
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel approach for obtaining α,β-diaminophosphonates bearing structurally diverse side chains and their interactions with transition metal ions studied by ITC†‡
Paweł Lenartowicz *a,
Danuta Witkowskab,
Beata Żyszka-Haberechta,
Błażej Dziukac,
Krzysztof Ejsmonta,
Jolanta Świątek-Kozłowskab and
Paweł Kafarskid
*a,
Danuta Witkowskab,
Beata Żyszka-Haberechta,
Błażej Dziukac,
Krzysztof Ejsmonta,
Jolanta Świątek-Kozłowskab and
Paweł Kafarskid
aFaculty of Chemistry, University of Opole, Oleska 48, 45-052 Opole, Poland. E-mail: pawel.lenartowicz@uni.opole.pl
bPublic Higher Medical Professional School in Opole, Katowicka 68, 45-060 Opole, Poland
cFaculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
dDepartment of Bioorganic Chemistry, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
First published on 23rd June 2020
Abstract
Aminophosphonates are an important group of building blocks in medicinal and pharmaceutical chemistry. Novel representatives of this class of compounds containing nontypical side chains are still needed. The aza-Michael-type addition of amines to phosphonodehydroalanine derivatives provides a simple and effective approach for synthesizing N′-substituted α,β-diaminoethylphosphonates and thus affords general access to aminophosphonates bearing structurally diverse side chains. Thermodynamic analysis of the chosen aminophosphonates at physiological pH proves that they serve as potent chelators for copper(II) ions and moderate chelators for nickel(II) ions.
Introduction
Aminophosphonates, which are broadly defined as phosphorus analogues of amino acids, are currently attracting interest in the areas of industrial, agricultural, and medicinal chemistry due to their biological and physical properties as well as their utility as synthetic intermediates. The presence of a characteristic N–C–P scaffold affords many opportunities for structural modifications, resulting in a broad range of bioactivity.1,2 Among aminophosphonate derivatives, a particular group containing a N′-substituted α,β-diaminoethyl motif (Fig. 1) has been reported. | ||
| Fig. 1 Structure of phosphonates with N′-substituted α,β-diaminoethyl motif. | ||
The presence of this structural fragment is responsible for the unique properties and chemistry of these phosphonates. Diaminophosphonate was used as a building block for the synthesis of integrin ligands that bind to metal ion-dependent adhesion sites. The additional amine function allows for extension of a side chain with an arginine-mimicking fragment via an amide linkage.3 Moreover, these compounds were successfully tested as inhibitors of various aminopeptidases,1,4,5 with selective activity regulation among two homologous human endoplasmic reticulum aminopeptidases (i.e., ERAP1 and ERAP2).6 The diaminophosphonic acids complex zinc ion per s via oxygen atoms of the phosphonic moiety in the active site these enzymes. The presence of an additional substituted amine group enhances the ionic interaction with glutamate- and aspartate-rich binding sites in the enzymes and provides specific interaction with the side chain in the binding pocket. In addition, the ionic fragment may be responsible for destabilization of the overall ligand-binding mode. These unique features provide an opportunity to synthesize ligands that can be used as specific enzyme inhibitors or molecular probes for activity visualization of specific enzymes. The preparation of α,β-diaminophosphonates is rarely described and involves multistep synthetic procedures,7–16 which are discussed in detail by Głowacka et al.13 One of the most popular methods involves regioselective ring-opening of aziridine phosphonic acid with amine nucleophiles.4,5,11,16 For this purpose, diethyl vinylphosphonate was converted to 1-bromo-2-amino-phosphonic acid followed by cyclization in boiling aqueous sodium hydroxide. Then, the resulting aziridine was reacted with an aqueous solution consisting of ammonia or benzylamine at 100 °C.16 This methodology has been recently improved by using benzyl or α-methylbenzyl N-substituted aziridine phosphonate obtained from a Gabriele–Cromwell reaction. Starting from diethyl vinylphosphonate, a large series of α,β-diaminophosphonic acids and dipeptide phosphinates were obtained.4 Alternatively, N-substituted aziridine intermediates can be prepared from diethyl acetyl phosphonate.17 It is important to note that this approach was successfully adopted for the preparation of enantiomerically pure α,β-diaminophosphonic acid derivatives using chiral α-methylbenzylamine followed by chromatographic separation of the aziridine diastereomers as a key step.5 Some of the other synthetic methods include a three-component Kabachnik–Fields reaction utilizing aziridine aldehydes with a chiral auxiliary,13 three-step conversion of cyclic phosphonothiourea derivatives obtained from a reaction of diethyl isothiocyanatomethylphosphonate with activated imines,7 reduction of the nitro group of N-protected β-amino-α-nitrophosphonates obtained from a reaction of nitrophosphonates with imines,8,15 conversion of the hydroxyl group from the corresponding α-hydroxyphosphonates by para-toluenesulfonamide.9 To the best of our knowledge, only two papers18,19 have reported preliminary mentions on the Michael-type addition of amines to phosphono α,β-dehydroamino acids. However, the authors did not develop this strategy as a convenient and general method.
α,β-Dehydroamino acids are unsaturated analogues of classic amino acids containing a conjugated double bond between the α and β carbon atoms. These analogues have been found in many bioactive peptides isolated from microorganisms, invertebrates and even higher plants.20 From a synthetic point of view, the presence of a dehydroamino acid residue in a peptide structure affords access to late-stage diversification of biomolecules through their chemical transformations.21 Although the chemistry of classic dehydroamino acid derivatives with nucleophiles, such as amines and thiols, is well known,22–31 the reactivity of their phosphonate counterparts is hardly ever described. Thus, we decided to demonstrate the utility of this synthetic approach as a general reaction for the preparation of N′-substituted α,β-diaminoethylphosphonates. Thus, the 1,4-addition of primary and secondary amines (aza-Michael-type addition) to three phosphonodehydroalanine derivatives is presented here as an alternative procedure for the preparation of this group of phosphonates. Further, the interactions between three model diaminophosphonates and Cu2+, Ni2+, Zn2+ metal ions were evaluated by isothermal titration calorimetry.
Results and discussion
Compounds 1 and 3 containing phosphonic acids analogues of dehydroalanine were obtained according to previously reported protocol including the condensation of appropriate amide or carbamate with phosphonic analogue of pyruvic acid under acidic catalyst. Preparation of dipeptide analogue 5 employed the 2-chloroacetamide as glycine precursor. Alternatively, the substrate 1 and 3 could be synthesized using the different routes.32,33 The structures of phosphonodehydroalanine 3 and its dipeptide derivative 5 were undoubtedly confirmed by crystallographic studies (Fig. 2). Relevant crystallographic data for these molecules and the full geometrical information are summarized in Tables S1–S3 of the ESI.‡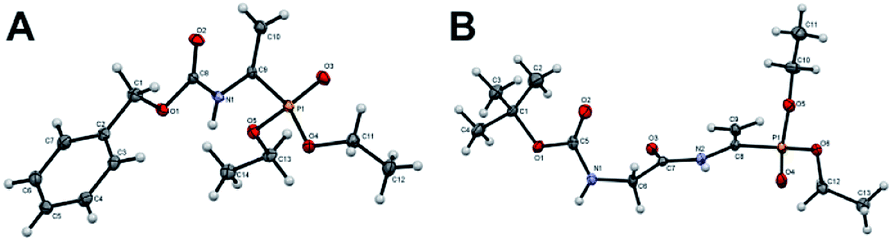 | ||
| Fig. 2 Molecular structures of 3 (A) and 5 (B), displacement ellipsoids are drawn at the 50% probability level (for details see ESI‡). | ||
With these substrates in hand, we decided to put insight into the reaction optimization of amine addition in terms of solvent as well reaction time. As a representative reaction, addition of piperidine to two model substrates (i.e., Ac-ΔAla-PO(OEt)2 and Z-ΔAla-PO(OEt)2) was chosen (Table 1). The yield of the reaction performed in dioxane was poor, or no product was isolated. The shift to methanol improves the yields and reduces the reaction time. Therefore, a protic solvent with a higher dielectric constant is beneficial in this reaction. We speculate that this behaviour may be due to stabilization of the phosphonodehydroalanine resonance structure with a partially positive charge that is localized on the β carbon atom, resulting in an increase in the electrophilicity of that site. Moreover, significant acceleration of product formation was achieved by adding water to the reaction mixtures. This acceleration was enhanced when water was used along with dioxane (entries 2 and 6). Similar effect has also been reported for amine addition to classic dehydroalanine amides.28 In 50% aqueous methanol, the N-acetyl substrate (entry 4) completely reacted in one hour. Similarly, the presence of water also reduces the reaction time with a simultaneous increase in yields to 82% for the Z-protected substrate (entry 8). In addition, a prolonged reaction time for Z-ΔAla-PO(OEt)2 resulted in the formation of side products and reduced the overall yield. Finally, the use of aqueous methanol and a reaction time that does not exceed 72 h were considered as optimal conditions.
|
||||
|---|---|---|---|---|
| Entry | Substrate | Solvent | Time (h) | Isolated yield (%) |
| a n.i. – product was not isolated from the reaction mixture. Reagent dosages: 1 or 3 (0.5 mmol); piperidine (2.5 mmol), RT. | ||||
| 1 | Ac-ΔAla-PO(OEt)2 | Dioxane | 24 | 4 |
| 2 | Dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1) :H2O (1:1) |
24 | 82 | |
| 3 | MeOH | 24 | 81 | |
| 4 | MeOH:H2O (1:1) |
1 | 79 | |
| 5 | Z-ΔAla-PO(OEt)2 | Dioxane | 72 | n.i. |
| 6 | Dioxane:H2O (1:1) |
72 | 67 | |
| 7 | MeOH | 24 | 69 | |
| 8 | MeOH:H2O (1:1) |
24 | 82 | |

With the optimal conditions in hand, we had studied the addition reactions using various amines as substrates. The preliminary studies with Z-ΔAla-PO(OEt)2 (Table 2, entries 13–16) revealed that the addition of secondary amines required a long reaction time and yielded complex reaction mixtures that were difficult to isolate by chromatography. The desired products were obtained with moderate yields up to 46% (after isolation). In addition, the geometric isomers of the desired product of the reaction with 2,6-dimethylmorpholine were separated. The NMR analysis of a partially purified fraction of Z-(β-N-(morpholine)Ala-PO(OEt)2 (entry 16)) revealed that the reaction mixtures also contain a residual amount of substrate, benzylcarbamate and an unidentified phosphorus-containing derivative (signal at 19.44 ppm in 31P NMR spectrum, see ESI‡). Due to the previously mentioned difficulties and low yields, we decided to abandon further reactions with the Z-protected substrate.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Amine fragment | Isolated yield (%) | Entry | Substrate | Amine fragment | Isolated yield (%) |
| a Yield of amine addition.b Yield of HCl hydrolysis. Reagent dosages: entries 3 and 4 – 1 (0.25 mmol); entries 1,2,6,9–16 – 1 or 3 (0.5 mmol); entry 5 – 1 (0.75 mmol); entries 7 and 8 – 1 (1.0 mmol); amine (5 eq. – 1.25, 2.5, 3.75, 5.0 mmol appropriately); time – up to 72 h; RT. | |||||||
| 1 | Ac-ΔAla-PO(OEt)2 |  |
82 | 9 | Ac-ΔAla-PO(OEt)2 |  |
70 |
| 2 | Ac-ΔAla-PO(OEt)2 |  |
85 | 10 | Ac-ΔAla-PO(OEt)2 |  |
81 |
| 3 | Ac-ΔAla-PO(OEt)2 |  |
70 | 11 | Ac-ΔAla-PO(OEt)2 |  |
84 |
| 4 | Ac-ΔAla-PO(OEt)2 |  |
63 | 12 | Ac-ΔAla-PO(OEt)2 |  |
91 |
| 5 | Ac-ΔAla-PO(OEt)2 |  |
88a/91b | 13 | Z-ΔAla-PO(OEt)2 | 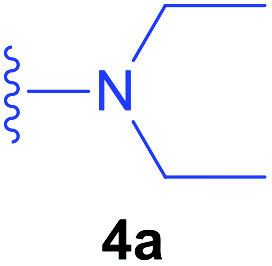 |
37 |
| 6 | Ac-ΔAla-PO(OEt)2 |  |
79a/95b | 14 | Z-ΔAla-PO(OEt)2 |  |
82 |
| 7 | Ac-ΔAla-PO(OEt)2 |  |
91a/94b | 15 | Z-ΔAla-PO(OEt)2 |  |
46 |
| 8 | Ac-ΔAla-PO(OEt)2 | 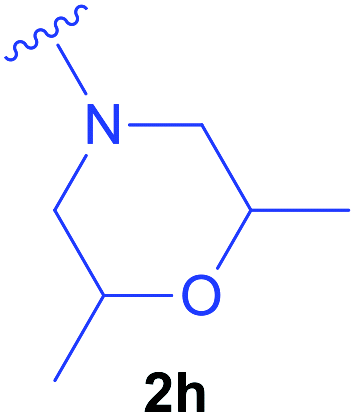 |
83 cis/trans | 16 | Z-ΔAla-PO(OEt)2 | 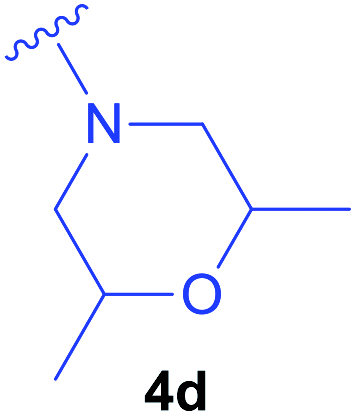 |
31 cis/trans |
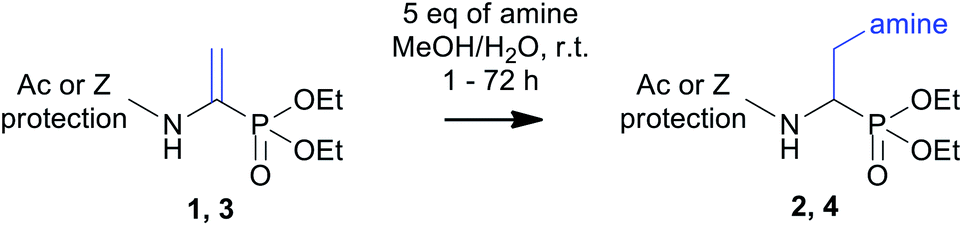
For Ac-ΔAla-PO(OEt)2, the reaction proceeded readily, and thus, we extended its scope by including aliphatic primary amines. The reaction appeared to be general and yielded aminophosphonates of structurally variable side chains (Table 2, entries 1–12). In most cases, the desired adducts were isolated in high yield. It is important to note that the presence of the propargyl group (entry 2) provides an opportunity to use this diaminophosphonate as a substrate for further elongation of the side chains (e.g., in click-type reactions with azides). In addition, the deprotected derivative of N-acetyl piperazine (entry 9) can be considered a starting point for further modifications. Finally, the total hydrolysis of three representative N-substituted α,β-diaminophosphonates (Table 2, entries 5–7) with aqueous 6 M HCl afforded unprotected aminophosphonates with high yield of 91–94%.
Our experience with synthesis of dipeptides containing a phosphonodehydroalanine fragment prompted us to use this substrate in the studied reaction (Table 3). Among all tested substrates, the addition of amines to Boc-Gly-ΔAla-PO(OEt)2 yielded the best results. The reactions performed under the optimized conditions were free of side products, and after chromatographic separation, the products were obtained with excellent yields up to 98% for piperidine addition (entry 3). Further N-terminal amine group deprotection was performed under standard conditions with trifluoroacetic acid. In contrast to the harsh HCl hydrolysis required for deprotection of N-acetyl products, the milder conditions for Boc group removal led to dipeptide building blocks, which can be further elongated at the N-terminal site.
|
|||
|---|---|---|---|
| Entry | Substrate | Amine fragment | Isolated yield (a/b %) |
| a Yield of amine addition.b Yield of Boc deprotection. Reagent dosages: substrate 5 (0.25 mmol); amine (1.25 mmol); time – up to 72 h; RT. | |||
| 1 | Boc-Gly-ΔAla-PO(OEt)2 |  |
97/99 |
| 2 | Boc-Gly-ΔAla-PO(OEt)2 |  |
83/99 |
| 3 | Boc-Gly-ΔAla-PO(OEt)2 |  |
98/99 |
| 4 | Boc-Gly-ΔAla-PO(OEt)2 |  |
95/98 |
| 5 | Boc-Gly-ΔAla-PO(OEt)2 |  |
95/98 |

Metal complexes with ligands possessing a broad spectrum of biological activities are important for living systems, as their incorporation into structurally more complex metallo-organic compounds offers new opportunities in the medicinal chemistry. Aminophosphonates were shown to be very effective and specific metal ion binding molecules forming biologically relevant metal complexes.34,35 Thus, we have checked whether compounds synthesized in this work posses these abilities for selected metal ions. We decided to evaluate the interaction of Cu2+, Ni2+ and Zn2+ ions with three model compounds, which side chains contain diethylamine (2e′), piperidinyl (2f′) and morpholinyl (2g′) moiety. For that, we have applied technique used to determine the thermodynamic parameters of interactions in solution, namely Isothermal Titration Calorimetry (ITC).
The dissociation constants (Kd) and binding enthalpies (ΔHITC) of interactions of the chosen aminophosphonates with the copper(II) ions were obtained directly from ITC experiments36 by fitting isotherm initially to a model that assumes a two sets of binding sites. However, the binding data of Cu2+ to 2f′ and 2e′ can be fitted also to a sequential binding sites model. As the “two sets of sites” model has given unusual binding stoichiometry (n = ca. 2.0 and 0.2 per each side) and we had excluded that it was because of ligand and metal concentrations inaccuracy, we decided to choose the “sequential binding sites” model.37 The best-fit of experimental values (nITC, KdITC, ΔHITC and entropy) in sodium cacodylate buffer have been summarized in Table 4. Fig. 3 (Panel A, last section) shows the ITC thermogram of titration of 2g′ by Cu2+ at 25 °C in cacodylate buffer. The copper ions are bound to two non-identical and independent ligand binding sites of 2g′ with moderate (Kd = 3.31 ± 0.20) × 10−6 M and high (Kd = 2.18 ± 0.10) × 10−10 M binding affinity (Table 4). That is binding with favorable enthalpy and entropy for both sets of sites.
| Ligand | KdITC [M] | ΔHITC [kcal mol−1] | −TΔSITC [kcal mol−1] | nITC |
|---|---|---|---|---|
| The interactions of Cu2+ with aminophosphonates | ||||
| 2e′ | (26.1 ± 3.38) × 10−6 | −5.30 ± 0.49 | −0.96 | 2 sequential binding sites |
| (3.43 ± 0.44) × 10−6 | 1.33 ± 0.53 | −8.79 | ||
| 2f′ | (16.20 ± 1.38) × 10−6 | −8.19 ± 0.53 | 1.65 | 2 sequential binding sites |
| (5.03 ± 0.43) × 10−6 | 4.50 ± 0.57 | −11.7 | ||
| 2g′ | (3.31 ± 0.20) × 10−6 | −1.72 ± 0.02 | −5.75 | 1.13 ± 0.01 |
| (2.18 ± 0.10) × 10−10 | −1.65 ± 0.01 | −11.5 | 1.00 ± 0.003 | |
|
||||
| The interactions of Ni2+ with aminophosphonates | ||||
| 2e′ | (631 ± 44.5) × 10−6 | 2.58 ± 0.14 | −6.95 | 1.15 ± 0.03 |
| 2f′ | (772 ± 39.6) × 10−6 | 4.16 ± 0.44 | −8.41 | 0.526 ± 0.05 |
| 2g′ | (281 ± 15.1) × 10−6 | 6.63 ± 0.17 | −11.5 | 1.23 ± 0.01 |
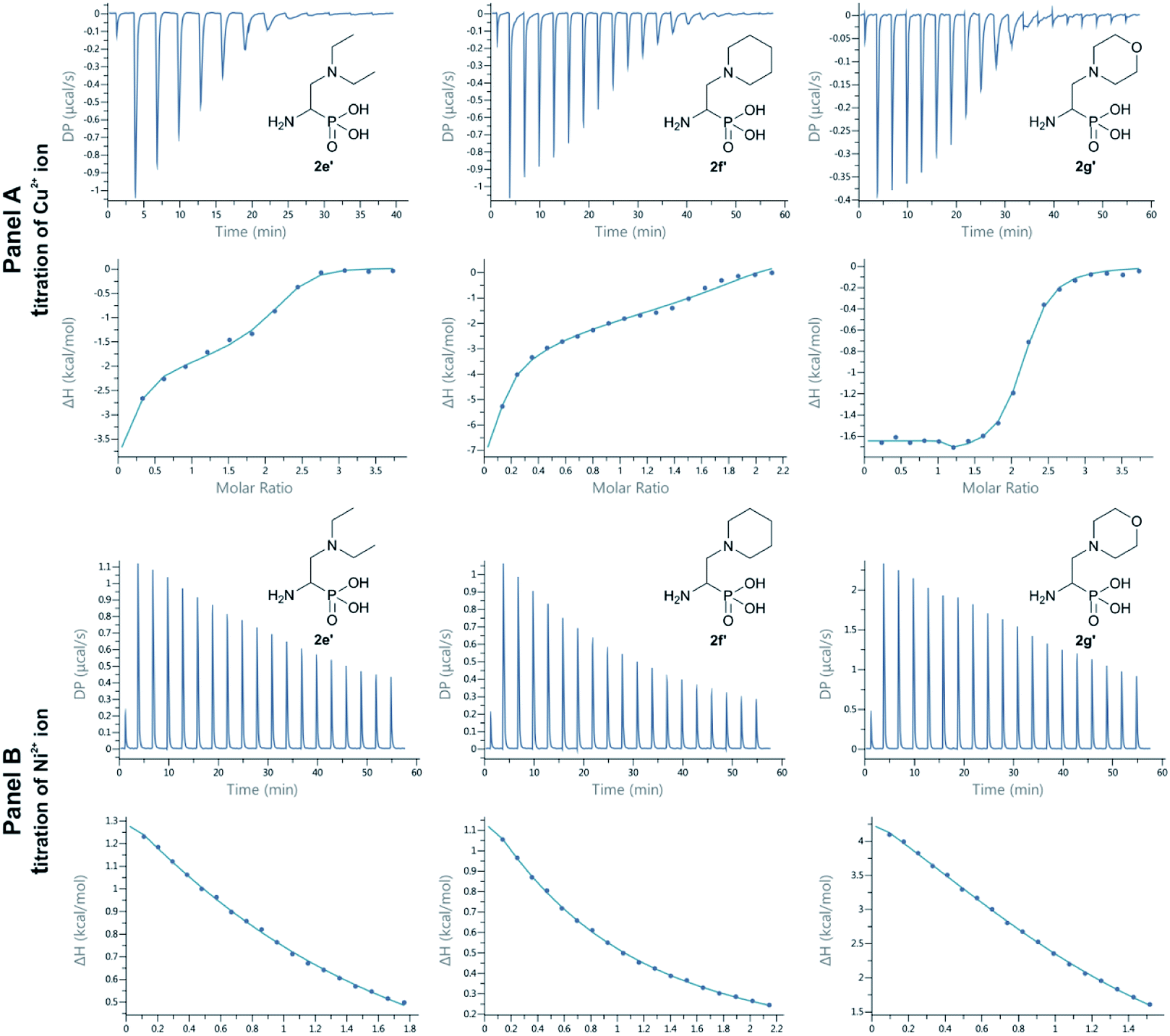 | ||
| Fig. 3 ITC titration (raw data above and corresponding plots below): Panel A: 2 mM Cu2+ 2e′, 2f′ and 2g′; Panel B: 5 mM Ni2+ to 2e′, 6 mM Ni2+ to 2f′ and 3 mM Ni2+ to 2g′. | ||
In the sequential binding sites model, dissociation constants do not correspond to intrinsic site-specific constants. The same applies to the binding enthalpies, so extreme care should be taken when interpreting that kind of data.38 Because the binding sites of 2e′ and 2f′ do not show considerably different affinities for copper ions (Table 4) all possible ligation states can coexist in equilibrium. However, in general these compounds exhibit weaker affinity towards copper(II) ions than 2g′.
The titration of aminophosphonates with 3 mM nickel ions gave reasonable fits only for 2g′ ligand. Also in this case 2g′ proved to be better chelator than compounds 2e′ and 2f′, for which two higher concentrations of 5 mM and 6 mM, respectively (Fig. 3, Panel B) were used. The reason is much lower affinity of Ni2+ to studied aminophosphonates than that for copper ions. For the data of nickel titration only one site-model could be successfully used and the values from best fits of the experimental data are found in Table 4.
The binding stoichiometry is close to the expected value of 1.0 for 2g′ and 2e′ for interaction with nickel ions, but is closer to 0.5 for 2f′ what suggests that two 2f′ molecules are bound to one nickel ion at given experimental conditions. On the other hand, the observed stoichiometry of 1.23 ± 0.01 for binding of nickel to 2g′, may also correspond to five nickel ions being coordinated by four ligands.39 As shown in Fig. 3 the nickel binding is an endothermic process for all three aminophosphonates, thus entropically-driven (Table 4), which suggests considerable solvation effects and/or enhanced ligand/metal flexibility upon complex formation. Since ITC measurements of binding of Cu2+, Ni2+ and Zn2+ to 2e′, 2f′ and 2g′ were obtained under identical experimental conditions, a direct comparison of the best fit parameters allows to determine the thermodynamic contribution of the aminophosphonate side-chains to the metal ion binding.40 Unfortunately, the affinity of Zn2+ ion to the tested compounds is too low to obtain any reasonable data for the range of the metal concentration of 3–5 mM. ITC titration plots for these reactions have been shown in Fig. SI1 in ESI.‡
Conclusions
In summary, the application of an aza-Michael-type addition of amines to phosphonodehydroalanine derivatives afforded N′-substituted α,β-diaminoethylphosphonates. High yields, mild reaction conditions and a simple protocol make this approach a good alternative for the preparation of these phosphonates. In addition, this reaction is of general value and may be used for the preparation of aminophosphonates and their short peptides with variously functionalized side chains. The results of ITC measurements revealed reasonable potency of the three chosen compounds for interactions with Cu2+ and Ni2+ ions. The most striking feature of that analysis is the impact of morpholinyl moiety on the affinity of aminophosphonates towards transitions metal ions with the binding strength of HCl·β-N-(morpholine)Ala-PO(OH)2 being higher for both ions.Experimental section
Synthesis
All amines, 2-chloracetamide, Boc anhydride, trifluoroacetic acid, hydrochloric acid 37% were purchased from Merck Poland. Methanol (MeOH), cyclohexane, aqueous ammonia 25%, isopropyl alcohol (purchased from Avantor Performance Materials Poland) were analytical grade and used without further purification. Ethyl acetate (EtOAc), chloroform (CHL) and dichloromethane (DCM) were refluxed over P2O5 and distilled. Reaction progress was monitored by thin-layer chromatography on Merck 60 silica plates using methanol/dichloromethane mixture as eluent. The spots were visualized by chlorine/o-tolidine reaction. Purification of synthesized compounds was performed using silica gel 60 (0.040–0.063 mm) from Merck. The NMR analyses were performed on a Bruker Ultrashield 400 MHz spectrometer operating at 400 MHz (1H), 162 MHz (31P) and 101 MHz (13C). The samples were dissolved in d6-DMSO (99.8 at% D) containing 0.03% TMS or D2O (99.9 at% D) and measured at 297 K. High-resolution mass spectra (HRMS) were recorded on a Waters LCT Premier XE mass spectrometer equipped with an ESI source in the positive ion mode (Waters, Milford, MA, USA). The preparation protocols of Ac-PO(OEt)2, Ac-ΔAla-PO(OEt)2 (1), Z-ΔAla-PO(OEt)2 (3) are described elsewhere.19,41,42 The detailed preparation of Boc-Gly-ΔAla-PO(OEt)2 (5) is presented in a recently published paper.43:1 v/v), MeOH, MeOH/H2O (1:1 v/v). For this purpose, the substrate (0.5 mmol) was dissolved in the appropriate solvent (2 ml) and piperidine (0.247 ml, 2.5 mmol, 5 eq.) was added. The reaction mixture was mixed at room temperature until the entire phosphonate was reacted or for 72 h. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography using an increasing gradient of methanol in chloroform from 0 to 5% (v/v). Time of the reaction and yields are given in Table 1.:1 v/v) mixtures (the amount of the substrate is indicated at appropriate product description). The reaction was mixed at room temperature until the entire phosphonate was reacted or for 72 h. Then volatile components were removed under reduced pressure and residue was co-evaporated two times with toluene and two times with DCM. The crude product was purified by flash chromatography using an increasing gradient of methanol in dichloromethane or chloroform.Diethyl 1-(N-acetylamino)-2-(N-n-propylamino)ethylphosphonate (2a) – Ac-(β-N-(propylamine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.111 g; 0.5 mmol) was used. Product 0.115 g (0.41 mmol) was obtained as colorless oil. Yield: 82%. 1H NMR (400 MHz, DMSO) δ 8.04 (d, J = 9.5 Hz, 1H, NH), 4.41–4.29 (m, 1H, CH), 4.07–3.92 (m, 4H, 2 × CH2CH3), 2.84–2.65, 2.47–2.33 (2 × m, 4H, CH2Ala and N(CH2CH2CH3)), 1.86 (d, J = 1.1 Hz, 3H, CH3CO), 1.41–1.31 (m, 2H, N(CH2CH2CH3)), 1.21 and 1.20 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped), 0.83 (t, J = 7.4 Hz, 3H, N(CH2CH2CH3)). 13C{1H} NMR (101 MHz, DMSO) δ 169.12 (d, J = 4.5 Hz), 61.91 and 61.53 (2 × d, J = 6.6 Hz), 50.22, 48.13 (d, J = 5.9 Hz), 44.78 (d, J = 153.0 Hz), 22.49, 22.43, 16.31 and 16.25 (2 × d, J = 5.5 Hz), 11.68. 31P{1H} NMR (162 MHz, DMSO) δ 25.68. HRMS (ESI) calcd for C11H26N2O4P [(M + H)+] 281.1625, found: 281.1639.
Diethyl 1-(N-acetylamino)-2-(N-propargylamino)ethylphosphonate (2b) – Ac-(β-N-(propargylamine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.111 g; 0.5 mmol) was used. Product 0.117 g (0.42 mmol) was obtained as colorless oil. Yield: 85%. 1H NMR (400 MHz, DMSO) δ 8.04 (d, J = 9.5 Hz, 1H, NH), 4.41–4.27 (m, 1HCHAla), 4.07–3.94 (m, 4H, 2 × CH2CH3), 3.29 and 3.28 (2 × d overlapped, J = 2.4 Hz, 2H, CH2prg.), 3.06 (t, J = 2.4 Hz, 1H,
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 2.94–2.85 (m, 1H, CHAHBAla), 2.78–2.68 (m, 1H, CHAHBAla), 1.86 (d, J = 1.0 Hz, 3H, CH3CO), 1.22 and 1.21 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 169.14 (d, J = 4.5 Hz), 82.47, 73.80, 61.96 and 61.61 (2 × d, J = 6.6 Hz), 46.85 (d, J = 6.5 Hz), 44.80 (d, J = 153.8 Hz), 36.76, 22.41, 16.28 and 16.22 (2 × d, J = 5.7 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.36. HRMS (ESI) calcd for C11H22N2O4P [(M + H)+] 277.1312, found: 277.1323.
CH), 2.94–2.85 (m, 1H, CHAHBAla), 2.78–2.68 (m, 1H, CHAHBAla), 1.86 (d, J = 1.0 Hz, 3H, CH3CO), 1.22 and 1.21 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 169.14 (d, J = 4.5 Hz), 82.47, 73.80, 61.96 and 61.61 (2 × d, J = 6.6 Hz), 46.85 (d, J = 6.5 Hz), 44.80 (d, J = 153.8 Hz), 36.76, 22.41, 16.28 and 16.22 (2 × d, J = 5.7 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.36. HRMS (ESI) calcd for C11H22N2O4P [(M + H)+] 277.1312, found: 277.1323.
Diethyl 1-(N-acetylamino)-2-(piperidin-1-yl-ethylamino)ethylphosphonate (2c) – Ac-(β-N-(ethylpiperidine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.055 g; 0.25 mmol) was used. Product 0.061 g (0.17 mmol) was obtained as colorless oil. Yield: 70%. 1H NMR (400 MHz, DMSO) δ 8.09 (d, J = 9.5 Hz, 1H, NH), 4.44–4.31 (m, 1H, CH), 4.07–3.92 (m, 4H, 2 × CH2CH3), 2.87–2.69 (m, 2H)*, 2.66–2.56 (m, 1H)*, 2.47–2.19 (m, 7H)*, 1.86 (d, J = 1.0 Hz, 3H, CH3CO), 1.52–1.41 (m, 4H)*, 1.41–1.31 (m, 2H)*, 1.21 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). (*) – partially overlapped multiplets, derived from protons (16H) of side chain. 13C{1H} NMR (101 MHz, DMSO) δ 169.19 (d, J = 4.2 Hz), 61.97 and 61.57 (2 × d, J = 6.7 Hz), 58.03, 53.99, 47.92 (d, J = 6.1 Hz), 44.66, 44.36 (d, J = 152.7 Hz), 25.53, 24.04, 22.42, 16.35 and 16.29 (2 × d, J = 5.7 Hz, overlapped). 31P{1H} NMR (162 MHz, DMSO) δ 25.63. HRMS (ESI) calcd for C15H33N3O4P [(M + H)+] 350.2203, found: 350.2210.
Diethyl 1-(N-acetylamino)-2-[(morpholin-2-yl)ethylamino)]ethylphosphonate (2d) – Ac-(β-N-(ethylmorpholine)Ala-PO(OEt)2.
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.055 g; 0.25 mmol) was used. Product 0.055 g (0.16 mmol) was obtained as colorless oil. Yield: 63%. 1H NMR (400 MHz, DMSO) δ 8.08 (d, J = 9.5 Hz, 1H, NH), 4.44–4.31 (m, 1H, CH), 4.07–3.94 (m, 4H, 2 × CH2CH3), 3.54 (t broad, J = 4.3 Hz, 4H)*, 2.86–2.69 (m, 2H)*, 2.66–2.57 (m, 1H)*, 2.47–2.23 (m, 7H)*, 1.86 (d, J = 0.7 Hz, 3H, CH3CO), 1.21 (2 × t, 6H, J = 6.8 Hz, 2 × CH2CH3 overlapped). (*) – partially overlapped multiplets, derived from protons (14H) of side chain. 13C{1H} NMR (101 MHz, DMSO) δ 169.22 (d, J = 4.3 Hz), 66.28, 61.98 and 61.58 (d, J = 6.6 Hz), 57.88, 53.30, 47.95 (d, J = 6.1 Hz), 44.37, 44.35 (d, J = 152.7 Hz), 22.43, 16.37 and 16.30 (2 × d, J = 5.6 Hz, overlapped). 31P{1H} NMR (162 MHz, DMSO) δ 25.60. HRMS (ESI) calcd for C14H31N3O5P [(M + H)+] 352.1996, found: 352.2010.
Diethyl 1-(N-acetylamino)-2-(N,N-diethylamino)ethylphosphonate (2e) – Ac-(β-N-(diethylamine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.168 g; 0.75 mmol) was used. Product 0.195 g (0.66 mmol) was obtained as colorless oil. Yield: 88%. 1H NMR (400 MHz, DMSO) δ 8.17 (d, J = 9.6 Hz, 1H, NH), 4.44–4.30 (m, 1H, CH), 4.05–3.93 (m, 4H, 2 × CH2CH3), 2.78–2.69 and 2.64–2.51 (2 × m, 2H, CH2Ala overlapped with DMSO), 2.48–2.35 (m, 4H, N(CH2CH3)2), 1.83 (d, J = 1.1 Hz, 3H, CH3CO), 1.20 (2 × t, 6H, 2 × CH2CH3 overlapped), 0.91 (t, J = 7.1 Hz, 6H, N(CH2CH3)2). 13C{1H} NMR (101 MHz, DMSO) δ 168.85 (d, J = 4.7 Hz), 61.99 and 61.51 (2 × d, J = 6.7 Hz), 51.66 (d, J = 8.2 Hz), 46.28, 43.45 (d, J = 152.7 Hz), 22.44, 16.36 and 16.31 (2 × d, J = 5.5 Hz), 11.71. 31P{1H} NMR (162 MHz, DMSO) δ 25.98. HRMS (ESI) calcd for C12H28N2O4P [(M + H)+] 295.1781, found: 295.1781.
Hydrochloride of 1-amino-2-(N,N-diethylamino)ethylphosphonic acid (2e′) – HCl·β-N-(diethylamine)Ala-PO(OH)2.
Synthesised as new compound. Ac-(β-N-(diethylamine)Ala-PO(OEt)2) (0.160 g; 0.54 mmol) was used. Product 0.114 g (0.49 mmol) was obtained as white solid. Yield: 91%. Mp = 135–138 °C, 1H NMR (400 MHz, D2O) δ 3.80–3.69 (m, 1H, CH), 3.63–3.44 (m, 2H, CH2Ala), 3.42–3.26 (m, 4H, N(CH2CH3)2), 1.31 (t, J = 7.3 Hz, 6H, N(CH2CH3)2). 13C{1H} NMR (101 MHz, D2O) δ 50.13, 48.05, 42.74 (d, J = 135.3 Hz), 7.71. 31P{1H} NMR (162 MHz, D2O) δ 10.44. HRMS (ESI) calcd for C6H18N2O3P [(M + H)+] 197.1050, found: 197.1053.
Diethyl 1-(N-acetylamino)-2-(piperidin-1-yl)ethylphosphonate (2f) – Ac-(β-N-(piperidine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.111 g; 0.5 mmol) was used. Product 0.122 g (0.40 mmol) was obtained as colorless oil. Yield: 79%. 1H NMR (400 MHz, DMSO) δ 8.15 (d, J = 9.5 Hz, 1H, NH), 4.49–4.36 (m, 1H, CH), 4.06–3.92 (m, 4H, 2 × CH2CH3), 2.58–2.50 (m, 2H, CH2Ala overlapped with DMSO), 2.43–2.30 (m, 2H)*, 2.30–2.15 (m, 2H)*, 1.83 (d, J = 1.1 Hz, 3H, CH3CO), 1.51–1.38 (m, 4H)*, 1.38–1.28 (m, 2H)*, 1.21 and 1.19 (2 × t, J = 7.1 Hz, 6H, 2 × CH2CH3 overlapped). (*) – signals derived from protons of piperidine ring (10H). 13C{1H} NMR (101 MHz, DMSO) δ 168.84 (d, J = 4.6 Hz), 61.99 and 61.61 (2 × d, J = 6.7 Hz), 57.69 (d, J = 7.5 Hz), 53.63, 42.90 (d, J = 154.2 Hz), 25.57, 23.92, 22.44, 16.36 and 16.31 (2 × d, J = 4.0 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.80. HRMS (ESI) calcd for C13H28N2O4P [(M + H)+] 307.1781, found: 307.1792.
Hydrochloride of 1-amino-2-(piperidin-1-yl)ethylphosphonic acid (2f′) – HCl·β-N-(piperidine)Ala-PO(OH)2. Ac-(β-N-(piperidine)Ala-PO(OEt)2) (0.250 g; 0.82 mmol) was used. Product 0.190 g (0.78 mmol) was obtained as white solid. Yield: 95%. Mp = 164–167 °C, 1H NMR (400 MHz, D2O) δ 3.72–3.62 (m, 1H, CH), 3.58–3.45 (m, 2H)*, 3.40 (dd, J = 11.1, 7.0 Hz, 2H, CH2Ala), 3.12–2.80 (m, 2H)*, 1.85 (m, 2H)*, 1.73–1.51 (m, 3H)*, 1.43–1.25 (m, 1H)*. (*) – signals derived from protons of piperidine ring (10H). 13C{1H} NMR (101 MHz, D2O) δ 54.76, 53.84 (d, J = 35.2 Hz), 42.74 (d, J = 135.0 Hz), 22.58, 20.32. 31P{1H} NMR (162 MHz, D2O) δ 9.99. HRMS (ESI) calcd for C7H18N2O3P [(M + H)+] 209.1050, found: 197.1056.
Diethyl 1-(N-acetylamino)-2-(morpholin-1-yl)ethylphosphonate (2g) – Ac-(β-N-(morpholine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.221 g; 1.00 mmol) was used. Product 0.280 g (0.91 mmol) was obtained as colorless oil. Yield: 91%. 1H NMR (400 MHz, DMSO) δ 8.19 (d, J = 9.5 Hz, 1H, NH), 4.51–4.37 (m, 1H, CH), 4.06–3.94 (m, 4H, 2 × CH2CH3), 3.57–3.46 (m, 4H, 2 × CH2morph.), 2.58–2.52, 2.45–2.36, 2.33–2.24 (3 × m, 6H, 2 × CH2morph. and CH2Ala), 1.85 (d, J = 1.1 Hz, 3H, CH3CO), 1.21 (2 × t, J = 7.1 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 168.98 (d, J = 4.8 Hz), 66.17, 62.09 and 61.69 (2 × d, J = 6.7 Hz), 57.31 (d, J = 7.8 Hz), 52.89, 42.64 (d, J = 155.0 Hz), 22.42, 16.36 and 16.30 (2 × d, J = 4.0 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.51. HRMS (ESI) calcd for C12H26N2O5P [(M + H)+] 309.1574, found: 309.1576.
Hydrochloride of 1-amino-2-(morpholin-1-yl)ethylphosphonic acid (2g′) – HCl·β-N-(morpholine)Ala-PO(OH)2. Ac-(β-N-(morpholine)Ala-PO(OEt)2) (0.246 g; 0.80 mmol) was used. Product 0.186 g (0.75 mmol) was obtained as white solid. Yield: 94%. 1H NMR (400 MHz, D2O) δ 3.97 (s broad, 4H, 2 × CH2morph.), 3.87–3.75 (m, 1H, CH), 3.66–3.56 (m, 2H, CH2Ala), 3.46 (d broad, 4H, 2 × CH2morph.). 13C{1H} NMR (101 MHz, D2O) δ 63.46, 55.01, 52.15, 42.39 (d, J = 134.7 Hz). 31P{1H} NMR (162 MHz, D2O) δ 9.76. HRMS (ESI) calcd for C6H16N2O4P [(M + H)+] 211.0842, found: 211.084.
Diethyl 1-(N-acetylamino)-2-(2,6-dimethylmorpholin-1-yl)ethylphosphonate (2h) – Ac-(β-N-(2,6-dimethylmorpholine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.221 g; 1.00 mmol) was used. Product: isomers trans 0.100 g (0.30 mmol), isomer cis 0.181 g (0.54 mmol) were obtained as colorless oils. Overal yield: 83%. Trans isomers: 1H NMR (400 MHz, DMSO) δ 8.14 and 8.13 (2 × d, J = 9.5 Hz, 1H, NH), 4.52–4.35 (m, 1H, CHAla), 4.06–3.94 (m, 4H, 2 × CH2CH3),3.88–3.76 (m, 2H, 2 × CHmorph.), 2.61–2.51 (m, 1H)*, 2.49–2.39 (m, 2H)*, 2.28 (dd, J = 10.8, 2.9 Hz, 1H)*, 2.13 (dd, J = 10.7, 5.7 Hz, 1H)*, 2.01 (dd, J = 10.8, 5.6 Hz, 1H)*, 1.84 (2 × d, J = 1.2 Hz, 3H, CH3CO), 1.22 and 1.20 (t overlapped, J = 7.0, 6H, 2 × CH2CH3), 1.08 and 1.06 (2 × d, J = 6.4 Hz, 6H, 2 × CH3morph.). (*) – partially overlapped signals, derived from protons CH2Ala and 2 × CH2morph. (6H). 13C{1H} NMR (101 MHz, DMSO) δ 169.11 and 168.97 (2 × d, J = 5.1 Hz), 65.76, 65.73, 62.13, 62.06, 61.65, 61.59 (4 × d (d, J = 6.7 Hz)), 58.06, 57.81, 57.25 and 56.77 (d, J = 8.3 Hz), 43.02 and 42.22 (2 × d, J = 155.1 Hz), 22.36, 22.34, 17.96, 17.88, 16.35 and 16.30 (2 × d, J = 5.0 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.52, 25.45. HRMS (ESI) calcd for C14H30N2O5P [(M + H)+] 337.1887, found: 337.1898. Cis isomers: 1H NMR (400 MHz, DMSO) δ 8.18 (d, J = 9.5 Hz, 1H, NH), 4.54–4.34 (m, 1H, CHAla), 4.07–3.92 (m, 4H, 2 × CH2CH3), 3.56–3.46 and 3.46–3.38 (2 × m, 2H, 2 × CHmorph.), 2.70 (d broad, J = 10.7 Hz, 1H)*, 2.62 (d broad, J = 10.8 Hz, 1H)*, 2.56–2.50 (m, 2H)*, 1.84 (d, J = 1.0 Hz, 3H, CH3CO), 1.70 (t, J = 10.5 Hz, 1H)*, 1.56 (t, J = 10.4 Hz, 1H)*, 1.21 and 1.19 (t overlapped, J = 7.3 Hz, 6H, 2 × CH2CH3), 1.02 and 1.01 (2 × d, J = 5.7 Hz, 6H, 2 × CH3morph.). (*) – partially overlapped signals, derived from protons CH2Ala and 2 × CH2morph. (6H). 13C{1H} NMR (101 MHz, DMSO) δ 168.97 (d, J = 4.8 Hz), 71.00, 70.93, 62.07 and 61.68 (2 × d, J = 6.7 Hz), 59.25, 58.09, 57.02 (d, J = 7.7 Hz), 42.62 (d, J = 154.8 Hz), 22.42, 18.99, 18.97, 16.37 and 16.31 (2 × d, J = 5.2 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.57. HRMS (ESI) calcd for C14H30N2O5P [(M + H)+] 337.1887, found: 337.1898.
Diethyl 1-(N-acetylamino)-2-(4-acetylpiperazin-1-yl)ethylphosphonate (2i) – Ac-(β-N-(acetylpiperazine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.111 g; 0.5 mmol) was used. Product 0.123 g (0.35 mmol) was obtained as colorless oil. Yield: 70%. 1H NMR (400 MHz, DMSO) δ 8.20 (d, J = 9.5 Hz, 1H, NH), 4.51–4.37 (m, 1H, CH), 4.09–3.92 (m, 4H, 2 × CH2CH3), 3.43–3.34 (m, 4H, 2 × CH2piper.), 2.63–2.53, 2.47–2.39, 2.39–2.22 (3 × m, 6H, 2 × CH2piper. and CH2Ala), 1.96 (s, 3H, CH3COpiper.), 1.85 (d, J = 1.1 Hz, 3H, CH3COAla), 1.22 and 1.20 (2 × t, J = 7.1 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 169.02 (d, J = 4.5 Hz), 168.13, 62.11 and 61.71 (2 × d, J = 6.6 Hz), 56.81 (d, J = 7.6 Hz), 52.47, 52.16, 45.64, 42.85 (d, J = 154.9 Hz), 40.81, 22.44, 21.21, 16.36 and 16.31 (2 × d, J = 3.8 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.43. HRMS (ESI) calcd for C14H29N3O5P [(M + H)+] 350.1839, found: 350.1850.
Diethyl 1-(N-acetamido)-2-[4-(2-(N,N,-dimethylamino)ethylpiperazin-1-yl]ethylphosphonate (2j) – Ac-(β-N-((dimethylamino)ethylpiperazine)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.111 g; 0.5 mmol) was used. Product 0.154 g (0.41 mmol) was obtained as colorless oil. Yield: 81%. 1H NMR (400 MHz, DMSO) δ 8.11 (d, J = 9.5 Hz, 1H, NH), 4.47–4.35 (m, 1H, CH), 4.05–3.93 (m, 4H, 2 × CH2CH3), 2.58–2.51 (m, 2H, CH2Ala), 2.45–2.22 (2 × m overlapped, 12H, 6 × CH2), 2.11 (s, 6H, 2 × CH3), 1.84 (d, J = 1.0 Hz, 3H, CH3CO), 1.22 and 1.20 (2 × t, J = 7.1 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 168.85 (d, J = 4.6 Hz), 61.97 and 61.63 (2 × d, J = 6.7 Hz), 56.91 (d, J = 7.6 Hz), 56.61, 55.86, 53.05, 52.38, 45.50, 42.92 (d, J = 154.6 Hz), 22.37, 16.28 and 16.24 (d, J = 3.9 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.63. HRMS (ESI) calcd for C16H36N4O4P [(M + H)+] 379.2469, found: 379.2473.
Diethyl 1-(N-acetylamino)-2-(4-pyrimidin-2-yl)-piperazin-1-yl)ethylphosphonate (2k) – Ac-(β-N-(4-(pyrimidin-2-yl)piperazine)Ala-PO(OEt)2.
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.111 g; 0.50 mmol) was used. Product 0.162 g (0.42 mmol) was obtained as colorless oil. Yield: 84%. 1H NMR (400 MHz, DMSO) δ 8.34 (d, J = 4.7 Hz, 2H, HAr), 8.23 (d, J = 9.5 Hz, 1H, NH), 6.61 (t, J = 4.7 Hz, 1H, HAr), 4.55–4.39 (m, 1H, CH), 4.07–3.94 (m, 4H, 2 × CH2CH3), 3.74–3.60 (m, 4H, 2 × CH2piper.), 2.65–2.58, 2.49–2.43 (overlapped with DMSO), 2.41–2.31 (3 × m, 6H, 2 × CH2piper. and CH2Ala), 1.86 (d, J = 1.1 Hz, 3H, CH3CO), 1.21 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 169.01 (d, J = 4.7 Hz), 161.16, 157.94, 110.13, 62.10 and 61.71 (2 × d, J = 6.7 Hz), 57.02 (d, J = 7.3 Hz), 52.17, 43.27, 42.86 (d, J = 154.8 Hz), 16.37 and 16.32 (2 × d, J = 3.8 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.52. HRMS (ESI) calcd for C16H29N5O4P [(M + H)+] 386.1952, found: 386.1965.
Diethyl 1-(N-acetylamino)-2-(3,4-dihydroisoquinolin-2(1H)-yl)ethylphosphonate (2l) – Ac-(β-N-(tetrahydroisoquinoline)Ala-PO(OEt)2).
Synthesised as new compound. Ac-ΔAla-PO(OEt)2 (0.111 g; 0.50 mmol) was used. Product 0.162 g (0.46 mmol) was obtained as colorless oil. Yield: 91%. 1H NMR (400 MHz, DMSO) δ 8.23 (d, J = 9.5 Hz, 1H, NH), 7.12–7.00 (m, 4H, HAr), 4.62–4.50 (m, 1H, CH), 4.07–3.96 (m, 4H, 2 × CH2CH3), 3.63 (d, J = 14.9 Hz, 1H, NCHAHBC), 3.51 (d, J = 14.9 Hz, 1H, NCHAHBC), 2.82–2.69 and 2.65–2.54 (2 × m, 6H, CH2Ala and 2 × CH2tetrahydroisoquinoline), 1.21 and 1.20 (2 × t, J = 7.0, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 168.98 (d, J = 4.7 Hz), 134.63, 134.00, 128.41, 126.36, 125.93, 125.47, 62.04 and 61.64 (2 × d, J = 6.7 Hz), 56.59 (d, J = 7.8 Hz), 55.03, 49.98, 43.09 (d, J = 154.6 Hz), 28.58, 22.41, 16.33 and 16.24 (2 × d, J = 5.5 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.54. HRMS (ESI) calcd for C17H28N2O4P [(M + H)+] 355.1781, found: 355.1782.
Diethyl 1-(N-benzyloxycarbonylamino)-2-(N,N-diethylamino)ethylphosphonate (4a) – Z-(β-N-(diethylamine)Ala-PO(OEt))2.
Synthesised as new compound. Z-ΔAla-PO(OEt)2 (0.157 g; 0.50 mmol) was used. Product 0.071 g (0.18 mmol) was obtained as colorless oil. Yield: 37%. 1H NMR (400 MHz, DMSO) δ 7.64 (d, J = 9.6 Hz, 1H, NH), 7.39–7.27 (m, 5H, HArZ group), 5.06 (s, 2H, CH2Z group), 4.05–3.89 (m, 5H, CH and 2 × CH2CH3 overlapped), 2.76–2.57 (m, 2H, CH2Ala), 2.56–2.36 (m, 4H, 2 × NCH2CH3 overlapped with DMSO), 1.20 and 1.16 (2 × t, J = 6.9 Hz, 6H, 2 × CH2CH3 overlapped), 0.91 (t, J = 7.1 Hz, 6H, 2 × NCH2CH3). 13C{1H} NMR (101 MHz, DMSO) δ 156.08 (d, J = 5.3 Hz), 137.25, 128.30, 127.77, 127.56, 65.39, 61.92 and 61.58 (2 × d, J = 6.6 Hz), 51.64 (d, J = 9.4 Hz), 46.48 (d, J = 153.5 Hz), 46.26, 16.31 and 16.26 (2 × d, J = 2.0 Hz), 11.67. 31P{1H} NMR (162 MHz, DMSO) δ 25.66 and 25.23 (2 × s, two conformers with ratio 0.89
:0.11). HRMS (ESI) calcd for C18H32N2O5P [(M + H)+] 387.2043, found: 387.2047.
Diethyl 1-(N-benzyloxycarbonylamino)-2-(piperidin-1-yl)ethylphosphonate (4b) – Z-(β-N-(piperidine)Ala-PO(OEt)2).
Synthesised as new compound. Z-ΔAla-PO(OEt)2 (0.157 g; 0.50 mmol) was used. Product 0.164 g (0.41 mmol) was obtained as colorless oil. Yield: 82%. 1H NMR (400 MHz, DMSO) δ 7.61 (d, J = 9.5 Hz, 1H, NH), 7.40–7.28 (m, 5H, HArZ group), 5.12–5.01 (m, 2H, CH2Z group), 4.14–4.04 (m, 1H, CH overlapped), 4.04–3.91 (m, 4H, 2 × CH2CH3 overlapped), 2.61–2.44 (m, 2H, CH2Ala overlapped with DMSO), 2.44–2.33 (m, 2H, CH2piper.), 2.30–2.18 (m, 2H, CH2piper.), 1.50–1.38 (m, 4H, 2 × CH2piper.), 1.38–1.29 (m, 2H, CH2piper.), 1.20 and 1.17 (2 × t, J = 6.3 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 156.13 (d, J = 5.3 Hz), 137.28, 128.32, 127.79, 127.57, 65.40, 61.99 and 61.71 (d, J = 6.6 Hz), 57.41 (d, J = 8.4 Hz), 53.69, 45.79 (d, J = 155.4 Hz), 25.58, 23.94, 16.34 and 16.29. 31P{1H} NMR (162 MHz, DMSO) δ 25.53 and 25.12 (2 × s, two conformers with ratio 0.88
:0.12). HRMS (ESI) calcd for C19H32N2O5P [(M + H)+] 399.2043, found: 399.2049.
Diethyl 1-(N-benzyloxycarbonylamino)-2-(morpholin-1-yl)ethylphosphonate (4c) – Z-(β-N-(morpholine)Ala-PO(OEt)2).
Synthesised as new compound. Z-ΔAla-PO(OEt)2 (0.157 g; 0.50 mmol) was used. Product 0.093 g (0.23 mmol) was obtained as colorless oil. Yield: 46%. 1H NMR (400 MHz, DMSO) δ 7.60 (d, J = 9.5 Hz, 1H, NH), 7.39–7.28 (m, 5H, HArZ group), 5.12–5.01 (m, 2H, CH2Z group), 4.16–4.04 (m, 1H, CH overlapped), 4.05–3.93 (m, 4H, 2 × CH2CH3 overlapped), 3.56–3.44 (m, 4H, 2 × CH2morph), 2.66–2.51 (m, 2H, CH2Ala), 2.48–2.38 (m, 2H, CH2morph), 2.34–2.23 (m, 2H, CH2morph), 1.21 and 1.18 (t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 156.11 (d, J = 5.4 Hz), 137.15, 128.25, 127.73, 127.51, 66.11, 65.42, 61.98 and 61.71 (2 × d, J = 6.7 Hz), 57.00 (d, J = 8.7 Hz), 52.87, 45.54 (d, J = 156.7 Hz), 16.24 and 16.19. 31P{1H} NMR (162 MHz, DMSO) δ 25.14 and 24.77 (2 × s, two conformers with ratio 0.92
:0.08). HRMS (ESI) calcd for C18H30N2O6P [(M + H)+] 401.1836, found: 401.1935.
Diethyl 1-(N-benzyloxycarbonylamino)-2-(2,6-dimethylmorpholin-1-yl)ethylphosphonate (4d) – Z-(β-N-(2,6-dimethylmorpholine)Ala-PO(OEt)2).
Synthesised as new compound. Z-ΔAla-PO(OEt)2 (0.157 g; 0.50 mmol) was used. Product: isomers trans 0.022 g (0.05 mmol), isomer cis 0.044 g (0.10 mmol) were obtained as colorless oils. Overall yield: 31%. The NMR spectra are characterized only for cis isomers. The interpretation of spectra for isomers trans is complicated due to presence of conformers. The 1H, 13C and 31P NMR spectra of isomers trans are given in the ESI.‡ Isomers: 1H NMR (400 MHz, DMSO) δ 7.64 (d, J = 9.5 Hz, 1H, NH), 7.38–7.28 (m, 5H, HArZ group), 5.14–4.99 (m, 2H, CH2Z group), 4.15–4.04 (m, 1H, CH overlapped), 4.04–3.91 (m, 4H, 2 × CH2CH3 overlapped), 3.52–3.44 and 3.43–3.37 (2 × m, 2H, 2 × CHmorph.), 2.72 (d broad, J = 10.6 Hz, 1H)*, 2.64–2.52 (m, 3H)*, 1.74 (t, J = 10.5 Hz, 1H)*, 1.57 (t, J = 10.4 Hz, 1H)*, 1.20 and 1.17 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped), 1.01 and 1.00 (2 × d, J = 3.9 Hz, 6H, 2 × CH3morph.). (*) – partially overlapped signals, derived from protons CH2Ala and 2 × CH2morph. (6H). 13C{1H} NMR (101 MHz, DMSO) δ 156.13 (d, J = 5.3 Hz), 137.24, 128.32, 127.82, 127.61, 71.00, 70.97, 65.46, 62.05 and 61.77 (2 × d, J = 6.5 Hz), 59.36, 57.94, 56.64 (d, J = 8.5 Hz), 45.53 (d, J = 155.8 Hz), 19.00, 18.95, 16.34 and 16.28. 31P{1H} NMR (162 MHz, DMSO) δ 25.22 and 24.80 (2 × s, two conformers with ratio 0.88
:0.12). HRMS (ESI) calcd for C20H34N2O6P [(M + H)+] 429.2149, found: 429.2159 for cis isomers, 429.2162 for trans isomers.
Diethyl 1-(N-tert-butyloxycarbonylglycylamino)-2-(N-n-propylamino)ethylphosphonate (6a) – Boc-Gly-(β-N-(popylamine)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-ΔAla-PO(OEt)2 (0.084 g; 0.25 mmol) was used. Product 0.096 g (0.24 mmol) was obtained as colorless oil. Yield: 97%. 1H NMR (400 MHz, DMSO) δ 7.97 (d, J = 9.3 Hz, 1H, NHAla), 7.00 and 6.56 (t and s broad, J = 5.7 Hz, 1H, NHGly two conformers), 4.45–4.31 (m, 1H, CH), 4.07–3.93 (m, 4H, 2 × CH2CH3), 3.64–3.50 (m, 2H, CH2Gly), 2.86–2.73, 2.49–2.35 (2 × m, 4H, CH2Ala and NH(CH2CH2CH3) overlapped with DMSO), 1.37 and 1.46–1.27 (s and m overlapped, 11H, 3 × CH3Boc and NH(CH2CH2CH3)), 1.21 and 1.20 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped), 0.84 (t, J = 7.4 Hz, 3H, NH(CH2CH2CH3)). 13C{1H} NMR (101 MHz, DMSO) δ 169.50 (d, J = 4.4 Hz), 155.77, 78.02, 62.18 and 61.70 (2 × d, J = 6.6 Hz), 50.06, 47.92 (d, J = 6.0 Hz), 44.60 (d, J = 153.5 Hz), 43.20, 28.15, 22.25, 16.33 and 16.24 (2 × d, J = 5.6 Hz), 11.64. 31P{1H} NMR (162 MHz, DMSO) δ 25.06 and 24.81 (2 × s, two conformers with ratio 0.16
:0.84). HRMS (ESI) calcd for C16H35N3O6P [(M + H)+] 396.2258, found: 396.2262.
Diethyl 1-(N-glycylamino)-2-(N-n-propylamino)ethylphosphonate trifluoroacetate (6a′) – 2TFA·Gly-(β-N-(propylamine)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-(β-N-(propylamine)Ala-PO(OEt)2) (0.074 g; 0.19 mmol) was used. Product 0.098 g (0.19 mmol) was obtained as colorless oil. Yield: 99%. 1H NMR (400 MHz, DMSO) δ 9.04 (d, J = 9.3 Hz, 1H, NHAla), 8.85 (s, 2H, (C3H7) NH2+), 8.19 (s, 3H, NH+3Gly), 4.76–4.57 (m, 1H, CH), 4.18–4.02 (m, 4H, 2 × CH2CH3), 3.65 (d broad, J = 5.2 Hz, 2H, CH2Gly), 3.30 and 3.12 (2 × s broad, 2H, CH2Ala), 2.93 (s, 2H, NH(CH2CH2CH3)), 1.70–1.50 (m, 2H, NH(CH2CH2CH3)), 1.26 and 1.25 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped), 0.89 (t, J = 7.4 Hz, 3H, NH(CH2CH2CH3)). 13C{1H} NMR (101 MHz, DMSO) δ 166.92 (d, J = 4.4 Hz), 158.52 (q, J = 34.5 Hz), 116.28 (q, J = 294.6 Hz), 63.05 and 62.76 (2 × d, J = 6.7 Hz), 48.51, 46.23 (d, J = 11.0 Hz), 43.05 (d, J = 157.4 Hz), 40.34, 18.82, 16.28 and 16.19 (2 × d, J = 5.3 Hz), 10.83. 31P{1H} NMR (162 MHz, DMSO) δ 20.47. HRMS (ESI) calcd for C11H27N3O4P [(M + H)+] 296.1734, found: 296.1733.
Diethyl 1-(N-tert-butyloxycarbonylglycylamino)-2-(N-propargylamino)ethylphosphonate (6b) –Boc-Gly-(β-N-(propargylamine)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-ΔAla-PO(OEt)2 (0.084 g; 0.25 mmol) was used. Product 0.081 g (0.21 mmol) was obtained as colorless oil. Yield: 83%. 1H NMR (400 MHz, DMSO) δ 7.91 (d, J = 9.5 Hz, 1H, NHAla), 6.92 and 6.49 (t and s broad, J = 5.8 Hz, 1H, NHGly two conformers), 4.40–4.28 (m, 1H, CH), 4.09–3.92 (m, 4H, 2 × CH2CH3), 3.64–3.51 (m, 2H, CH2Gly), 3.29 (d, J = 2.4 Hz, 1H, NHCHAHBC
), 3.28 (d, J = 2.4 Hz, 1H, NHCHAHBC), 3.05 (t, J = 2.4 Hz, 1H, CH), 2.94–2.83 and 2.83–2.72 (2 × m, 2H, CH2Ala), 1.38 (s, 9H, 3 × CH3Boc), 1.22 and 1.20 (2 × t, J = 7.1 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 169.37 (d, J = 4.6 Hz), 155.72, 82.39, 78.00, 73.78, 62.11 and 61.68 (2 × d, J = 6.7 Hz), 46.84 (d, J = 5.9 Hz), 44.87 (d, J = 153.9 Hz), 43.12, 36.77, 28.13, 16.27 and 16.19 (2 × d, J = 5.5 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 24.94 and 24.81 (2 × s, two conformers with ratio 0.11:0.89). HRMS (ESI) calcd for C16H31N3O6P [(M + H)+] 392.1945, found: 392.1957.
Diethyl 1-(N-glycylamino)-2-(N-propargylamino)ethylphosphonate trifluoroacetate (6b′) – 2TFA·Gly-(β-N-(popargylamine)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-(β-N-(propylamine)Ala-PO(OEt)2) (0.060 g; 0.15 mmol) was used. Product 0.079 g (0.15 mmol) was obtained as colorless oil. Yield: 99%. 1H NMR (400 MHz, DMSO) δ 9.56 (s broad, 2H, NH+2prg.), 9.04 (d, J = 9.4 Hz, 1H, NHAla), 8.19 (s, 3H, NH+3Gly), 4.73–4.59 (m, 1H, CH overlapped with water), 4.13–4.05 (m, 4H, 2 × CH2CH3), 3.99 and 3.98 (2 × d, J = 2.4 Hz, 2H, NHCHAHBC
), 3.77 (t, J = 2.4 Hz, 1H, CH), 3.64 (s broad, 2H, CH2Gly), 3.40 and 3.19 (2 × m, 2H, CH2Ala), 1.26 and 1.25 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 166.90 (d, J = 4.6 Hz), 158.48 (q, J = 33.7 Hz), 116.47 (q, J = 295.4 Hz), 79.94, 74.67, 63.10 and 62.81(2 × d, J = 6.7 Hz), 45.57 (d, J = 10.7 Hz), 42.98 (d, J = 157.7 Hz), 40.33, 35.92, 16.26 and 16.18 (2 × d, J = 5.3 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 20.28. HRMS (ESI) calcd for C11H23N3O4P [(M + H)+] 292.1421, found: 292.1430.
Diethyl 1-(N-tert-butyloxycarbonylglycylamino)-2-(piperidin-1-yl)ethylphosphonate (6c) – Boc-Gly-(β-N-(piperidine)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-ΔAla-PO(OEt)2 (0.084 g; 0.25 mmol) was used. Product 0.103 g (0.24 mmol) was obtained as colorless oil. Yield: 98%. 1H NMR (400 MHz, DMSO) δ 8.03 (d, J = 9.4 Hz, 1H, NHAla), 6.95 and 6.55 (t and s broad, J = 6.1 Hz, 1H, NHGly two conformers), 4.48–4.33 (m, 1H, CH), 4.07–3.92 (m, 4H, 2 × CH2CH3), 3.62–3.47 (m, 2H, CH2Gly), 2.61–2.50 (m, 2H, CH2Ala overlapped with DMSO), 2.43–2.30 (m, 2H, CH2piper.), 2.30–2.16 (m, 2H, CH2piper.), 1.49–1.39 (m, 4H, 2 × CH2piper.), 1.37 (s, 9H, 3 × CH3Boc), 1.36–1.29 (m, 2H, CH2piper.), 1.21 and 1.19 (2 × t, J = 7.1 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 169.16 (d, J = 4.6 Hz), 155.70, 77.93 62.15 and 61.66 (2 × d, J = 6.6 Hz), 57.60 (d, J = 5.9 Hz), 53.61, 43.14 (d, J = 154.5 Hz), 43.03, 28.16, 25.53, 23.87, 16.34 and 16.27 (2 × d, J = 5.8 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.54 and 25.37 (2 × s, two conformers with ratio 0.13
:0.87). HRMS (ESI) calcd for C18H37N3O6P [(M + H)+] 422.2414, found: 422.2426.
Diethyl 1-(N-glycylamino)-2-(piperidin-1-yl)ethylphosphonate trifluoroacetate (6c′) – 2TFA·Gly-(β-N-(piperidine)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-(β-N-(piperidine)Ala-PO(OEt)2) (0.095 g; 0.23 mmol) was used. Product 0.128 g (0.23 mmol) was obtained as colorless oil. Yield: 99%. 1H NMR (400 MHz, DMSO) δ 9.79 (s, 1H, NH+piper.), 9.16 (d, J = 9.5 Hz, 1H, NHAla), 8.19 (s, 3H, NH+3Gly), 4.87–4.72 (m, 1H, CH overlapped with water), 4.17–4.02 (m, 4H, 2 × CH2CH3), 3.67 (s broad, 2H, CH2Gly), 3.53–3.24 (m, 4H, 2 × CH2piper.), 3.01 and 2.92 (2 × s broad and overlapped, 2H, CH2Ala), 1.89–1.52 and 1.45–1.30 (2 × m, 6H, 3 × CH2piper.), 1.27 and 1.25(2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). 13C NMR (101 MHz, DMSO) δ 166.72 (d, J = 4.1 Hz), 158.45 (q, J = 34.1 Hz), 116.33 (q, J = 295.5 Hz), 63.21 and 62.91 (2 × d, J = 6.7 Hz), 54.73 (d, J = 12.2 Hz), 53.38, 41.43 (d, J = 156.3 Hz), 40.28, 22.27, 21.07, 16.28 and 16.18 (2 × d, J = 5.3 Hz). 31P NMR (162 MHz, DMSO) δ 20.55. HRMS (ESI) calcd for C13H29N3O4P [(M + H)+] 322.1890, found: 322.1894.
Diethyl 1-(N-tert-butyloxycarbonylglycylamino)-2-(morpholin-1-yl)ethylphosphonate (6d) – Boc-Gly-(β-N-(morpholine)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-ΔAla-PO(OEt)2 (0.084 g; 0.25 mmol) was used. Product 0.100 g (0.24 mmol) was obtained as colorless oil. Yield: 95%. 1H NMR (400 MHz, DMSO) δ 8.06 (d, J = 9.4 Hz, 1H, NHAla), 6.95 and 6.55 (t and s broad, J = 5.7 Hz, 1H, NHGly two conformers), 4.49–4.35 (m, 1H, CH), 4.08–3.95 (m, 4H, 2 × CH2CH3), 3.60–3.46 (m, 6H, CH2Gly and 2 × CH2morph. overlapped), 2.62–2.53, 2.44–2.35, 2.34–2.25 (3 × m, 6H, CH2Ala and 2 × CH2morph.), 1.37 (s, 9H, 3 × CH3Boc), 1.22 and 1.20 (2 × t, J = 7.2 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 169.36 (d, J = 4.7 Hz), 155.74, 77.98, 66.16, 62.26 and 61.77 (2 × d, J = 6.6 Hz), 57.18 (d, J = 7.5 Hz), 52.90, 43.05, 42.98 (d, J = 155.1 Hz), 28.19, 16.36 and 16.29 (2 × d, J = 5.8 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.18 and 25.02 (2 × s, two conformers with ratio 0.13
:0.87). HRMS (ESI) calcd for C17H35N3O7P [(M + H)+] 424.2207, found: 424.2214.
Diethyl 1-(N-glycylamino)-2-(morpholin-1-yl)ethylphosphonate trifluoroacetate (6d′) – 2TFA·Gly-(β-N-(morpholine)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-(β-N-(morpholine)Ala-PO(OEt)2) (0.078 g; 0.18 mmol) was used. Product 0.099 g (0.18 mmol) was obtained as colorless oil. Yield: 98%. 1H NMR (400 MHz, DMSO) δ 9.18 (d, J = 9.5 Hz, 1H, NHAla), 8.17 (s, 3H, NH+3Gly), 4.89–4.69 (m, 1H, CH), 4.19–4.01 (m, 4H, 2 × CH2CH3), 3.80 (s broad, 4H, 2 × CH2morph.), 3.66 (d broad, J = 4.7 Hz 2H, CH2Gly), 3.51 (d broad, J = 13.7 Hz, 1H, CHAHBAla), 3.42–3.13 (m, 5H, CHAHBAla and 2 × CH2morph.), 1.27 and 1.26 (2 × t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). 13C NMR (101 MHz, DMSO) δ 166.79 (d, J = 4.2 Hz), 158.50 (q, J = 34.8 Hz), 116.15 (q, J = 293.8 Hz), 63.25, 63.19 and 62.90 (2 × d, J = 6.7 Hz), 54.91 (d, J = 11.5 Hz), 51.30, 41.32 (d, J = 155.9 Hz), 40.25, 16.29 and 16.19 (2 × d, J = 5.3 Hz). 31P NMR (162 MHz, DMSO) δ 20.60. HRMS (ESI) calcd for C12H27N3O5P [(M + H)+] 324.1683, found: 324.1688.
Diethyl 1-(N-tert-butyloxycarbonylglycylamino)-2-(3,4-dihydroisoquinolin-2(1H)-yl)ethylphosphonate (6e) – Boc-Gly-(β-N-(tetrahydroisoquinoline)Ala-PO(OEt)2).
Synthesised as new compound. Boc-Gly-ΔAla-PO(OEt)2 (0.084 g; 0.25 mmol) was used. Product 0.112 g (0.24 mmol) was obtained as colorless oil. Yield: 95%. 1H NMR (400 MHz, DMSO) δ 8.11 (d, J = 9.4 Hz, 1H, NHAla), 7.11–7.01 (m, 4H, HAr THI), 6.91 and 6.50 (t and s broad, J = 6.0 Hz, 1H, NHGly two conformers), 4.63–4.47 (m, 1H, CH), 4.09–3.94 (m, 4H, 2 × CH2CH3), 3.65–3.46 (m, 4H, CH2Gly and CH2THI), 2.84–2.70, 2.65–2.55 (2 × m, 6H, CH2Ala and 2 × CH2THI), 1.36 and 1.26 (2 × s, 9H, 3 × CH3Boc two conformers), 1.21 and 1.20 (2 × t, J = 7.0 Hz, 5H), 1.20 (t, J = 7.0 Hz, 6H, 2 × CH2CH3 overlapped). 13C{1H} NMR (101 MHz, DMSO) δ 169.25 (d, J = 4.4 Hz), 155.69, 134.65, 134.01, 128.35, 126.36, 125.87, 125.39, 77.93, 62.20 and 61.71 (2 × d, J = 6.5 Hz), 56.55 (d, J = 7.3 Hz), 49.98, 43.42 (d, J = 154.8 Hz), 43.02, 28.58, 28.13, 16.32 and 16.19 (d, J = 5.6 Hz). 31P{1H} NMR (162 MHz, DMSO) δ 25.21 and 25.05 (2 × s, two conformers with ratio 0.15
:0.85). HRMS (ESI) calcd for C22H37N3O6P [(M + H)+] 470.2414, found: 470.2423.
Diethyl 1-(N-glycylamino)-2-(3,4-dihydroisoquinolin-2(1H)-yl)ethylphosphonate trifluoroacetate (6e′) – 2TFA·Gly-(β-N-tetrahydroisoquinoline)Ala-PO(OEt)2.
Synthesised as new compound. Boc-Gly-(β-N-(tetrahydroisoquinoline)Ala-PO(OEt)2) (0.098 g; 0.21 mmol) was used. Product 0.122 g (0.20 mmol) was obtained as colorless oil. Yield: 98%. 1H NMR (400 MHz, DMSO) δ 9.21 (d, J = 9.4 Hz, 1H, NHAla), 8.19 (s, 3H, NH+3Gly), 7.31–7.14 (m, 4H, HAr), 5.02–4.85 (m, 1H, CH), 4.52–4.38 (m, 2H, CH2THI overlapped with water), 4.19–4.06 (m, 4H, 2 × CH2CH3), 3.67 (s broad, 2H, CH2Gly), 3.63–3.19 (m broad, 4H, CH2THI and CH2Ala), 3.07 (s, 2H, CH2THI), 1.27 and 1.26 (2 × t, J = 7.0 Hz, 6H). 13C NMR (101 MHz, DMSO) δ 166.95 (d, J = 4.6 Hz), 158.41 (q, J = 32.9 Hz), 116.68 (q, J = 297.0 Hz), 63.17 and 62.90 (2 × d, J = 6.6 Hz), 54.30 (d, J = 8.2 Hz), 41.67 (d, J = 155.5 Hz), 40.31, 24.88, 16.27 and 16.18 (2 × d, J = 5.2 Hz). 31P NMR (162 MHz, DMSO) δ 20.56. HRMS (ESI) calcd for C17H29N3O4P [(M + H)+] 370.1890, found: 370.1890.
Crystallography
The single-crystal X-ray diffraction experiments were performed at 100.0(1) K on Xcalibur diffractometer (Rigaku Oxford Diffraction, Sevenoaks, Kent, UK), equipped with a CCD detector and a graphite monochromator (Rigaku Oxford Diffraction) with MoKα radiation and furnished with an Oxford Cryosystem N2 gas stream device. The reciprocal space was explored by ω scans. The reflections were measured with a radiation exposure time from 4 to 25 s, according to diffraction intensities. The detector was positioned at a 60 mm distance from the crystal. Procession of the diffraction data was performed using the CrysAlis CCD.44,45 Structures of compounds 3 and 5 were solved in the monoclinic crystal system, P21/c for 3 and P21/n for 5 space group respectively (Table S2.1‡), by direct methods and refined by a full-matrix least squares method using SHELXL14 program.46,47 Lorentz and polarization corrections were applied. Non-hydrogen atoms were refined anisotropically. In structures, H atoms were refined using a riding model. The structure drawings were prepared using the Mercury program.48 The crystallographic data for all compounds have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1935968 for 3 and CCDC 1935969 for 5.‡Isothermal titration calorimetry (ITC)
ITC measurements were carried out at 25 °C on a MicroCal PEAQ Isothermal Titration Calorimeter. All reagents were obtained from Sigma-Aldrich and were >99% pure. Metal salts (copper(II) nitrate trihydrate, nickel(II) nitrate hexahydrate and zinc(II) nitrate hexahydrate) and each of the three tested aminophosphonates (2e′, 2f′, 2g′) were dissolved directly into 25 mM buffer solution of sodium cacodylate (Caco). The pH of the buffer solution was adjusted to 7.4 using 0.3 M NaOH and 0.2 mM HClO4. After stabilizing the instrument at 25 °C, 40 μL of metal ion solutions (concentration of ca. 2 mM for Cu2+ and 3–6 mM for Ni2+ and Zn2+) were used for titration of 200 μL of aminophosphonate solutions (concentration initially ten times smaller than that of metal ion) by 13 or 19 successive injections with an interval of 180 s between each drop (each assay had been repeated few times). A background titration was performed after each assay using identical titrant in the syringe with the buffer solution placed in the sample cell. The result was subtracted from each experimental titration to account for the heat of dilution. The stirring rate was set at 750 rpm during the experiments. The reference cell was filled with distilled water. The data were processed with MicroCal PEAQ-ITC Analysis Software. An initial 0.4 μL injection was discarded from each data set in order to remove the effect of titrant diffusion across the syringe tip during the equilibration process. The CaCl2-EDTA titration was performed to check the apparatus and the results were compared with those obtained for the same samples (test kit) at MicroCal.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This work was supported by the National Science Centre, Poland (NCN) grant number 2016/21/B/ST5/00115. We wish to thank Dorota Wieczorek (NMR), Gabriela Maciejewska (HRMS) for assistance with the acquisition of analytical data. The authors would like to acknowledge valuable discussions with Dariusz Wyrzykowski regarding the ITC assays.Notes and references
- B. Lejczak, P. Kafarski and J. Zygmunt, Biochemistry, 1989, 28, 3549–3555 CrossRef CAS PubMed.
- A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed.
- M. Bollinger, F. Manzenrieder, R. Kolb, A. Bochen, S. Neubauer, L. Marinelli, V. Limongelli, E. Novellino, G. Moessmer, R. Pell, W. Lindner, J. Fanous, A. Hoffman and H. Kessler, J. Med. Chem., 2012, 55, 871–882 CrossRef CAS PubMed.
- E. Węglarz-Tomczak, Ł. Berlicki, M. Pawełczak, B. Nocek, A. Joachimiak and A. Mucha, Eur. J. Med. Chem., 2016, 117, 187–196 CrossRef PubMed.
- E. Węglarz-Tomczak, K. Staszewska, M. Talma and A. Mucha, Tetrahedron Lett., 2016, 57, 4812–4814 CrossRef.
- E. Węglarz-Tomczak, S. Vassiliou and A. Mucha, Bioorg. Med. Chem. Lett., 2016, 26, 4122–4126 CrossRef PubMed.
- R. Błaszczyk and T. Gajda, Tetrahedron Lett., 2007, 48, 5859–5863 CrossRef.
- R. Błaszczyk, A. Gajda, S. Zawadzki, E. Czubacka and T. Gajda, Tetrahedron, 2010, 66, 9840–9848 CrossRef.
- T. Cytlak, M. Skibinska, P. Kaczmarek, M. Kaźmierczak, M. Rapp, M. Kubicki and H. Koroniak, RSC Adv., 2018, 8, 11957–11974 RSC.
- C. De Risi, D. Perrone, A. Dondoni, G. P. Pollini and V. Bertolasi, Eur. J. Org. Chem., 2003, 2003, 1904–1914 CrossRef.
- E. K. Dolence and J. B. Roylance, Tetrahedron: Asymmetry, 2004, 15, 3307–3322 CrossRef CAS.
- E. K. Dolence, G. Mayer and B. D. Kelly, Tetrahedron: Asymmetry, 2005, 16, 1583–1594 CrossRef CAS.
- I. E. Głowacka, D. G. Piotrowska, A. E. Wroblewski and A. Trocha, Tetrahedron: Asymmetry, 2017, 28, 1602–1607 CrossRef.
- Y. J. Seo, J. A. Kim and H. K. Lee, J. Org. Chem., 2015, 80, 8887–8902 CrossRef CAS PubMed.
- J. C. Wilt, M. Pink and J. N. Johnston, Chem. Commun., 2008, 4177–4179 RSC.
- J. Zygmunt, Tetrahedron, 1985, 41, 4979–4982 CrossRef CAS.
- O. Dogan, H. Babiz, A. G. Gozen and S. Budak, Eur. J. Med. Chem., 2011, 46, 2485–2489 CrossRef CAS PubMed.
- B. Vanderhoydoncky and C. V. Stevens, Tetrahedron, 2007, 63, 7679–7689 CrossRef.
- P. Lenartowicz, B. Dziuk, B. Zarychta, M. Makowski and P. Kafarski, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 706–712 CrossRef CAS.
- D. Siodłak, Amino Acids, 2015, 47, 1–17 CrossRef PubMed.
- J. W. Bogart and A. A. Bowers, Org. Biomol. Chem., 2019, 17, 3653–3669 RSC.
- D. Chol and H. Kohn, Tetrahedron Lett., 1995, 36, 7371–7374 Search PubMed.
- M. Perez and R. Pleixats, Tetrahedron, 1995, 51, 8355–8362 CrossRef CAS.
- G. Boschin, L. Scaglioni and A. Arnoldi, J. Agric. Food Chem., 1999, 47, 939–944 CrossRef CAS PubMed.
- P. M. T. Ferreira, H. L. S. Maia and L. S. Monteiro, Tetrahedron Lett., 1999, 40, 4099–4102 CrossRef CAS.
- P. M. T. Ferreira, H. L. S. Maia, L. S. Monteiro and J. Sacramento, J. Chem. Soc., Perkin Trans. 1, 2001, 3167–3173 RSC.
- P. M. T. Ferreira, H. L. S. Maia and L. S. Monteiro, Eur. J. Org. Chem., 2003, 2003, 2635–2644 CrossRef.
- B. N. Naidu, M. E. Sorenson, T. P. Connolly and Y. Ueda, J. Org. Chem., 2003, 68, 10098–10102 CrossRef CAS PubMed.
- A. M. Freedy, M. J. Matos, O. Boutureira, F. Corzana, A. Guerreiro, P. Akkapeddi, V. J. Somovilla, T. Rodrigues, K. Nicholls, B. Xie, G. Jimenez-Oses, K. M. Brindle, A. A. Neves and G. J. L. Bernardes, J. Am. Chem. Soc., 2017, 139, 18365–18375 CrossRef CAS PubMed.
- A. M. King, C. Salome, J. Dinsmore, E. Salome-Grosjean, M. De Ryck, R. Kaminski, A. Valade and H. Kohn, J. Med. Chem., 2011, 54, 4815–4830 CrossRef CAS PubMed.
- P. Lenartowicz, M. Makowski, B. Oszywa, K. Haremza, R. Latajka, M. Pawełczak and P. Kafarski, Biochimie, 2017, 139, 46–55 CrossRef CAS PubMed.
- M. M. Jimenez-Andreu, J. Bueno-Moron, F. J. Sayago, C. Cativiela, T. Tejero and P. Merino, Eur. J. Org. Chem., 2019, 2019, 1268–1272 CrossRef CAS.
- A. Kuznik, R. Mazurkiewicz, M. Zieba and K. Erfurt, Tetrahedron Lett., 2018, 59, 3307–3310 CrossRef CAS.
- T. Kiss, E. Farkas, H. Kozlowski, Z. Siatecki, P. Kafarski and B. Lejczak, J. Chem. Soc., Dalton Trans., 1989, 1053–1057 RSC.
- T. Kiss, M. Jezowska-Bojczuk, H. Kozlowski, P. Kafarski and K. Antczak, J. Chem. Soc., Dalton Trans., 1991, 2275–2279 RSC.
- D. Witkowska and M. Rowinska-Zyrek, J. Inorg. Biochem., 2019, 199, 110783 CrossRef CAS PubMed.
- D. Witkowska, H. L. Cox, T. C. Hall, G. C. Wildsmith, D. C. Machin and M. E. Webb, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 254–263 CrossRef CAS PubMed.
- S. Vega, O. Abian and A. Velazquez-Campoy, J. Therm. Anal. Calorim., 2019, 138, 3229–3248 CrossRef CAS.
- M. Ostrowska, I. A. Golenya, M. Haukka, I. O. Fritsky and E. Gumienna-Kontecka, New J. Chem., 2019, 43, 10237–10249 RSC.
- Y. Zhang, S. Akilesh and D. E. Wilcox, Inorg. Chem., 2000, 39, 3057–3064 CrossRef CAS.
- J. Zoń, Synthesis, 1981, 324 CrossRef.
- N. Lefevre, J. L. Brayer, B. Folleas and S. Darses, Org. Lett., 2013, 15, 4274–4276 CrossRef CAS PubMed.
- H. Fernandez-Perez, P. Lenartowicz, L. Carreras Vinent, A. Grabulosa, P. Kafarski and A. Vidal-Ferran, J. Org. Chem., 2020 DOI:10.1021/acs.joc.0c00914.
- CrysAlis CCD, Version 1.171.33.57, Oxford Diffraction Ltd., Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- CrysAlis RED, Version 1.171.33.57, Oxford Diffraction Ltd., Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnotes |
| † This article is dedicated in honour of Professor Maurizio Peruzzini on his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: 1935968 and 1935969. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03764h |
| This journal is © The Royal Society of Chemistry 2020 |