
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal-free domino acyl substitution/Michael addition of alkenyl Grignard reagents to lactam esters: synthesis of lactam-bearing homoallylic ketones†
Abdikani Omar Farah,
Muhannad Rabah and
Timothy K. Beng *
*
Department of Chemistry, Central Washington University, Ellensburg, WA 98926, USA. E-mail: Timothy.beng@cwu.edu
First published on 11th June 2020
Abstract
A solvent-controlled protocol for the direct and transition metal-free addition of alkenyl Grignard reagents to vicinally functionalized sp3-rich morpholinones has been developed, leading to the chemo and regioselective synthesis of lactam-bearing homoallylic ketones. The addition of lithium chloride proved to be essential. In cases where a new stereocenter is generated, the doubly branched homoallylic ketones are obtained in unexpectedly high diastereoselectivities. Efforts to extend the methodology to other heterosubstituted lactams revealed some important reactivity and selectivity differences.
Introduction
Primarily due to the continuous search for new therapeutic agents for current difficult-to-cure diseases, the assembly of libraries of nitrogen- and oxygen-containing drug-like cyclic molecules with diversity has become a responsibility entrusted on synthetic organic chemists.1 However, designing selective, efficient, cost-effective and modular strategies for accessing N-, N,O- and N,S-heterocycles can be quite daunting mainly due to conformational constraints. Meanwhile, the homoallylic ketone motif is an excellent synthon for accessing a plethora of medicinally pertinent entities, including quinolines,2 isoquinolines,3 pyrroles,4 pyridines,5 dehydropyrrolidines,6 and homopiperazinones.7 A synthetic strategy that merges a functionalized lactam and a homoallylic ketone would likely enhance the potential for the discovery of new small molecules with medicinal value given that the lactam topology is prevalent in natural products, ligands, and pharmaceuticals, including antibacterial, antioxidants, antitumor and antibiotics.8 One way to reduce cost is through the implementation of domino/cascade reactions given that they are inherently step and atom-economical. We previously disclosed the synthesis of vicinally functionalized morpholinonates of type 1 (Fig. 1)9 and demonstrated their synthetic versatility through the preparation of alkenols such as 2, which smoothly underwent Cu-catalyzed dehydrogenative coupling to furnish dihydropyran-fused morpholinones of type 3.10 Following insightful work by Lubell and co-workers,11 organic chemists are beginning to embrace the notion that the direct (i.e., copper-free) addition of excess vinylmagnesium bromide to simple oxoesters can afford the expected 1,2-addition products as well as 1,4-addition products (see 4) as contaminants. Indeed, during our studies on the preparation and dehydrogenative alkoxylation of 2, we noticed that the addition of vinylmagnesium bromide to 1 using tetrahydrofuran (THF) as the solvent, yielded very small amounts of the homoallylic ketone side product (<5%).10 Seeking a transition metal- and directing group-free cascade approach to lactam-bearing homoallylic ketones from ester 1, we became interested in tuning the selectivity of the aforementioned reaction with the aim of overriding the innate tendency of 1 to form the 1,2-addition product (see 5). In order to prepare homoallylic ketone 4 in an efficient and selective manner, we recognized that several other obstacles would have to be circumvented. For example, as has been demonstrated by Fleischer and co-workers using inherently more reactive thioesters (Fig. 1C),12 the displaced methoxide nucleophile could out-compete the C-nucleophile, leading to products of type 12 (Fig. 1D). Additionally, electronically-suitable amides are known to react with excess vinylmagnesium bromide to afford β-aminoketones, following attack of the transient enone by the displaced amine (see 13).13 We have surmounted most of these barriers and herein demonstrate that 1 is amenable to transition metal- and directing group-free addition of alkenyl Grignard reagents, under solvent-controlled conditions. The best results are achieved with bulky solvents, consistent with literature reports that steric congestion tends to promote 1,4-addition of vinyl Grignard reagents to esters.11 The addition of LiCl proved to be beneficial. The homoallylic ketones (4) are formed via a cascade process whereby conjugate addition of a second equivalent of the alkenyl Grignard reagent to transient enone 11 out-competes 1,2-addition as well as 1,4-addition of the displaced methoxide nucleophile.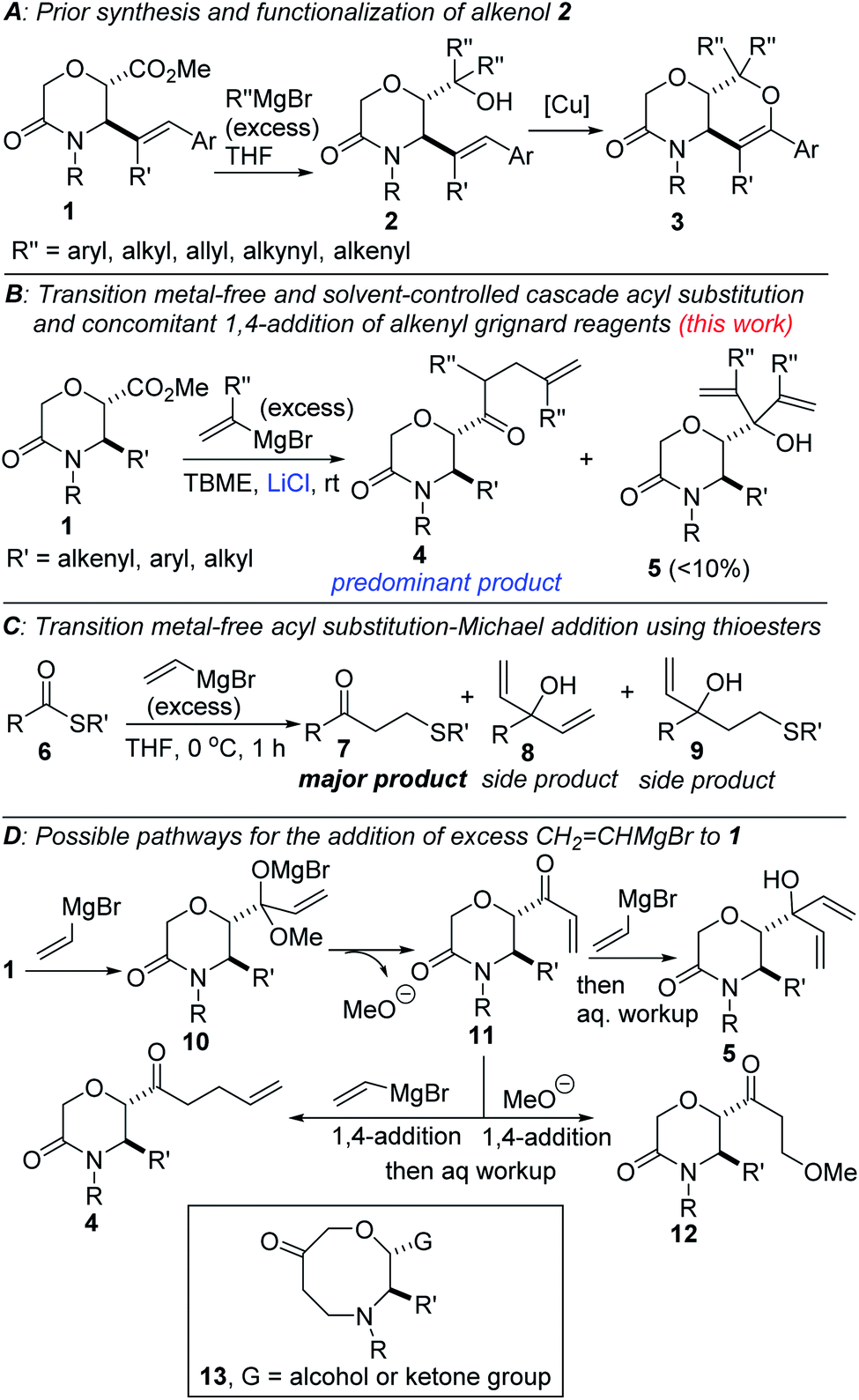 | ||
| Fig. 1 (A) Prior work on morpholinone esters, (B) proposed plan for the construction of lactam-bearing homoallylic ketones, (C) first detailed report on transition metal-free cascade addition of vinylmagnesium bromide to thioesters, (D) possible reaction pathways. | ||
Results and discussion
We initiated studies on the selective synthesis of lactam-bearing homoallylic ketones by subjecting model ester 1a to the reaction conditions described in Table 1. After cooling a solution of 1a in THF to −20 °C, excess vinylmagnesium bromide was introduced over a 1 minute period. Upon warming to room temperature and stirring for 30 min, subsequent hydrolytic workup afforded a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 ratio of bis-homoallylic ketone 4a and trienol 5a (as judged by NMR and GC-MS, Table 1, entry 1). This result served as a control experiment since we already knew that strongly coordinating and unhindered THF favors the overaddition pathway. Of note, it is necessary to precool the reaction mixture prior to the addition of the Grignard reagent since attack of the lactam moiety is competitive at elevated temperatures. When environmentally benign 2-methyltetrahydrofuran (2-MeTHF)14 was employed as the solvent, the reaction proceeded smoothly and the product ratio changed (entry 2). The nucleophilic addition proceeded recalcitrantly when less coordinating ethereal solvents such as diethyl ether (Et2O) and tert-butylmethyl ether (TBME) were employed (entries 3 & 4). Importantly, in the case of TBME, a reversal in selectivity was observed and the desirable 1,4-addition product was mostly obtained. This outcome is probably a reflection of the significant steric encumbrance of TBME since steric effects are known to influence the regio and chemoselectivity of addition of vinyl Grignard reagents to esters.11 Ultimately, we found that when the reaction is aged for 12 h at room temperature in the presence of 0.5 equivalents of LiCl,15 4a is isolated in 82% yield (entry 8). This level of efficiency is noteworthy for a cascade reaction. Another hindered solvent, cyclopentylmethyl ether (CPME), was evaluated but it did not perform as well as TBME. Such a solvent-controlled addition of Grignard reagents to carbonyl compounds has previously been observed.16 Mechanistically speaking, two equivalents of vinylmagnesium bromide suffice for this transformation, but the third equivalent aided to maximize efficiency.
:9 ratio of bis-homoallylic ketone 4a and trienol 5a (as judged by NMR and GC-MS, Table 1, entry 1). This result served as a control experiment since we already knew that strongly coordinating and unhindered THF favors the overaddition pathway. Of note, it is necessary to precool the reaction mixture prior to the addition of the Grignard reagent since attack of the lactam moiety is competitive at elevated temperatures. When environmentally benign 2-methyltetrahydrofuran (2-MeTHF)14 was employed as the solvent, the reaction proceeded smoothly and the product ratio changed (entry 2). The nucleophilic addition proceeded recalcitrantly when less coordinating ethereal solvents such as diethyl ether (Et2O) and tert-butylmethyl ether (TBME) were employed (entries 3 & 4). Importantly, in the case of TBME, a reversal in selectivity was observed and the desirable 1,4-addition product was mostly obtained. This outcome is probably a reflection of the significant steric encumbrance of TBME since steric effects are known to influence the regio and chemoselectivity of addition of vinyl Grignard reagents to esters.11 Ultimately, we found that when the reaction is aged for 12 h at room temperature in the presence of 0.5 equivalents of LiCl,15 4a is isolated in 82% yield (entry 8). This level of efficiency is noteworthy for a cascade reaction. Another hindered solvent, cyclopentylmethyl ether (CPME), was evaluated but it did not perform as well as TBME. Such a solvent-controlled addition of Grignard reagents to carbonyl compounds has previously been observed.16 Mechanistically speaking, two equivalents of vinylmagnesium bromide suffice for this transformation, but the third equivalent aided to maximize efficiency.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time (h) | Ratio 4a:5a |
% conversion |
| a Isolated yield of 4a in parentheses.b 0.5 equiv. LiCl added.c 0.2 equiv. LiCl added.d 1 equiv. LiCl added. | ||||
| 1 | THF | 0.5 | ∼10:90 |
≥99 |
| 2 | 2-MeTHF | 0.5 | ∼25:75 |
≥99 |
| 3 | Et2O | 3 | ∼40:60 |
<50 |
| 4 | TBME | 3 | ∼80:20 |
<50 |
| 5 | TBME | 12 | ∼80:20 |
80 |
| 6 | TBME | 18 | ∼80:20 |
≥99 (69)a |
| 7 | TBME | 18 | >95:5 |
≥99 (78)a,b |
| 8 | TBME | 12 | >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :5 :5 |
≥99 (82)a,b |
| 9 | TBME | 8 | >95:5 |
≥99 (75)a,b |
| 10 | TBME | 12 | >95:5 |
≥99 (70)a,c |
| 11 | TBME | 12 | >95:5 |
≥99 (82)a,d |
| 12 | CBME | 12 | >95:5 |
≥99 (71)a,b |
 |
||||

With satisfactory conditions for transition metal-free cascade acyl substitution and 1,4-addition of vinylmagnesium bromide to 1a in hand (entry 8), the scope of the transformation with respect to the steric and electronic effects of the morpholinone ester was next explored (Scheme 1). Morpholinonates bearing N-alkyl and aryl substituents were surveyed as were those harboring α-amino alkenyl, aryl, and alkyl groups. Pleasingly, several homoallylic ketones were obtained in synthetically attractive yields when vinylmagnesium bromide was added to diversely functionalized lactam esters (see 4a–f).
 | ||
| Scheme 1 Synthesis of homoallylic ketones by solvent-controlled direct 1,4-addition of vinyl Grignard reagents to morpholinonates. | ||
It is a testament to the mitigated reactivity of alkenyl Grignard reagents that they react with highly activated N-aryl-substituted lactam esters exclusively at the ester terminus to afford products such as 4e given that alkyl and allyl Grignard reagents react non-chemoselectively. Prior studies on the cascade acyl substitution-conjugate addition of alkenyl Grignard reagents to oxoesters11 and thioesters12 have mostly been centered around vinylmagnesium bromide.17 We were therefore pleased to find that the addition of excess isopropenylmagnesium bromide to these vicinally functionalized morpholinonates proceeds highly stereoselectively and furnishes predominantly a single diastereomer (see 4g–r). The relative configurations of these doubly branched homoallylic ketones are yet to be fully established at this point (mainly due to overlap of signals in the 1H NMR spectrum). As with all existing synthetic methodologies, limitations are bound to exist. In our case, we find that due to steric encumbrance, alkenyl Grignard reagents bearing an external substituent fail to participate in this cascade process, even at elevated temperatures (see 4s–u). The mass balance in these cases is mostly accounted for by the recovered ester precursors. Heteroaryl groups are well tolerated as exemplified through the synthesis of branched homoallylic ketones 4p/q.
Congruent with morpholinonates, the transition metal-free addition of vinylmagnesium bromide to sp3-rich piperidinonates9 proceeds efficiently and furnishes homoallylic ketones (Scheme 2, see 15a–c). The LiCl additive was not necessary in these cases. Indeed, these valerolactam esters display a strong preference for 1,4-addition even when THF is employed as the solvent. Under the THF conditions, dienoate 16 reacts satisfactorily to afford trienone 17. Although in TBME, vicinally functionalized morpholinonates and piperidinonates display 1,4-selectivity when treated with excess alkenyl Grignard reagents, we have found that piperazinonates18 instead undergo exclusive 1,2-addition (Scheme 3). Substrate-dependent regioselectivies of the type described herein have also been observed on trifluoromethylated α-bromoenones.19 These α-amino esters react much faster than their α-alkoxy congeners (i.e., morpholinonates). Presumably, coordination of the proximal nitrogen to magnesium brings the carbonyl group in close proximity and facilitates 1,2-addition.
 | ||
| Scheme 2 Synthesis of δ-valerolactam-bearing homoallylic ketones. | ||
 | ||
| Scheme 3 Overaddition of alkenyl Grignard reagents to ketopiperazine esters in TBME or THF. | ||
Primarily due to conformational differences, it is now fully appreciated that extending reactivity trends from one class of N-heterocycle to another can be quite daunting and even foolhardy at times. This salient point is highlighted by observations that thiomorpholinate 20 (ref. 20) undergoes a novel domino reaction featuring overaddition of vinylmagnesium bromide, branch-regioselective intramolecular hydroalkylation,21 and concomitant deconstructive elimination to arrive at highly functionalized unsaturated caprolactam 21. Three new contiguous stereocenters are generated with impeccable stereocontrol and the two pre-existing stereocenters are obliterated. A full investigation of the scope and mechanistic underpinnings of this formal one-carbon homologation process is underway.
Finally, we note in passing that when electronically diverse α-alkoxy esters and α-amino esters of types 1 and 18 were exposed to alkynyl Grignard reagents (e.g., ethynylmagnesium bromide), only the overaddition products were obtained (see the ESI† for details).
Conclusions
In summary, a solvent-controlled and transition metal-free addition of alkenyl Grignard reagents to vicinally functionalized morpholinonates and piperidinonates has been successfully implemented, leading to the selective synthesis of lactam-bearing branched homoallylic ketones. The transformation involves a tandem two-step process featuring acyl substitution of esters and concomitant conjugate addition of vinyl or isopropenylmagnesium bromide to the transient enone. The heteroatom proximal to the ester group appears to influence the selectivity of this domino process as highlighted by observations that α-amino esters (i.e., piperazinonates) deliver exclusively the overaddition products whereas α-thioalkoxy esters (i.e., thiomorpholinonates) undergo a one-pot four-step cascade process to furnish unsaturated caprolactams. The divergent nature of the transformation bodes well for future late-stage assembly of complex azaheterocycles. Efforts to expand the scope of the ring expansion of thiomorpholinonates to caprolactams are underway.Experimental
All experiments involving air and moisture sensitive reagents were carried out under an inert atmosphere of nitrogen and using freshly distilled solvents. Column chromatography was performed on silica gel (230–400 mesh). Thin-layer chromatography (TLC) was performed using Silicycle SiliaplateTM glass backed plates (250 μm thickness, 60 Å porosity, F-254 indicator) and visualized using UV (254 nm) or KMnO4 stain. Unless otherwise indicated, 1H, 13C, and DEPT-135 NMR, and NOESY spectra were acquired using CDCl3 solvent at room temperature. Chemical shifts are quoted in parts per million (ppm). HRMS-EI+ data were obtained using either electrospray ionization (ESI) or electron impact (EI) techniques. High-resolution ESI was obtained on an LTQ-FT (ion trap; analyzed using Excalibur). High resolution EI was obtained on an Autospec (magnetic sector; analyzed using MassLynx). Representative GC-MS traces are provided to substantiate the diastereomeric ratios.General procedure A: direct addition of alkenyl Grignard reagents
To the lactamoyl ester (1.0 mmol) dissolved in freshly purchased TBME (5 mL), LiCl (0.5 mmol, 0.5 equiv.) was added vinylmagnesium bromide or isopropenylmagnesium bromide (3 mmol, 3 equiv.) under nitrogen at −20 °C (over a 1 minute period). The mixture was warmed slowly to room temperature. After complete consumption of the ester (as indicated by TLC and GC-MS), the mixture was cooled to 0 °C, diluted with Et2O and quenched by slow addition of sat. aq NH4Cl. The layers were separated and the aqueous layer was extracted twice with EtOAc. The combined organic layers were dried over Na2SO4 for 30 min, filtered, and concentrated under reduced pressure to give the desired product. Purification: flash chromatography on silica eluting with hexane/EtOAc.Synthesis of ketone 4a
Prepared in 1 mmol scale using General procedure A. Purification: flash chromatography on silica eluting with hexane/EtOAc (80:20). Oily substance. Yield = 280 mg, 82%, 95:5 dr. 1H NMR (400 MHz, Chloroform-d) δ 7.35–7.24 (m, 5H), 6.47 (s, 1H), 5.80 (ddt, J = 16.9, 10.2, 6.6 Hz, 1H), 5.03 (dd, J = 19.8, 13.5 Hz, 2H), 4.47 (d, J = 7.0 Hz, 2H), 4.38–4.27 (m, 3H), 2.82 (dt, J = 18.0, 7.3 Hz, 1H), 2.64 (dt, J = 18.0, 7.3 Hz, 1H), 2.36 (p, J = 9.3, 8.2 Hz, 2H), 1.96 (s, 3H), 1.25 (d, J = 6.3 Hz, 3H), 1.20 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 208.40, 165.76, 136.73, 136.57, 129.41, 128.96, 128.77, 128.47, 128.37, 127.13, 115.96, 80.85, 65.28, 58.61, 47.66, 38.37, 27.36, 19.71, 19.58, 15.41. FTIR (KBr): 2976.0754, 2927.2335, 1721.7979, 1650.1792, 1492.0415, 1438.4625, 1362.2698, 1320.5399, 1290.1484, 1206.364, 1180.3512, 1146.7618, 1132.397, 995.8166, 918.8793, 700.1334. HRMS calc. for C21H27NO3 341.1991, found 341.1996.
Note: all other homoallylic ketones depicted in Scheme 1 were prepared as described above. Spectroscopic data can be found in the ESI.†
Synthesis of ketone 15a
Prepared in 1 mmol scale using General procedure A but without LiCl. Purification: flash chromatography on silica eluting with hexane/EtOAc (60:40). Oily substance. Yield = 331.1 mg, 85%, 95:5 dr. 1H NMR (400 MHz, Chloroform-d) δ 7.32–7.18 (m, 7H), 6.86 (d, J = 8.3 Hz, 2H), 6.31 (d, J = 15.8 Hz, 1H), 6.15 (dd, J = 15.8, 7.2 Hz, 1H), 5.82–5.75 (m, 1H), 5.05–4.96 (m, 2H), 4.82–4.67 (m, 1H), 3.75 (s, 3H), 2.97 (dt, J = 7.3, 4.7 Hz, 1H), 2.79–2.46 (m, 4H), 2.42–2.33 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 208.77, 169.48, 158.44, 136.71, 135.93, 134.80, 134.35, 133.32, 129.05, 128.71, 128.17, 128.01, 126.52, 115.80, 114.41, 63.33, 55.39, 51.56, 40.75, 30.62, 27.54, 21.49. HRMS calc. for C25H27NO3 389.1991, found 389.1996.
Note: all homoallylic ketones depicted in Scheme 2 were prepared as described above. Spectroscopic data can be found in the ESI.†
Synthesis of alcohol 19a
Prepared in 1 mmol scale using General procedure A but without LiCl. Purification: flash chromatography on silica eluting with hexane/EtOAc (50:50). Oily substance. Yield = 308.4 mg, 87%, 95:5 dr. 1H NMR (400 MHz, Chloroform-d) δ 7.42–7.28 (m, 5H), 6.42 (d, J = 16.0 Hz, 1H), 6.18–6.04 (m, 3H), 5.48 (d, J = 17.2 Hz, 1H), 5.37 (d, J = 17.2 Hz, 1H), 5.27 (d, J = 10.8 Hz, 1H), 5.18 (d, J = 10.6 Hz, 1H), 4.74 (d, J = 6.6 Hz, 1H), 3.44 (d, J = 15.1 Hz, 1H), 3.33 (d, J = 15.0 Hz, 1H), 3.02 (s, 1H), 2.83 (s, 1H), 2.49 (s, 3H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 169.14, 141.88, 139.72, 136.24, 132.01, 130.92, 130.56, 128.81, 128.03, 126.62, 126.39, 114.37, 113.78, 78.66, 72.22, 58.88, 58.12, 55.58, 47.54, 28.88. FTIR (KBr): 3384.5506, 2924.8333, 1642.2515, 1494.9545, 1448.8548, 1427.0419, 1393.4602 1361.6968, 1328.7144, 1289.7737, 1223.6425, 1198.9141, 1130.0001, 1074.1578, 1030.4745, 988.561, 966.1662, 925.5022, 741.6755, 693.4562. HRMS calc. for C22H30N2O2 354.2307, found 354.2304.
Note: all other alcohols depicted in Scheme 3 were prepared as described above. Spectroscopic data can be found in the ESI.†
Synthesis of unsaturated caprolactam 21
Prepared in 1 mmol scale using General procedure A but without LiCl. Purification: flash chromatography on silica eluting with hexane/EtOAc (80:20). Oily substance. Yield = 256.7 mg, 71%, 95:5 dr. 1H NMR (400 MHz, Chloroform-d) δ 7.35 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 8.4 Hz, 2H), 6.52 (s, 1H), 6.03 (s, 1H), 5.93 (s, 1H), 5.81–5.62 (m, 2H), 5.43–5.34 (m, 1H), 3.83–3.76 (m, 4H), 2.34 (p, J = 7.2 Hz, 1H), 1.43–1.24 (m, 10H), 1.09 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 171.77, 158.78, 140.29, 131.72, 129.87, 120.84, 116.15, 114.13, 113.75, 85.69, 55.76, 55.37, 55.32, 52.50, 47.40, 28.62, 9.54. FTIR (KBr): 3391.475, 2971.495, 2923.9815, 1644.4038, 1491.4705, 1446.8961, 1429.3142, 1391.6056, 1362.4703 1318.9377, 1292.671, 1268.871, 1223.0439, 1199.5844, 1151.3831, 1117.7388, 993.1353, 905.3314, 744.2027, 699.8784. HRMS calc. for C20H27NO3S 361.1712, found 361.1718.
Note: all other methyl ethers depicted in Scheme 4 were prepared as described above. Spectroscopic data can be found in the ESI.†
 | ||
| Scheme 4 Overaddition of vinylmagnesium bromide to 20 and concomitant intramolecular hydroalkylation-elimination. | ||
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Central Washington University for financial support through startup funds. The school of graduate studies is thanked for research fellowships to T. K. B and A. O. F. We also thank the Office of Undergraduate Studies (OUR) for a summer fellowship to M. R.Notes and references
- C.-V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809–2815 CrossRef CAS PubMed.
- C. Crifar, A. A. Dorr and W. D. Lubell, Tetrahedron Lett., 2015, 56, 3451–3453 CrossRef CAS.
- H. Tsutsui and K. Narasaka, Chem. Lett., 2001, 526–527 CrossRef CAS.
- H. Tsutsui and K. Narasaka, Chem. Lett., 1999, 45–46 CrossRef CAS.
- M. Tingoli, M. Tiecco, L. Testaferri, R. Andrenacci and R. Balducci, J. Org. Chem., 1993, 58, 6097–6102 CrossRef CAS.
- M. Yoshida, M. Kitamura and K. Narasaka, Chem. Lett., 2002, 144–145 CrossRef CAS.
- H. S. Iden and W. D. Lubell, J. Org. Chem., 2007, 72, 8980–8983 CrossRef CAS PubMed.
- (a) F. J. R. Rombouts, G. Tresadern, O. Delgado, C. Martinez-Lamenca, M. Van Gool, A. Garcia-Molina, S. A. Alonso de Diego, D. Oehlrich, H. Prokopcova, J. M. Alonso, N. Austin, H. Borghys, S. Van Brandt, M. Surkyn, M. De Cleyn, A. Vos, R. Alexander, G. Macdonald, D. Moechars, H. Gijsen and A. A. Trabanco, J. Med. Chem., 2015, 58, 8216–8235 CrossRef CAS PubMed; (b) T. J. Sindhu, D. Paul, M. Chandran, A. R. Bhat and K. Krishnakumar, World J. Pharm. Pharm. Sci., 2014, 3, 1655–1662 CAS; (c) V. A. Pal'chikov, Russ. J. Org. Chem., 2013, 49, 787–814 CrossRef; (d) J. W. Corbett, S. S. Ko, J. D. Rodgers, L. A. Gearhart, N. A. Magnus, L. T. Bacheler, S. Diamond, S. Jeffrey, R. M. Klabe, B. C. Cordova, S. Garber, K. Logue, G. L. Trainor, P. S. Anderson and S. K. Erickson-Viitanen, J. Med. Chem., 2000, 43, 2019–2030 CrossRef CAS PubMed; (e) J. Nozulak, J. M. Vigouret, A. L. Jaton, A. Hofmann, A. R. Dravid, H. P. Weber, H. O. Kalkman and M. D. Walkinshaw, J. Med. Chem., 1992, 35, 480–489 CrossRef CAS PubMed.
- H. Braunstein, S. Langevin, M. Khim, J. Adamson, K. Hovenkotter, L. Kotlarz, B. Mansker and T. K. Beng, Org. Biomol. Chem., 2016, 14, 8864–8872 RSC.
- M. Bauder, M. J. Rodriguez, A. Antonio and T. K. Beng, New J. Chem., 2018, 42, 16451–16455 RSC.
- (a) K. A. Hansford, J. E. Dettwiler and W. D. Lubell, Org. Lett., 2003, 5, 4887–4890 CrossRef CAS PubMed; (b) A. Douchez, A. Geranurimi and W. D. Lubell, Acc. Chem. Res., 2018, 51, 2574–2588 CrossRef CAS PubMed and references cited therein.
- V. Hirschbeck, M. Boldl, P. H. Gehrtz and I. Fleischer, Org. Lett., 2019, 8, 2578–2582 CrossRef PubMed.
- A. Gomtsyan, Org. Lett., 2000, 2, 11–13 CrossRef CAS PubMed.
- A. R. Alcantara and P. D. de Maria, Curr. Green Chem., 2018, 5, 88 Search PubMed.
- H. Zong, H. Huang, J. Liu, G. Bian and L. Song, J. Org. Chem., 2012, 77, 4645–4652 CrossRef CAS PubMed.
- M. Sassian and A. Tuulmets, Helv. Chim. Acta, 2003, 86, 82–90 CrossRef CAS.
- For the use of other alkenyl Grignard reagents, see (a) T. Lama, S. E. Del Valle, N. Genest and W. D. Lubell, Int. J. Pept. Res. Ther., 2007, 13, 355–366 CrossRef CAS; (b) A. I. A. Dörr and W. D. Lubell, Can. J. Chem., 2007, 85, 1006–1017 CrossRef.
- A. Moreno and T. K. Beng, New J. Chem., 2020, 44, 4257–4261 RSC.
- A. R. Romanov, D. Cahard and A. Y. Rulev, Eur. J. Org. Chem., 2019, 11, 2143–2149 CrossRef.
- D. Dar'in, O. Bakulina, M. Chizhova and M. Krasavin, Org. Lett., 2015, 17, 3930–3933 CrossRef PubMed.
- For a review on the hydroalkylation of unactivated olefins with enolates, see F. Dénès, A. Pérez-Luna and F. Chemla, Chem. Rev., 2010, 110, 2366–2447 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: 10.1039/d0ra03885g |
| This journal is © The Royal Society of Chemistry 2020 |