
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafine Pd nanoparticles loaded benzothiazole-linked covalent organic framework for efficient photocatalytic C–C cross-coupling reactions†
Yongliang Yangab,
Hongyun Niu a,
Weijia Zhaoab,
Lin Xua,
Hui Zhanga and
Yaqi Cai*acd
a,
Weijia Zhaoab,
Lin Xua,
Hui Zhanga and
Yaqi Cai*acd
aState Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing, 100085, China. E-mail: caiyaqi@rcees.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cInstitute of Environment and Health, Jianghan University, Wuhan 430056, China
dInstitute of Environment and Health, Hangzhou Institute For Advanced Study, UCAS, China
First published on 10th August 2020
Abstract
We proposed a strategy that a benzothiazole-linked covalent organic framework (TTT-COF) was used as a substrate to prepare metal composite photocatalyst Pd NPs@TTT-COF. Firstly, benzothiazole linked TTT-COF exhibited superior chemical stability and photoresponse. Moreover, a finer particle size (2.01 nm) and more uniform distribution of Pd NPs were observed in Pd NPs@TTT-COF owing to the binding interaction between Pd NPs and S in benzothiazole groups. Pd NPs@TTT-COF exhibited superior efficiency and reusability in photocatalytic C–C cross-coupling reactions. Mechanism study suggested that photogenerated electrons and holes on TTT-COF played important roles in these reactions.
Introduction
Covalent Organic Frameworks (COFs) are a class of conjugated porous organic materials constructed with organic building units via strong covalent bonds.1–3 To date, most research on the design of COFs has been focused on regulating the chemical structure of the organic modules. It's worth noting that the linkage of COFs is also crucial to the properties of COFs. Dynamic covalent linkages between building blocks are necessary for the construction of crystalline and ordered COFs architectures, but are not conducive to the robustness of COFs,2 which is a major impediment to the applications. Recently, the exploration of novel linkages has become the focus in the development of COFs, a series of novel covalent linkages such as olefin linkage,4 benzimidazole5/benzoxazole6/benzothiazole7 linkages, amide linkage,8 dioxin linkage9 have been reported, which could not only enhance the chemical stability compared to the COFs based on boronate ester linkage and imine linkage, adopted by a majority of known COFs, but also exhibited more fascinating and unique properties.Metal nanopaticles (NPs) are widely used as heterogeneous catalysts in chemical synthesis due to their high surface area and ultrafine size distribution. However, the ultrafine metal clusters tend to gradually accumulate on account of their high surface energy and result in the deactivation of the catalysts.10 Therefore, stable and uniform supporting substrates are necessary for metal NPs catalysts, and COFs are suitable for being adopted as a kind of substrates. On the one hand, on the basis of regular and tunable pore structure as well as high surface area of COFs, ultrafine metal clusters can be embedded on the pores of COFs with confined nano sizes uniformly. On the other hand, heteroatoms can be artificially introduced into the COFs frameworks owing to the designability of COFs building blocks, thus metal clusters can be anchored in the pores via the binding interactions between metal atoms and heteroatoms. Zhang's group has reported some metal NPs-COFs hybrid materials as stable and efficient heterogeneous catalysts,11,12 whose COFs substrates were synthesized from the precursors containing heteroatoms (S, P).
Here in, we propose an ingenious strategy to load Pd NPs on photoactive benzothiazole-linked TTT-COF as a Mott–Schottky heterojunction. Heteroatoms were introduced into the COFs linkage bonds through a post-synthesis modification scheme. The as-designed hybrid materials Pd NPs@TTT-COF exhibited remarkable photocatalytic activities as well as high stability and recyclability in a series of C–C cross-coupling reactions (Suzuki–Miyaura, Stille, Heck and Sonogashira) (Scheme 1).
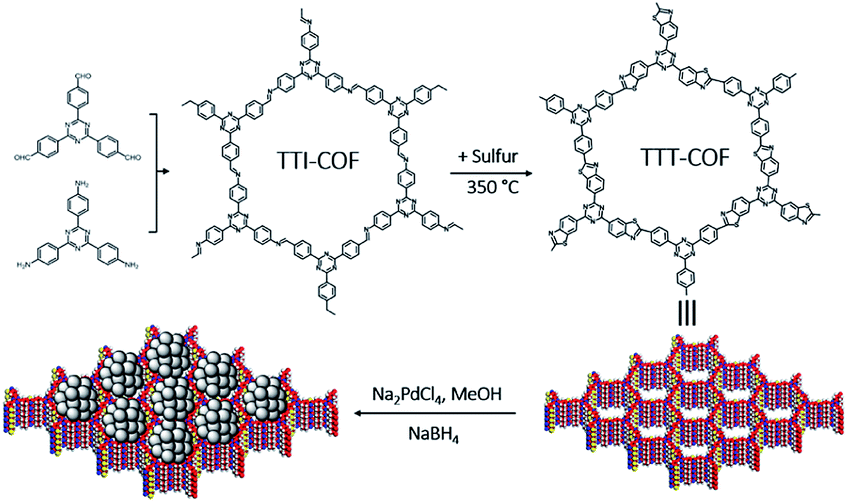 | ||
| Scheme 1 Graphical representation of the synthesis of Pd NPs@TTT-COF. | ||
Results and discussion
Imine-linked TTI-COF was converted to benzothiazole-linked TTT-COF by a post-synthetic sulfuration strategy,7 Pd NPs were introduced to the TTT-COF substrates subsequently via an impregnation-reduction method to obtain Pd NPs@TTT-COF. The content of Pd in the composite material is 6.01 wt% determined by inductively coupled plasma mass spectrometry (ICP-MS).As shown in Fig. 1a and b, the powder X-ray diffraction (PXRD) patterns exhibited six distinct diffraction peaks at 4.08°, 6.88°, 8.16°, 10.62°, 14.54° and 25.64° for TTI-COF and 4.16°,7.14°, 8.32°, 10.96°, 14.44° and 25.82° for TTT-COF, corresponding to the crystal planes of (100), (110), (200), (210), (310) and (001). The structure models of TTI-COF and TTT-COF were simulated by Material Studio (v. 7.0), and the experimental patterns of both showed good agreement with the each simulated PXRD pattern of eclipsed AA stacking models. (Fig. S1†) After Pawley refinement of experimental PXRD patterns, a unit cell with parameters a = b = 25.54 Å, c = 3.51 Å, α = β = 90°, γ = 120° and agreement factors Rwp = 6.42%, Rp = 4.67% for TTI-COF and a = b = 24.88 Å, c = 3.51 Å, α = β = 90°, γ = 120° with Rwp = 3.87% and Rp = 2.68% for TTT-COF was obtained. An ordered sliding of the PXRD patterns was observed after sulfuration (Fig. 1c), which could be explained by the decrease of cell parameters (25.54 Å vs. 24.88 Å) from TTI-COF to TTT-COF according to Bragg's law. An almost undifferentiated PXRD pattern for Pd NPs@TTT-COF confirmed the retention of the crystal structure of TTT-COF after reduction by NaBH4 and loading of Pd NPs (Fig. S2†).
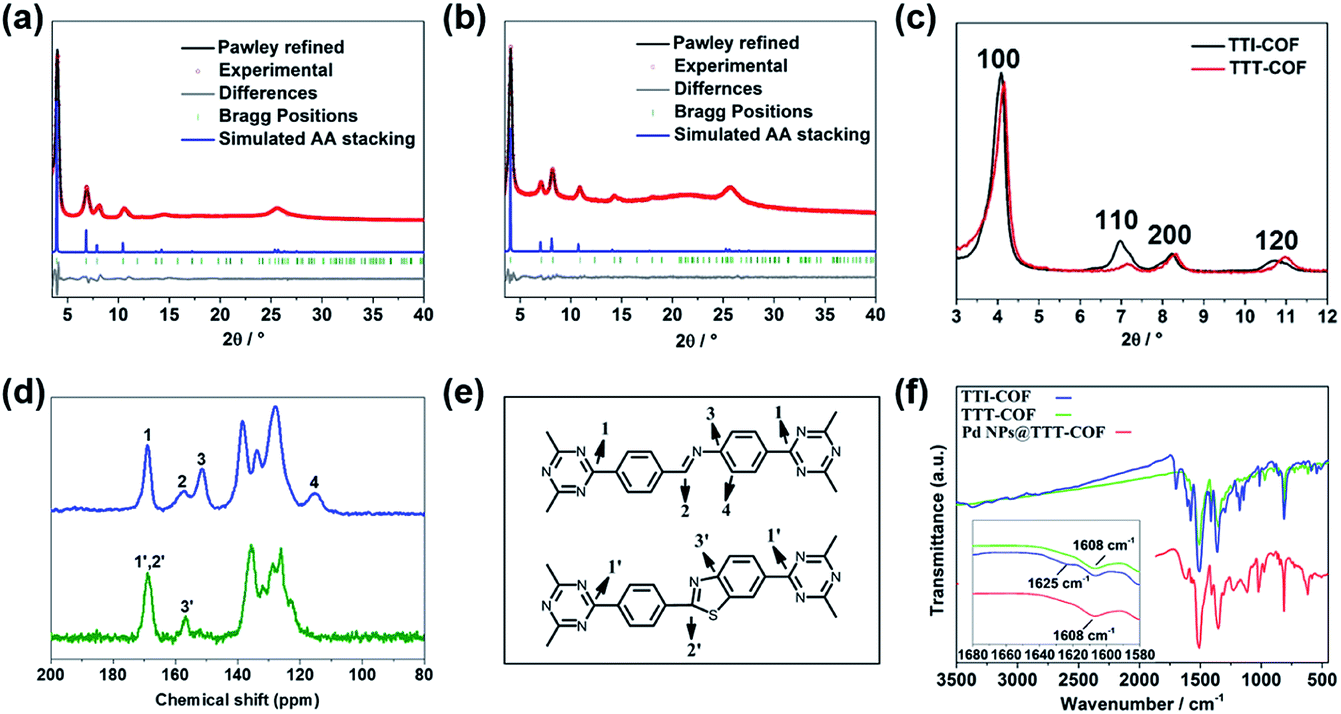 | ||
| Fig. 1 Comparison of the experimental PXRD patterns of TTI-COF (a) and TTT-COF (b) with the Pawley refined pattern (black), simulated pattern for AA stacking (blue) and difference plot (green). Comparison of PXRD (c) and 13C NMR (d) patterns between TTI-COF and TTT-COF. (e) Assignment of the 13C signals to the respective 13C nuclei in the structures. (f) Comparison of IR patterns between TTI-COF, TTT-COF and Pd NPs@TTT-COF. | ||
The chemical transformation of TTI-COF to TTT-COF was verified by solid-state 13C cross-polarization magic angle spinning nuclear magnetic resonance (CP-MAS NMR) spectroscopy and Fourier transform infrared (FT-IR) spectroscopy. As shown in Fig. 1d, the imine carbon 2 signal at 158 ppm was shifted to 169 ppm corresponding to thiazole carbon 2′ signal and merged into one peak with triazine carbon 1′ signal. Carbon 3 signal at 151 ppm was shifted to 3′ signal at 158 ppm and carbon 4 signal at 115 ppm disappeared due to integration of 4′ signal into the benzene rings range of 120–140 ppm. All these variations confirmed the formation of thiazole structures. A weak 151 ppm signal indicated trace amounts of unreacted imine structures. In the FT-IR spectra (Fig. 1f), the disappearance of the characteristic vibration of imine HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N at 1625 cm−1 in TTI-COF and the presence of CN vibration at 1608 cm−1 corresponding to thiazole units in TTT-COF confirmed successful conversion of TTI-COF to TTT-COF.13 All characteristic peaks in TTT-COF were preserved in Pd NPs@TTT-COF, which manifested the retention of chemical structure after treatment.
N at 1625 cm−1 in TTI-COF and the presence of CN vibration at 1608 cm−1 corresponding to thiazole units in TTT-COF confirmed successful conversion of TTI-COF to TTT-COF.13 All characteristic peaks in TTT-COF were preserved in Pd NPs@TTT-COF, which manifested the retention of chemical structure after treatment.
The thermogravimetric analysis (TGA) curves (Fig. S3†) showed that benzothiazole-linked TTT-COF exhibited better thermal stability in comparison to imine-linked TTI-COF. Additionally, the chemical stability was significantly enhanced by the transformation of linkage. Both TTI-COF and TTT-COF can resist neutral and strong acidic (10 M HCl) conditions, however, structural collapses of TTI-COF were observed under strongly alkaline (10 M NaOH) and strongly reductive (NaBH4) conditions whereas TTT-COF exhibited high tolerance to such extreme conditions (Fig. 2a and b). Considering that the reduction process via NaBH4 was essential to prepare Pd NPs@TTT-COF and alkaline conditions were generally used in cross-coupling reactions, it is of great significance to improve the chemical stability of the substrates by locking the reversible imine linkage.
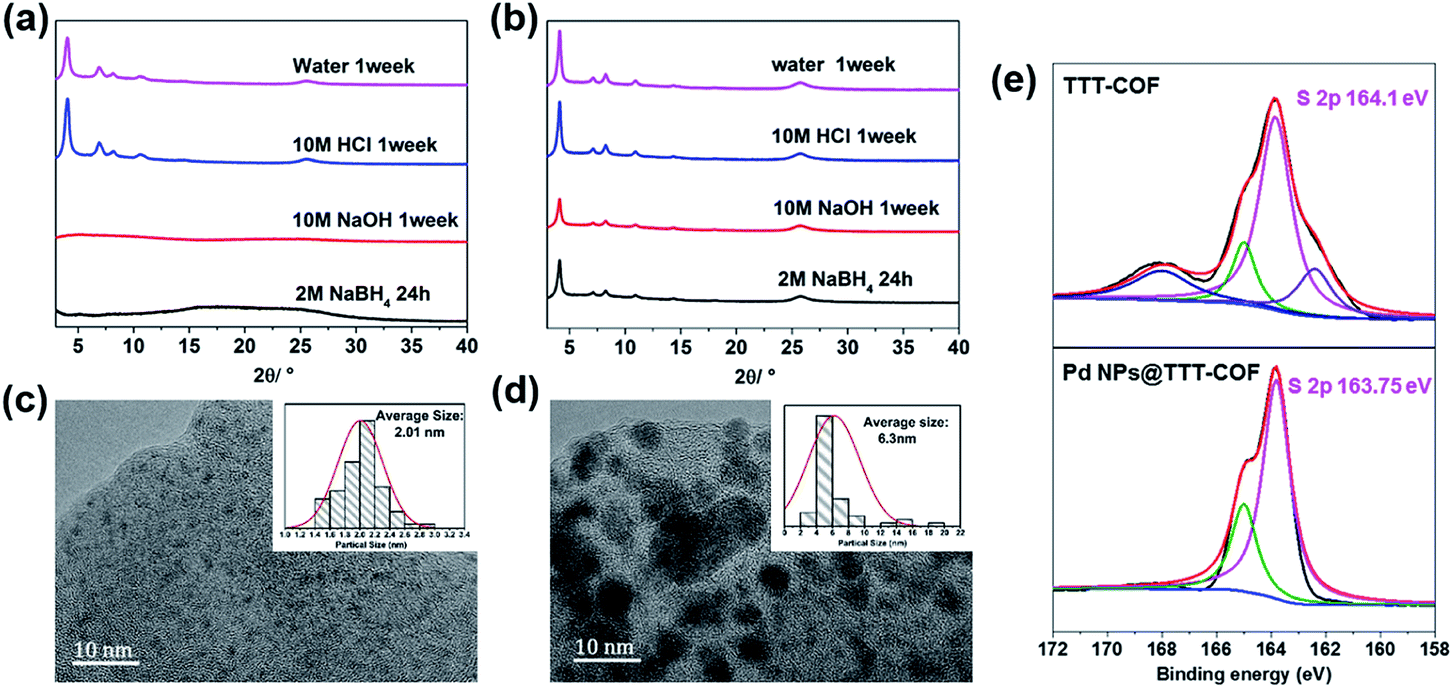 | ||
| Fig. 2 PXRD patterns of TTI-COF (a) and TTT-COF (b) after processing under relevant conditions. HR-TEM images of Pd NPs@TTT-COF (c) and Pd NPs@TTI-COF (d), the insets are the size distribution profiles of Pd NPs. (e) S 2p region in the XPS spectra of TTT-COF before and after Pd loading. | ||
The permanent porosities of COFs were assessed by measuring nitrogen adsorption isotherms at 77 K. All of TTI-COF, TTT-COF and Pd NPs@TTT-COF showed typical type-IV isotherms, which are characteristic of mesoporous materials. The Brunauer–Emmett–Teller (BET) surface areas of TTI-COF, TTT-COF and Pd NPs@TTT-COF were found to be 1659, 1640 and 1025 m2 g−1, with total pore volumes of 0.7024, 0.6989, and 0.434 cm3 g−1, respectively (Fig. S4†). The similar BET surface areas of TTI-COF and TTT-COF indicated that sulfuration process did not significantly change the porous structure, and an evident decrease of that in Pd NPs@TTT-COF confirmed that Pd NPs were distributed in pores within COFs. Pore size distribution (PSD) of TTI-COF and TTT-COF were derived from NLDFT as 2.34 and 2.16 nm, respectively, which were consistent with the reduction of cell parameters after sulfuration deduced by PXRD.
Scanning Electron Microscopy (SEM) was used to characterize the morphology of COFs. The characteristic petal-like morphology of TTI-COF formed by π–π stacking of the COF layers was maintained in Pd NPs@TTT-COF after sulfuration under high temperature, reduction by NaBH4 and loading of Pd NPs (Fig. S5†). SEM-EDX elemental mapping exhibited homogeneous distribution of sulfur and palladium in COF substrate (Fig. S6†). Long-ordered channels of TTT-COF can be clearly observed in the high resolution transmission electron microscope (HR-TEM) image (Fig. S7†). The characteristic d-spacing of π–π stacking, which is equal to the distance between face-to face 2D TTO-COF plane. Another d-spacing of 2.16 nm is observed in the vertical direction. The d-spacing of 0.34 and 2.16 nm are corresponding to the diffraction peaks of (001) and (100) facets in the PXRD pattern according to Bragg's law (nλ = 2d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ). In the Fig. 2c, Pd NPs were distributed in TTT-COF substrate uniformly with an average particle size of 2.01 nm. The interplanar spacing of 0.23 nm in the region of Pd NPs corresponded to Pd (111) crystal plane (Fig. S7†). However, a larger and uneven size of Pd NPs (average size: 6.3 nm) was observed in Pd NPs@TTI-COF (Fig. 2d), which was similar to Au NPs and Pd NPs supported by TpPa-1 previously reported.14,15 In addition, Pd NPs in Pd NPs@TTI-COF exhibited higher contrast and clearer lattice fringes in HR-TEM images than those in Pd NPs@TTT-COF, revealing that Pd NPs tend to be distributed on the surface of the TTI-COF substrate, whereas in Pd NPs@TTT-COF, more Pd NPs were encapsulated in the pores and interlayer of TTT-COF. This could be attributed to the addition of sulfur element. Sulfur atoms in benzothiazole groups could boost the anchoring effect of COFs substrates on Pd NPs and confine them in the internal pores of COFs to restrict their aggregation, thus Pd NPs can be distributed in the substrates with a finer and more uniform size.
sinθ). In the Fig. 2c, Pd NPs were distributed in TTT-COF substrate uniformly with an average particle size of 2.01 nm. The interplanar spacing of 0.23 nm in the region of Pd NPs corresponded to Pd (111) crystal plane (Fig. S7†). However, a larger and uneven size of Pd NPs (average size: 6.3 nm) was observed in Pd NPs@TTI-COF (Fig. 2d), which was similar to Au NPs and Pd NPs supported by TpPa-1 previously reported.14,15 In addition, Pd NPs in Pd NPs@TTI-COF exhibited higher contrast and clearer lattice fringes in HR-TEM images than those in Pd NPs@TTT-COF, revealing that Pd NPs tend to be distributed on the surface of the TTI-COF substrate, whereas in Pd NPs@TTT-COF, more Pd NPs were encapsulated in the pores and interlayer of TTT-COF. This could be attributed to the addition of sulfur element. Sulfur atoms in benzothiazole groups could boost the anchoring effect of COFs substrates on Pd NPs and confine them in the internal pores of COFs to restrict their aggregation, thus Pd NPs can be distributed in the substrates with a finer and more uniform size.
X-ray photoelectron spectroscopy (XPS) was used to investigate the oxidation state of the Pd NPs and interactions between Pd species and COFs substrates. In the XPS spectra of Pd NPs@TTT-COF, the characteristic peaks of Pd 3d5/2 and 3d3/2 were located at 335.9 eV and 341.2 eV respectively (Fig. S8†), which were slightly higher than the binding energy (BE) values of metallic Pd (335.0 eV for Pd 3d5/2 and 340.3 eV for Pd 3d3/2) but much lower than the BE values of Pd(II) species, confirming that Pd(II) precursor was successfully reduced to Pd(0) by NaBH4. The BE center of S 2p in Pd NPs@TTT-COF was located at 163.75 eV, exhibiting a negative shift of 0.3 eV compared with that in TTT-COF. Such positive and negative shift of BE for Pd 3d and S 2p were probably caused by the charge transfer from Pd NPs to S atoms, which confirmed the metal–ligands interactions between Pd NPs and S atoms of TTT-COF substrates.16 The S 2p XPS spectra of TTT-COF exhibited two extra peaks in 168.3 and 162.8 eV, which is caused by the residual sulfur in TTT-COF after the sulfuration reactions. However, no changes were observed in BE of N 1s after Pd NPs loading in TTT-COF. These results revealed that sulfur atoms have a stronger interaction with Pd NPs than N atoms (Fig. S9†), which is an important factor for finer particle size of the Pd NPs in benzothiazole-linked TTT-COF than that in imine-linked TTI-COF.
In the UV-DRS spectrums, TTT-COF exhibited a broader absorption band in the visible region than TTI-COF (Fig. 3a) with a band narrowing of 0.3 eV (Fig. S10†), revealing that the sulfuration of COFs can improve the light-harvesting efficiency in the visible region and narrow the optical bandgap. Compared with TTI-COF, a significant decrease of fluorescence of TTT-COF meant lower recombination rate of photogenerated electron–hole pairs (Fig. 3b), and a shorter transient fluorescence lifetime of TTT-COF confirmed higher conduction efficiency of photocarriers17,18 (Fig. 3c). In addition, higher photocurrent response of TTT-COF also indicated better separation and migration of photocarriers in COFs with benzothiazole linkages (Fig. 3d), which could be attribute to the enhanced π-conjugation within the COF planes from imine linkages to benzothiazole linkages. A similar result was also confirmed in previous report of benzoxazole-linked COFs.5 The valence band X-ray photoelectron spectroscopy (VB XPS) was used to investigate the variation of band structure in TTI-COF after sulfuration. The VB edges of TTI-COF and TTT-COF were located at 2.38 and 1.38 eV (Fig. 3e), and the conduction band (CB) potential were calculated as −0.27 and −0.96 eV, respectively, according to the formula ECB = EVB − Eg.
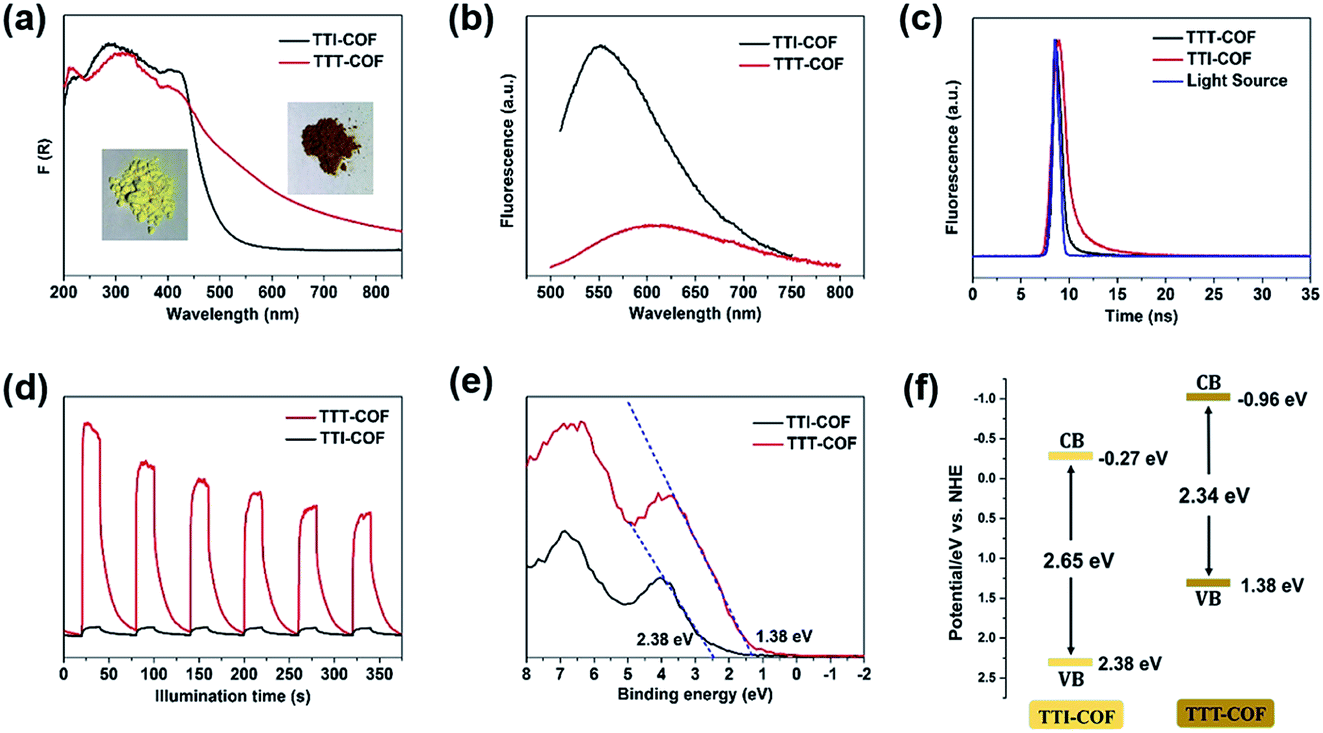 | ||
| Fig. 3 (a) UV-DRS patterns of TTI-COF and TTT-COF, the insets are the photographs of the samples. (b) Steady-state photoluminescence (PL) spectra (b) and transient fluorescence spectra (c) of TTI-COF and TTT-COF. (d) Photocurrent measurements of TTI-COF and TTT-COF. (e) VB XPS spectra of TTI-COF and TTT-COF. (f) Band alignment of TTI-COF and TTT-COF. | ||
Palladium catalysts have been widely used in C–C cross-coupling reactions, but the conditions of heating and reflux are necessary for most of these reactions. Considering that NPs with ultrafine size would generally exhibit better catalytic performance and TTT-COF is a photosensitive semiconductor, the Suzuki–Miyaura cross-coupling reaction of iodotoluene and phenylboronic acid under visible light was used as a model reaction to investigate the photocatalytic activity of Pd NPs@TTT-COF. The reaction system included iodotoluene (0.3 mmol), phenylboronic acid (0.4 mmol), excess potassium carbonate and 5 mg photocatalyst in ethanol/H2O, an excellent yield (99%) of desired product biphenyl was obtained within an hour under visible light. The turnover frequency (TOF) of the composite photocatalyst for the model reaction was calculated as 344 h−1, which is significantly higher than that of previous Pd NPs loaded photocatalysts (Table S3†). A series of control experiments were carried out (Table 1), which confirmed that illumination, Pd NPs and TTT-COF substrates were all necessary for this photocatalytic Suzuki–Miyaura reaction. Additionally, p-benzoquinone (BQ, an electron scavenger) and diisopropylethylamine (iPr2NEt, a hole scavenger) were added to the reaction system respectively to investigate the mechanism of the photocatalytic reaction. The photocatalytic cross-coupling reaction was greatly blocked in the presence of BQ and iPr2NEt, respectively. When both were added to the reaction system, the reaction was almost blocked. It can be seen that photogenerated electrons and holes were both significant to the photocatalytic Suzuki–Miyaura cross-coupling reaction.
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst | Light | Additiveb | Yieldc/% |
| a Reaction conditions: 4-iodotoluene (0.3 mmol), phenylboronic acid (0.35 mmol), K2CO3 (0.6 mmol), 6 mL ethanol/H2O, Xe lamp (>420 nm), room temperature, N2 atmosphere.b The amount of additives was 1 mmol.c Yields were determined by GC-MS. | ||||
| 1 | Pd NPs@TTT-COFa | ✓ | 99 | |
| 2 | TTT-COF | ✓ | N.D. | |
| 3 | - | ✓ | N.D. | |
| 4 | Pd NPs@TTT-COF | ✗ | 22 | |
| 5 | Pd NPs@TTT-COF | ✓ | BQ | 31 |
| 6 | Pd NPs@TTT-COF | ✓ | iPr2NEt | 37 |
| 7 | Pd NPs@TTT-COF | ✓ | BQ + iPr2NEt | Trace |

In the light of the above results and relevant studies,19 we proposed a possible reaction mechanism of this photocatalytic Suzuki–Miyaura reaction as shown in Scheme 2. Upon the irradiation of visible light, electrons on the VB of TTT-COF were excited to jump to the CB and migrate to Pd NPs which act as electron sink. C–I bonds of the adsorbed iodotoluene will become longer when excited electrons on the surface of the electron-rich Pd NPs enter the unoccupied orbital of iodotoluene, which facilitated the cleavage of the C–I bond to generate phenyl radicals and forming the intermediate of organometallic complex. The B(OH)2 groups of phenylboronic acid were converted into B(OH)3− groups under the alkaline atmosphere, and the C–B bonds were cleaved by photogenerated holes to generate another phenyl radical. After the process of reductive elimination, the desired product biphenyl was generated and Pd catalysts were regenerated.
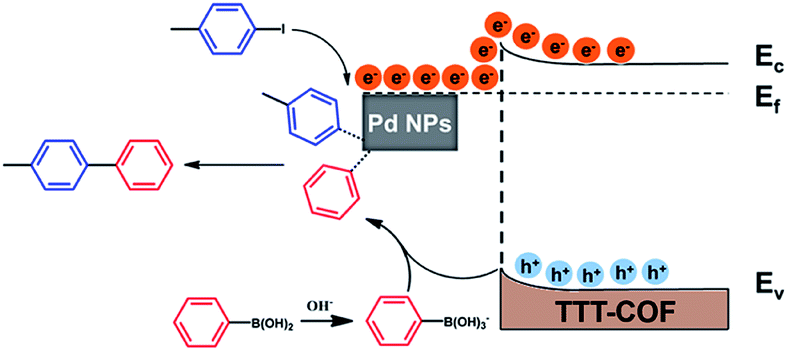 | ||
| Scheme 2 Proposed mechanism for the photocatalytic model. | ||
Pd NPs@TTT-COF exhibited superior stability and reusability. Almost no loss of activity was found in the model reaction among the four recyclings of photocatalyst (Fig. S11†). No significant changes were observed in the PXRD and IR patterns of Pd NPs@TTT-COF before and after the fourth run of the photocatalytic reaction (Fig. S12†) which benefited from the good chemical stability of benzothiazole-linked TTT-COF. Moreover, the porosity of Pd NPs@TTT-COF was also well maintained after four cycles, the BET surface areas was found to be 1003.2 cm3 g−1. As shown in the TEM images, there is no significant increase in the size of Pd NPs after the fourth run, (Fig. S13†) whereas in previous reports of metal NPs@TpPa-1, the size of NPs was increased with the number of cycles.14 The loading rate of Pd in the composite photocatalyst (5.88 wt%) dropped slightly after four cycles determined by ICP-MS. These results can be attributed to the high chemical stability of TTT-COF and the strong anchoring effect of the benzothiazole groups in TTT-COF to Pd NPs.
The photocatalytic performance of Pd NPs@TTT-COF in photocatalytic C–C cross-coupling reactions was further investigated (Table 2). Satisfactory conversions (82–99%) of various substrates were observed in the photocatalytic Suzuki–Miyaura reactions. Iodobenzenes with electron-donating substituents showed lower conversions compared to the electron-withdrawing ones, which can be explained by the fact that the electron-rich aryl iodides are not easily attacked by the electrons on the Pd NPs to result in the cleavage of C–I bond. On the contrary, phenylboronic acid with electron-withdrawing substituents exhibited lower conversion because electron-deficient phenylboronic acids were not favorable to react with positive holes (h+). In addition, meta-methyl substituted iodobenzene and phenylboronic acid showed lower conversions which could be attribute to electronic effect and steric hindrance.20 Surprisingly, Pd NPs@TTT-COF has also shown excellent efficiency of photocatalysis in a series of other classic C–C cross-coupling reactions (Stille, Heck and Sonogashira). The reactants with electron-withdrawing substituents still exhibited higher conversions in such reactions, suggesting that the series of C–C cross-coupling reactions could probably follow similar reaction mechanisms. The TOF of each reaction was calculated to be higher than that of other reported photocatalysts, (Table S3†) which was mainly attributed to two factors: (1) the ultrafine size and uniform distribution of Pd NPs resulted in higher contact area and more catalytic sites of Pd catalysts; (2) benzothiazole-linked π-conjugated COFs frameworks exhibited a strong photoelectric response, indicating high photoelectron density on Pd NPs and the increased the catalytic efficiency. Interestingly, it is noted that cis-stilbene accounted for a large part of the products of the photocatalytic Heck reactions whereas trans-stilbene was the main product in previous reports.15 Control experiments have been carried out to prove that isomerization of stilbene from trans (E) form to cis (Z) form can be catalyzed by Pd NPs@TTT-COF under the visible light irradiation (Table S4†). Recently, Banerjee's group reported a triazine functionalized COF (TpTt) for the E to Z isomerization of trans-stilbene via a process of triplet state excitation,21 and triazine groups have also been found to play an important role in the photoisomerization of stilbene.22 Hereby, we believed that the cis products of the photocatalytic Heck reactions were obtained by photoisomerization of the original trans products catalyzed by triazine-based TTT-COF substrate via a similar mechanism.
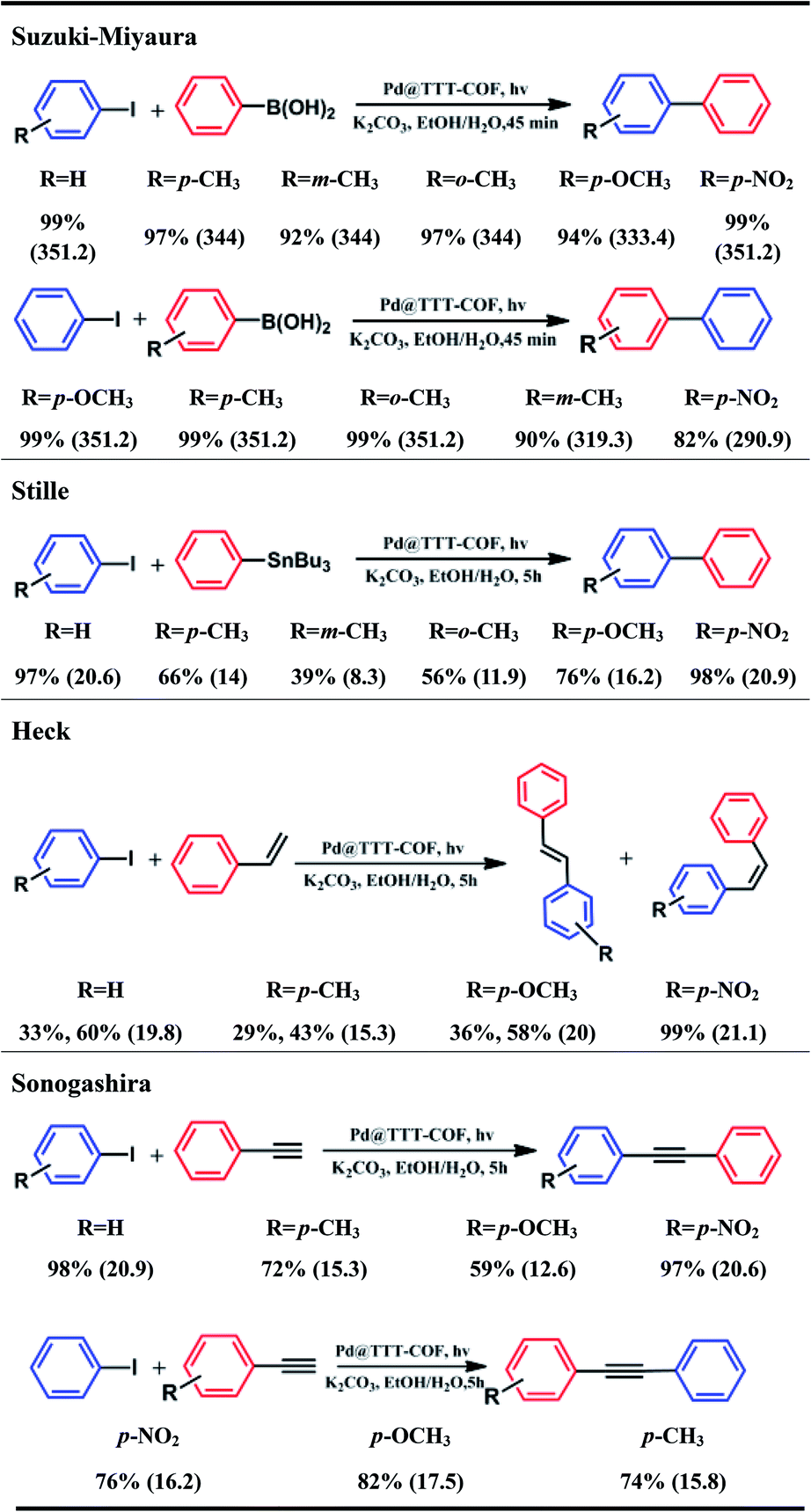 |
Conclusions
In summary, we have developed a hybrid material Pd NPs@TTT-COF photocatalyst. Several advantages of benzothiazole-linked COFs substrate were proved as follows: (1) superior chemical stability makes the COFs substrate resistant to extreme conditions in the preparation and application of catalysts; (2) benzothiazole-linked conjugated framework exhibited higher conduction efficieney of photocarriers; (3) Pd NPs were anchored by S atoms of benzothiazole groups in the pores of COFs with an ultrafine and uniform particle size. Pd NPs@TTT-COF showed superior photocatalytic performance and reusability in a series of C–C cross-coupling reactions. Considering the designability of COFs as well as the diversity of metal NPs, this strategy will provide great opportunities for the design and synthesis of photocatalysts for a wide range of applications. This work would inspire researchers that the linkage could also bring some fascinating properties to COFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program (2016YFA0203100), the National Natural Science Foundation of China (21537004, 21621064, 21777169).References
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355 Search PubMed.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673 CrossRef CAS PubMed.
- P. F. Wei, M. Z. Qi, Z. P. Wang, S. Y. Ding, W. Yu, Q. Liu, L. K. Wang, H. Z. Wang, W. K. An and W. Wang, J. Am. Chem. Soc., 2018, 140, 4623 CrossRef CAS PubMed.
- P. Das and S. K. Mandal, Chem. Mater., 2019, 31, 1584 CrossRef CAS.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Dorfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Nat. Commun., 2018, 9, 2600 CrossRef PubMed.
- P. J. Waller, S. J. Lyle, T. M. Osborn Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15519 CrossRef CAS PubMed.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715 CrossRef CAS PubMed.
- Q. Gu, Q. Jia, J. Long and Z. Gao, ChemCatChem, 2019, 11, 669–683 CrossRef CAS.
- S. Lu, Y. Hu, S. Wan, R. McCaffrey, Y. Jin, H. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082 CrossRef CAS PubMed.
- R. Tao, X. Shen, Y. Hu, K. Kang, Y. Zheng, S. Luo, S. Yang, W. Li, S. Lu, Y. Jin, L. Qiu and W. Zhang, Small, 2020, 1906005 CrossRef CAS PubMed.
- M. G. Rabbani, T. Islamoglu and H. M. El-Kaderi, J. Mater. Chem. A, 2017, 5, 258 RSC.
- P. Pachfule, S. Kandambeth, D. D. Diaz and R. Banerjee, Chem. Commun., 2014, 50, 3169 RSC.
- P. Pachfule, M. K. Panda, S. Kandambeth, S. M. Shivaprasad, D. Diaz Diaz and R. Banerjee, J. Mater. Chem. A, 2014, 2, 7944 RSC.
- H. I. Lee, S. H. Joo, J. H. Kim, D. J. You, J. M. Kim, J.-N. Park, H. Chang and C. Pak, J. Mater. Chem., 2009, 19, 5934 RSC.
- Q. Jia, S. Zhang and Q. Gu, J. Energy Chem., 2019, 30, 152–161 CrossRef.
- Q. Jia, S. Zhang, X. Jia, X. Dong, Z. Gao and Q. Gu, Catal. Sci. Technol., 2019, 9, 5077–5089 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Mater., 2015, 27, 1921 CrossRef CAS.
- D. Sun and Z. Li, J. Phys. Chem. C, 2016, 120, 19744–19750 CrossRef CAS.
- M. Bhadra, S. Kandambeth, M. K. Sahoo, M. Addicoat, E. Balaraman and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 6152 CrossRef CAS PubMed.
- K. Ohara, Y. Inokuma and M. Fujita, Angew. Chem., Int. Ed., 2010, 49, 5507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03739g |
| This journal is © The Royal Society of Chemistry 2020 |