
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification and bioactivity evaluation of secondary metabolites from Antarctic-derived Penicillium chrysogenum CCTCC M 2020019†
Imran Khan ab,
Haibo Zhang*ac,
Wei Liuad,
Liping Zhangac,
Fang Penge,
Yuchan Chenf,
Qingbo Zhangac,
Guangtao Zhangac,
Weimin Zhangf and
Changsheng Zhang*ac
ab,
Haibo Zhang*ac,
Wei Liuad,
Liping Zhangac,
Fang Penge,
Yuchan Chenf,
Qingbo Zhangac,
Guangtao Zhangac,
Weimin Zhangf and
Changsheng Zhang*ac
aKey Laboratory of Tropical Marine Bio-resources and Ecology, Guangdong Key Laboratory of Marine Materia Medica, RNAM Center for Marine Microbiology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, 164 West Xingang Road, Guangzhou 510301, China. E-mail: zhanghb@scsio.ac.cn; czhang2006@gmail.com
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSouthern Marine Science and Engineering Guangdong Laboratory (Guangzhou), 1119 Haibin Rd., Nansha District, Guangzhou 511458, China
dSouth China Sea Resource Exploitation and Protection Collaborative Innovation Center (SCS-REPIC), School of Marine Sciences, Sun Yat-sen University, Guangzhou 510006, China
eWuhan University, China Center for Type Culture Collection, Wuhan 430072, China
fState Key Laboratory of Applied Microbiology Southern China, Guangdong Institute of Microbiology, 100 Central Xianlie Road, Guangzhou 510070, China
First published on 2nd June 2020
Abstract
Extracts from Antarctic-derived Penicillium chrysogenum CCTCC M 2020019 showed potent antibacterial bioactivities. We report herein the isolation of chrysonin (1), a new compound containing a pair of enantiomers 6S- and 6R-chrysonin (1a and 1b) featuring an unprecedented eight-membered heterocycle fused with a benzene ring. Compound 2, a mixture consisting of a new zwitterionic compound chrysomamide (2a) and N-[2-trans-(4-hydroxyphenyl) ethenyl] formamide (2b) in a ratio around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8, was isolated together with seven known compounds 3–9. Chemical structures of all compounds were determined by comprehensive spectroscopic analyses. The isolated compounds were evaluated for antimicrobial, cytotoxic and alpha-glucosidase inhibition activities. Chrysonin (1) showed moderate alpha-glucosidase inhibitory activity. The dominant product xanthocillin X (4) displayed potent inhibition activities against Gram-negative pathogens Acinetobacter baumannii, Klebsiella pneumoniae, and Pseudomonas aeruginosa with MIC values at 0.125 μg mL−1. Xanthocillins X (4) and Y1 (5) also showed significant cytotoxicities against four cancer cell lines with IC50 values ranging from 0.26 to 5.04 μM. This study highlights that microorganisms from polar regions are emerging as a new resource for the discovery of natural products combating human pathogens.
:2.8, was isolated together with seven known compounds 3–9. Chemical structures of all compounds were determined by comprehensive spectroscopic analyses. The isolated compounds were evaluated for antimicrobial, cytotoxic and alpha-glucosidase inhibition activities. Chrysonin (1) showed moderate alpha-glucosidase inhibitory activity. The dominant product xanthocillin X (4) displayed potent inhibition activities against Gram-negative pathogens Acinetobacter baumannii, Klebsiella pneumoniae, and Pseudomonas aeruginosa with MIC values at 0.125 μg mL−1. Xanthocillins X (4) and Y1 (5) also showed significant cytotoxicities against four cancer cell lines with IC50 values ranging from 0.26 to 5.04 μM. This study highlights that microorganisms from polar regions are emerging as a new resource for the discovery of natural products combating human pathogens.
Introduction
Besides the inner cytoplasmic membrane, Gram-negative bacteria have an extra outer membrane which consists of lipopolysaccharide, phospholipid and lipoprotein.1 Although human beings have developed several generations of β-lactams, quinolones or other antibiotics to overcome antimicrobial resistance,2 the unique structure of the outer membrane of Gram-negative bacteria prevents antibiotics from entering into the cell.3 Thus, Gram-negative bacteria are resistant to most clinically used antibiotics and are more dangerous as disease-causing organisms.4 Currently, the ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) pathogens account for the majority of hospital infections worldwide with an increasing incidence of drug resistance.4 Five members of ESKAPE are Gram-negative bacteria except for Staphylococcus aureus. Historically, natural products have provided streptomycin, kanamycin and tetracycline as antibiotics to combat against Gram-negative bacteria for several decades,5 however, no antibiotics with novel chemical skeletons are approved for marketing over the last fifty years.6,7 Polar regions consist of the Arctic, the Antarctic and their sub regions. They comprise a third of global geography and conserve many untapped bio-resources.8Microorganisms from polar regions produced various natural products with high chemodiversity and promising biological properties,9,10 such as antimicrobial, antifungal, anti-inflammatory, immunosuppressive, and other bioactivities.11,12 From a point view of natural product chemistry, the secondary metabolites derived from antarctic fungi could be categorized into polyketides, peptides, alkaloids, terpenoids, and sterols.13–15 Several polar fungi-derived compounds showed activities against Gram-negative pathogens.16–20 For example, the cyclic tetrapeptides pseudoxylallemycins A–D from fungus Pseudoxylaria sp. X802 showed moderate activity against Pseudomonas aeruginosa.20 In this study, we report the isolation, structure elucidation and bioactivity evaluation of nine compounds (Fig. 1) from the polar fungus Penicillium chrysogenum CCTCC M 2020019, including chrysonin (1), a novel eight-membered heterocyclic compound containing a pair of enantiomers 6S-chrysonin (1a) and 6R-chrysonin (1b); compound 2, isolated as a mixture consisting of a new zwitterionic component chrysomamide (2a) and N-[2-trans-(4-hydroxyphenyl)ethenyl]formamide (2b); N-[2-cis-(4-hydroxyphenyl)ethenyl]formamide (3); xanthocillins X (4) and Y1 (5); N-acetylquestiomycin A (6); 2-aminophenoxazin-3-one (7); cyclo(L-Phe–L-Pro) (8); and N-(2-hydroxyphenyl)acetamide (9). Chrysonin (1) showed moderate alpha-glucosidase inhibitory activity, and xanthocillins X (4) and Y1 (5) displayed potent inhibition activities against the Gram-negative pathogens Acinetobacter baumannii, Klebsiella pneumoniae, and Pseudomonas aeruginosa.
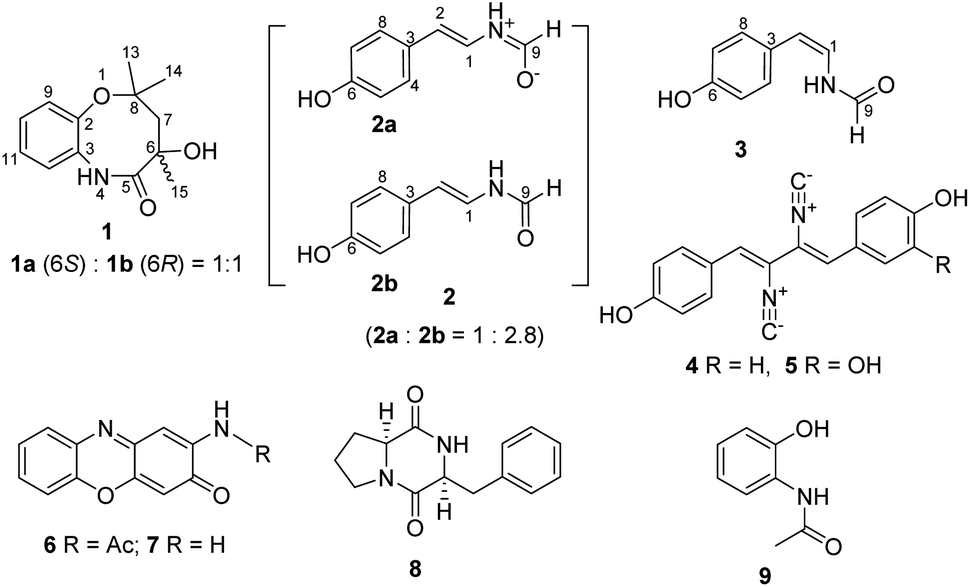 | ||
| Fig. 1 Chemical structures of 1–9. | ||
Results and discussion
In our search for bioactive metabolites from polar region microorganisms, Penicillium chrysogenum CCTCC M 2020019 (classified on the basis of 28S rDNA sequence, GenBank accession number MT229078, Fig. S1†), a strain isolated and identified from the excrement sample of the Adélie penguins living on Antarctic area, was found to exhibit potent antibacterial activities (Table S1†). Subsequently, crude extracts from 10 L fermentation broths of P. chrysogenum CCTCC M 2020019 in Czapek-Dox media were subjected to multiple chromatographic methods, resulting in the isolation of nine compounds (1–9). The known compounds 3–9 were characterized by carefully comparing the NMR and MS data with those reported in literatures, and were respectively identified as N-[2-cis-(4-hydroxyphenyl)ethenyl]formamide (3),21 xanthocillin X (4),22 xanthocillin Y1 (5),23 N-acetylquestiomycin A (6),24 2-aminophenoxazin-3-one (7),25 cyclo(L-Phe–L-Pro) (8),26 and N-(2-hydroxyphenyl)acetamide (9).27 Xanthocillin X (4) was found to be the dominant product with a titer of more than 0.9 g L−1.Compound 1 was purified as a greenish-yellow powder. The molecular formula of 1 was determined as C13H18NO3 (m/z 236.1293 [M + H]+, calcd for 236.1281, Fig. S2†) by high resolution electrospray ionization mass spectroscopy (HRESIMS), suggesting 6 degrees of unsaturation. Analysis of the 1H and 13C-NMR spectra of 1 (Table 1 and Fig. S2†) revealed the existence of an ortho-disubstituted benzene ring moiety (Fig. 1), which was supported by COSY correlations of H-9/H-10/H-11/H-12 and coupling constants (J = 7.7 Hz). Two non-protonated aromatic carbons (δC 151.1, C-2; 130.4, C-3) were assigned to the benzene ring by HMBC correlations from H-9/H-11 to C-3 and H-10/H-12 to C-2 (Fig. 2A). In addition, the chemical shifts of C-2 (δC 151.1) and C-3 (δC 130.4) were highly similar to their counterparts in N-(2-hydroxyphenyl)acetamide (9) (Table S2 and Fig. S3†),27 suggesting that C-2 and C-3 should be substituted by an oxygen atom and a nitrogen atom, respectively, to form the 2-aminophenol-like substructure (unit A) in 1 (Fig. 2A). Furthermore, an aliphatic chain (unit B) in 1, containing two oxygenated non-protonated carbons (δC 62.6, C-6; 81.6, C-8), was established by HMBC correlations from H3-13/H3-14 to C-8/C-7, H2-7 to C-8/C-6 and H3-15 to C-7/C-5. The benzene ring and the carbonyl group (C-5, δC 180.2) accounted for 5 unsaturation degrees. To satisfy 6 unsaturation degrees of 1, unit A and unit B were supposed to be connected by another ring system in 1. The long-range HMBC signal of H3-15 to C-3 and the NOESY correlation of 6-OH/4-NH supported the connection of unit B with unit A by the C-5–N-4 amide bond. Since C-8 was the only left oxygenated non-protonated carbon, unit A and unit B should also be connected by an ether bond (C-2–O–C-8). Therefore, the planar structure of 1 was determined to feature an eight-membered heterocycle fused with a benzene ring, designated chrysonin (Fig. 1).
| No. | 1a | 2ab | 2bb | 3b | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (mult, J in Hz) | δC | δH (mult, J in Hz) | δC | δH (mult, J in Hz) | δC | δH (mult, J in Hz) | |
| a Recorded in CDCl3, 1H at 700 MHz, 13C at 175 MHz.b Recorded in DMSO-d6, 1H at 700 MHz, 13C at 175 MHz. | ||||||||
| 1 | 123.5, CH | 7.16 (dd, 10.5, 14.7), overlap | 118.9, CH | 7.21 (dd, 10.5, 14.7) | 117.8, CH | 6.64 (t, 9.8) | ||
| 2 | 151.1, C | 110.6, CH | 5.93 (d, 14.7) | 113.2, CH | 6.14 (d, 14.7) | 110.9, CH | 5.60 (d, 9.8) | |
| 3 | 130.4, C | 127.2, C | 126.9, C | 126.1, C | ||||
| 4 | 126.2, CH | 7.12 (d, 8.4) | 126.6, CH | 7.16 (d, 8.4) | 129.6, CH | 7.19 (d, 8.4) | ||
| 5 | 180.2, C | 115.5, CH | 6.67 (d, 8.4) | 115.6, CH | 6.69 (d, 8.4) | 115.5, CH | 6.75 (d, 8.4) | |
| 6 | 62.6, C | 155.9, C | 156.3, C | 156.3, C | ||||
| 7 | 44.5, CH2 | 2.07 (d, 14.0), 2.25 (d, 14.0) | 115.5, CH | 6.67 (d, 8.4) | 115.6, CH | 6.69 (d, 8.4) | 115.5, CH | 6.75 (d, 8.4) |
| 8 | 81.6, C | 126.2, CH | 7.12 (d, 8.4) | 126.6, CH | 7.16 (d, 8.4) | 129.6, CH | 7.19 (d, 8.4) | |
| 9 | 115.0, CH | 6.85 (d, 7.7) | 163.6, CH | 8.36 (d, 10.5) | 158.6, CH | 8.06 s | 160.1, CH | 8.10 s |
| 10 | 125.3, CH | 6.95 (t, 7.7) | ||||||
| 11 | 120.9, CH | 6.78 (d, 7.7) | ||||||
| 12 | 123.2, CH | 6.94 (t, 7.7) | ||||||
| 13 | 29.7, CH3 | 1.13, s | ||||||
| 14 | 29.9, CH3 | 1.43, s | ||||||
| 15 | 27.7, CH3 | 1.62, s | ||||||
| NH | 6.49, brs | 10.04 (t, 10.5) | 10.15 (d, 10.5) | 9.80 (d, 10.5) | ||||
| OH | 3.70, brs | 9.40, brs | 9.40, brs | 9.52, brs | ||||
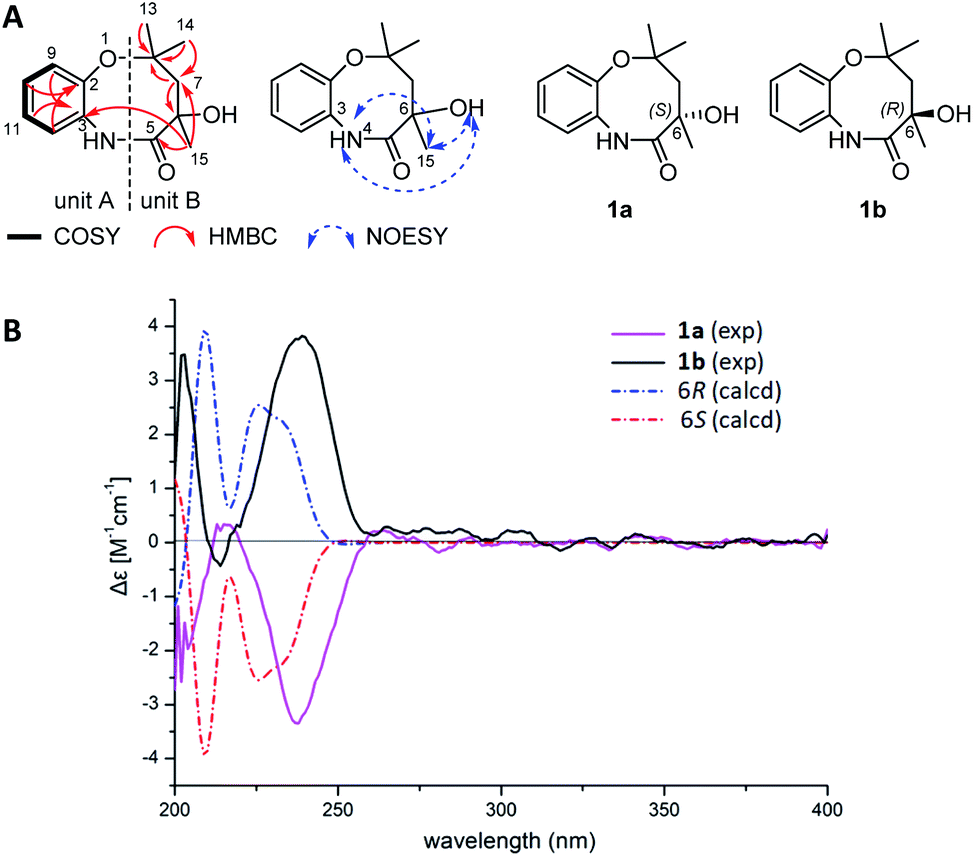 | ||
| Fig. 2 Selected key COSY, HMBC and NOESY correlations of 1 (A) and comparison of experimental ECD spectra of 1a and 1b with those calculated for 6R or 6S configurations (B). | ||
Although having a chiral center at C-6, 1 appeared to be optically inactive, given a very low specific rotation value ([α]D20 +0.001, c 0.10, MeOH). This observation indicated that 1 might be an enantiomeric mixture. Indeed, a pair of enantiomers (1a:1b ≈ 1:1) were separated from each other using a chiral column (Fig. S4†). 1H and 13C-NMR spectra of 1a and 1b were almost identical with those of 1 (Fig. S5 and S6†). However, the ECD spectra of 1a and 1b showed opposite cotton effects (Fig. 2B). The experimental ECD spectra of 1a and 1b were in good agreement with those calculated for 6R and 6S configurations by application of the Boltzmann-weighted solution TDDFT-ECD protocol at the B3LYP/6-31+G(d, p) level (Tables S3–S5†). Thus, compounds 1a and 1b were determined as 6S-chrysonin and 6R-chrysonin, respectively. The eight-membered heterocycle ring in 1, fused by a 2-aminophenol with an aliphatic chain, was unprecedented in natural product scaffolds. The most similar compound, bearing a 2-aminophenol-derived seven-membered heterocycle ring, was described as a synthetic compound (Fig. S7†).
The chemical formula of 2 was determined as C9H9NO2 (m/z 164.0719 [M + H]+, calcd 164.0706, Fig. S8†) by HRESIMS. Careful inspection of the 1H and 13C-NMR spectra of 2 indicated the presence of two sets of spectra which were similar to each other in a ratio of around 1:2.8 (2a:2b). A tiny but diffractable crystal was formed from the MeOH solution of 2 at room temperature in two days. The crystal was then subjected to the X-ray diffraction analysis (Fig. 3A, CCDC 1987282, Table S6†) to establish its structure as N-[2-trans-(4-hydroxyphenyl)ethenyl]formamide,28 consistent with the NMR data of the major component 2b in 2 (Table 1, Fig. S9†).
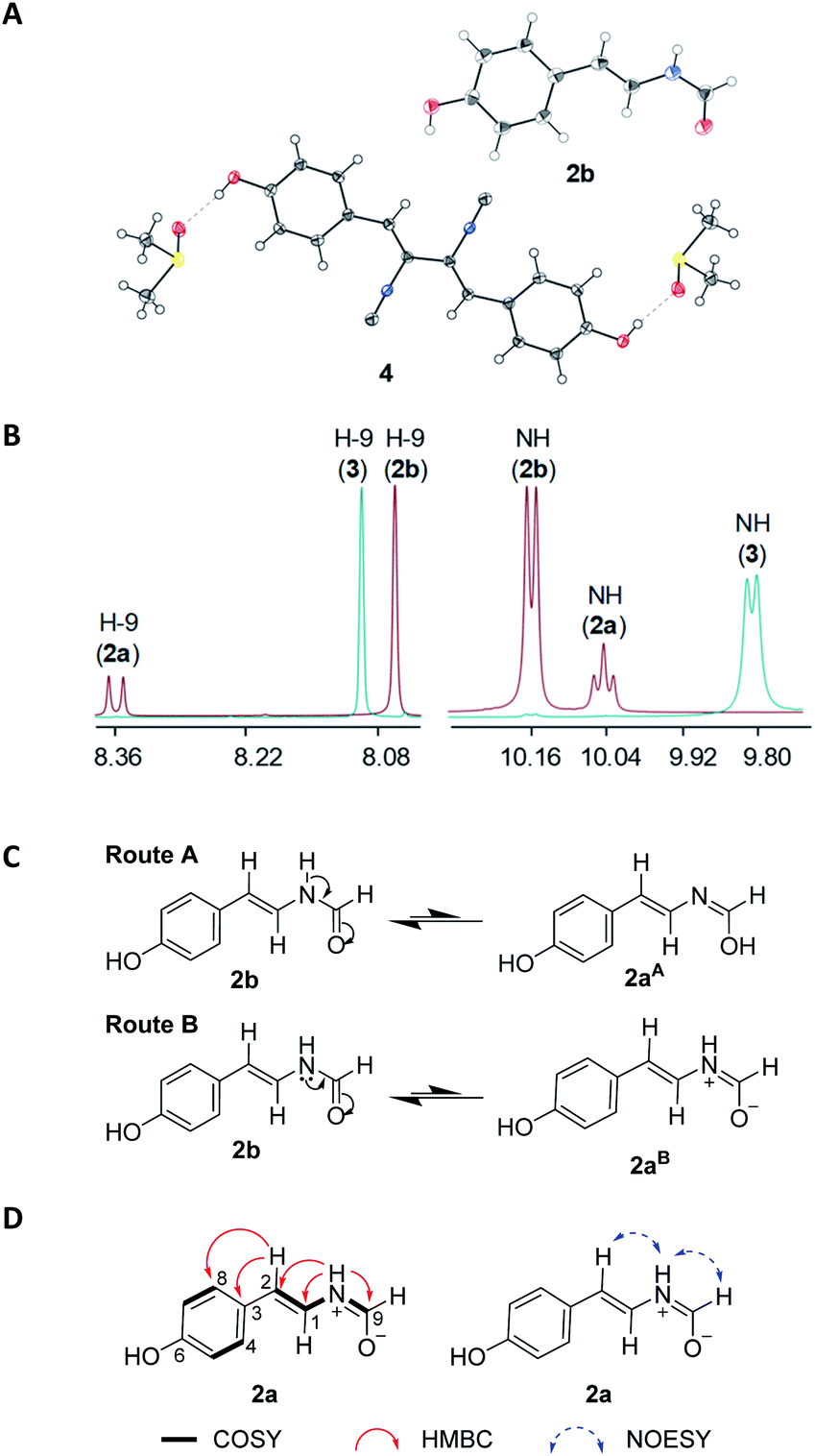 | ||
| Fig. 3 (A) Single crystal X-ray structures of compounds 2b and 4 (in DMSO). The ellipsoids of non-hydrogen atoms are shown at 50% probability levels. (B) Comparison of different types of proton signals of compounds 2a, 2b and 3. (C) Two alternative routes A and B to generate putative structures of 2a from 2b. (D) Selected key COSY, HMBC and NOESY correlations of 2a. | ||
NMR data of the minor component 2a in 2 were found to be mainly different from those of 2b in chemical shifts and splitting patterns at H-9 (Fig. 3B, S9,† Tables 1 and S7†), which were also different from those of N-[2-cis-(4-hydroxyphenyl)ethenyl]formamide (3, ZΔ1,2) (Fig. S10†), an isomer of 2b (EΔ1,2). A careful analysis of the 1H-NMR data of 2a, 2b and 3 revealed the presence of a doublet signal of H-9 (δH 8.36, d, J = 10.5 Hz) in 2a, which was distinct from the counterpart singlets in 2b (H-9, δH 8.06) and 3 (H-9, δH 8.10) (Fig. 3B, Fig. S11†). Compounds 2 and 3 could be separated by a reversed phase HPLC analysis (Fig. S12†), however, 2a and 2b in 2 could not be resolved from each other, even when trying with six kinds of chiral columns (data not shown). Thus, these data indicated that 2a should be structurally different from both 2b and 3. Two possible structures of 2a, putatively derived from 2b via two routes (Fig. 3C), could explain the difference in splitting patterns of H-9 in 2a and 2b. In route A, conversion of 2b to the enolate 2aA shared a common mechanism with lactam-lactim tautomerization.29 In route B, the formation of a zwitterion 2aB was proposed. A triplet NH signal (δH 10.04, t, J = 10.5 Hz) was observed in 2a, in contrast to the doublet NH signals in both 2b (δH 10.15, d, J = 10.5 Hz) and 3 (δH 9.80, d, J = 10.5 Hz) (Table 1). These data supported that 2a should be derived from 2b via the route B (Fig. 3C). The zwitterion 2aB kept a proton on the cationized nitrogen atom, which could interact with two protons H-9 and H-1 to explain the coupling constants and the multiplicity of the NH signal. This assignment was further confirmed by COSY correlations of NH/H-9 and NH/H-1 in 2a (Fig. 3D). In contrast, only the COSY correlation of NH/H-1 was found in 2b (Fig. S8†). In addition, the zwitterion structure of 2a was supported by HMBC correlations from the NH signal (δH 10.04) to C-1/C-2/C-9 and from H-2 (δH 5.93) to C-3/C-4/C-8 (Fig. 3D). Thus, compound 2a (designated chrysomamide) was elucidated as a new zwitterionic isomer of 2b. It should be noted that the previously reported N-[2-trans-(4-hydroxyphenyl)ethenyl]formamide,28 displayed the same two sets of 1H-NMR spectra signals as those of 2 (Fig. S9†), indicating that it should be a mixture of 2a/2b as well. The component 2a belongs to a class of rarely reported zwitterionic natural products. The antifungal drug amphotericin B can exist in both neutral and zwitterionic forms.30 Recent examples include the potent α-glucosidase inhibitor salacinol,31 the analgesic active aconicatisulfonines A and B,32 and the pancreatic lipase inhibitor flavipesides A–C.33
Compounds 1–9 were evaluated for antibacterial activities by paper-disc diffusion method on Mueller Hinton Agar (Huankai Microbial Sci. & Tech. Co., Ltd. Guangdong Province, China).34 Four Gram-negative bacteria Acinetobacter baumannii ATCC 19606, Escherichia coli ATCC 25922, Klebsiella pneumoniae ATCC 13883, and Pseudomonas aeruginosa ATCC 27853, and three Gram-positive bacteria Staphylococcus aureus ATCC 29213, methicillin-resistant S. aureus (MRSA) shhs-A1, and Micrococcus luteus SCSIO ML01 were selected as the indicator strains. Compounds 4, 5 and 7 showed potent antibacterial activity. Xanthocillin X (4) and Y1 (5) showed strong inhibition activities against the seven indicator strains (Table 2). Especially, xanthocillin X (4) was found highly active against the four Gram-negative pathogens with MIC (minimal inhibition concentration) values ranging from 0.125 to 0.25 μg mL−1. Xanthocillin X (4) was the first reported natural product containing the isocyanide functional group as an antibacterial, antiviral and antitumor agent.22,35,36 In this study, the structure of 4 was confirmed by X-ray diffraction analysis (Fig. 3A, CCDC 1987281). More importantly, xanthocillin X (4) and Y1 (5) were reported for the first time of their activities against the Gram-negative pathogens A. baumannii, K. pneumoniae, and P. aeruginosa.
| A. baumannii, ATCC 19606 | E. coli, ATCC 25922 | K. pneumoniae, ATCC 13883 | P. aeruginosa, ATCC 27853 | M. luteus, SCSIO ML01 | MRSA, shhs-A1 | S. aureus, ATCC 29213 | |
|---|---|---|---|---|---|---|---|
| a Vancomycin.b Streptomycin. | |||||||
| 1 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 2 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 3 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 4 | 0.125 | 0.25 | 0.125 | 0.125 | 0.125 | 0.25 | 0.25 |
| 5 | 0.5 | 0.5 | 4 | 8 | 1 | 8 | 16 |
| 6 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 7 | 64 | 64 | >64 | >64 | 32 | 64 | >64 |
| 8 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| 9 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| Vma | 32 | 0.25 | >64 | >64 | 0.25 | 0.25 | 0.5 |
| Smb | 2 | 2 | |||||
Compounds 1–9 were also evaluated for in vitro cytotoxicity against four human cancer cell lines (SF-268, MCF-7, HepG2, and A549) by the SRB method.37 Xanthocillin X (4) displayed comparable or better inhibitory activities against the four cancer cell lines than the positive control amycin (Table 3). Xanthocillin Y1 (5) was also cytotoxic to the four cancer cell lines. However, other compounds showed weak inhibitory activity or no activities. Compounds 1, 3, 8 and 9 were nontoxic with IC50 values greater than 100 μM.
| SF-268 | MCF-7 | HepG-2 | A549 | |
|---|---|---|---|---|
| a Values are expressed as the means ± SD from three independent experiments. | ||||
| 2 | 32.75 ± 3.25 | 58.07 ± 5.54 | 50.44 ± 2.07 | 42.87 ± 1.95 |
| 4 | 1.23 ± 0.13 | 0.26 ± 0.03 | 1.34 ± 0.05 | 0.38 ± 0.03 |
| 5 | 2.11 ± 0.01 | 3.65 ± 0.07 | 4.50 ± 0.03 | 5.04 ± 0.22 |
| 6 | 25.31 ± 0.86 | 76.74 ± 2.34 | 46.81 ± 2.07 | 52.61 ± 1.30 |
| 7 | 7.12 ± 0.57 | 35.23 ± 3.94 | 6.79 ± 0.57 | 25.60 ± 2.38 |
| Amycin | 1.06 ± 0.06 | 1.47 ± 0.14 | 1.21 ± 0.01 | 1.36 ± 0.01 |
Finally, compounds 1–9 were assayed as alpha-glucosidase inhibitors using acarbose as a positive control (Table S9†). The function of α-glucosidase is to produce glucose through hydrolyzing disaccharide, inhibition of which will decrease the concentration of blood glucose to benefit patients with non-insulin-dependent type II diabetes.38,39 At the concentration of 10 μM, compounds 7 and 9 displayed significant inhibitory activities with inhibition ratios of 85.4% and 82.4%, respectively (Table S9†). These data indicated that they might be useful in treating type II diabetes, for which acarbose was the first-line clinical drug.40,41 Compounds 1 and 2 showed moderate α-glucosidase inhibitory activity with inhibition ratios about 54% at the concentration of 10 μM, which were quite lower than that of acarbose (99%) at the same concentration.
It has been reported that 858 novel marine natural products of various scaffolds were found from marine Penicillium strains during 1991–2018, including alkaloids, terpenoids, steroids, polyketide, and lipopeptides, about 50% of which showed antibacterial, antiviral, antitumor or anti-inflammatory biological activities.42–46 Our data suggested that microorganisms from polar regions are emerging as new resources for discovering natural products combating with human-being pathogens.
Experimental section
General procedures
Optical rotations were measured using a 341 Polarimeter (PerkinElmer, Inc., Norwalk, CT, USA). The CD spectra were measured on a Chirascan circular dichroism spectrometer (Applied Photophysics, Ltd., Surrey, UK). UV spectra were recorded on UV-2600 spectrophotometer (Shimadzu). IR spectra were recorded on Affinity-1 FT-IR spectrometer (Shimadzu). NMR spectra were recorded on a AMC instrument (Bruker Biosciences Corporation, USA) operating at 700 MHz (1H-NMR) or 175 MHz (13C-NMR), respectively. Mass spectrometric data were obtained on a time-of-flight mass spectrometry (Bruker maxis 4G) for HRESIMS. Materials for column chromatography (CC) were silica gel (100–200 mesh; 300–400 mesh; Jiangyou Silica gel development Inc.), Sephadex LH-20 (40–70 μm; Amersham Pharmacia Biotech), and YMC*GEL ODS-A (12 nm S-50 μm; YMC Company Ltd.). Thin-Layer-Chromatography (TLC, 0.1–0.2 mm or 0.3–0.4 mm) was conducted with precoated glass plates (silica gel GF254, 10–40 nm, Jiangyou Silica gel development, Inc.). Medium pressure liquid chromatography (MPLC) was performed on CHEETAH flash system (Bonna-Agela technologies Inc.). Compounds were detected with a 1260 Infinity HPLC (Agilent Inc.). Semi-preparative HPLC was carried out on Gemini C18 column (5 μm; Phenomenex Company Ltd.) except for specific indication.Fungal material
Penicillium chrysogenum CCTCC M 2020019 was isolated from excrements of the Adélie penguins living on Antarctic area. The fungus was streaked on fresh potato dextrose agar (PDA) Petri dishes and incubated at 28 °C for 7 days to produce spores. The spores were inoculated into potato dextrose broth (PDB) and cultured for three days. Genomic DNA of P. chrysogenum CCTCC M 2020019 was extracted from the mycelia cultured in PDB and used as the template to produce 28S rDNA fragment by polymerase chain reaction (PCR) amplification. Then the phylogenetic tree of P. chrysogenum CCTCC M 2020019 was constructed by neighbor joining method. The phylogenetic tree showed 100% similarity of the strain CCTCC M 2020019 to Penicillium chrysogenum (Fig. S1†).Fermentation, extraction and isolation
P. chrysogenum CCTCC M 2020019 was cultured on PDA Petri dishes (i.d. 90 mm) for one week at 28 °C to produce fresh spores. The mycelia plugs (5 mm in diameter) were transferred to two 500 mL Erlenmeyer flasks containing 100 mL of Czapek-Dox media (NaNO3 3 g L−1, K2HPO4 1 g L−1, MgSO4 0.5 g L−1, KCl 1 g L−1, FeSO4 0.01 g L−1, saccharose 20 g L−1, deionized water 1 L) and incubated on a rotary shaker (200 rpm) at 28 °C for one week. Around 1% inoculums were transferred into 200 mL Czapek-Dox media in 1000 mL Erlenmeyer flasks, and were subsequently incubated on a rotary shaker at 200 rpm, 28 °C for 20 days. After 20 days, 10 L fermentation broths were centrifuged at 4000 rpm to provide the supernatants and a mycelia cake. The supernatants were extracted with equal volume of ethyl acetate (EtOAc) for three times. The mycelia cake was extracted with 70% acetone–water for three times. After removal of the acetone, the residues from the mycelia cake, was also extracted with equal volume of EtOAc for three times. All EtOAc extracts were combined and yielded 8.7 g crude extracts after drying by vacuum. The crude extracts were subjected to reverse phase C18 column of MPLC and eluted with gradient methanol (MeOH)-water system (0%→100% MeOH) for 120 min. After gradient elution the column was washed with 100% MeOH for 20 min. The MPLC process produced twelve fractions F1–F12. Fraction F7 was purified again by semi-preparative HPLC (ACE 5-C18, 250 × 10 mm) and gave compound 1 (5.2 mg). Compound 1 was further treated with a chiral column (Lux 5 μm i-cellulose-5, 250 × 4.6 mm) by isocratic elution (27% acetonitrile-water) to produce isomers 1a and 1b. Fraction F4 (121 mg) was loaded onto a Sephadex LH-20 column and eluted with MeOH. A total of 21 sub-fractions (ea. 2.5 mL) were collected. Subfracions F4-4–F4-9 were combined to give 31 mg residue after evaporation. The 31 mg residue was purified again by semi-preparative HPLC to yield compounds 2 (6.5 mg) and 9 (22 mg). F5 was applied to semi-preparative HPLC to give compound 3 (3 mg). Compound 4 (181 mg) was crystallized from the MeOH–water solution of fraction F10. From fraction F11 (the major fraction containing 4), 4.1 gram of 4 was isolated. Fraction F9 was purified by semi-preparative HPLC (ACE-5 C18, 250 × 10 mm) to provide two compounds 5 (8.7 mg) and 6 (3 mg). F8 (98 mg) was fractioned again by Sephadex LH-20 column to produce 31 sub-fractions. Sub-fractions F8-12–F8-23 were combined to afford 11.5 mg residue. The 11.5 mg residue was purified again by semi-preparative HPLC to produce compound 7 (3.8 mg). The strain CCTCC M 2020019 was also cultured on the rice medium for 28 days. After extraction with 1 L EtOAc, the extracts were fractionated with MPLC on C18 column. Compound 8 (2.6 mg) was obtained from MPLC fraction (RF-5A) by semi-preparative HPLC.ε) 206 nm (2.97), 238 nm (2.35), 290 nm (1.903); CD (c 0.025, MeOH) λmax (Δε) 210 (−0.580), 240 (−3.144) nm; IR νmax 3389, 2922, 2850, 1747, 1647, 1456, 1022 cm−1. 1H and 13C NMR data, see Table 1 in main text; HRESIMS m/z 236.1293 [M + H]+, (calcd for C13H18NO3, 236.1281).ε) 206 nm (2.97), 238 nm (2.35), 290 nm (1.903); CD (c 0.025, MeOH) λmax (Δε) 210 (0.0509), 240 (3.804) nm; IR νmax 3389, 2922, 2850, 1747, 1647, 1456, 1022 cm−1. 1H and 13C NMR data, see Table 1 in main text; HRESIMS m/z 236.1293 [M + H]+, (calcd for C13H18NO3, 236.1281).ε) 218 nm (3.85), 284 nm (4.12); IR νmax 3277, 1638, 1508, 1393, 1254, 947, 717 cm−1. 1H and 13C NMR data, see Table 1 in main text; HRESIMS m/z 164.0719 [M + H]+, (calcd for C9H9NO2, 164.0706).Bioactivity evaluations
Conclusions
In summary, chrysonin (1), bearing an unprecedented eight-membered heterocycle fused with a benzene ring, was isolated from the Antarctic-derived fungus P. chrysogenum CCTCC M 2020019. In addition, compound 2 was identified as a mixture that contained an interesting zwitterionic component chrysomamide (2a) and N-[2-trans-(4-hydroxyphenyl)ethenyl]formamide (2b). The strain P. chrysogenum CCTCC M 2020019 could also produce another 7 known compounds with Xanthocillin X (4) as the dominant product. Xanthocillin X (4) exhibited potent inhibition activities against Acinetobacter baumannii, Klebsiella pneumoniae, and Pseudomonas aeruginosa, and thus should be worth of further investigation as a hit against Gram-negative pathogens. Xanthocillins X (4) and Y1 (5) also showed significant cytotoxicity against four cancer cell lines. This study disclosed that microbial resources from polar regions were of promising potential for discovery of natural products combating with human-being pathogens. Xanthocillin X (4) was found to be the most potent compound for both antibacterial and cytotoxic activities. Compound 7 displayed the best alpha glucosidase inhibition among the 9 compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the National Key Research & Development Program of China (2018YFC1406705, 2018YFC1406704), National Natural Science Foundation of China (NSFC) (81872778, 41406183), and Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou) (GML2019ZD0406). I. K. is an awardee of the UCAS Scholarship for International Students. We thank Professor T. Kurtán (University of Debrecen) for his valuable discussions. We are grateful to Z. Xiao, C. Li, A. Sun, Y. Zhang and X. Ma in analytical facilities of SCSIO for recording spectroscopic data.Notes and references
- C. Walsh, Antibiotics: Actions, Origins, Resistance, ASM Press, 2003 Search PubMed.
- I. Karaiskos, S. Lagou, K. Pontikis, V. Rapti and G. Poulakou, Frontiers in Public Health, 2019, 7, 151 CrossRef PubMed.
- A. H. Delcour, Biochim. Biophys. Acta, 2009, 1794, 808–816 CrossRef CAS PubMed.
- R. Domalaon, T. Idowu, G. G. Zhanel and F. Schweizer, Clin. Microbiol. Rev., 2018, 31, e00077-00017 CrossRef PubMed.
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 CrossRef CAS PubMed.
- H. Lu, D. Zhang and H. Huang, Acta Pharm. Sin. B, 2019, 54, 1554–1563 Search PubMed.
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- S. Soldatou and B. J. Baker, Nat. Prod. Rep., 2017, 34, 585–626 RSC.
- I. F. Santiago, M. A. Soares, C. A. Rosa and L. H. Rosa, Extremophiles, 2015, 19, 1087–1097 CrossRef PubMed.
- M. E. Rateb and R. Ebel, Nat. Prod. Rep., 2011, 28, 290–344 RSC.
- J. Macheleidt, D. J. Mattern, J. Fischer, T. Netzker, J. Weber, V. Schroeckh, V. Valiante and A. A. Brakhage, Annu. Rev. Genet., 2016, 50, 371–392 CrossRef CAS PubMed.
- N. P. Keller, G. Turner and J. W. Bennett, Nat. Rev. Microbiol., 2005, 3, 937–947 CrossRef CAS PubMed.
- R. Chen, Z. B. Cheng, J. Huang, D. Liu, C. M. Wu, P. Guo and W. H. Lin, RSC Adv., 2017, 7, 49235–49243 RSC.
- A. Debbab, A. H. Aly, W. H. Lin and P. Proksch, Microb. Biotechnol., 2010, 3, 544–563 CrossRef CAS PubMed.
- H. Zhou, L. Li, W. Wang, Q. Che, D. Li, Q. Gu and T. Zhu, J. Nat. Prod., 2015, 78, 1442–1445 CrossRef CAS PubMed.
- Y. Li, B. Sun, S. Liu, L. Jiang, X. Liu, H. Zhang and Y. Che, J. Nat. Prod., 2008, 71, 1643–1646 CrossRef CAS PubMed.
- J. Ren, C. Xue, L. Tian, M. Xu, J. Chen, Z. Deng, P. Proksch and W. Lin, J. Nat. Prod., 2009, 72, 1036–1044 CrossRef CAS PubMed.
- J. T. Liu, B. Hu, Y. Gao, J. P. Zhang, B. H. Jiao, X. L. Lu and X. Y. Liu, Chem. Biodiversity, 2014, 11, 800–806 CrossRef CAS.
- X. L. Lu, J. T. Liu, X. Y. Liu, Y. Gao, J. P. Zhang, B. H. Jiao and H. Zheng, J. Antibiot., 2014, 67, 171–174 CrossRef CAS PubMed.
- H. J. Guo, N. B. Kreuzenbeck, S. Otani, M. Garcia-Altares, H. M. Dahse, C. Weigel, D. K. Aanen, C. Hertweck, M. Poulsen and C. Beemelmanns, Org. Lett., 2016, 18, 3338–3341 CrossRef CAS PubMed.
- Q. Peng, L. Peipei, F. Peng, W. Yi and Z. Weiming, Acta Microbiol. Sin., 2012, 52, 1103–1112 Search PubMed.
- Z. Shang, X. M. Li, L. Meng, C. S. Li, S. S. Gao, C. G. Huang and B. G. Wang, Chin. J. Oceanol. Limnol., 2012, 30, 305–314 CrossRef CAS.
- H. Achenbach, H. Strittmatter and W. Kohl, Chem. Ber., 1972, 105, 3061–3066 CrossRef CAS PubMed.
- S. Imai, A. Shimazu, K. Furihata, K. Furihata, Y. Hayakawa and H. Seto, J. Antibiot., 1990, 43, 1606–1607 CrossRef CAS PubMed.
- E. Graf, K. Schneider, G. Nicholson, M. Strobele, A. L. Jones, M. Goodfellow, W. Beil, R. D. Sussmuth and H. P. Fiedler, J. Antibiot., 2007, 60, 277–284 CrossRef CAS PubMed.
- K. Strom, J. Sjogren, A. Broberg and J. Schnurer, Appl. Environ. Microbiol., 2002, 68, 4322–4327 CrossRef CAS PubMed.
- Z. Pei, L. Chen, J. Xu and C. Shao, Chin. J. Mar. Drugs, 2017, 36, 59–64 Search PubMed.
- T. Jiang, L. Tian, Y. B. Lao, L. I. Jun and W. H. Lin, Chin. J. Mar. Drugs, 2001, 20, 40–42 CAS.
- T. V. Sycheva, N. M. Rubtsov, Y. N. Sheinker and L. N. Yakhontov, Chem. Heterocycl. Compd., 1987, 23, 82–87 CrossRef.
- G. Cuddihy, E. K. Wasan, Y. Di and K. M. Wasan, Pharmaceutics, 2019, 11, 99, DOI:10.3390/pharmaceutics11030099.
- M. Yoshikawa, T. Murakami, H. Shimada, H. Matsuda, J. Yamahara, G. Tanabe and O. Muraoka, Tetrahedron Lett., 1997, 38, 8367–8370 CrossRef CAS.
- Y. Z. Wu, S. Shao, Q. L. Guo, C. B. Xu, H. Xia, T. T. Zhang and J. G. Shi, Org. Lett., 2019, 21, 6850–6854 CrossRef CAS PubMed.
- W. H. Jiao, Q. H. Xu, G. B. Ge, R. Y. Shang, H. R. Zhu, H. Y. Liu, J. Cui, F. Sun and H. W. Lin, Org. Lett., 2020, 22, 1825–1829 CrossRef CAS PubMed.
- Clinical and Laboratory Standards Institute (CLSI), Performance Standards for Antimicrobial Susceptibility Testing; Seventeenth Informational Supplement, CLSI document M100-S117, CLSI, Wayne, PA, 2007 Search PubMed.
- N. Kaniss, Pharmazie, 1954, 9, 203–209 CAS.
- A. Takatsuki, S. Suzuki, K. Ando, G. Tamura and K. Arima, J. Antibiot., 1968, 21, 671–675 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- J. F. Wang, L. M. Zhou, S. T. Chen, B. Yang, S. R. Liao, F. D. Kong, X. P. Lin, F. Z. Wang, X. F. Zhou and Y. H. Liu, Fitoterapia, 2018, 125, 49–54 CrossRef CAS PubMed.
- M. S. Hedrington and S. N. Davis, Expert Opin. Pharmacother., 2019, 20, 2229–2235 CrossRef PubMed.
- U. F. Wehmeier and W. Piepersberg, Appl. Microbiol. Biotechnol., 2004, 63, 613–625 CrossRef CAS PubMed.
- Q. Zhao, Y. Luo, X. Zhang, Q. Kang, D. Zhang, L. Zhang, L. Bai and Z. Deng, Nat. Commun., 2020, 11, 1468 CrossRef CAS PubMed.
- H. G. Ma, Q. Liu, G. L. Zhu, H. S. Liu and W. M. Zhu, J. Asian Nat. Prod. Res., 2016, 18, 92–115 CrossRef CAS PubMed.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2017, 34, 235–294 RSC.
- J. W. Blunt, A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2018, 35, 8–53 RSC.
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2019, 36, 122–173 RSC.
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2020, 37, 175–223 RSC.
- H. Nakabayashi, K. Taketa, K. Miyano, T. Yamane and J. Sato, Cancer Res., 1982, 42, 3858–3863 CAS.
- W. Liu, W. J. Zhang, H. B. Jin, Q. B. Zhang, Y. C. Chen, X. D. Jiang, G. T. Zhang, L. P. Zhang, W. M. Zhang, Z. G. She and C. S. Zhang, Mar. Drugs, 2019, 17, 663 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures and tables, characterization data and original spectra (NMR, MS, IR, UV, and ECD) of 1a, 1b and 2. CCDC 1987281 and 1987282. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03529g |
| This journal is © The Royal Society of Chemistry 2020 |