
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbonylative Suzuki–Miyaura cross-coupling by immobilized Ni@Pd NPs supported on carbon nanotubes
Liu Nana,
Cai Yalanb,
Li Jixiang*b,
Ouyang Dujuana,
Duan Wenhuia,
Jalal Rouhi *c and
Mazli Mustaphad
*c and
Mazli Mustaphad
aChina Key Laboratory of Light Industry Pollution Control and Recycling, Department of Material and Chemical Engineering, Zhengzhou University of Light Industry, Zhengzhou 450001, PR China
bShanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, PR China. E-mail: lijixiang@sari.ac.cn
cFaculty of Physics, University of Tabriz, Tabriz 51566, Iran. E-mail: jalalrouhi@gmail.com
dCentre for Corrosion Research, Department of Mechanical Engineering, Universiti Teknologi PETRONAS, 32610 Bandar Seri Iskandar, Perak Darul Ridzuan, Malaysia
First published on 27th July 2020
Abstract
In this study, a novel carbon nanotube (CNT) based nanocatalyst (Ni@Pd/CNT) was synthesized by modifying CNTs using Ni@Pd core–shell nanoparticles (NPs). Ni@Pd/CNT was used in catalytic carbonylative cross-coupling between 4-iodoanisole and phenylboronic acid. The Ni@Pd NPs possessed a magnetic nickel (Ni) core with a palladium (Pd) structural composite shell. Thus, the use of Ni had led to a reduced consumption of Pd without sacrificing the overall catalytic performance, simultaneously making it reusable as it could be conveniently recovered from the reaction mixture by using an external magnetic field. Immobilization of the Ni@Pd NPs on carbon nanotubes not only prevented their aggregation, but also significantly enhanced the accessibility of the catalytically active sites. The abovementioned approach based on carbon nanotubes and Ni@Pd NPs provided a useful platform for the fabrication of noble-metal-based nanocatalysts with easy accessibility and low cost, which may allow for an efficient green alternative for various catalytic reductions.
Introduction
Recently, noble metal nanoparticles have been given significant consideration as catalysts due to their appropriate catalytic activity in organic catalytic reactions. The catalytic activity of noble metal nanoparticles was usually based on size, crystallinity, or shape and also the surface zone.1–5 Moreover, the development of nano-sized metal catalysts in the case of catalyst supports has been dramatically investigated. Many supporting materials have been utilized to suggest Pd nanoparticles (NPs) as catalysts for organic catalytic reactions, like Pd/SBA-15, Pd/Al2O3, or Pd/C nanospheres.6–9 The pores of considered materials along with large particular surface zones prevent the aggregation of NPs, allowing homogeneous scattering in the catalytic spaces and improving the catalytic activity of the approach. However, poor accessibility of these active zones into the pores limits their applications and significant mass transport is known to be necessary for them. Therefore, silica supports and the simple availability of high surface zones were usually desirable.It should be noted that many reactions of noble metal nanoparticle catalysts are commonly performed only on the nanoparticle site and a wide fraction of atoms within the core can be inactive catalytically. However, to make a considerable percentage of noble metal atoms available in catalysis and to reduce their utilization, the inner atoms of a noble metal can be varied with other non-noble metals. The existence of a different metal core usually allows control over catalytic activity, selectivity, and stability, because of “synergistic influences” coming from core–shell metal interactions.10 Transition Fe-group metals such as Fe, Ni, and Co, among distinct non-noble metals, are generally hybridized using noble metals for creating magnetic NPs that can be simply reproduced utilizing an external magnetic area.11–16 However, cost-effective catalysts of noble metal NPs, which possess great catalytic activities, inactive cores, and effective recoverable properties are extremely rare.
In organic chemistry, the production of biaryl ketones was attended with an impressive reaction. Biaryl ketones can be placed on central construction blocks prepared in a wide diversity of molecules, which include pharmaceutical drugs and natural products as well as sunscreen agents. The production of biaryl ketones is known to be one of the most important and customary applications through Friedel–Crafts acylation.17 It can also utilize stoichiometric amounts of Lewis acid, and tolerates adaptability issues with different functional groups. This can lead to the production of significant amounts of waste, so causing poor atom economy. Producing the isomers of ortho and para that has low regioselectivity, their separation and availability biaryl ketones and meta substitution are extra issues accompanied with the acylation of Friedel–Crafts. With different aims, the transition metal catalyzed distinct component carbonylative cross-coupling between aryl organometallic and aryl halides reagents, using carbon monoxide (as the C1 origin), is known to be a direct protocol.18 Carbonylative Suzuki–Miyaura cross-coupling was particularly proposed because of its broad functional group tolerance along with the fact that aryl boronic acids are nontoxic, ease of controlling air and moisture and thermal strength.
The development of supported catalysts is the central step in a lot of work because of increasing demands in terms of ecologically-viable and economic procedures. Carbonaceous materials are considered to be appropriate candidates in terms of synthesis supported catalysts.19–22 Carbonaceous materials are commonly used supports, yet they have many disadvantages, such as the presence of distinct impurities, which can act in micropore construction or poison the catalyst leading to mass-transfer restrictions. In addition, the problem of spectroscopic characteristics may result in a lack of basic comprehension. Nano-carbons are encountered as a promising option in the layout of supported catalysts to overcome these problems.23,24 Carbon nanotubes (CNTs) have been studied as catalyst supports due to their outstanding characteristics, such as high surface zone, and high thermal as well as chemical stability.25,26
In this paper, for the nanoscale composition of Ni@Pd NPs, carbon nanotubes (CNTs) were chosen as supports. Due to the richness in the level of functional groups and deficiencies, the interest in utilizing CNTs as supports is illustrated for distributing metal catalyst nanoparticles. The single electronic structure of CNTs shows a significant charge transfer, which kinetically decreases the diffusion resistance apart from their accessibility for the active part and appropriate chemical consistency in aggressive media. In addition, active stages, which forcefully interact with CNTs, can lead to structural deficiencies in CNTs along with more influenced over products and reactant transfer outside or inside CNTs.27–36 Given our consecutive interest in catalyst expansion for carbonylation reactions and nanocatalysis, Ni@Pd nanoparticles supported on CNTs were therefore used as a promising catalyst for carbonylative Suzuki–Miyaura cross-coupling (refer to Scheme 1).
 | ||
| Scheme 1 Carbonylative Suzuki–Miyaura coupling in the presence of a catalyst. | ||
Experimental
Materials and methods
High-purity chemicals were procured from Fluka and Merck. Electrothermal 9100 apparatus was utilized for the determination of uncorrected melting points in open capillaries. A VERTEC 70 spectrometer (Bruker) in transmission mode was used for the determination of FTIR spectra. Samples were pulverized and pelletized with spectroscopic-grade KBr. The determination of the size and structure of nanoparticles was done with a transmission electron microscope (TEM) (Phillips CM10) operated at 100 kV. The crystallographic structures of nanoparticles were determined using powder X-ray diffraction (Bruker D8 Advance model) with Cu Kα radiation. Thermogravimetric analysis (TGA) (NETZSCH STA449F3) was utilized under a nitrogen atmosphere with a heating rate of 10 °C min−1. NMR spectra of 1H and 13C were determined with a BRUKER DRX-300 AVANCE spectrometer and a BRUKER DRX-400 AVANCE spectrometer. The NMR spectra for both elements were recorded at 300.13 and 75.46 MHz; and 400.22 and 100.63 MHz, respectively. A Heraeus CHN–O-Rapid analyzer was used to perform elemental analyses for carbon, hydrogen and nitrogen. Thin later chromatography (TLC) was done on silica gel polygram SILG/UV 254 plates which were used in determining the product purity and monitoring the reaction. A Shimadzu GCMS-QP5050 mass spectrometer was used to record the mass spectra.General approach in the case of the preparation of Ni@Pd NPs
Ni@Pd core–shell NPs were synthesized by a high-temperature one-port solution phase production process, like the consecutive reduction of palladium(II) and nickel(II) into oleylamine (OAm), as suggested by Metin et al.37 We combined about 49.7 mg of nickel(II) acetate tetrahydrate (Ni(ac)2·4H2O, 0.2 mmol) and 53.4 mg of palladium(II) bromide (PdBr2, 0.2 mmol) with 0.3 mL of TOP and 18.0 mL of OAm at room temperature. Under a flow of mild argon (with a 5 °C min−1 rate), the reaction was heated to a temperature of 245 °C. Expansion of nucleation and related particles was indicated by a gradual change in color from yellow-green to dark brown that was specified in the production stage. The reaction mixture was kept at a temperature of 245 °C for 60 min and was later left to cool to room temperature. The NP product was obtained from the reaction mixture by utilizing 40 mL of isopropanol and using centrifugation at 8500 rpm for 8 min. The nanoparticles were further refined and later gathered by scattering them in 5 mL of hexane, utilizing an equivalent mixture of isopropanol and ethanol, and separating them for 8 min under 8500 rpm. The production gathered Ni/Pd core/shell NPs, which had Ni/Pd = 3/2. 62.2 mg of Ni(ac)2·4H2O (0.25 mmol) and also 53.4 mg of PdBr2 (0.2 mmol) prepared 49.7 mg of Ni(ac)2·4H2O (yielding 0.2 mmol) and Ni/Pd (Ni/Pd = 7/3) as well as 80 mg of PdBr2 (0.3 mmol), which led to the production of Ni/Pd (Ni/Pd = 2/3) for similar reaction states.Usual approaches for the production of Ni@Pd/CNT NPs
A constant amount of CNT (0.40 g) was ultrasonically scattered in 50 mL of water, and after that 80 mg of Ni@Pd nanoparticles was added. Since the mixture was being stirred as well as ultrasonically scattered for about 120 min, Ni@Pd/CNT was prepared by utilizing an external magnet. After that the mixture was left to dry in a vacuum.Common method for synthesis of the carbonylative Suzuki reaction of aryl iodides
Aryl boronic acid (1.2 mmol), anisole (10 mL), aryl iodide (1.0 mmol), Ni@Pd/CNT MNPs (2 mg), and K2CO3 (3 mmol) were added to a 100 mL autoclave. The autoclave was sealed, purified twice with CO, pressurized under 1.5 MPa of CO and heated under reflux for 30 min. When the reaction had been performed, the reaction compound was cooled to 25 °C and the residual CO gas was carefully vented. After that, the reactor was opened. The progress of the reaction was analyzed by taking advantage of TLC. When the reaction was done, EtOH was released to the reaction compound and the catalyst was removed using an external magnet. In this way, the solvent was removed from the solution at a decreased pressure and the final product was purified using recrystallization by n-hexane/ethyl acetate.Usual approaches for the production of the carbonylative Suzuki reaction of aryl iodides
1.0 mmol of aryl iodide, 10 mL of anisole, 3 mmol of K2CO3, 1.2 mmol of aryl boronic acid, and 2 mg of Ni@Pd/CNT MNPs were added to a 100 mL autoclave. The autoclave was sealed, purged twice using carbon monoxide, pressurized under a pressure of 1.5 MPa with a CO atmosphere and heated under reflux for 30 min. When the reaction had occurred, the product of the reaction was cooled to a temperature of 25 °C and the residual CO was carefully vented. Then, the reactor was opened. The progress of the reaction was analyzed using TLC. When the reaction had been performed, EtOH was released to the reaction compound. After that the catalyst was removed by an external magnet, after it had performed the reaction. In addition, the solvent was separated from the solution at a reduced pressure and the obtained product was purified using recrystallization by adding n-hexane/ethyl acetate.Results and discussion
Ni@Pd NPs were derived from the decomposition of PdBr2 over the surface of Ni NP cores. The Ni core was firstly produced from Ni(ac)2 as the starting material by adding seeds of Pd to the reaction admixture at a temperature of 25 °C. After that, the PdBr2 was decomposed and the shell of Pd was formed at a temperature of 245 °C. The CNT was functionalized with mercaptopropyl groups in order to produce a Ni@Pd/CNT nanocatalyst. The Ni@Pd NPs can be simply anchored on the CNT (see Scheme 2). | ||
| Scheme 2 Preparation of the Ni@Pd/CNT nanocatalyst. | ||
Fig. 1a shows the SEM picture of the unutilized Ni@Pd/CNT catalyst. An extremely tangled rope-like CNT construction is clear. The TEM picture, as shown in Fig. 1b, suggests that CNT was properly modified using the Ni@Pd catalyst. The obvious black spots suggest that the Ni@Pd catalyst was used in the support and the CNTs were open-ended and relatively short, which was likely due to acid pre-treatment that was performed in providing surface functional groups. Such groups confirmed that Ni@Pd was added to CNT and also the metal was properly dispersed. No dark spots were found in the background to Fig. 1b, suggesting that the metal particles were completely used up by the support. As can be seen in the TEM and SEM pictures, in the cases of raw CNTs and metal, full CNTs show a similar nature.
 | ||
| Fig. 1 (a) SEM image of Ni@Pd/CNT, and (b) TEM image of Ni@Pd/CNT. | ||
XRD analysis demonstrated the existence of the mixture of Ni@Pd/CNT nanocatalyst. As can be observed in Fig. 2, the broad hump that is located in the range of 2θ from 15 to 30° indicates the CNT properties. A comparison with the information from JCPDS with referral number 65-5788 clearly indicated the existence of Ni–Pd NPs for more validation. It was found that NiO and PdO did not exist. The XRD pattern of the Ni@Pd/CNT nanocatalyst, presented after the Ni@Pd nanoparticle correction on CNT, specified the existence of all the feasible characteristic peaks of Ni@Pd nanoparticles and the peak with respect to CNT, indicating the rich immobilization of Ni@Pd nanoparticles onto CNT. In addition, XPS was utilized to study the chemical elements upon the Ni@Pd/CNT MNP surface. Fig. 3 shows a full-scan XPS spectrum for the as-prepared catalyst. Peaks relating to Pd, O, Ni, and C can obviously be seen.
 | ||
| Fig. 2 XRD pattern of Ni@Pd/CNT MNPs. | ||
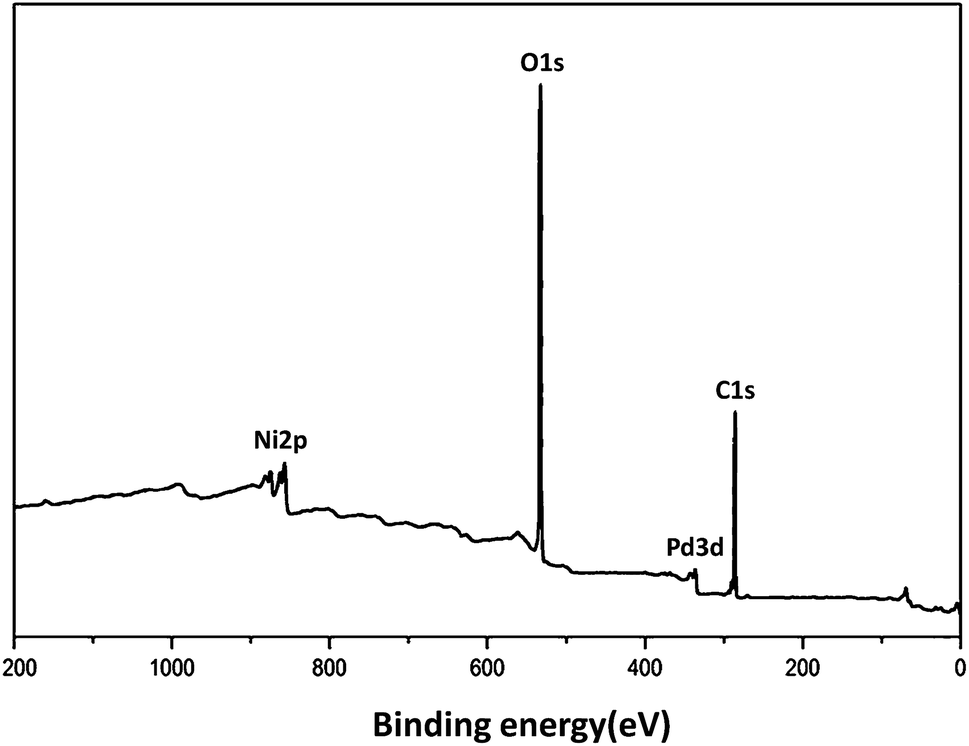 | ||
| Fig. 3 XPS spectrum of Ni@Pd/CNT MNPs. | ||
A vibrating sample magnetometer (VSM) was used for the determination of the magnetic properties of the nanoparticles with the magnetization curves of the obtained nanocomposite registered at 300 K. In Fig. 4, it is shown that no residual magnetism was detected; thus paramagnetic characteristics were exhibited by the nanocomposites. Saturation magnetization values of 47.4, and 23.6 emu g−1 were determined for Ni@Pd, and Ni@Pd/CNT MNPs, respectively. Responsivity towards an external magnetic field and the ability for quick redispersion upon the removal of the magnetic field are characteristic of paramagnetic nanocomposites with high magnetization values. Hence, the resultant nanocomposite exhibited good magnetic responsivity, suggesting potential applications for targeting and separation. The EDS pattern was used for the detection of the chemical composition of Ni@Pd/CNT MNPs. Fig. 5 shows the peaks related to Ni and Pd indicating the compositions of Ni@Pd NPs as well as C and O indicating the compositions of CNT. The copper (Cu) peak in the TEM pattern is from the Cu grid.
 | ||
| Fig. 4 Room-temperature magnetization curves of (a) Ni@Pd, and (b) Ni@Pd/CNT MNPs. | ||
 | ||
| Fig. 5 EDS spectra of Ni@Pd/CNT MNPs. | ||
In order to improve the reaction states, the carbonylative cross-coupling reaction of iodobenzene as well as phenyl boronic acid in the presence of Ni@Pd/CNT MNPs (a catalyst) was selected as the procedure. The effect of different factors like base, time and solvent (refer to Table 1) were tested for the model reaction. The effect of solvents on the carbonylative Suzuki coupling reaction was investigated (Table 1, Entries 1–15). Based on the obtained outcomes of solvents, when polar protic solvents, like water, ethanol, isopropanol or methanol were used, no quantity of the desirable product was created. Nevertheless, the product of the cross-coupling yield was relatively poor in polar aprotic solvents, including EtOAc, DMSO, DMF, CHCl3, CH2Cl2, and anisole. When the reaction was done in less polar solvents, such as toluene, appropriately high amounts of product resulting from carbonylative cross-coupling were isolated. Anisole was determined to be the most appropriate selection. Nevertheless, in polar solvents, the carbonylative cross-coupling product is established in low yield. In the present paper, it was determined that common heating under anisole was more efficient in comparison with using organic solvents (refer to Table 1, Entry 8). It was noted that bases play a dramatic role in coupling reactions; therefore the reaction was conducted with the existence of distinct organic and also inorganic bases (refer to Table 1, Entries 16–26). As an outcome, the most utilized base K2CO3 is a suitable base for generating carbonylative cross-coupling yield. Under the optimized states, the reaction advance for the shortest time essential in the attendance of 3 mg of Ni@Pd/CNT MNPs is detected using GC, that great products of carbonylative Suzuki coupling reaction may be obtained data of 30 min (refer to Table 1, Entry 26).
| Entry | Solvent | Base | Time (min) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: iodobenzene (1 mmol), phenyl boronic acid (1.2 mmol), Ni@Pd/CNT MNPs (3 mg), K2CO3 (3 mmol), solvent (10 mL), CO pressure = 3 MPa.b Isolated yields. | ||||
| 1 | EtOH | K2CO3 | 60 | — |
| 2 | H2O | K2CO3 | 60 | — |
| 3 | CH3CN | K2CO3 | 60 | 49 |
| 4 | DMF | K2CO3 | 60 | 62 |
| 5 | CH2Cl2 | K2CO3 | 60 | 35 |
| 6 | EtOAc | K2CO3 | 60 | 24 |
| 7 | THF | K2CO3 | 60 | 33 |
| 8 | Toluene | K2CO3 | 60 | 60 |
| 9 | n-Hexane | K2CO3 | 60 | — |
| 10 | CHCl3 | K2CO3 | 60 | 38 |
| 11 | DMSO | K2CO3 | 60 | 45 |
| 12 | MeOH | K2CO3 | 60 | — |
| 13 | Dioxane | K2CO3 | 60 | — |
| 14 | i-PrOH | K2CO3 | 60 | — |
| 15 | Anisole | K2CO3 | 60 | 98 |
| 16 | Solvent-free | K2CO3 | 60 | — |
| 17 | Anisole | — | 60 | — |
| 18 | Anisole | CsF | 60 | — |
| 19 | Anisole | Na2CO3 | 60 | 63 |
| 20 | Anisole | Et3N | 60 | — |
| 21 | Anisole | NaOAc | 60 | — |
| 22 | Anisole | KOH | 60 | 45 |
| 23 | Anisole | K3PO4 | 60 | 73 |
| 24 | Anisole | Cs2CO3 | 60 | 65 |
| 25 | Anisole | tBuOK | 60 | — |
| 26 | Anisole | K2CO3 | 30 | 98 |
| 27 | Anisole | K2CO3 | 15 | 62 |
We investigated the effect of temperature in terms of the production of the carbonylative Suzuki–Miyaura reaction in the presence of catalyst Ni@Pd/CNT MNPs. The most appropriate temperature for the introduced heating of the reaction for reflux led to variations in the efficiency of the reaction. It was demonstrated that the catalytic activity can be changed by varying the reaction temperature. The catalyst loading is considered to be another significant factor in the common reaction for carbonylative cross-coupling (refer to Fig. 6). As indicated in Fig. 6, a low amount of product from the carbonylative Suzuki–Miyaura reaction was obtained when a model reaction was undertaken without a catalyst. While 0.2–1.8 mg of Ni@Pd/CNT MNPs are weighted on sample reaction, moderate products of carbonylative Suzuki coupling reaction are established. The most appropriate outcome was obtained when the model reaction was done in the presence of 2 mg of Ni@Pd/CNT MNPs. An increase in the catalyst value had proved no enhancement in the reaction model. No outcome was achieved with the existence of the catalyst. The influence of pressurized CO in the presence of Ni@Pd/CNT MNPs, phenyl boronic acid and iodobenzene for half an hour is indicated in Fig. 7. The catalyst compound attained a mean > 98% conversion under a pressure of 1.5 Mpa. The carbonylative Suzuki–Miyaura reaction was achieved with 98% product and no by-products were detected using GC in any of the measurements.
 | ||
| Fig. 6 Effect of increasing the amount of Ni@Pd/CNT MNPs on the carbonylative Suzuki–Miyaura reaction. | ||
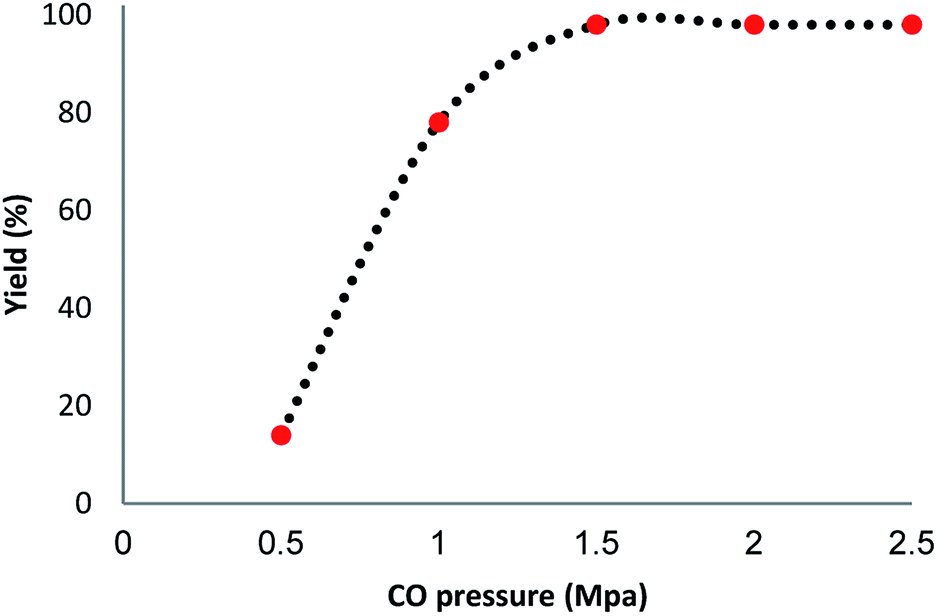 | ||
| Fig. 7 Effect of pressure on the yield of carbonylative Suzuki–Miyaura reaction. | ||
To further study the performance of the catalyst, various control measurements were done and the results achieved are shown in Table 2. In the first step, a standard reaction was performed by utilizing CNT and the result indicated that no value of the desired outcome was produced after a reaction time of 30 min (refer to Table 2, Entry 1). Furthermore, when Ni/CNT was utilized (as the catalyst), no reaction was shown (refer to Table 2, Entry 2). The Ni cannot admit the middle catalytic activity at mild reactions. In order to enhance the yield, Pd was added according to these disappointing outcomes. These results indicated that the reaction cycle was particularly catalyzed using Pd NPs. It should be noted that, in the reaction yields, there was no considerable variation when the reaction was performed with Ni@Pd/CNT, Pd/CNT or a Ni@Pd NPs catalyst (refer to Table 3, Entries 5–7). The activity of Ni/Pd was analyzed by changing the ratio of Ni to Pd. The most appropriate efficiency was achieved at a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 of Ni to Pd, as can be observed in Table 2, Entries 3–5. In addition, Ni@Pd was not simply reusable and recoverable for the next runs. CNT was utilized to prevent the dispersion of Ni@Pd nanoparticles with easy separation to solve this issue.
:2 of Ni to Pd, as can be observed in Table 2, Entries 3–5. In addition, Ni@Pd was not simply reusable and recoverable for the next runs. CNT was utilized to prevent the dispersion of Ni@Pd nanoparticles with easy separation to solve this issue.



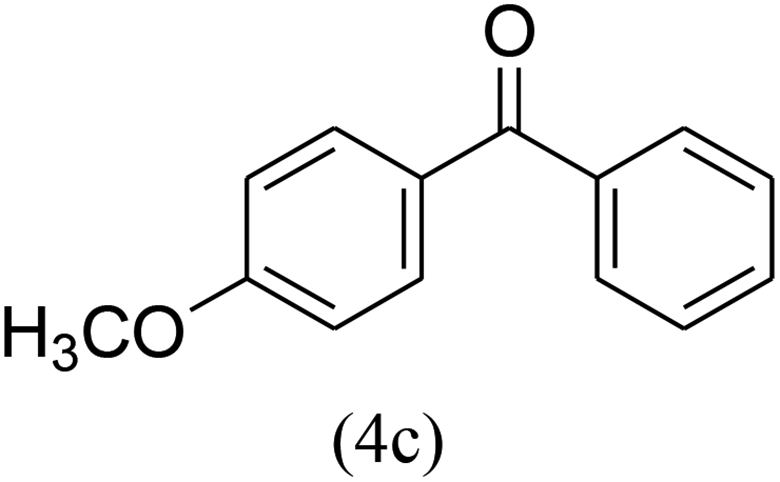



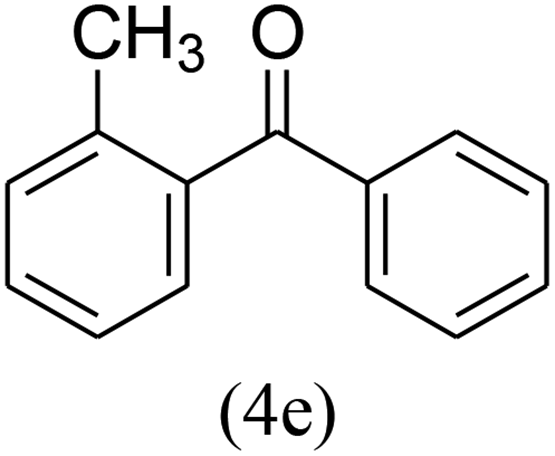



With the improved reaction states, we developed the area of the carbonylative Suzuki reaction for a diversity of distinct aryl iodides by phenyl boronic acids. Different electron-donating and electron-withdrawing groups, such as –NO2, –COCH3, –CH3, –OCH3, –NH2, –Cl and –Br upon both aryl iodide as well as aryl boronic acid smoothly tolerate the carbonylative Suzuki coupling reaction, presenting desirable outcomes with appreciable products, as determined in Table 3. Iodobenzene reacted with phenyl boronic acid, presenting a 98% product of benzophenone (refer to Table 3, Entry 1). It is important to note that aryl iodides, including electron-withdrawing groups, like –Br, –COCH3, and –Cl are determined to be more active compared to aryl iodides including electron-donating groups, like –CH3, –OCH3 and –NH2.
In this paper, the catalytic efficiency of the suggested catalyst compared with catalysts related to the carbonylative Suzuki coupling reaction was investigated (refer to Table 4). Table 4 obviously displays that the most desirable parameters are needed in the case of the carbonylative Suzuki coupling reaction, utilizing Ni@Pd/CNT MNPs, when a suitable, excellent, efficiency of the available catalyst is considered for the reaction.
| Entry | Catalyst | Experimental conditions | Yield (%) | Ref. |
|---|---|---|---|---|
| a Reaction conditions: iodobenzene (1.0 mmol), phenylboronic acid (1.0 mmol), with different temperatures, times, and pressures of CO. | ||||
| 1 | Pd/C | 100 °C, 8 h, 4 bar CO | 90 | 38 |
| 2 | P(DVB-NDIIL)-Pd | 120 °C, 12 h, 30 bar CO | 86 | 39 |
| 3 | Fe3O4@SiO2-2N-Pd | 80 °C, 8 h, 1 bar CO | 91 | 40 |
| 4 | PS-Pd-NHC | 100 °C, 10 h, 7 bar CO | 94 | 41 |
| 5 | HMMS-SH-Pd | 80 °C, 9 h, 1 bar CO | 94 | 42 |
| 6 | MCM-41-2N-Pd | 80 °C, 8 h, 1 bar CO | 91 | 43 |
| 7 | Ni@Pd/CNT | Reflux, 0.5 h, 1.5 bar CO | 98 | This work |
In the catalyst, the values of Pd achieved after and before the reaction were about 2.6% and 2.7%, as detected by ICP-MS. After accomplishing the reaction, as observed in Table 5, this indicated that most of the Pd species leaching in solution were retaken on the CNT fibers. The most considerable characteristics of a catalyst involve determining the stability as well as activity of a nanocatalyst upon being recycled. However, recycling experiments were done by taking into account the ten different periods under mild conditions. The catalyst was recovered from the reaction solution and after that it was cleaned with DI water (deionized water) and dried at a temperature of 80 °C. Then, for the next period, the carbonylative Suzuki coupling reaction was tested, and the residual concentration was analyzed singly after each period. The catalyst activity experienced a 4% decrease after accomplishing ten implementations. After ten implementations, we observed that the catalyst had undergone deactivation, as observed in Fig. 8. The result displays the reusability of this heterogeneous approach.
| Entry | Catalyst | wt% |
|---|---|---|
| 1 | Ni@Pd/CNT MNPs | 6.1 |
| 2 | Ni@Pd/CNT MNPs after ten reuses | 5.9 |
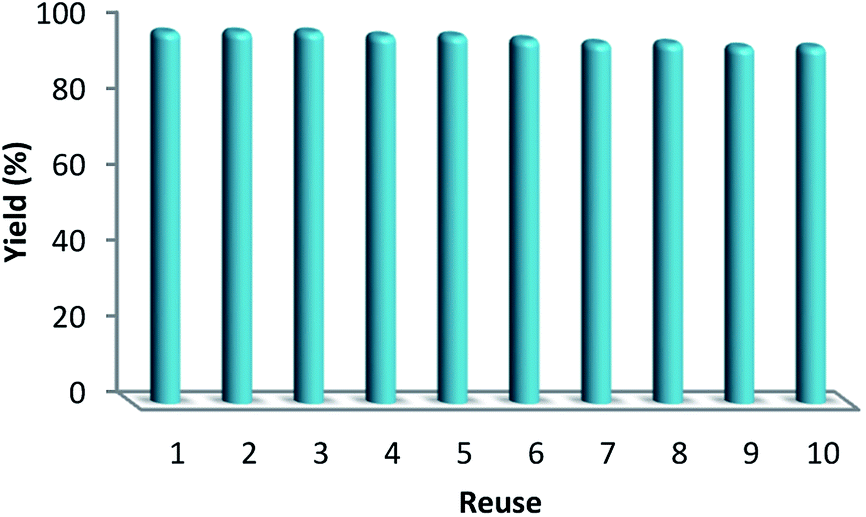 | ||
| Fig. 8 Reuse performance of the catalysts. | ||
Moreover, a complete investigation was undertaken to specify the heterogeneous nature of the catalyst. In the first step, a hot filtration experiment was performed on the carbonylative Suzuki–Miyaura reaction under mild conditions and it was found that about 62% of the catalyst was removed magnetically in situ after 15 min of removal. Moreover, the reactants were able to tolerate more reactions. The results indicated that the remainder of the free catalyst was feebly active, after removing the heterogeneous catalyst, and a conversion of 64% was achieved after 30 min of the Suzuki–Miyaura reaction. We had proved that the catalyst acted heterogeneously in the reaction and there was only slight leaching during the reaction. In the second step, to ensure the heterogeneous type of catalyst, mercury poisoning analysis was further undertaken. Mercury(0) was introduced as a metal or by synthesis and it considerably deactivated the metal catalyst over the active surface. Therefore, it can neutralize the catalyst activity. Our experiments proved that the catalyst is homogeneous. This analysis was done by the above-mentioned reaction model under optimized conditions. Around 300 moles mercury were released into the reaction compound after 15 min of reaction. The reaction area was stirred for more than 30 min and also, since the catalyst was poisoned, no more conversion was observed after 30 min. A kinetic approach to the reaction with the existence of Hg(0) is proved in Fig. 9. The negative results achieved from all the heterogeneity experiments (Hg(0) hot filtration along with poisoning) suggested that the solid catalyst was particularly heterogeneous and there was no Pd leaching obtained over the carbonylative Suzuki–Miyaura reaction.
 | ||
| Fig. 9 Reaction kinetics, Hg(0) poisoning, and hot filtration studies for the carbonylative Suzuki–Miyaura reaction. | ||
Eventually, to ensure the recovered catalyst structure was maintained for the carbonylative Suzuki coupling reaction, we investigated it after the 10th passage in the determined premium states, as observed in Fig. 10. The EDX pattern of the resynthesized catalyst determined the presence of the attendant parameters, so vouching for the catalyst's consistency in the reactions, as can be seen in Fig. 10a. As can be observed in Fig. 10b, the XRD pattern of the reproduced catalyst demonstrated that the catalyst structure remained completely intact after recycling. The XPS pattern demonstrated that the Pd and Ni elements shown in the catalyst were retained entirely after the passage of the 10th run when their oxidation condition was the same as in the fresh catalyst. As can be seen in Fig. 10c and d, no other oxidation states were specified for the catalyst. The TEM picture demonstrated that the generic gray and white dots placed over the straight chain of CNT after the 10th run are Ni@Pd (refer to Fig. 10e). Note that no morphological changes in the nanocatalyst were indicated in the FE-SEM pictures, which were captured from the recovered catalyst (refer to Fig. 10f).
 | ||
| Fig. 10 (a) EDX, (b) VSM, (c) XRD, (d) XPS spectra of Ni, (e) XPS spectra of Pd, (f) TEM, and (g) FE-SEM images of the recovered Ni@Pd/CNT MNPs after the 10th run for the carbonylative Suzuki–Miyaura reaction. | ||
Conclusions
In the present paper, a new class of functionalized CNTs was produced for selectively including Ni@Pd, that exhibited great catalytic activity for the carbonylative Suzuki coupling reaction with good products. EDS, TEM, VSM, SEM and XRD analyses suggested the functionalization of Ni@Pd on the CNT. Moreover, the catalyst was quickly retrievable and reusable. A rational plan for single-site catalysts with total usage of each Ni@Pd active zone and also great recyclability and insignificant catalyst leaching can coincide with the requirements of green chemistry. Therefore, the investigation of the Ni@Pd/CNT nanocatalyst can suggest a possible platform for the construction of other nanocatalysts with easy availability that could be highly impressive for different catalytic reactions. This method can be expanded in the future for further nanocatalysts, which have desirable characteristics, like performance and ease of reuse.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the support of the National Key R&D Program of China (2017YFE0116300); the Key Research Programs of Henan Educational Committee (19A610011); the National Natural Science Foundation of China (51878646); and the Natural Science Foundation of Henan Province (182300410102).Notes and references
- M. Garcia-Melchor, M. C. Pacheco, C. Najera, A. Lledos and G. Ujaque, ACS Catal., 2012, 2, 135–144 CrossRef CAS.
- A. L. Isfahani, I. Mohammadpoor-Baltork, V. Mirkhani, A. R. Khosropour, M. Moghadam, S. Tangestaninejad and R. Kia, Adv. Synth. Catal., 2013, 355, 957–972 CrossRef CAS.
- Y. P. Shang, X. M. Jie, J. Zhou, P. Hu, S. J. Huang and W. P. Su, Angew. Chem., Int. Ed., 2013, 52, 1299–1303 CrossRef CAS PubMed.
- H. Kamisaki, T. Nanjo, C. Tsukano and Y. Takemoto, Chem.–Eur. J., 2011, 17, 626–633 CrossRef CAS.
- H. Li and J. J. Cooper-White, Nanoscale, 2013, 5, 2915–2920 RSC.
- M. Cobo, A. Orrego and J. A. Conesa, Appl. Catal., A, 2012, 445, 83–91 CrossRef.
- W. Hu, B. Liu, Q. Wang, Y. Liu, Y. Liu, P. Jing, S. Yu, L. Liu and J. Zhang, Chem. Commun., 2013, 49, 7596–7598 RSC.
- J.-H. Park, S.-K. Kim, H. S. Kim, Y. J. Cho, J. Park, K. E. Lee, C. W. Yoon, S. W. Nam and S. O. Kang, Chem. Commun., 2013, 49, 10832–10834 RSC.
- S. Wang, W. C. Li, G. P. Hao, Y. Hao, Q. Sun, X. Q. Zhang and A. H. Lu, J. Am. Chem. Soc., 2011, 133, 15304–15307 CrossRef CAS PubMed.
- O. Metin, S. F. Ho, C. Alp, H. Can, M. N. Mankin, M. S. Gultekin, M. Chi and S. Sun, Nano Res., 2013, 6, 10–18 CrossRef CAS.
- S. M. Sadeghzadeh, R. Zhiani and S. Emrani, RSC Adv., 2017, 7, 24885–24894 RSC.
- M. Li, X. Chen, J. Guan, X. Wang, J. Wang, C. T. Williams and C. Liang, J. Mater. Chem., 2012, 22, 609–616 RSC.
- S. M. Sadeghzadeh, R. Zhiani and S. Emrani, Appl. Organomet. Chem., 2018, 32, e3941 CrossRef.
- H. Bae, V. Burungale, J. B. Park, S. W. Bang, H. Rho, S. H. Kang, S. W. Ryu and J. S. Ha, Appl. Surf. Sci., 2020, 510, 145389 CrossRef CAS.
- T. C. Lin and B. R. Huang, Sens. Actuators, B, 2012, 162, 108–113 CrossRef CAS.
- A. Chaichi, S. K. Sadrnezhaad and M. Malekjafarian, Int. J. Hydrogen Energy, 2018, 43, 1319–1336 CrossRef CAS.
- P. H. Gore, Chem. Rev., 1955, 55, 229–281 CrossRef CAS.
- J.-J. Brunet and R. Chauvin, Chem. Soc. Rev., 1995, 24, 89–95 RSC.
- V. Calvino-Casilda, A. J. López-Peinado, C. J. Durán-Valle and R. M. Martín-Aranda, Catal. Rev., 2010, 52, 325–380 CrossRef CAS.
- S. M. Sadeghzadeh, RSC Adv., 2015, 5, 68947–68952 RSC.
- S. M. Sadeghzadeh, RSC Adv., 2015, 5, 17319–17324 RSC.
- E. Auer, A. Freund, J. Pietsch and T. Tacke, Appl. Catal., A, 1998, 173, 259–271 CrossRef CAS.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS.
- A. Schaetz, M. Zeltner and W. J. Stark, ACS Catal., 2012, 2, 1267–1284 CrossRef CAS.
- D. Vairavapandian, P. Vichchulada and M. D. Lay, Anal. Chim. Acta, 2008, 626, 119–129 CrossRef CAS PubMed.
- Y. Yan, J. Miao, Z. Yang, F.-X. Xiao, H. B. Yang, B. Liu and Y. Yang, Chem. Soc. Rev., 2015, 44, 3295–3346 RSC.
- J. Zhang, C. Xu, D. Zhang, J. Zhao, S. Zheng, H. Su, F. Wei, B. Yuan and C. Fernandez, J. Electrochem. Soc., 2017, 164, B92–B96 CrossRef CAS.
- B. Singh, L. Murad, F. Laffir, C. Dickinson and E. Dempsey, Nanoscale, 2011, 3, 3334–3349 RSC.
- S. S. Munjewar, S. B. Thombre and R. K. Mallick, Renewable Sustainable Energy Rev., 2017, 67, 1087–1104 CrossRef CAS.
- A. M. Al-Enizi, A. A. Elzatahry, A. M. Abdullah, A. Vinu, H. Iwai and S. S. Al-Deyab, Appl. Surf. Sci., 2017, 401, 306–313 CrossRef CAS.
- Y. Mu, H. Liang, J. Hu, L. Jiang and L. Wan, J. Phys. Chem. B, 2005, 109, 22212–22216 CrossRef CAS PubMed.
- C.-K. Cheng, T.-K. Yeh, M.-C. Tsai, H.-Y. Chou, H.-C. Wu and C.-K. Hsieh, Surf. Coat. Technol., 2017, 320, 536–541 CrossRef CAS.
- J. Tang, D. Chen, C. Li, X. Yang, H. Liu and J. Yang, RSC Adv., 2017, 7, 3455–3460 RSC.
- G. Wu and B.-Q. Xu, J. Power Sources, 2007, 174, 148–158 CrossRef CAS.
- R. Nie, J. Wang, L. Wang, Y. Qin, P. Chen and Z. Hou, Carbon, 2012, 50, 586–596 CrossRef CAS.
- S. H. Chae, Y. Jin, T. S. Kim, D. S. Chung, H. Na, H. Nam, H. Kim, D. J. Perello, H. Y. Jeong, T. H. Ly and Y. H. Lee, ACS Nano, 2016, 10, 1309–1316 CrossRef CAS PubMed.
- O. Metin, S. F. Ho, C. Alp, H. Can, M. N. Mankin, M. S. Gultekin, M. Chi and S. Sun, Nano Res., 2013, 6, 10–18 CrossRef CAS.
- M. V. Khedkar, P. J. Tambade, Z. S. Qureshi and B. M. Bhanage, Eur. J. Org. Chem., 2010, 6981–6986 CrossRef CAS.
- N. Jiao, Z. Li, Y. Wang, J. Liu and C. Xia, RSC Adv., 2015, 5, 26913–26922 RSC.
- X. Sun, Y. Zheng, L. Sun, Q. Lin, H. Su and C. Qi, Nano-Struct. Nano-Objects, 2016, 5, 7–14 CrossRef CAS.
- M. Deshmukh, P. J. Tambade and B. M. Bhanage, Synthesis, 2011, 2, 243–250 Search PubMed.
- J. Niu, M. Liu, P. Wang, Y. Long, M. Xie, R. Li and J. Ma, New J. Chem., 2014, 38, 1471–1476 RSC.
- M. Cai, J. Peng, W. Hao and G. Ding, Green Chem., 2011, 13, 190–196 RSC.
| This journal is © The Royal Society of Chemistry 2020 |