the obviously reduced conduction electron density for the dilute intercalant cases. These calculated results suggest that the essential electronic properties are relevant to the concentrations of intercalant alkali metal atoms. The radii of the distinct alkali metal atoms cause the very different interlayer distances, e.g., 3.8/4.3/5.3/5.8/6.0 Å for stage-1 Li/Na/K/Rb/Cs, respectively [Table 1]. Therefore, the smallest alkali metal atom, Li, must have the strongest orbital hybridization between the alkali metal atom and carbons. The charge transfers from the alkali metal to carbons decrease quickly with the increase of the n number, and the lithium systems possess the highest ratio of charge transfer. The lithium systems also have the largest blue shift of the Fermi level of ∼1.8 eV.
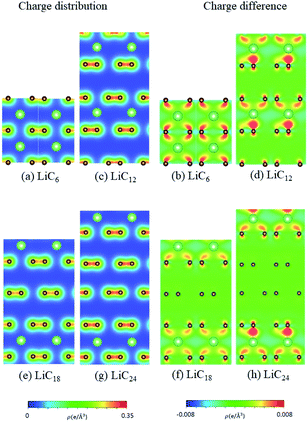 |
| | Fig. 8 The spatial charge distribution before/after Li intercalation: (a and b) LiC6, (c and d) LiC12, (e and f) LiC18, (g and h) LiC24. | |
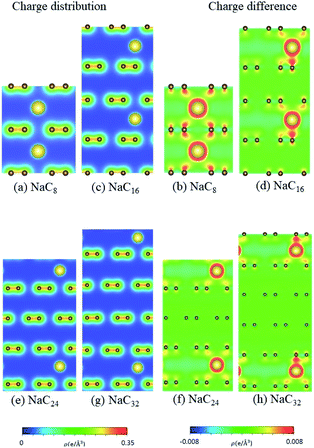 |
| | Fig. 9 The spatial charge distribution before/after Na intercalation: (a and b) NaC8, (c and d) NaC16, (e and f) NaC24, (g and h) NaC32. | |
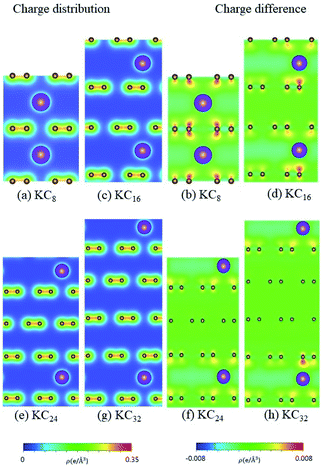 |
| | Fig. 10 The spatial charge distribution before/after K intercalation: (a and b) KC8, (c and d) KC16, (e and f) KC24, (g and h) KC32. | |
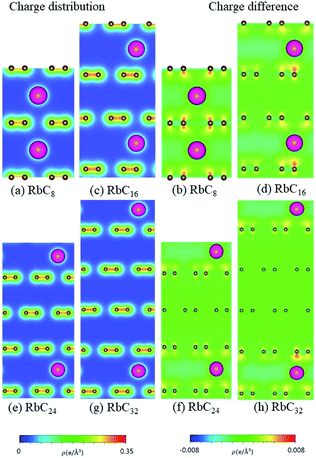 |
| | Fig. 11 The spatial charge distribution before/after Rb intercalation: (a and b) RbC8, (c and d) RbC16, (e and f) RbC24, (g and h) RbC32. | |
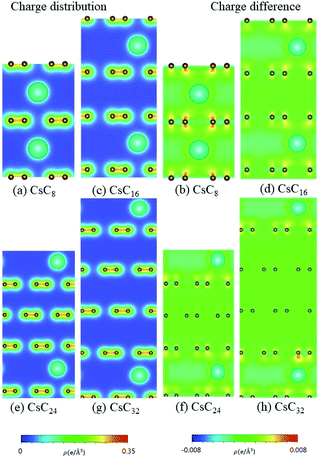 |
| | Fig. 12 The spatial charge distribution before/after Cs intercalation: (a and b) CsC8, (c and d) CsC16, (e and f) Cs C24, (g and h) CsC32. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Hi-GEM Research Center and the Taiwan Ministry of Science and Technology under grant numbers MOST 108-2212-M-006-022-MY3 and MOST 108-3017-F-006-003.
References
- M. Matthew and B. Regan, Phys. Rev. Lett., 2011, 106, 116803 CrossRef PubMed; M. Rafique, N. H. Mirjat, A. M. Soomro, S. Khokhar and Y. Shuai, Phys. Lett. A, 2018, 16, 1108–1119 CrossRef.
- S.-M. Lee, D.-S. Kang and J.-S. Roh, Carbon Lett., 2015, 16, 135–146 CrossRef.
- N. T. T. Tran, S. Y. Lin, C. Y. Lin and M. F. Lin, Geometric and Electronics Properties of Graphene Related Systems, CRC Press, 2017, ISBN 9781351368483 Search PubMed.
- G. Gómez-Santos, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 245424 CrossRef.
- D. D. L. Chung, J. Mater. Sci., 2002, 37, 1475–1489 CrossRef CAS.
- Y. Wang, J. E. Panzik, B. Kiefer and K. K. Lee, Sci. Rep., 2012, 2, 520 CrossRef CAS PubMed; J. H. Ho, C. P. Chang and M. F. Lin, Phys. Lett. A, 2006, 352, 446–450 CrossRef; C. Y. Lin, R. B. Chen, Y. H. Ho and M. F. Lin, Electronic and Optical Properties of Graphite-Related Systems, CRC Press, 2017, ISBN 9781138571068 Search PubMed.
- Z. Zhang, H. Huang, X. Yang and L. Zang, J. Phys. Chem. Lett., 2011, 2, 2897–2905 CrossRef CAS.
- Q. Cheng, et al., Sci. Rep., 2017, 7, 14782 CrossRef PubMed.
- C.-Y. Lin, J.-Y. Wu, C.-W. Chiu and M.-F. Lin, Work function of alkali-atom adsorbed graphite, CRC Press, 2019, ISBN: 9780429277368 Search PubMed.
- M. Aoki and H. Amawashi, Solid State Commun., 2007, 142, 123–127 CrossRef CAS.
- R. Matsumoto, Y. Okabe and N. Akuzawa, J. Electron. Mater., 2015, 44, 399–406 CrossRef CAS; X. Meng, S. Tongay, J. Kang, Z. Chen, F. Wu, S.-S. Li, J.-B. Xia, J. Li and J. Wu, Carbon, 2013, 53, 507–514 CrossRef.
- A. Natori, T. Ohno and A. Oshiyama, J. Phys. Soc. Jpn., 1985, 54, 3042–3050 CrossRef CAS.
- Y. Li and Y. Qu, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 425, 72–77 CrossRef CAS.
- C. Mao, et al., Selecting the Best Graphite for Long-Life, High-Energy Li-Ion Batteries, J. Electrochem. Soc., 2018, 165, A1837–A1845 CrossRef CAS.
- Q. Cheng, Sci. Rep., 2017, 7, 14782 CrossRef CAS PubMed; S. Byun, J. Park, W. A. Appiah, M.-H. Ryou and Y. M. Lee, RSC Adv., 2017, 7, 10915 RSC; M. Steinhauer, S. Risse, N. Wagner and K. Andreas Friedrich, Electrochim. Acta, 2017, 228, 652–658 CrossRef; F. M. Kindermann, P. J. Osswald, S. Klink, G. Ehlert, J. Schuster, A. Noel, S. V. Erhard, W. Schuhmann and A. Jossen, J. Power Sources, 2017, 342, 638–642 CrossRef; M. Bauer, B. Rieger, S. Schindler, P. Keil, M. Wachtler, M. A. Danzer and A. Jossen, J. Energy Storage, 2017, 10, 1–10 CrossRef; H. P. T. Sasanka Hewathilake, N. Karunarathne, A. Wijayasinghe and N. W. B. Balasooriya, Ionics, 2017, 23, 1417–1422 CrossRef.
- S.-Y. Lin, N. T. T. Tran, S.-L. Chang, W.-P. Su and M. F. Lin, Structure- and Adatom-Enriched Essential Properties of Graphene Nanoribbons, CRC
Press, 2018, ISBN: 9780367002299 Search PubMed.
- J. H. Ho, Y. H. Lai, S. J. Tsai, J. Hwang, C. Chang and M. F. Lin, J. Phys. Soc. Jpn., 2006, 75, 72–77 Search PubMed; Y. K. Huang, S. C. Chen, Y. H. Ho, C. Y. Lin and M. F. Lin, Sci. Rep., 2014, 4, 7509 CrossRef CAS PubMed; M. Koshino and E. McCann, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 165443 CrossRef; Z. F. Wang, F. Liu and M. Y. Chou, Nano Lett., 2012, 12, 3833–3838 CrossRef PubMed.
- J. H. Ho, C. P. Chang and M. F. Lin, Phys. Lett. A, 2006, 352, 446–450 CrossRef CAS; M. F. Lin, Y. C. Chuang and J. Y. Wu, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 125434 CrossRef; J. Y. Wu, S. C. Chen, O. Roslyak, G. Gumbs and M. F. Lin, ACS Nano, 2011, 5, 1026–1032 CrossRef PubMed; Y. E. Lozovik and A. A. Sokolik, Nanoscale Res. Lett., 2012, 7, 134 CrossRef PubMed; T. Ohta, et al., Phys. Rev. Lett., 2007, 98, 206802 CrossRef PubMed.
- L. M. Roth and G. W. Pratt Jr, J. Phys. Chem. Solids, 1959, 8, 47–49 CrossRef CAS; K. Schrüfer, C. Metzner, M. C. Hofmann and G. H. Döhler, Superlattices Microstruct., 1997, 21, 223–230 CrossRef; Z. L. Mišković, P. Sharma and F. O. Goodman, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 115437 CrossRef.
- Q. T. Ain, A. Al-Modlej, A. Alshammari and M. N. Anjum, Mater. Res. Express, 2018, 5, 035017 CrossRef.
- O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 125416 CrossRef.
- E. Ziambaras, J. Kleis, E. Schröder and P. Hyldgaard, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 155425 CrossRef.
- K. Persson, Y. Hinuma, Y. S. Meng, A. Van der Ven and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125416 CrossRef.
- Z. Wang, M. S. Sverre and T. Grande, RSC Adv., 2014, 4, 4069 CAS.
- S.-Y. Lin, et al., Phys. Chem. Chem. Phys., 2015, 39, 26443–26450 RSC.
- M. Rafique, M. A. Unar, I. Ahmed, A. R. Chachar and Y. Shuai, J. Phys. Chem. Solids, 2018, 118, 114–125 CrossRef CAS.
- M. Rafique, Y. Shuaia, H.-P. Tan and M. Hassan, Appl. Surf. Sci., 2017, 408, 21–33 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- R. Juza and V. Wehle, Naturwissenschaften, 1965, 52, 560 CrossRef CAS.
- E. Ziambaras, J. Kleis, E. Schröder and P. Hyldgaard, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 155425 CrossRef.
- G. E. Pascal and F. Mireille, Solid State Ionics, 1988, 28–30, 1172–1175 Search PubMed.
- G. Yoon, H. Kim, I. Park and K. Kang, Adv. Energy Mater., 2017, 7, 1601519 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac and
Kuang-I Lind
*ac and
Kuang-I Lind