
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow synthesis of aryl aldehydes by Pd-catalyzed formylation of phenol-derived aryl fluorosulfonates using syngas†
Manuel Köckinger ab,
Paul Hanselmannc,
Guixian Huc,
Christopher A. Hone*ab and
C. Oliver Kappe*ab
ab,
Paul Hanselmannc,
Guixian Huc,
Christopher A. Hone*ab and
C. Oliver Kappe*ab
aCenter for Continuous Flow Synthesis and Processing (CCFLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, 8010 Graz, Austria
bInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: christopher.hone@rcpe.at; oliver.kappe@uni-graz.at
cR&D Chemistry, LPBN, Lonza AG, CH-3930 Visp, Switzerland
First published on 11th June 2020
Abstract
This communication describes the palladium-catalyzed reductive carbonylation of aryl fluorosulfonates (ArOSO2F) using syngas as an inexpensive and sustainable source of carbon monoxide and hydrogen. The conversion of phenols to aryl fluorosulfonates can be conveniently achieved by employing the inexpensive commodity chemical sulfuryl fluoride (SO2F2) and base. The developed continuous flow formylation protocol requires relatively low loadings for palladium acetate (1.25 mol%) and ligand (2.5 mol%). Good to excellent yields of aryl aldehydes were obtained within 45 min for substrates containing electron withdrawing substituents, and 2 h for substrates containing electron donating substituents. The optimal reaction conditions were identified as 120 °C temperature and 20 bar pressure in dimethyl sulfoxide (DMSO) as solvent. DMSO was crucial in suppressing Pd black formation and enhancing reaction rate and selectivity.
Aryl aldehydes are ubiquitous intermediates and building blocks in the chemical and pharmaceutical industry. As such, their convenient and cost-efficient synthesis is of high interest. One of the simplest ways of forming aldehydes is through the Pd-catalyzed formylation of Ar–X (where X = I, Br, or OTf) using syngas (CO and H2).1 The main limitations in the case of aryl iodides and bromides are the price and availability of the starting materials. In the case of triflates, trifluoromethanesulfonic anhydride (Tf2O) is the reagent most commonly employed to prepare triflates from alcohols, but it is relatively expensive, prone to hydrolysis and atom uneconomic.2 Fluorosulfonates (–OSO2F) have been proposed as an alternative leaving group to triflates because their reactivity is considered largely the same,2,3 or placed in-between bromides and chlorides.4 The fluorosulfonate leaving group is an emerging chemical motif5 that has been used in many types of chemical transformations including reduction,6 metal catalyzed cross-coupling,3,5,7 deoxyfluorination,8 amination,9 and methoxycarbonylation.10 Aryl fluorosulfonates can be conveniently and inexpensively prepared by treating readily available phenols with the commodity chemical sulfuryl fluoride (SO2F2) and base.
Previously, Beller and co-workers successfully demonstrated the Pd-catalyzed formylation of aryl triflates with syngas as a sustainable and cost effective reagent under batch conditions (Scheme 1a).11 We were interested in the synthesis of aryl aldehydes from aryl fluorosulfonates, which could be derived from their corresponding phenols. Our group previously reported the continuous flow Pd-catalyzed formylation of (hetero)aryl bromides within a continuous flow system.12 Carbonylation reactions benefit from the highly efficient mixing, enhanced mass and heat transfer characteristics, precise residence time, accessibility to high pressure and temperature regimes, and the high operational safety of continuous flow reactors.13–16 Herein, we describe the development of a continuous flow protocol for the synthesis of aryl aldehydes from their corresponding aryl fluorosulfonates (Scheme 1b).
 | ||
| Scheme 1 Pd-catalyzed reductive carbonylation using syngas for (a) triflates (Beller)11 and (b) fluorosulfonates (this work) as leaving groups. | ||
We commenced our investigation by evaluating the catalytic system palladium(II) acetate (Pd(OAc)2) and 1,3-bis(diphenylphosphino)propane (dppp), which was utilized by Beller and co-workers for the batch formylation of aryl triflates.11,17 The reaction was optimized within a continuous flow reactor for 4-methoxyphenyl sulfurofluoridate (1a) as model substrate. We used a Uniqsis FlowSyn flow system consisting of two HPLC pumps and a heated reactor coil (32 mL). Two sample loops were used to deliver the substrate solution (2 mL) and catalyst feed solution (3 mL). The gases, H2 and CO, were introduced into a four-way mixer by using two mass flow controllers (MFCs). The flow system was pressurized to 10 bar by using a back pressure regulator (BPR). Substrate 1a was mixed with triethylamine (Et3N) as base and diphenyl ether (Ph2O) as internal standard for preparation of the substrate feed. Pd(OAc)2 and dppp were used as the catalyst feed. Initially, we evaluated toluene (PhMe), tetrahydrofuran (THF), acetonitrile (MeCN) and dimethylformamide (DMF) as solvents for the carbonylation reaction (Table 1).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp. [°C] | Conv. 1ab [%] | Sel.b [%] | Yieldb 2a [%] |
a General conditions: feed 1: 0.2 M 4-methoxy sulfurofluoridate (1a), 1.0 equiv. Et3N and 0.15 equiv. Ph2O in solvent; feed 2: 5 mol% Pd(OAc)2 and 10 mol% dppp in solvent. Feed 1/feed 2/H2/CO = 0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1:5:5 mL min−1 resulting in a 27 min residence time (tres). 10 bar system pressure.b Analyzed by GC-FID.c 13.5 min tres.d 1 equiv. of pyridine as base.e Flow rates for feed 1/feed 2/H2/CO = 0.3:0.3:1.87:1.87 mL min−1, tres 35 min. :0.1:5:5 mL min−1 resulting in a 27 min residence time (tres). 10 bar system pressure.b Analyzed by GC-FID.c 13.5 min tres.d 1 equiv. of pyridine as base.e Flow rates for feed 1/feed 2/H2/CO = 0.3:0.3:1.87:1.87 mL min−1, tres 35 min. |
|||||
| 1 | PhMe | 100 | 7.7 | 0 | 0 |
| 2 | PhMe | 120 | 17.1 | 2.2 | 0.4 |
| 3 | THF | 100 | 9.6 | 6.9 | 0.7 |
| 4 | THF | 120 | 17.0 | 4.9 | 0.8 |
| 5 | MeCN | 100 | 20.7 | 3.0 | 5.9 |
| 6 | MeCN | 120 | 42.7 | 5.9 | 3.4 |
| 7 | DMF | 100 | 67.1 | 19.1 | 12.8 |
| 8 | DMF | 120 | 79.1 | 22.1 | 17.5 |
| 9 | DMFc | 120 | 50.3 | 22.6 | 11.4 |
| 10 | DMFd | 120 | 36.7 | 59.0 | 21.6 |
| 11 | DMFd,e | 120 | 32.8 | 50.0 | 16.4 |

Higher conversion was typically observed when using 120 °C instead of 100 °C. Using PhMe and THF as solvent gave only minimal conversion and trace amount of product (Table 1, entries 1–4). MeCN as solvent gave higher conversions but still very low selectivity towards product (Table 1, entry 5 & 6). Using DMF gave the best preliminary results (entry 7–9). By using pyridine as base, selectivity could be improved, from approximately 20% to 60%, albeit with a drop in conversion (entry 10). We selected to optimize conditions using pyridine since higher selectivity is more desirable. The throughput of the reaction could be almost tripled by using equimolar amounts of gas (1.4 equiv.) and higher liquid flow rates without high loss in yield (entry 11).
During the initial trials with DMF as solvent, we noticed that the obtained results were irreproducible without pre-washing the reactor coil with 20% aqueous nitric acid (HNO3) at 60 °C in-between experimental runs. Subsequent reaction runs without a wash run in-between resulted in a drop of 4-methoxybenzaldehyde (2a) yield and ultimately resulted in a black output solution (Fig. 1). The drop in yield and black output solution indicates a well-known phenomenon of Pd0 – agglomerating and forming clusters.18 These clusters then irreversibly precipitate as Pd black particles. These Pd black particles coat the reactor wall and can catalyze further Pd black formation.12,19 In order to tackle this issue, we looked for an additive or a method to prevent or slow the rate of Pd black agglomeration.
 | ||
| Fig. 1 Drop in 4-methoxybenzaldehyde (2a) GC yield after repeated reaction runs without wash runs in-between. Conditions used are provided in Table 1, entry 11. | ||
In 2005, Zierkiewicz and Privalov studied the Pd(OAc)2/dimethyl sulfoxide (DMSO) system and identified that DMSO can coordinate to free Pd0, which can help the re-oxidation of Pd0 to PdII.20 When applied to our reaction system using 4-chloro sulfurofluoridate (1b) as substrate, the use of DMSO as co-solvent alleviated the issues associated with Pd black formation (Fig. 2). Furthermore, we could drastically increase the yield of desired product 2b, due to reduced catalyst decomposition. The only side product in all cases was the hydrogenated product 3b, with the loss of the fluorosulfonate group (-OSO2F). Increased pressure (20 bar) and base (1.5 equiv.) resulted in higher selectivity to the aldehyde product 2b (Fig. S1†). The fraction of DMSO solvent could be increased to no more than 80% owing to catalyst solubility.
 | ||
| Fig. 2 Influence of DMSO on conversion and yield for Pd-catalyzed formylation of 4-chloro sulfurofluoridate (1b) with syngas measured by GC-FID (Ph2O as internal standard). For 0–80 vol% DMSO, 0.2 M 4-chloro sulfurofluoridate (1b), 1.0 equiv. pyridine and 0.15 equiv. Ph2O in DMF/DMSO; feed 2: 5 mol% Pd(OAc)2 and 10 mol% dppp in DMF/DMSO. 120 °C temperature and 10 bar system pressure. Flow rates for feed 1/feed 2/H2/CO = 0.3:0.3:1.87:1.87 mL min−1, corresponding to tres 35 min. 100 vol% DMSO experiment was performed using 1.25 mol% Pd(OAc)2 and 2.5 mol% dppp. | ||
The reaction conditions in terms of residence time and catalyst loading were then re-optimized for using DMSO as solvent. The catalyst loading was lowered to 1.25 mol% Pd(OAc)2 and 2.5 mol% dppp to ensure full dissolution of the catalyst system. A further decrease in loading did not provide satisfactory results, with a drop in yield observed (Fig. S2†). Under these conditions 40 min of residence time was sufficient to achieve a high yield of 4-chlorobenzaldehyde (2b) (Fig. S3†). With these new conditions (Scheme 2a), a long run was successfully operated which was stable for 4 hours with no apparent drop in yield, thus demonstrating no or minimal catalyst decomposition (Scheme 2b).
 | ||
| Scheme 2 (a) Continuous flow setup for the Pd-catalyzed formylation of 4-chloro sulfurofluoridate (1b) long run. (b) 4-Chlorobenzaldehyde (2b) GC yield at 30 min intervals over 4 h operation time. 0.15 equiv. Ph2O was used as an internal standard. | ||
The applicability of the optimized conditions, shown in Scheme 2a, was demonstrated on a range of substrates. In most cases, substrates bearing an electron withdrawing group (EWG) in the para position underwent full conversion and the corresponding aryl aldehyde was formed in moderate to excellent yields (Fig. 3). The hydrogenated product was the sole side product observed. However, the reaction of substrates 1e and 1f resulted in the formation of a mixture of double formylation and hydrogenated products. The formylation of 4-nitro sulfurofluoridate (1 m) was problematic because of deactivation of the catalyst.
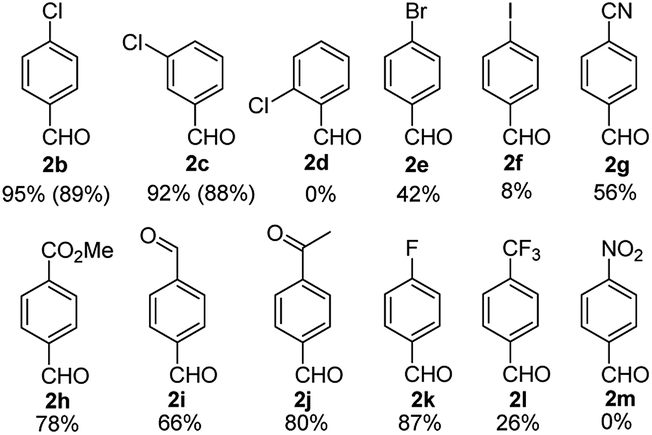 | ||
| Fig. 3 Substrate scope for aryl fluorosulfonates containing electron withdrawing groups. Yields were determined by GC-FID using Ph2O as internal standard. Yields in parentheses were isolated yield. Conditions are the same as given in Scheme 2a. | ||
Less reactive substrates, 1k and 1l, were only fully converted when modified reaction conditions were applied. In these cases, the liquid flow rates were lowered, which prolonged the residence time to approximately 2 h (Fig. 4a). Moreover, the H2 flow rate was modified to deliver 4.2 equiv. The same modified conditions were also used for substrates containing electron donating groups (EDG). The formation of the hydrogenated side product was not a significant problem for substrates containing EDG substituents with the aldehyde products obtained in good to excellent yields (Fig. 4b). Gratifyingly, 6-methoxy-2-naphthalaldehyde (2p) could be isolated in 82% yield after purification by column chromatography. Substrate 2b is a potential precursor to naproxen, an important non-steroidal anti-inflammatory active pharmaceutical ingredient.
 | ||
| Fig. 4 (a) Unreactive substrates containing electron withdrawing groups; (b) substrates containing electron donating groups. For 43 min conditions, see Scheme 2a. Conditions: feed 1: 0.2 M fluorosulfonate, 1.5 equiv. pyridine and 0.15 equiv. Ph2O in DMSO; feed 2: 1.25 mol% Pd(OAc)2 and 2.5 mol% dppp in DMSO. 120 °C temperature and 10 bar system pressure. Feed 1/feed 2/H2/CO = 0.08:0.08:1.5:0.5 mL min−1, corresponding to a tres = 123 min. | ||
Substrates bearing a substituent in ortho position to the fluorosulfonate group afforded the corresponding aldehyde either in low yield, for 1r and 1u, or no yield, for 1d and 1o. The drop in yield could be caused by steric hindrance from the ortho-substituent. The steric block on the catalytic center will favor the addition of the much smaller hydrogen molecule instead of a larger CO molecule, resulting in the hydrogenated product.
In conclusion, we have described a continuous flow method for the Pd-catalyzed synthesis of valuable aryl aldehyde building blocks from aryl fluorosulfonates and syngas as an inexpensive, atom-economic, and environmentally friendly source of CO and H2. The continuous flow approach enabled the precise addition of gas by using mass flow controllers. Meta and para substituted aryl fluorosulfonates could be converted to their corresponding aldehydes in good to excellent yields. Catalyst decomposition was successfully avoided by using DMSO as solvent. DMSO coordinates to Pd0 and facilitates the re-oxidation to PdII. Additionally, reaction rates and selectivity could be enhanced by the use of DMSO. Starting materials for the reductive carbonylation could be conveniently derived in excellent yields from readily-available phenols and the commodity chemical sulfuryl fluoride. The developed process is especially appealing for the chemical industry, where reagent cost and availability are important factors in process feasibility.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The CCFLOW project (Austrian Research Promotion Agency FFG No. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT), the Austrian Federal Ministry of Digital and Economic Affairs (BMDW), and the State of Styria (Styrian Funding Agency SFG).Notes and references
- (a) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed; (b) M. Beller and X.-F. Wu, in Transition Metal Catalyzed Carbonylation Reactions, Reductive Carbonylations, Springer, Berlin, Heidelberg, 2013, ch. 3 CrossRef; (c) C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS.
- L. Revathi, L. Ravindar, J. Leng, K. P. Rakesh and H.-L. Qin, Asian J. Org. Chem., 2018, 7, 662–682 CrossRef CAS.
- C. E. Fuller, J. Org. Chem., 1991, 56, 3493–3496 CrossRef.
- E. Zhang, J. Tang, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Chem.–Eur. J., 2016, 22, 5692–5697 CrossRef CAS PubMed.
- (a) J. Dong, L. Krasnova, M. G. Finn and K. Barry Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed; (b) J. Dong, K. B. Sharpless, L. Kwisnek, J. S. Oakdale and V. V. Fokin, Angew. Chem., Int. Ed., 2014, 53, 9466–9470 CrossRef CAS PubMed.
- X. Y. Wang, J. Leng, S. M. Wang, A. M. Asiri, H. M. Marwani and H. L. Qin, Tetrahedron Lett., 2017, 58, 2340–2343 CrossRef CAS.
- (a) Y. Gan, G. Wang, X. Xie and Y. Liu, J. Org. Chem., 2018, 83, 14036–14048 CrossRef CAS PubMed; (b) V. Bieliū Nas and W. M. De Borggraeve, J. Org. Chem., 2019, 84, 15706–15717 CrossRef PubMed.
- (a) S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. J. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 1452–1455 CrossRef CAS PubMed; (b) S. D. Schimler, R. D. J. Froese, D. C. Bland and M. S. Sanford, J. Org. Chem., 2018, 83, 11178–11190 CrossRef CAS PubMed.
- (a) T. Lim, S. Byun and B. M. Kim, Asian J. Org. Chem., 2017, 6, 1222–1225 CrossRef CAS; (b) P. S. Hanley, T. P. Clark, A. L. Krasovskiy, M. S. Ober, J. P. O'Brien and T. S. Staton, ACS Catal., 2016, 6, 3515–3519 CrossRef CAS.
- M. A. McGuire, E. Sorenson, F. W. Owings, T. M. Resnick, M. Fox and N. H. Baine, J. Org. Chem., 1994, 59, 6683–6686 CrossRef CAS.
- A. Brennführer, H. Neumann and M. Beller, Synlett, 2007, 2537–2540 Search PubMed.
- C. A. Hone, P. Lopatka, R. Munday, A. O'Kearney-McMullan and C. O. Kappe, ChemSusChem, 2019, 12, 326–337 CrossRef CAS PubMed.
- (a) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (b) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (c) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC; (d) C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 327–360 CrossRef CAS.
- For reviews on the use of CO and syngas within continuous flow, see: (a) T. Fukuyama, T. Totoki and I. Ryu, Green Chem., 2014, 16, 2042–2050 RSC; (b) M. Ramezami, M. A. Kashfipour and A. Abolhasani, J. Flow Chem., 2020, 10, 93–101 CrossRef.
- For examples of carbonylation reactions with CO gas performed within continuous flow, see: (a) M. T. Rahman, T. Fukuyama, N. Kamata, M. Sato and I. Ryu, Chem. Commun., 2006, 2236–2238 RSC; (b) E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734–1737 CrossRef CAS PubMed; (c) X. Gong, P. W. Miller, A. D. Gee, N. J. Long, A. J. Mello and R. Vilar, Chem.–Eur. J., 2012, 18, 2768–2772 CrossRef CAS PubMed; (d) P. W. Miller, N. J. Long, A. J. de Mello, R. Vilar, J. Passchier and A. Gee, Chem. Commun., 2006, 546–548 RSC; (e) P. W. Miller, N. J. Long, A. J. de Mello, R. Vilar, H. Audrain, D. Bender, J. Passchier and A. Gee, Angew. Chem., Int. Ed., 2007, 46, 2875–2878 CrossRef CAS PubMed; (f) P. W. Miller, L. E. Jennings, A. J. de Mello, A. D. Gee, N. J. Long and R. Vilar, Adv. Synth. Catal., 2009, 351, 3260–3268 CrossRef CAS; (g) S. Kasinathan, S. L. Bourne, P. Tolstoy, P. Koos, M. Obrien, R. W. Bates, I. R. Baxendale and S. V. Ley, Synlett, 2011, 2648–2651 CAS; (h) U. Gross, P. Koos, M. O'Brien, A. Polyzos and S. V. Ley, Eur. J. Org. Chem., 2014, 6418–6430 CrossRef CAS; (i) Y. Chen, C. A. Hone, B. Gutmann and C. O. Kappe, Org. Process Res. Dev., 2017, 21, 1080–1087 CrossRef CAS; (j) A. Mata, C. A. Hone, B. Gutmann, L. Moens and C. O. Kappe, ChemCatChem, 2019, 11, 997–1001 CrossRef CAS PubMed.
- For utility of flow reactors in terms of safety, see: (a) N. Kockmann, P. Thenée, C. Fleischer-Trebes, G. Laudadio and T. Noël, React. Chem. Eng., 2017, 2, 258–280 RSC; (b) C. A. Hone, D. M. Roberge and C. O. Kappe, ChemSusChem, 2017, 10, 32–41 CrossRef CAS PubMed.
- Other ligands were screened but all displayed inferior performance to dppp (see ESI,† section 5.5).
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS PubMed.
- C. A. Hone, A. O'Kearney-McMullan, R. Munday and C. O. Kappe, ChemCatChem, 2017, 9, 3298–3302 CrossRef CAS.
- W. Zierkiewicz and T. Privalov, Organometallics, 2005, 24, 6019–6028 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, further optimization data and characterization of all compounds. See DOI: 10.1039/d0ra04629a |
| This journal is © The Royal Society of Chemistry 2020 |