
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected cyclization of ortho-nitrochalcones into 2-alkylideneindolin-3-ones†
Nicolai A. Aksenov *a,
Dmitrii A. Aksenova,
Nikolai A. Arutiunova,
Daria S. Aksenovaa,
Alexander V. Aksenova and
Michael Rubin*ab
*a,
Dmitrii A. Aksenova,
Nikolai A. Arutiunova,
Daria S. Aksenovaa,
Alexander V. Aksenova and
Michael Rubin*ab
aDepartment of Chemistry, North Caucasus Federal University, 1a Pushkin St., Stavropol 355009, Russian Federation
bDepartment of Chemistry, University of Kansas, 1567 Irving Hill Rd., Lawrence, KS 66045-7582, USA. E-mail: mrubin@ku.edu; Tel: +1-785-864-5071
First published on 14th May 2020
Abstract
An original, facile, and highly efficient method for the preparation of 2-(3-oxoindolin-2-ylidene)acetonitriles from ortho-nitrochalcones is described. The featured transformation is a triggered Michael addition of the cyanide anion to the chalcone followed by a cascade cyclization mechanistically related to the Baeyer–Drewson reaction.
Introduction
It would be hard to overstate the importance of 2-alkylideneindolin-3-one derivatives in modern medicinal chemistry. The bis-indole indirubin, a main component of “Tyrian purple” dye, is also a known active component of a traditional Chinese herbal medicine, while its numerous synthetic derivatives show potent and highly selective pharmacological inhibition of glycogen synthase kinases and cycline-dependent kinases.1–6 These molecules induce apoptosis of human cancer cells and have promising potential for applications in the treatment of several neurodegenerative conditions, such as Alzheimer's disease.1–6 Indirubin, as well as other related dyes, can be easily prepared via base-assisted condensation of ortho-nitrobenzaldehydes with acetone according to the classical Baeyer–Drewson reaction.7–9 2-Alkylideneindolin-3-one derivatives possessing a single indoline subunit (or two remotely positioned subunits) also occur in nature and also exhibit a wide spectrum of important biological properties (Fig. 1).10–17 Normally, preparation of such compounds relies heavily on the chemistry of isatins, which makes synthetic approaches to certain substitution patterns hardly accessible. An alternative synthetic platform for assembling indoline alkaloids and related non-natural, biologically active target molecules has also emerged, relying on the chemistry of 2-alkylidene-3-oxindoles.18–23 Various synthetic approaches to these synthons have been developed based on the aldol condensation of 3H-indol-3-ones with carbonyl compounds (path A, Scheme 1),24–28 transition metal-catalyzed carbonylative coupling of ortho-iodoanilines to acetylenes (path B, Scheme 1),29–32 and cascade reactions of anilines with α-ketoesters involving an electrophilic aromatic substitution step (path C, Scheme 1)10,33 Herein we wish to report on our recent serendipitous discovery of the unexpected one-pot cascade transformation of ortho-nitrochalcones 1 via a Baeyer–Drewson-like pathway, but affording 2-alkylideneindolin-3-ones 2 rather than indigo-like dimers (Scheme 1).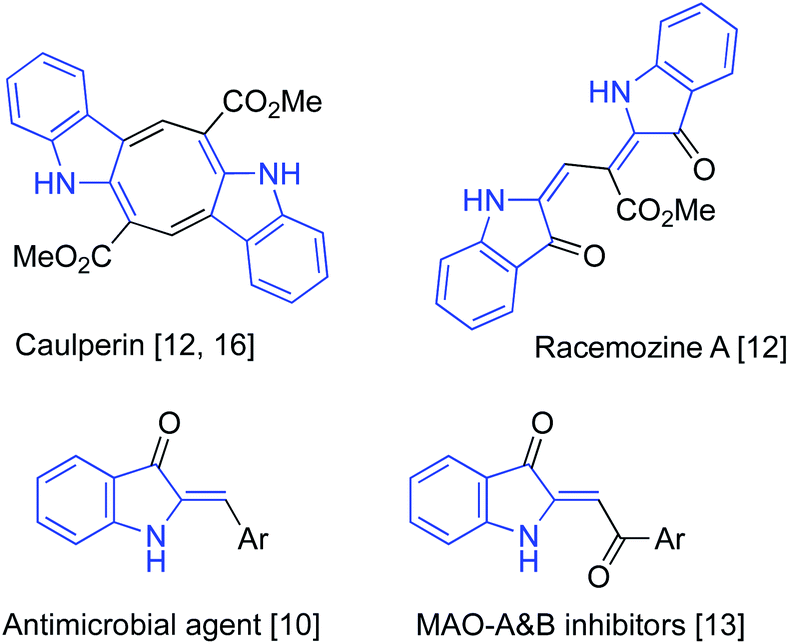 | ||
| Fig. 1 Biologically active 2-alkylideneindolin-3-ones. | ||
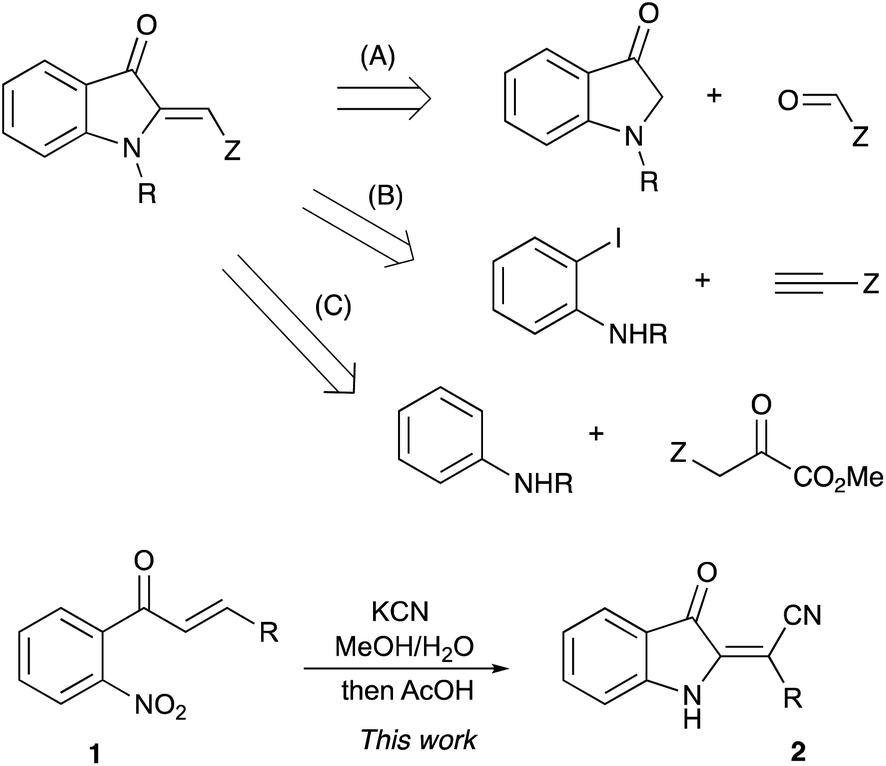 | ||
| Scheme 1 Synthetic approaches to 2-alkylideneindolin-3-ones. | ||
Results and discussion
In the frame of our ongoing project dealing with the synthesis of nitrogen-based heterocyclic compounds and evaluation of their biological activity, we were interested in the preparation of a series of minaprine analogs 4,34–37 possessing an additional amino-group handle at C-2′.38,39 To tackle this task, we decided to employ a routine cyclocondensation of hydrazine with 3-cyanoketone 3 bearing an ortho-nitro group, which was supposed to be routinely reduced and properly modified in subsequent steps (Scheme 2). We planned to access precursor 3 via conjugate addition of hydrogen cyanide to α,β-unsaturated ketones 1.40–43 Although hydrocyanation of conjugate carbonyl compounds is unknown for specific substrates of type 1, possessing ortho-nitro functionality, we did not initially expect any problems with this well-established chemistry.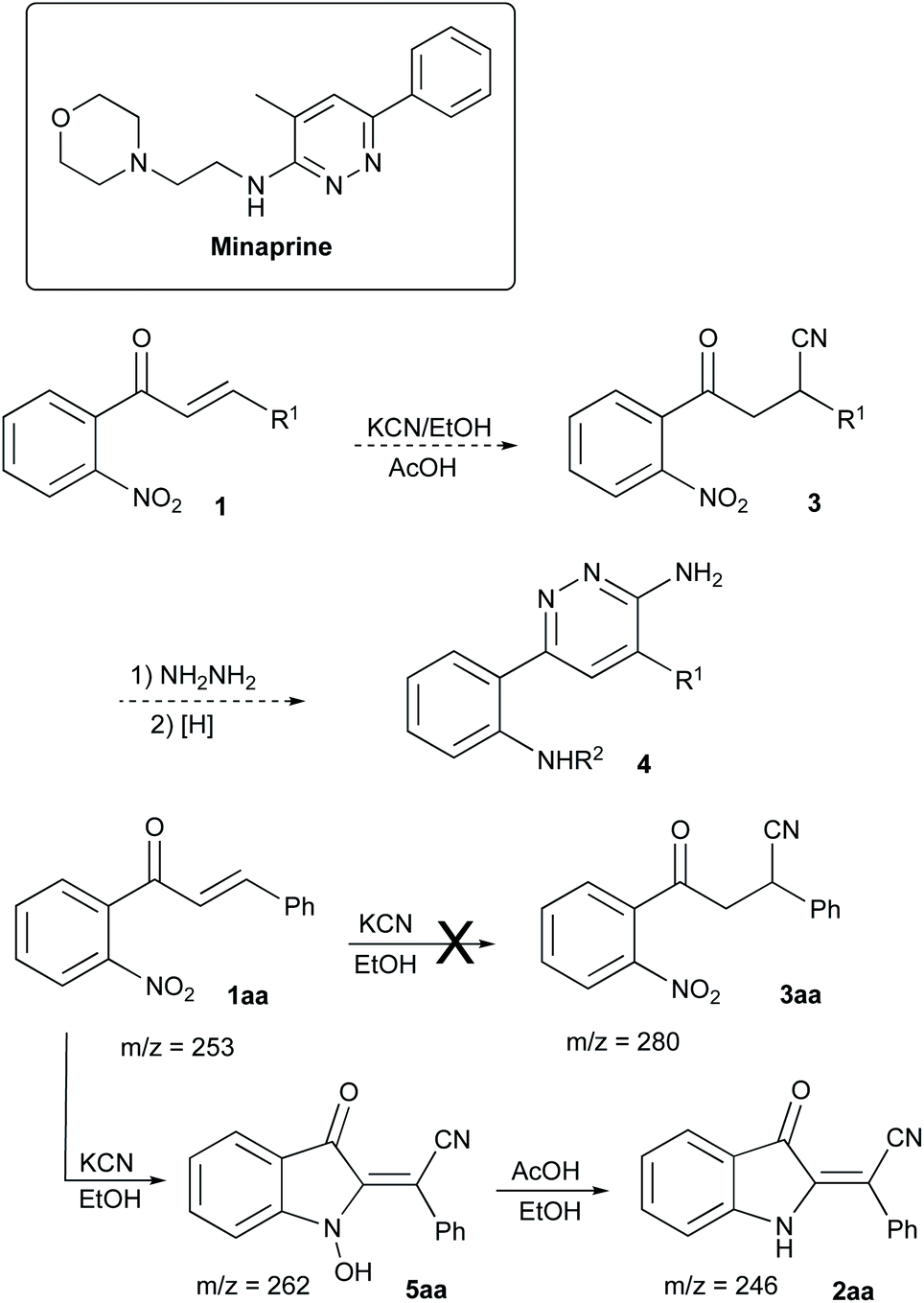 | ||
| Scheme 2 Unexpected assembly of 2-benzylideneindolin-3-one 2a. | ||
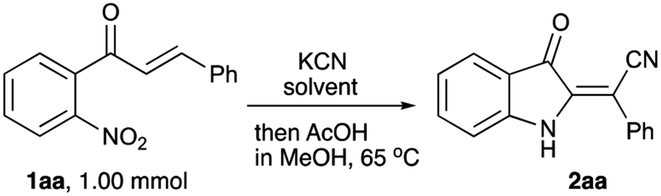
To evaluate the planned synthetic route, chalcone 1aa – prepared by aldol condensation of ortho-nitroacetophenone (6a) with benzaldehyde (7a) – was treated with KCN in EtOH in the presence of acetic acid (1.3 equiv.) at room temperature. Unexpectedly, this reaction provided only marginal yields, which was initially attributed to poor solubility of 1aa in ethanol. To address this situation, we tried to perform this reaction in methanol at elevated temperature, which also failed (Table 1, entry 1). In one of the trial experiments, a mixture of chalcone 1aa and KCN was pre-heated in MeOH to reflux prior to addition of the acetic acid. To our great surprise, within 15 min the reaction mixture turned emerald green. The starting material (m/z 276, M + Na) disappeared, but the expected product 3aa (m/z 303, M + Na) did not form, while cyclic hydroxylamine product 5aa was detected in MS (m/z 285, M + Na) and NMR spectra of the crude reaction mixture instead. The following treatment with acetic acid in boiling methanol led to the conversion of 5aa into indoline 2aa (m/z 269, M + Na), which was isolated in 57% yield (entry 2) as a yellowish-orange crystalline solid with properties identical to those reported in the literature.44 Next, we attempted to increase the loading of KCN, which had a significant positive effect – although not dramatic (entry 3). Nearly the same efficiency was achieved in a test in which the second stage of the reaction was carried out at room temperature for 12 h (entry 4). The best results were obtained in the experiment involving the initial treatment of chalcone 1aa with KCN in methanol in the presence of water (entry 5), which improves the solubility of cyanide. It is important to mention, that ideal homogenization of the reaction mixture seems to be crucial for achieving good yields of 2aa. Indeed, KCN reagent did not dissolve in the mixtures when the test reactions were carried out in THF or acetone even in the presence of additional water. In these cases, product 2aa did not form at all (Table 1, entries 6–9). The reaction in polar aprotic solvents, such as DMSO and DMF, was also tested. It was found that the outcome of these reactions also improves in the presence of water, but the overall performance in these solvents remains relatively poor (entries 10–13).
|
||||
|---|---|---|---|---|
| # | KCN, mg | Solvent, 1.5 mL | H2O, mg | Yield of 2aaa, % |
| a NMR yields are reported.b All reagents were mixed in one pot and the reaction was carried out at reflux for 1 h.c The second stage of the reaction was carried out at RT for 12 h.d KCN is insoluble in this reaction mixture. | ||||
| 1 | 65 | MeOH | 0 | 0b |
| 2 | 40 | MeOH | 0 | 57 |
| 3 | 65 | MeOH | 0 | 65 |
| 4 | 40 | MeOH | 0 | 62c |
| 5 | 40 | MeOH | 200 | 78 |
| 6 | 40 | THF | 0 | 0d |
| 7 | 40 | THF | 200 | 0d |
| 8 | 40 | Acetone | 0 | 0d |
| 9 | 40 | Acetone | 200 | 0d |
| 10 | 40 | DMSO | 0 | 24 |
| 11 | 40 | DMSO | 200 | 50 |
| 12 | 40 | DMF | 0 | 0d |
| 13 | 40 | DMF | 200 | 44 |
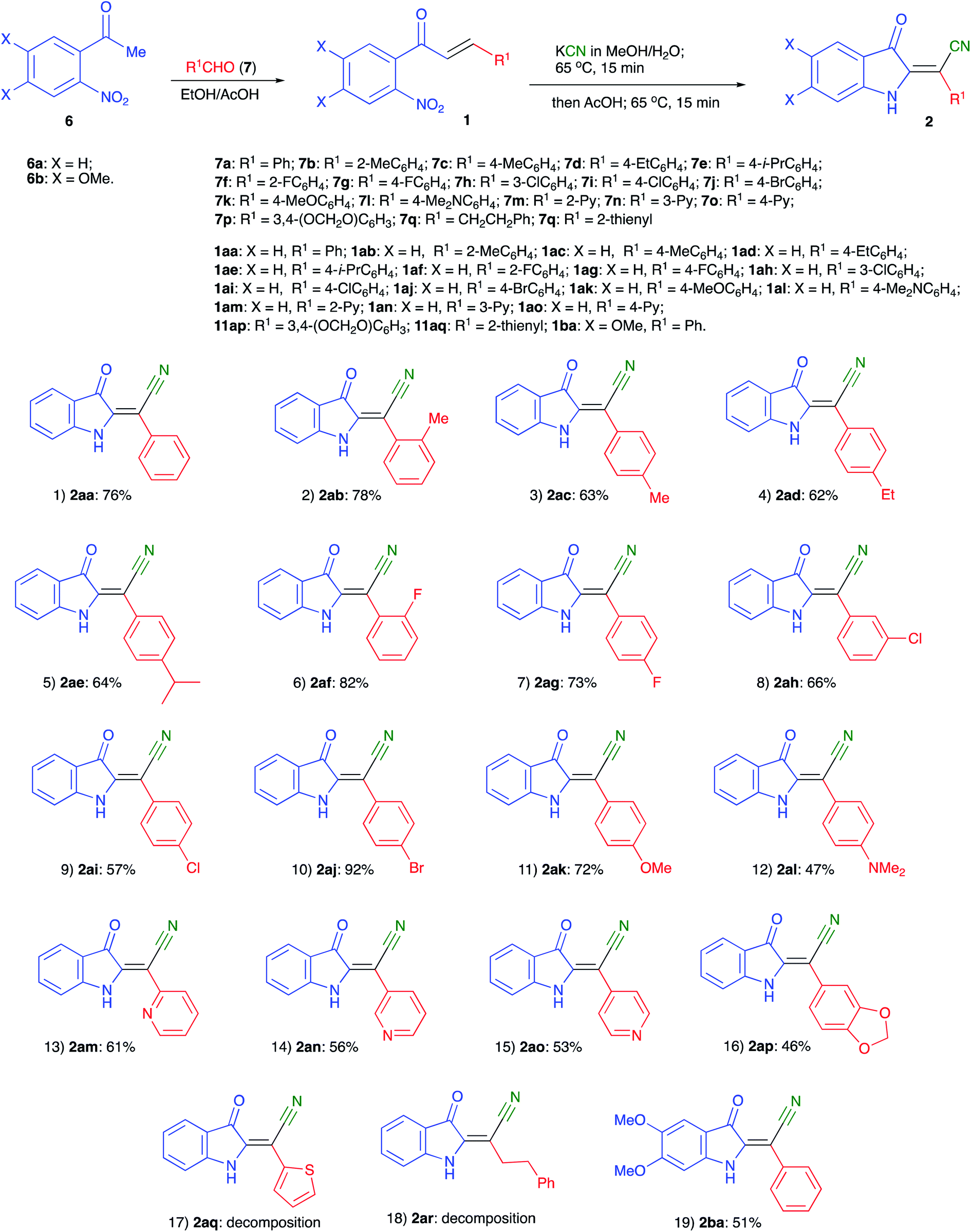
With optimized conditions in hand we decided to evaluate the scope of the reaction of various chalcones and with respect to the nature of substituent R1 (originated from an aldehyde precursor). To this end, a series of chalcones 1 were prepared from o-nitroacetophenones 6 and aldehydes 7. These chalcones were subjected to the reaction with KCN under the optimized reaction conditions. The results are presented in Scheme 3. The preparative reaction of chalcone 1aa proceeded uneventfully affording product 1aa in 76% isolated yield (entry 1). Reactions of chalcones 1ab–1ae, derived from benzaldehydes 7b–e bearing alkyl substituents also proceeded smoothly to yield the corresponding indolines 2ab–2ae in good yields (Scheme 3, entries 2–5). Next, the tolerance to substitution with halogenes was tested. We were pleased to find that the corresponding products 2af–2aj formed in good to high yields (entries 6–10). The reactivity of chalcones 1ak and 2ak derived from electron-rich benzaldehydes 7k,l was also examined (entries 11 and 12). These materials also reacted smoothly, although isolation of product 2al bearing NMe2 substituent proved to be more challenging due to the partial decomposition, which reduced the overall efficiency of the process (entry 12). The same problem was encountered in the attempt to employ pyridine carboxaldehyde derivatives 1am–1ao. The corresponding indolines 2am–2ao formed smoothly, but were isolated in moderate yields (entries 13–15). Reaction of piperonal derivative 1ap was accompanied by a notable decomposition of the target product 2ap, which was isolated in quite marginal yield (entry 16). Such decomposition became much greater issue in the experiments involving chalcones 1aq and 1ar, derived from thiophene-2-carbaldehyde and hydrocinnamic aldehyde, respectively. The corresponding products 2aq and 2ar were not isolated (entries 17 and 18). Finally, the reaction of chalcone 1ba, derived from 1-(4,5-dimethoxy-2-nitrophenyl)ethan-1-one (6b) and benzaldehyde (7a), was also tested. The corresponding product 2ba was isolated in 51% yield (Scheme 3, entry 19), thus confirming the possibility for the installation of additional substituents onto the aromatic ring of the indoline. Formation of the (E)-2-(3-oxoindolin-2-ylidene)-2-arylacetonitrile moiety was unambiguously confirmed by single crystal X-ray diffraction of compound 2ad (CCDC #1992506, Fig. 2).
 | ||
| Scheme 3 Preparation of (E)-2-(3-oxoindolin-2-ylidene)-2-arylacetonitriles via featured cyanide-induced cyclization of chalcones. | ||
 | ||
| Fig. 2 ORTEP drawing of crystal structure of compound 2ad (CCDC #1992506) showing 50% probability thermal ellipsoids and atom numbering scheme. | ||
The putative mechanistic rationale proposed for the featured transformation is shown in Scheme 4. It is assumed that the reaction begins with the Michael-type addition of the CN-anion across the conjugate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of chalcone 1 to afford enolate 8. This enolate triggers a 5-exo-trig cyclization involving the ortho-nitro group in the substrate molecule. Mechanistically related to the Baeyer–Drewson reaction, this step affords cyclic nitronate 9, which should exist in equilibrium with tautomeric cyclic enolate form 10. Subsequent elimination of water would afford 3-oxo-3H-indole N-oxide 11, which should quickly transform into the thermodynamically more stable 1-hydroxy-2-methyleneindolin-3-one form 5. It should be pointed out, that this intermediate was detected in MS and 1H NMR spectra of the crude reaction mixture involving chalcone 1aa (R = Ph). Evidently, the formation of this structure is responsible for the intense color of the reaction mixtures. Finally, upon acidification with acetic acid, emerald-green 5 is reduced into orange-red product 2. Although the precise mechanism of this reduction was not elucidated, we believe it could involve the methanol used as a solvent. Since the product 2 is an enamine, it should exist in tautomeric equilibrium between E and Z forms. Only E-tautomers were observed, suggesting that they are thermodynamically much more favored. This stereochemical outcome could be easily rationalized taking into account greater steric hindrance provided by aryl substituent as compared to nitrile functional group.
C bond of chalcone 1 to afford enolate 8. This enolate triggers a 5-exo-trig cyclization involving the ortho-nitro group in the substrate molecule. Mechanistically related to the Baeyer–Drewson reaction, this step affords cyclic nitronate 9, which should exist in equilibrium with tautomeric cyclic enolate form 10. Subsequent elimination of water would afford 3-oxo-3H-indole N-oxide 11, which should quickly transform into the thermodynamically more stable 1-hydroxy-2-methyleneindolin-3-one form 5. It should be pointed out, that this intermediate was detected in MS and 1H NMR spectra of the crude reaction mixture involving chalcone 1aa (R = Ph). Evidently, the formation of this structure is responsible for the intense color of the reaction mixtures. Finally, upon acidification with acetic acid, emerald-green 5 is reduced into orange-red product 2. Although the precise mechanism of this reduction was not elucidated, we believe it could involve the methanol used as a solvent. Since the product 2 is an enamine, it should exist in tautomeric equilibrium between E and Z forms. Only E-tautomers were observed, suggesting that they are thermodynamically much more favored. This stereochemical outcome could be easily rationalized taking into account greater steric hindrance provided by aryl substituent as compared to nitrile functional group.
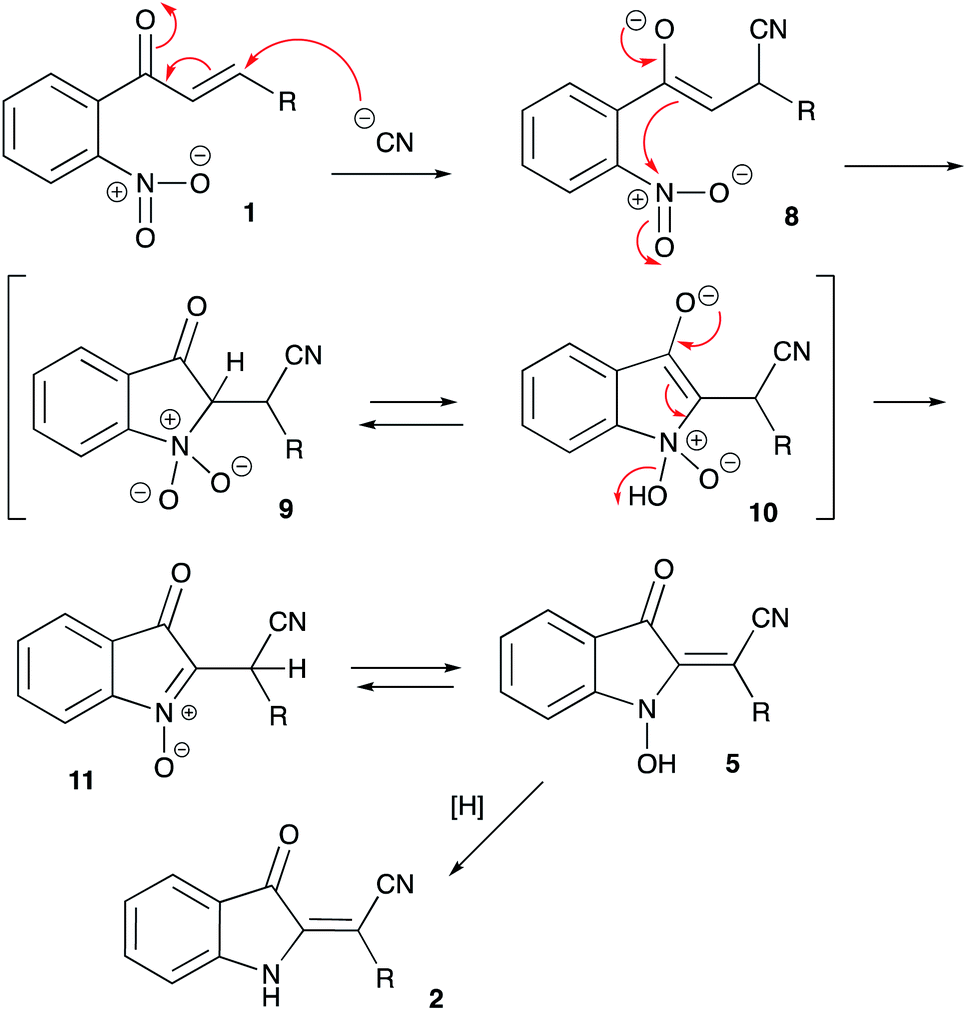 | ||
| Scheme 4 Proposed mechanistic rationale. | ||
In order to avoid utilization of highly toxic KCN reagent, other cyanide ion sources were also tested, such as Me3SiCN and K4[Fe(CN)6]. In both cases, however, formation of the (E)-2-(3-oxoindolin-2-ylidene)-2-arylacetonitrile products was not detected. Evidently, the reaction requires a high concentration of nucleophile, which cannot be achieved in the presence of reagents, slowly releasing free cyanide.
Conclusion
In conclusion, an unusual cascade cyclization triggered by the conjugate addition of the cyanide anion to ortho-nitrosubstituted chalcones was unexpectedly discovered. This novel transformation involves an intramolecular 5-exo-trig attack of an enolate on the electrophilic nitro-group, which is mechanistically related to the Baeyer–Drewson reaction. A series of (E)-2-(3-oxoindolin-2-ylidene)-2-arylacetonitriles was efficiently, obtained in good to excellent yield.Experimental part
General information. 1H and 13C NMR spectra were recorded on a Bruker Avance-III spectrometer (400 or 100 MHz, respectively) equipped with a BBO probe in CDCl3 or DMSO-d6 using TMS as an internal standard. High-resolution mass spectra were registered with a Bruker Maxis spectrometer (electrospray ionization, in MeCN solution, using HCO2Na–HCO2H for calibration). Melting points were measured with a Stuart smp30 apparatus. Unless specified otherwise, all reactions were performed in 5 mL round-bottomed flasks equipped with reflux condensers. The reaction progress and purity of isolated compounds were controlled by TLC on Silufol UV-254 plates, with hexanes/EtOAc mixtures used as eluents. 1-(4,5-Dimethoxy-2-nitrophenyl)ethan-1-one was prepared according to the known procedure45 and had physical and spectral properties identical to those reported in literature. (E)-1-(2-Nitrophenyl)-5-phenylpent-2-en-1-one was obtained according to the known procedure and was identical to the material described in literature.46 All other reagents and solvents were purchased from commercial vendors and used as received.Preparation of chalcones
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4). 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.1 Hz, 1H), 7.77 (t, J = 7.3 Hz, 1H), 7.66 (t, J = 7.4 Hz, 1H), 7.50 (dd, J = 7.7, 4.2 Hz, 3H), 7.39 (d, J = 6.5 Hz, 3H), 7.24 (d, J = 15.9 Hz, 1H), 7.01 (d, J = 16.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.2, 146.9, 146.5, 136.5, 134.2, 134.1, 131.2, 130.7, 129.2 (2C), 129.0, 128.7 (2C), 126.4, 124.7; FTIR (KBr, cm−1): 3741, 3298, 3092, 1647, 1531, 1340, 1277, 1107; HRMS (ES TOF) calc'd for C15H11NNaO3 (M + Na)+ 276.0631, found 276.0634 (1.0 ppm).:2); 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 7.7 Hz, 1H), 7.82–7.72 (m, 1H), 7.71–7.63 (m, 1H), 7.61–7.56 (m, 2H), 7.54 (dd, J = 7.5, 1.0 Hz, 1H), 7.33–7.14 (m, 3H), 6.91 (d, J = 16.1 Hz, 1H), 2.30 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.9, 147.0, 143.8, 138.2, 136.5, 134.1, 133.0, 131.0, 130.9, 130.8, 129.0, 127.1, 126.8, 126.6, 124.6, 19.7; FTIR (KBr, cm−1): 2931, 2363, 1673, 1561, 1525, 1363, 1327, 1218, 1030; HRMS (ES TOF) calc'd for C16H13NNaO3 (M + Na)+ 290.0788, found 290.0783 (1.6 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.2 Hz, 1H), 7.76 (t, J = 7.4 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.50 (d, J = 7.4 Hz, 1H), 7.39 (d, J = 7.9 Hz, 2H), 7.24–7.16 (m, 3H), 6.97 (d, J = 16.2 Hz, 1H), 2.37 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.2, 146.9, 146.7, 141.9, 136.6, 134.1, 131.3, 130.6, 129.9 (2C), 129.0, 128.7 (2C), 125.5, 124.7, 21.7; FTIR (KBr, cm−1): 3067, 1668, 1591, 1524, 1364, 1320, 1230, 1210, 1029; HRMS (ES TOF) calc'd for C16H13N1NaO3 (M + Na)+ 290.0788, found 290.0790 (0.8 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.1 Hz, 1H), 7.77 (dd, J = 7.4, 6.8 Hz, 1H), 7.72–7.62 (m, 1H), 7.59–7.47 (m, 2H), 7.41–7.33 (m, 2H), 7.16 (t, J = 7.5 Hz, 1H), 7.13–7.00 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 193.0, 161.5 (d, J = 254.6 Hz), 146.8, 138.5 (d, J = 3.3 Hz), 136.2, 134.2, 132.7 (d, J = 8.9 Hz), 130.8, 129.1 (d, J = 2.5 Hz), 128.9, 128.3 (d, J = 5.9 Hz), 124.72 (d, J = 3.8 Hz), 124.68, 122.2 (d, J = 11.5 Hz), 116.3 (d, J = 21.7 Hz); FTIR (KBr, cm−1): 3289, 3065, 1651, 1603, 1531, 1340, 1290, 1215; HRMS (ES TOF) calc'd for C15H10FNNaO3 (M + Na)+ 294.0537, found 294.0538 (0.4 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.2 Hz, 1H), 7.82–7.72 (m, 1H), 7.70–7.60 (m, 1H), 7.55–7.44 (m, 3H), 7.21 (d, J = 16.3 Hz, 1H), 7.07 (t, J = 8.6 Hz, 2H), 6.92 (d, J = 16.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.8, 164.4 (d, J = 252.8 Hz), 146.8, 145.0, 136.4, 134.2, 130.8, 130.7 (d, J = 8.7 Hz, 2C), 130.3 (d, J = 3.2 Hz), 128.9, 126.1 (d, J = 2.2 Hz), 124.7, 116.3 (d, J = 22.0 Hz, 2C); FTIR (KBr, cm−1): 3302, 3052, 1651, 1522, 1353, 1286, 1232, 1112; HRMS (ES TOF) calc'd for C15H10FNNaO3 (M + Na)+ 294.0537, found 294.0538 (0.4 ppm).:4), 0.52 (EtOAc/Hex, 1:2); 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.0 Hz, 1H), 7.82–7.73 (m, 1H), 7.71–7.62 (m, 1H), 7.50 (dd, J = 7.5, 1.0 Hz, 1H), 7.46 (s, 1H), 7.34 (tt, J = 15.2, 7.4 Hz, 3H), 7.18 (d, J = 16.3 Hz, 1H), 6.98 (d, J = 16.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.7, 146.8, 144.4, 136.2, 135.9, 135.1, 134.3, 130.93, 130.89, 130.4, 128.9, 128.4, 127.5, 126.7, 124.7; FTIR (KBr, cm−1): 3050, 1647, 1514, 1340, 1284, 1254, 1205, 1099; HRMS (ES TOF) calc'd for C15H10Cl1N1Na1O3 (M + Na)+ 310.0241, found 310.0246 (−1.5 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.28–8.14 (m, 1H), 7.77 (t, J = 7.1 Hz, 1H), 7.70–7.63 (m, 1H), 7.50 (dd, J = 7.6, 1.5 Hz, 1H), 7.43 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 7.20 (d, J = 16.3 Hz, 1H), 6.96 (d, J = 16.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.7, 146.8, 144.7, 137.1, 136.3, 134.3, 132.6, 130.8, 129.8 (2C), 129.4 (2C), 128.9, 126.7, 124.7; FTIR (KBr, cm−1): 3258, 3056, 2868, 1906, 1638, 1527, 1343; HRMS (ES TOF) calc'd for C15H10ClNNaO3 (M + Na)+ 310.0241, found 310.0246 (1.5 ppm).:4); 1H NMR (400 MHz, DMSO) δ 8.21 (d, J = 8.0 Hz, 1H), 7.91 (t, J = 7.3 Hz, 1H), 7.81 (t, J = 7.4 Hz, 1H), 7.76–7.68 (m, 3H), 7.62 (d, J = 8.3 Hz, 2H), 7.44–7.27 (m, 2H); 13C NMR (101 MHz, DMSO) δ 192.2, 146.7, 144.6, 135.3, 134.5, 133.3, 132.0 (2C), 131.5, 130.8 (2C), 129.1, 126.4, 124.6, 124.6; FTIR (KBr, cm−1): 3255, 3061, 1638, 1584, 1527, 1487, 1347, 1307, 1290; HRMS (ES TOF) calc'd for C15H10BrNNaO3 (M + Na)+ 353.9736, found 353.9739 (0.9 ppm).:4), 0.56 (EtOAc/Hex, 1:1). 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.2 Hz, 1H), 7.79–7.71 (m, 1H), 7.69–7.60 (m, 1H), 7.50 (dd, J = 7.5, 1.1 Hz, 1H), 7.45 (d, J = 8.7 Hz, 2H), 7.20 (d, J = 16.2 Hz, 1H), 6.94–6.82 (m, 3H), 3.83 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.1, 162.2, 146.9, 146.5, 136.7, 134.1, 130.6 (2C), 130.5, 129.0, 126.8, 124.7, 124.1, 114.6 (2C), 55.6; FTIR (KBr, cm−1): 3288, 2939, 1654, 1521, 1347, 1251, 1172, 1026; HRMS (ES TOF) calc'd for C16H13NNaO4 (M + Na)+ 306.0737, found 306.0730 (2.3 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.1 Hz, 1H), 7.71 (t, J = 7.1 Hz, 1H), 7.63–7.54 (m, 1H), 7.53–7.43 (m, 1H), 7.36 (d, J = 8.8 Hz, 2H), 7.19 (d, J = 16.0 Hz, 1H), 6.80 (d, J = 16.0 Hz, 1H), 6.61 (d, J = 8.8 Hz, 2H), 3.00 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 192.9, 152.4, 148.0, 146.9, 137.0, 133.8, 130.7, 130.2, 129.0, 124.5, 121.5, 120.8, 111.8 (2C), 40.1 (2C); FTIR (KBr, cm−1): 3096, 3029, 2900, 2820, 1598, 1522, 1433, 1348, 1299, 1237, 1179, 1112; HRMS (ES TOF) calc'd for C17H16N2NaO3 (M + Na)+ 319.1053, found 319.1053 (0.1 ppm).:2). Yield 1.330 g (4.25 mmol, 85%). 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 7.52–7.44 (m, 2H), 7.42–7.32 (m, 3H), 7.20 (d, J = 16.2 Hz, 1H), 6.95 (d, J = 16.2 Hz, 1H), 6.85 (s, 1H), 4.01 (s, 3H), 3.98 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.1, 153.9, 149.7, 145.4, 139.4, 134.1, 131.0, 130.8, 129.1 (2C), 128.6 (2C), 126.7, 110.0, 107.1, 56.8, 56.7; FTIR (KBr, cm−1): 3036, 2977, 1650, 1571, 1514, 1445, 1337, 1287, 1217, 1122; HRMS (ES TOF) calc'd for C17H15NNaO5 (M + Na)+ 336.0842, found 336.0849 (1.9 ppm).:2). Yield 2.525 g (8.5 mmol, 85%). 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.1 Hz, 1H), 7.75 (t, J = 7.1 Hz, 1H), 7.69–7.61 (m, 1H), 7.49 (dd, J = 7.5, 0.9 Hz, 1H), 7.16 (d, J = 16.1 Hz, 1H), 7.03 (d, J = 1.2 Hz, 1H), 6.95 (dd, J = 8.0, 1.2 Hz, 1H), 6.87–6.75 (m, 2H), 6.01 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 192.9, 150.5, 148.6, 146.9, 146.4, 136.6, 134.1, 130.6, 128.9, 128.5, 125.7, 124.7, 124.4, 108.8, 106.8, 101.9; FTIR (film, NaCl, cm−1): 3262, 3101, 3014, 2913, 1645, 1524, 1498, 1447, 1337, 1243, 1106; HRMS (ES TOF) calc'd for C16H11NNaO5 (M + Na)+ 320.0529, found 320.0527 (0.7 ppm).:Hex 1:4) the reaction mixture was diluted with cold water (20 mL) and extracted with EtOAc (4 × 15 mL). Combined organic extracts were washed consecutively with water (3 × 15 mL) and brine (15 mL). After concentration in vacuo the crude product was recrystallized from EtOH to afford the titled compound as colorless solid, mp 101.2–103.5 °C (EtOH), lit.48 mp 102–105 (isopropanol), Rf 0.40 (EtOAc/Hex, 1:1). Yield 1.143 g (0.45 mmol, 90% yield). 1H NMR (400 MHz, CDCl3) δ 8.62 (d, J = 4.1 Hz, 1H), 8.18 (d, J = 8.2 Hz, 1H), 7.81–7.69 (m, 2H), 7.69–7.61 (m, 1H), 7.55–7.46 (m, 2H), 7.41 (d, J = 16.1 Hz, 1H), 7.33–7.22 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 193.1, 152.7, 150.3, 146.7, 144.6, 137.0, 136.4, 134.3, 130.9, 129.7, 128.9, 124.8, 124.7, 124.5; FTIR (KBr, cm−1): 3074, 1752, 1661, 1528, 1431, 1337, 1277, 1247; HRMS (ES TOF) calc'd for C14H10N2NaO3 (M + Na)+ 277.0584, found 277.0593 (3.4 ppm).:4. Yield 1.306 g (4.65 mmol, 93%), pale brown oil, Rf 0.51 (EtOAc/Hex, 1:4); 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 8.2 Hz, 1H), 7.75 (td, J = 7.5, 0.9 Hz, 1H), 7.67–7.61 (m, 1H), 7.49 (dd, J = 7.5, 1.2 Hz, 1H), 7.41 (d, J = 8.1 Hz, 2H), 7.26–7.17 (m, 3H), 6.97 (d, J = 16.3 Hz, 1H), 2.65 (q, J = 7.6 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 193.1, 148.1, 146.8, 146.7, 136.4, 134.1, 131.5, 130.6, 128.9, 128.8 (2C), 128.6 (2C), 125.4, 124.6, 28.9, 15.3; FTIR (KBr, cm−1): 3035, 2970, 2873, 1652, 1596, 1531, 1350, 1210; HRMS (ES TOF) calc'd for C17H15NNaO3 (M + Na)+ 304.0944, found 304.0948 (1.1 ppm).:4. Yield 1.292 g (4.4 mmol, 88%), yellow oil, Rf 0.38 (EtOAc/Hex, 1:4); 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 8.2 Hz, 1H), 7.75 (td, J = 7.5, 0.9 Hz, 1H), 7.67–7.61 (m, 1H), 7.50 (dd, J = 7.5, 1.2 Hz, 1H), 7.44 (d, J = 8.2 Hz, 2H), 7.24 (dd, J = 12.1, 3.8 Hz, 3H), 6.99 (d, J = 16.3 Hz, 1H), 2.92 (septet, J = 6.9 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 193.1, 152.7, 146.8, 146.6, 136.4, 134.1, 131.6, 130.6, 128.9, 128.8 (2C), 127.2 (2C), 125.4, 124.6, 34.2, 23.7 (2C); FTIR (KBr, cm−1): 2958, 1742, 1653, 1591, 151, 1360, 1300, 1280, 1244, 1201, 1109; HRMS (ES TOF) calc'd for C18H17N1Na1O3 (M + Na)+ 318.1101, found 318.1101 (0.1 ppm).:1); 1H NMR (400 MHz, CDCl3) δ 8.66 (d, J = 1.4 Hz, 1H), 8.60 (dd, J = 4.7, 1.2 Hz, 1H), 8.19 (d, J = 8.2 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.82–7.72 (m, 1H), 7.72–7.58 (m, 1H), 7.55–7.42 (m, 1H), 7.33 (dd, J = 7.9, 4.8 Hz, 1H), 7.24 (d, J = 16.4 Hz, 1H), 7.04 (d, J = 16.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.4, 151.7, 150.3, 146.7, 142.1, 136.1, 134.5, 134.4, 131.0, 129.9, 128.9, 128.1, 124.8, 124.0; FTIR (KBr, cm−1): 3308, 3034, 1661, 1581, 1524, 1350, 1256, 1102; HRMS (ES TOF) calc'd for C14H10N2NaO3 (M + Na)+ 277.0584, found 277.0585 (−0.6 ppm).:1); 1H NMR (400 MHz, CDCl3) δ 8.63 (d, J = 5.1 Hz, 2H), 8.19 (d, J = 8.1 Hz, 1H), 7.78 (t, J = 7.4 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.50 (d, J = 7.3 Hz, 1H), 7.32 (d, J = 5.0 Hz, 2H), 7.13 (q, J = 16.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 192.3, 150.8 (2C), 146.7, 142.4, 141.2, 135.9, 134.5, 131.1, 130.1, 128.8, 124.7, 122.1 (2C); FTIR (KBr, cm−1): 3308, 3060, 1661, 1598, 1524, 1413, 1343, 1286, 1109; HRMS (ES TOF) calc'd for C14H10N2NaO3 (M + Na)+ 277.0584, found 277.0576 (2.8 ppm).:4). Yield 1.062 g (4.1 mmol, 82%). 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 8.0 Hz, 1H), 7.75 (t, J = 7.3 Hz, 1H), 7.64 (t, J = 7.5 Hz, 1H), 7.54–7.33 (m, 3H), 7.29–7.17 (m, 1H), 7.12–6.99 (m, 1H), 6.78 (d, J = 15.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.4, 146.7, 139.3, 138.8, 136.3, 134.1, 132.5, 130.7, 130.2, 128.8, 128.5, 124.8, 124.7; FTIR (KBr, cm−1): 3107, 2859, 1651, 1604, 1521, 1420, 1343, 1280, 1253, 1193; HRMS (ES TOF) calc'd for C13H9N1Na1O3S1 (M + Na)+ 282.0195, found 282.0195 (0.0 ppm).
:4). 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.1 Hz, 1H), 7.77 (t, J = 7.3 Hz, 1H), 7.66 (t, J = 7.4 Hz, 1H), 7.50 (dd, J = 7.7, 4.2 Hz, 3H), 7.39 (d, J = 6.5 Hz, 3H), 7.24 (d, J = 15.9 Hz, 1H), 7.01 (d, J = 16.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.2, 146.9, 146.5, 136.5, 134.2, 134.1, 131.2, 130.7, 129.2 (2C), 129.0, 128.7 (2C), 126.4, 124.7; FTIR (KBr, cm−1): 3741, 3298, 3092, 1647, 1531, 1340, 1277, 1107; HRMS (ES TOF) calc'd for C15H11NNaO3 (M + Na)+ 276.0631, found 276.0634 (1.0 ppm).:2); 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 7.7 Hz, 1H), 7.82–7.72 (m, 1H), 7.71–7.63 (m, 1H), 7.61–7.56 (m, 2H), 7.54 (dd, J = 7.5, 1.0 Hz, 1H), 7.33–7.14 (m, 3H), 6.91 (d, J = 16.1 Hz, 1H), 2.30 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.9, 147.0, 143.8, 138.2, 136.5, 134.1, 133.0, 131.0, 130.9, 130.8, 129.0, 127.1, 126.8, 126.6, 124.6, 19.7; FTIR (KBr, cm−1): 2931, 2363, 1673, 1561, 1525, 1363, 1327, 1218, 1030; HRMS (ES TOF) calc'd for C16H13NNaO3 (M + Na)+ 290.0788, found 290.0783 (1.6 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.2 Hz, 1H), 7.76 (t, J = 7.4 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.50 (d, J = 7.4 Hz, 1H), 7.39 (d, J = 7.9 Hz, 2H), 7.24–7.16 (m, 3H), 6.97 (d, J = 16.2 Hz, 1H), 2.37 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.2, 146.9, 146.7, 141.9, 136.6, 134.1, 131.3, 130.6, 129.9 (2C), 129.0, 128.7 (2C), 125.5, 124.7, 21.7; FTIR (KBr, cm−1): 3067, 1668, 1591, 1524, 1364, 1320, 1230, 1210, 1029; HRMS (ES TOF) calc'd for C16H13N1NaO3 (M + Na)+ 290.0788, found 290.0790 (0.8 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.1 Hz, 1H), 7.77 (dd, J = 7.4, 6.8 Hz, 1H), 7.72–7.62 (m, 1H), 7.59–7.47 (m, 2H), 7.41–7.33 (m, 2H), 7.16 (t, J = 7.5 Hz, 1H), 7.13–7.00 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 193.0, 161.5 (d, J = 254.6 Hz), 146.8, 138.5 (d, J = 3.3 Hz), 136.2, 134.2, 132.7 (d, J = 8.9 Hz), 130.8, 129.1 (d, J = 2.5 Hz), 128.9, 128.3 (d, J = 5.9 Hz), 124.72 (d, J = 3.8 Hz), 124.68, 122.2 (d, J = 11.5 Hz), 116.3 (d, J = 21.7 Hz); FTIR (KBr, cm−1): 3289, 3065, 1651, 1603, 1531, 1340, 1290, 1215; HRMS (ES TOF) calc'd for C15H10FNNaO3 (M + Na)+ 294.0537, found 294.0538 (0.4 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.2 Hz, 1H), 7.82–7.72 (m, 1H), 7.70–7.60 (m, 1H), 7.55–7.44 (m, 3H), 7.21 (d, J = 16.3 Hz, 1H), 7.07 (t, J = 8.6 Hz, 2H), 6.92 (d, J = 16.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.8, 164.4 (d, J = 252.8 Hz), 146.8, 145.0, 136.4, 134.2, 130.8, 130.7 (d, J = 8.7 Hz, 2C), 130.3 (d, J = 3.2 Hz), 128.9, 126.1 (d, J = 2.2 Hz), 124.7, 116.3 (d, J = 22.0 Hz, 2C); FTIR (KBr, cm−1): 3302, 3052, 1651, 1522, 1353, 1286, 1232, 1112; HRMS (ES TOF) calc'd for C15H10FNNaO3 (M + Na)+ 294.0537, found 294.0538 (0.4 ppm).:4), 0.52 (EtOAc/Hex, 1:2); 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 8.0 Hz, 1H), 7.82–7.73 (m, 1H), 7.71–7.62 (m, 1H), 7.50 (dd, J = 7.5, 1.0 Hz, 1H), 7.46 (s, 1H), 7.34 (tt, J = 15.2, 7.4 Hz, 3H), 7.18 (d, J = 16.3 Hz, 1H), 6.98 (d, J = 16.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.7, 146.8, 144.4, 136.2, 135.9, 135.1, 134.3, 130.93, 130.89, 130.4, 128.9, 128.4, 127.5, 126.7, 124.7; FTIR (KBr, cm−1): 3050, 1647, 1514, 1340, 1284, 1254, 1205, 1099; HRMS (ES TOF) calc'd for C15H10Cl1N1Na1O3 (M + Na)+ 310.0241, found 310.0246 (−1.5 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.28–8.14 (m, 1H), 7.77 (t, J = 7.1 Hz, 1H), 7.70–7.63 (m, 1H), 7.50 (dd, J = 7.6, 1.5 Hz, 1H), 7.43 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 7.20 (d, J = 16.3 Hz, 1H), 6.96 (d, J = 16.3 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.7, 146.8, 144.7, 137.1, 136.3, 134.3, 132.6, 130.8, 129.8 (2C), 129.4 (2C), 128.9, 126.7, 124.7; FTIR (KBr, cm−1): 3258, 3056, 2868, 1906, 1638, 1527, 1343; HRMS (ES TOF) calc'd for C15H10ClNNaO3 (M + Na)+ 310.0241, found 310.0246 (1.5 ppm).:4); 1H NMR (400 MHz, DMSO) δ 8.21 (d, J = 8.0 Hz, 1H), 7.91 (t, J = 7.3 Hz, 1H), 7.81 (t, J = 7.4 Hz, 1H), 7.76–7.68 (m, 3H), 7.62 (d, J = 8.3 Hz, 2H), 7.44–7.27 (m, 2H); 13C NMR (101 MHz, DMSO) δ 192.2, 146.7, 144.6, 135.3, 134.5, 133.3, 132.0 (2C), 131.5, 130.8 (2C), 129.1, 126.4, 124.6, 124.6; FTIR (KBr, cm−1): 3255, 3061, 1638, 1584, 1527, 1487, 1347, 1307, 1290; HRMS (ES TOF) calc'd for C15H10BrNNaO3 (M + Na)+ 353.9736, found 353.9739 (0.9 ppm).:4), 0.56 (EtOAc/Hex, 1:1). 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.2 Hz, 1H), 7.79–7.71 (m, 1H), 7.69–7.60 (m, 1H), 7.50 (dd, J = 7.5, 1.1 Hz, 1H), 7.45 (d, J = 8.7 Hz, 2H), 7.20 (d, J = 16.2 Hz, 1H), 6.94–6.82 (m, 3H), 3.83 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.1, 162.2, 146.9, 146.5, 136.7, 134.1, 130.6 (2C), 130.5, 129.0, 126.8, 124.7, 124.1, 114.6 (2C), 55.6; FTIR (KBr, cm−1): 3288, 2939, 1654, 1521, 1347, 1251, 1172, 1026; HRMS (ES TOF) calc'd for C16H13NNaO4 (M + Na)+ 306.0737, found 306.0730 (2.3 ppm).:4); 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.1 Hz, 1H), 7.71 (t, J = 7.1 Hz, 1H), 7.63–7.54 (m, 1H), 7.53–7.43 (m, 1H), 7.36 (d, J = 8.8 Hz, 2H), 7.19 (d, J = 16.0 Hz, 1H), 6.80 (d, J = 16.0 Hz, 1H), 6.61 (d, J = 8.8 Hz, 2H), 3.00 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 192.9, 152.4, 148.0, 146.9, 137.0, 133.8, 130.7, 130.2, 129.0, 124.5, 121.5, 120.8, 111.8 (2C), 40.1 (2C); FTIR (KBr, cm−1): 3096, 3029, 2900, 2820, 1598, 1522, 1433, 1348, 1299, 1237, 1179, 1112; HRMS (ES TOF) calc'd for C17H16N2NaO3 (M + Na)+ 319.1053, found 319.1053 (0.1 ppm).:2). Yield 1.330 g (4.25 mmol, 85%). 1H NMR (400 MHz, CDCl3) δ 7.69 (s, 1H), 7.52–7.44 (m, 2H), 7.42–7.32 (m, 3H), 7.20 (d, J = 16.2 Hz, 1H), 6.95 (d, J = 16.2 Hz, 1H), 6.85 (s, 1H), 4.01 (s, 3H), 3.98 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.1, 153.9, 149.7, 145.4, 139.4, 134.1, 131.0, 130.8, 129.1 (2C), 128.6 (2C), 126.7, 110.0, 107.1, 56.8, 56.7; FTIR (KBr, cm−1): 3036, 2977, 1650, 1571, 1514, 1445, 1337, 1287, 1217, 1122; HRMS (ES TOF) calc'd for C17H15NNaO5 (M + Na)+ 336.0842, found 336.0849 (1.9 ppm).:2). Yield 2.525 g (8.5 mmol, 85%). 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 8.1 Hz, 1H), 7.75 (t, J = 7.1 Hz, 1H), 7.69–7.61 (m, 1H), 7.49 (dd, J = 7.5, 0.9 Hz, 1H), 7.16 (d, J = 16.1 Hz, 1H), 7.03 (d, J = 1.2 Hz, 1H), 6.95 (dd, J = 8.0, 1.2 Hz, 1H), 6.87–6.75 (m, 2H), 6.01 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 192.9, 150.5, 148.6, 146.9, 146.4, 136.6, 134.1, 130.6, 128.9, 128.5, 125.7, 124.7, 124.4, 108.8, 106.8, 101.9; FTIR (film, NaCl, cm−1): 3262, 3101, 3014, 2913, 1645, 1524, 1498, 1447, 1337, 1243, 1106; HRMS (ES TOF) calc'd for C16H11NNaO5 (M + Na)+ 320.0529, found 320.0527 (0.7 ppm).:Hex 1:4) the reaction mixture was diluted with cold water (20 mL) and extracted with EtOAc (4 × 15 mL). Combined organic extracts were washed consecutively with water (3 × 15 mL) and brine (15 mL). After concentration in vacuo the crude product was recrystallized from EtOH to afford the titled compound as colorless solid, mp 101.2–103.5 °C (EtOH), lit.48 mp 102–105 (isopropanol), Rf 0.40 (EtOAc/Hex, 1:1). Yield 1.143 g (0.45 mmol, 90% yield). 1H NMR (400 MHz, CDCl3) δ 8.62 (d, J = 4.1 Hz, 1H), 8.18 (d, J = 8.2 Hz, 1H), 7.81–7.69 (m, 2H), 7.69–7.61 (m, 1H), 7.55–7.46 (m, 2H), 7.41 (d, J = 16.1 Hz, 1H), 7.33–7.22 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 193.1, 152.7, 150.3, 146.7, 144.6, 137.0, 136.4, 134.3, 130.9, 129.7, 128.9, 124.8, 124.7, 124.5; FTIR (KBr, cm−1): 3074, 1752, 1661, 1528, 1431, 1337, 1277, 1247; HRMS (ES TOF) calc'd for C14H10N2NaO3 (M + Na)+ 277.0584, found 277.0593 (3.4 ppm).:4. Yield 1.306 g (4.65 mmol, 93%), pale brown oil, Rf 0.51 (EtOAc/Hex, 1:4); 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 8.2 Hz, 1H), 7.75 (td, J = 7.5, 0.9 Hz, 1H), 7.67–7.61 (m, 1H), 7.49 (dd, J = 7.5, 1.2 Hz, 1H), 7.41 (d, J = 8.1 Hz, 2H), 7.26–7.17 (m, 3H), 6.97 (d, J = 16.3 Hz, 1H), 2.65 (q, J = 7.6 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 193.1, 148.1, 146.8, 146.7, 136.4, 134.1, 131.5, 130.6, 128.9, 128.8 (2C), 128.6 (2C), 125.4, 124.6, 28.9, 15.3; FTIR (KBr, cm−1): 3035, 2970, 2873, 1652, 1596, 1531, 1350, 1210; HRMS (ES TOF) calc'd for C17H15NNaO3 (M + Na)+ 304.0944, found 304.0948 (1.1 ppm).:4. Yield 1.292 g (4.4 mmol, 88%), yellow oil, Rf 0.38 (EtOAc/Hex, 1:4); 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 8.2 Hz, 1H), 7.75 (td, J = 7.5, 0.9 Hz, 1H), 7.67–7.61 (m, 1H), 7.50 (dd, J = 7.5, 1.2 Hz, 1H), 7.44 (d, J = 8.2 Hz, 2H), 7.24 (dd, J = 12.1, 3.8 Hz, 3H), 6.99 (d, J = 16.3 Hz, 1H), 2.92 (septet, J = 6.9 Hz, 1H), 1.24 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 193.1, 152.7, 146.8, 146.6, 136.4, 134.1, 131.6, 130.6, 128.9, 128.8 (2C), 127.2 (2C), 125.4, 124.6, 34.2, 23.7 (2C); FTIR (KBr, cm−1): 2958, 1742, 1653, 1591, 151, 1360, 1300, 1280, 1244, 1201, 1109; HRMS (ES TOF) calc'd for C18H17N1Na1O3 (M + Na)+ 318.1101, found 318.1101 (0.1 ppm).:1); 1H NMR (400 MHz, CDCl3) δ 8.66 (d, J = 1.4 Hz, 1H), 8.60 (dd, J = 4.7, 1.2 Hz, 1H), 8.19 (d, J = 8.2 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.82–7.72 (m, 1H), 7.72–7.58 (m, 1H), 7.55–7.42 (m, 1H), 7.33 (dd, J = 7.9, 4.8 Hz, 1H), 7.24 (d, J = 16.4 Hz, 1H), 7.04 (d, J = 16.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.4, 151.7, 150.3, 146.7, 142.1, 136.1, 134.5, 134.4, 131.0, 129.9, 128.9, 128.1, 124.8, 124.0; FTIR (KBr, cm−1): 3308, 3034, 1661, 1581, 1524, 1350, 1256, 1102; HRMS (ES TOF) calc'd for C14H10N2NaO3 (M + Na)+ 277.0584, found 277.0585 (−0.6 ppm).:1); 1H NMR (400 MHz, CDCl3) δ 8.63 (d, J = 5.1 Hz, 2H), 8.19 (d, J = 8.1 Hz, 1H), 7.78 (t, J = 7.4 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.50 (d, J = 7.3 Hz, 1H), 7.32 (d, J = 5.0 Hz, 2H), 7.13 (q, J = 16.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 192.3, 150.8 (2C), 146.7, 142.4, 141.2, 135.9, 134.5, 131.1, 130.1, 128.8, 124.7, 122.1 (2C); FTIR (KBr, cm−1): 3308, 3060, 1661, 1598, 1524, 1413, 1343, 1286, 1109; HRMS (ES TOF) calc'd for C14H10N2NaO3 (M + Na)+ 277.0584, found 277.0576 (2.8 ppm).:4). Yield 1.062 g (4.1 mmol, 82%). 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 8.0 Hz, 1H), 7.75 (t, J = 7.3 Hz, 1H), 7.64 (t, J = 7.5 Hz, 1H), 7.54–7.33 (m, 3H), 7.29–7.17 (m, 1H), 7.12–6.99 (m, 1H), 6.78 (d, J = 15.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.4, 146.7, 139.3, 138.8, 136.3, 134.1, 132.5, 130.7, 130.2, 128.8, 128.5, 124.8, 124.7; FTIR (KBr, cm−1): 3107, 2859, 1651, 1604, 1521, 1420, 1343, 1280, 1253, 1193; HRMS (ES TOF) calc'd for C13H9N1Na1O3S1 (M + Na)+ 282.0195, found 282.0195 (0.0 ppm).Synthesis of (E)-2-(3-oxoindolin-2-ylidene)-2-arylacetonitriles
:2–1:1. Additional purification can be performed by recrystallization from ethanol. The titled compound was obtained as red crystals, mp 233.1–235.9 °C (EtOH), lit.44 mp 236–237 °C, Rf 0.32 (EtOAc/hexanes 1:2), Rf 0.65 (EtOAc/hexanes 1:1). Yield 187 mg (0.76 mmol, 76%). 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1H), 7.70–7.43 (m, 7H), 7.09 (d, J = 7.9 Hz, 1H), 7.02 (t, J = 7.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 184.2, 152.5, 142.5, 137.5, 132.1, 129.3 (2C), 129.1, 128.8 (2C), 124.9, 121.5, 119.5, 118.0, 112.7, 88.8; FTIR (KBr, cm−1): 3308, 3060, 2222, 1708, 1601, 1470, 1447, 1391, 1340, 1213; HRMS (ES TOF) calc'd for C16H10N2NaO (M + Na)+ 269.0685, found 269.0692 (−2.3 ppm).:2. Yield 203 mg (0.78 mmol, 78%), red crystals, mp 201.8–203.5 °C (EtOH), Rf 0.29 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H), 7.65 (d, J = 7.5 Hz, 1H), 7.55 (dd, J = 11.2, 4.1 Hz, 1H), 7.44–7.28 (m, 4H), 7.08–6.87 (m, 2H), 2.31 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 184.0, 152.5, 143.9, 137.6, 136.9, 131.0, 130.8, 130.0, 129.5, 126.8, 124.9, 121.3, 119.6, 117.6, 112.4, 87.7, 19.3; FTIR (KBr, cm−1): 3336, 2215, 1708, 1598, 1377, 1330, 1220, 1139, 1082, 965; HRMS (ES TOF) calc'd for C17H12N2NaO (M + Na)+ 283.0842, found 283.0840 (0.5 ppm).:3. Yield 164 mg (0.63 mmol, 63%), orange crystals, mp 231–234 °C (EtOH), lit.44 mp 236–240 °C, Rf 0.46 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.56 (dd, J = 16.7, 7.8 Hz, 3H), 7.38 (d, J = 7.9 Hz, 2H), 7.09 (d, J = 8.0 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 2.38 (s, 3H); 13C NMR (101 MHz, DMSO) δ 184.2, 152.5, 142.2, 139.0, 137.4, 129.9 (2C), 129.2, 128.8 (2C), 124.9, 121.4, 119.5, 118.0, 112.7, 89.2, 20.9; FTIR (KBr, cm−1): 3296, 2208, 1705, 1591, 1471, 1307, 1243, 811; HRMS (ES TOF) calc'd for C17H12N2NaO+ (M + Na)+ 283.0842, found 283.0844 (0.8 ppm).:3. Yield 170 mg (0.62 mmol, 62%), red crystals, mp 223.2–225.7 °C (EtOH), Rf 0.56 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 7.65 (d, J = 7.5 Hz, 1H), 7.60–7.50 (m, 3H), 7.41 (d, J = 8.1 Hz, 2H), 7.09 (d, J = 8.0 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 2.68 (q, J = 7.5 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 184.2, 152.5, 145.2, 142.1, 137.4, 129.4, 128.9 (2C), 128.8 (2C), 124.9, 121.4, 119.5, 118.0, 112.7, 89.2, 28.0, 15.4; FTIR (KBr, cm−1): 3429, 2933, 2255, 2134, 1665, 1585, 1461, 1370, 1243, 1022, 998, 828; HRMS (ES TOF) calc'd for C18H14N2NaO (M + Na)+ 297.0998, found 297.0997 (0.4 ppm).:3–1:2. Yield 184 mg (0.64 mmol, 64%), red crystals, mp 223.0–226.6 °C (EtOH), lit.44 mp 228–230 °C, Rf 0.47 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 7.64 (d, J = 7.4 Hz, 1H), 7.57 (d, J = 7.5 Hz, 3H), 7.44 (d, J = 7.8 Hz, 2H), 7.09 (d, J = 7.9 Hz, 1H), 7.01 (t, J = 7.3 Hz, 1H), 2.96 (septet, J = 6.7 Hz, 1H), 1.24 (d, J = 6.7 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 184.2, 152.5, 149.7, 142.1, 137.4, 129.6, 128.9 (2C), 127.4 (2C), 124.9, 121.4, 119.5, 118.0, 112.8, 89.2, 33.4, 23.7 (2C); FTIR (KBr, cm−1): 3349, 2959, 2872, 2208, 1708, 1591, 1524, 1464, 1333, 1210; HRMS (ES TOF) calc'd for C19H16N2NaO (M + Na)+ 311.1155, found 311.1155 (0.0 ppm).:2. Yield 216 mg (0.82 mmol, 82%), red crystals, mp 211.1–212.6 °C (EtOH), lit.44 mp 215–216 °C, Rf 0.26 (EtOAc/hexanes 1:2), Rf 0.63 (EtOAc/hexanes 1:1); 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 7.70–7.50 (m, 4H), 7.47–7.33 (m, 2H), 7.12–6.96 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 183.9, 159.4 (d, J = 249.6 Hz), 152.3, 144.2, 137.8, 131.8 (d, J = 8.5 Hz), 131.5 (d, J = 1.8 Hz), 125.4 (d, J = 3.4 Hz), 125.1, 121.6, 119.43 (d, J = 14.7 Hz), 119.40, 117.3, 116.7 (d, J = 20.8 Hz), 112.4, 81.9; FTIR (KBr, cm−1): 3296, 3047, 2222, 1718, 1598, 1454, 1340, 1306; HRMS (ES TOF) calc'd for C16H9FN2NaO (M + Na)+ 287.0591, found 287.0597 (1.9 ppm).:3. Yield 192 mg (0.73 mmol, 73%), orange crystals, mp 281.1–282.8 °C (EtOH), lit.44 mp 282–284 °C, Rf 0.54 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 7.78–7.62 (m, 3H), 7.58 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 8.4 Hz, 2H), 7.08 (d, J = 7.9 Hz, 1H), 7.02 (t, J = 7.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 184.2, 162.1 (d, J = 247.8 Hz), 152.5, 142.7, 137.5, 131.3 (d, J = 8.7 Hz, 2C), 128.5 (d, J = 3.1 Hz), 124.9, 121.5, 119.5, 118.0, 116.4 (d, J = 22.0 Hz, 2C), 112.7, 87.8; FTIR (KBr, cm−1): 3302, 2222, 1715, 1608, 1511, 1468, 1330, 1240, 1213, 1103, 965, 844; HRMS (ES TOF) calc'd for C16H9FN2NaO (M + Na)+ 287.0591, found 287.0590 (0.5 ppm).:3. Yield 185 mg (0.66 mmol, 66%), orange solid, mp 267–269 °C (EtOH), Rf 0.57 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 7.66 (d, J = 7.5 Hz, 2H), 7.64–7.51 (m, 4H), 7.09 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 7.4 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) 184.2, 152.4, 143.2, 137.6, 134.2, 133.9, 131.2, 129.0, 128.5, 127.6, 125.0, 121.7, 119.4, 117.7, 112.7, 87.0; FTIR (KBr, cm−1): 3289, 2215, 1709, 1458, 1243, 1096, 1016; HRMS (ES TOF) calc'd for C16H9ClN2NaO+ (M + Na)+ 303.0296, found 303.0293 (1.0 ppm).:2–1:1. Yield 160 mg (0.57 mmol, 57%), orange crystals, mp 285.9–287.8 °C (EtOH), Rf 0.22 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 10.55 (s, 1H), 7.74–7.51 (m, 6H), 7.13–6.94 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 184.2, 152.4, 142.9, 137.6, 133.7, 131.0, 130.7 (2C), 129.4 (2C), 125.0, 121.6, 119.5, 117.8, 112.7, 87.5; FTIR (KBr, cm−1): 3282, 2222, 1715, 1601, 1468, 1407, 1336, 1250, 1096, 1015, 841; HRMS (ES TOF) calc'd for C16H9ClN2NaO (M + Na)+ 303.0296, found 303.0297 (0.5 ppm).:4–1:2. Yield 297 mg (0.92 mmol, 92%), red crystals, mp 278.8–282.5 °C (EtOH), Rf 0.66 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 10.55 (s, 1H), 7.76 (d, J = 8.1 Hz, 2H), 7.65 (d, J = 7.5 Hz, 1H), 7.58 (d, J = 8.1 Hz, 3H), 7.07 (d, J = 8.0 Hz, 1H), 7.03 (t, J = 7.5 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 184.2, 152.4, 142.9, 137.6, 132.3 (2C), 131.4, 130.9 (2C), 125.0, 122.4, 121.6, 119.4, 117.7, 112.7, 87.5; FTIR (KBr, cm−1): 3282, 3060, 2215, 1712, 1602, 1487, 1464, 1407, 1333, 1243; HRMS (ES TOF) calc'd for C16H9BrN2NaO (M + Na)+ 346.9790, found 346.9790 (0.2 ppm).:2–1:1. Yield 199 mg (0.72 mmol, 72%), red crystals, mp 250.1–251.1 °C (EtOH), lit.44 mp 245–247 °C, Rf 0.23 (EtOAc/hexanes 1:2), Rf 0.43 (EtOAc/hexanes 1:1); 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 7.60 (td, J = 15.9, 7.6 Hz, 4H), 7.11 (dd, J = 14.9, 8.4 Hz, 3H), 7.01 (t, J = 7.4 Hz, 1H), 3.84 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 184.1, 159.8, 152.4, 141.6, 137.3, 130.4 (2C), 124.8, 124.1, 121.3, 119.6, 118.0, 114.8 (2C), 112.7, 89.4, 55.5; FTIR (KBr, cm−1): 3289, 3060, 2208, 1705, 1594, 1300, 1246, 1176; HRMS (ES TOF) calc'd for C17H12N2NaO2 (M + Na)+ 299.0791, found 299.0794 (1.0 ppm).:1. Yield 135 mg (0.47 mmol, 47%), violet crystals, mp 234.8–237.6 °C (EtOH), lit.44 mp 220–225 °C, Rf 0.20 (EtOAc/hexanes 1:2), Rf 0.69 (EtOAc/hexanes 1:1); 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 7.63 (d, J = 7.4 Hz, 1H), 7.55 (t, J = 9.7 Hz, 3H), 7.12 (d, J = 8.0 Hz, 1H), 7.00 (t, J = 7.3 Hz, 1H), 6.87 (d, J = 8.6 Hz, 2H), 3.01 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 183.7, 152.2, 150.5, 139.7, 136.8, 130.0 (2C), 124.5, 121.0, 119.8, 118.6, 118.0, 112.8, 112.3 (2C), 91.4, 39.8 (2C); FTIR (KBr, cm−1): 3315, 2899, 2798, 2201, 1698, 1608, 1521, 1364, 1323, 1199; HRMS (ES TOF) calc'd for C18H15N3NaO (M + Na)+ 312.1107, found 312.1110 (−0.8 ppm).:2. Yield 151 mg (0.61 mmol, 61%), purple crystals, mp 202.6–204.9 °C (EtOH), Rf 0.36 (EtOAc/hexanes 1:2); 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.76 (d, J = 3.9 Hz, 1H), 8.00 (t, J = 7.2 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.72–7.54 (m, 2H), 7.49–7.28 (m, 2H), 7.05 (t, J = 7.4 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 185.2, 152.4, 151.8, 149.2, 143.2, 138.0, 137.6, 125.0, 122.6, 122.5, 122.2, 119.1, 116.6, 113.5, 85.0; FTIR (KBr, cm−1): 3228, 3121, 2214, 1708, 1591, 1464, 1434, 1343, 1236; HRMS (ES TOF) calc'd for C15H9N3NaO (M + Na)+ 270.0638, found 270.0640 (0.9 ppm).:3. Yield 131 mg (0.53 mmol, 53%), red crystals, mp 266.3–268.4 °C (EtOH), Rf 0.17 (EtOAc), 0.65 (EtOH/EtOAc 1:3); 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 8.74 (d, J = 5.7 Hz, 2H), 7.67 (d, J = 7.5 Hz, 1H), 7.60 (t, J = 7.1 Hz, 3H), 7.10 (d, J = 8.0 Hz, 1H), 7.06 (t, J = 7.4 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 184.3, 152.3, 150.5 (2C), 143.8, 140.1, 137.8, 125.2, 122.9 (2C), 122.1, 119.3, 117.2, 112.8, 85.3; FTIR (KBr, cm−1): 2993, 2221, 1742, 1718, 1598, 1557, 1517, 1373, 1250, 1206; HRMS (ES TOF) calc'd for C15H9N3NaO (M + Na)+ 270.0638, found 270.0630 (2.7 ppm).:2). Yield 133 mg (0.46 mmol, 46%). 1H NMR (400 MHz, DMSO-d6) δ 10.41 (s, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.23–7.07 (m, 4H), 7.01 (t, J = 7.4 Hz, 1H), 6.14 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 184.1, 152.4, 148.1, 148.0, 141.9, 137.4, 125.7, 124.8, 123.6, 121.4, 119.6, 118.0, 112.7, 109.2, 108.9, 101.9, 89.2; FTIR (film, NaCl, cm−1): 3316, 2926, 2208, 1712, 1595, 1481, 1350, 1247, 1206, 1046; HRMS (ES TOF) calc'd for C17H10N2NaO3 (M + Na)+ 313.0584, found 313.0586 (0.8 ppm).:1. Yield 156 mg (0.51 mmol, 51%), purple crystals, mp 205.2–207.6 °C (EtOH), Rf 0.47 (EtOAc/hexanes 1:1); 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 7.62 (d, J = 7.5 Hz, 2H), 7.55 (t, J = 7.5 Hz, 2H), 7.47 (t, J = 7.1 Hz, 1H), 7.07 (s, 1H), 6.62 (s, 1H), 3.85 (s, 3H), 3.75 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 181.9, 157.8, 150.2, 144.9, 143.9, 132.3, 129.3 (2C), 129.0, 128.8 (2C), 118.0, 110.4, 105.8, 95.9, 88.5, 56.1, 55.9; FTIR (KBr, cm−1): 3282, 3000, 2839, 2215, 1682, 1598, 1491, 1441, 1323, 1203, 1172; HRMS (ES TOF) calc'd for C18H14N2NaO3 (M + Na)+ 329.0897, found 329.0901 (1.3 ppm).It should be pointed out, that preparation of basic compounds, containing dimethylamine functionality (2al) or pyridine ring (2am–2ao) requires twice more acetic acid (80 mg) at the second stage of the procedure. It is also worth mentioning that these compounds slowly decompose in solutions of ethyl acetate or acetone, but perfectly shelf-stable in crystalline form.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Science Foundation (grant #19-73-00091).Notes and references
- R. Hoessel, S. Leclerc, J. A. Endicott, M. E. M. Nobel, A. Lawrie, P. Tunnah, M. Leost, E. Damiens, D. Marie, D. Marko, E. Niederberger, W. Tang, G. Eisenbrand and L. Meijer, Nat. Cell Biol., 1999, 1, 60–67 CrossRef CAS PubMed.
- S. Leclerc, M. Garnier, R. Hoessel, D. Marko, J. A. Bibb, G. L. Snyder, P. Greengard, J. Biernat, Y.-Z. Wu, E.-M. Mandelkow, G. Eisenbrand and L. Meijer, J. Biol. Chem., 2001, 276, 251–260 CrossRef CAS PubMed.
- L. Meijer, A.-L. Skaltsounis, P. Magiatis, P. Polychronopoulos, M. Knockaert, M. Leost, X. P. Ryan, C. A. Vonica, A. Brivanlou, R. Dajani, C. Crovace, C. Tarricone, A. Musacchio, S. M. Roe, L. Pearl and P. Greengard, Chem. Biol., 2003, 10, 1255–1266 CrossRef CAS PubMed.
- P. Polychronopoulos, P. Magiatis, A.-L. Skaltsounis, V. Myrianthopoulos, E. Mikros, A. Tarricone, A. Musacchio, S. M. Roe, L. Pearl, M. Leost, P. Greengard and L. Meijer, J. Med. Chem., 2004, 47, 935–946 CrossRef CAS PubMed.
- J. Seidler, S. L. McGovern, T. N. Doman and B. K. Shoichet, J. Med. Chem., 2003, 46, 4477–4486 CrossRef CAS PubMed.
- L. Wang, G.-B. Zhou, P. Liu, J.-H. Song, Y. Liang, X.-J. Yan, F. Xu, B.-S. Wang, J.-H. Mao, Z.-X. Shen, S.-J. Chen and Z. Chen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4826–4831 CrossRef CAS PubMed.
- A. Einhorn, Ber. Dtsch. Chem. Ges., 1884, 17, 2026–2028 CrossRef.
- L. E. Hinkel, E. E. Ayling and W. H. Morgan, J. Chem. Soc., 1932, 985–987 CAS.
- J. R. McKee and M. Zanger, J. Chem. Educ., 1991, 68, A242–A244 CrossRef CAS.
- V. L. Gein, V. V. Tatarinov, N. A. Rassudikhina, M. I. Vakhrin and E. V. Voronina, Pharm. Chem. J., 2011, 45, 231–232 CrossRef CAS.
- N. A. Lack, P. Axerio-Cilies, P. Tavassoli, F. G. Han, K. H. Chan, C. Feau, E. LeBlanc, E. T. Guns, R. K. Guy, P. S. Rennie and A. Cherkasov, J. Med. Chem., 2011, 54, 8563–8573 CrossRef CAS PubMed.
- N. A. Lack, P. Axerio-Cilies, P. Tavassoli, F. Q. Han, K. H. Chan, C. Feau, E. LeBlanc, E. T. Guns, R. K. Guy, P. S. Rennie and A. Cherkasov, J. Med. Chem., 2012, 55, 565 CrossRef CAS.
- D.-Q. Liu, S.-C. Mao, H.-Y. Zhang, X.-Q. Yu, M.-T. Feng, B. Wang, L.-H. Feng and Y.-W. Guo, Fitoterapia, 2013, 91, 15–20 CrossRef CAS PubMed.
- A. E. Medvedev, A. S. Ivanov, N. S. Kamyshanskaya, A. Z. Kirkel, T. A. Moskvitina, V. Z. Gorkin, N. Y. Li and V. Y. Marshakov, Biochem. Mol. Biol. Int., 1995, 36, 113–122 CAS.
- A. E. Medvedev, A. S. Ivanov, A. V. Veselovsky, V. S. Skvortsov and A. I. Archakov, J. Chem. Inf. Comput. Sci., 1996, 36, 664–671 CrossRef CAS PubMed.
- L. Ornano, Y. Donno, C. Sanna, M. Ballero, M. Serafini and A. Bianco, Nat. Prod. Res., 2014, 28, 1795–1799 CrossRef CAS PubMed.
- H. M. Roaiah, K. M. Ahmed, N. M. Fawzy, J. Wietrzyk, A. Pawlik, M. M. Ali and A. M. Soliman, Int. J. Pharm. Sci. Rev. Res., 2016, 36, 129–136 CAS.
- P. Langer, J. T. Anders, K. Weisz and J. Jaehnchen, Chem.–Eur. J., 2003, 9, 3951–3964 CrossRef CAS PubMed.
- S. Blechert, R. Knier, H. Schroers and T. Wirth, Synthesis, 1995, 592–604, DOI:10.1055/s-1995-3950.
- E. Wenkert and S. Liu, Synthesis, 1992, 323–327, DOI:10.1055/s-1992-26101.
- A. Buzas and J. Y. Merour, Synthesis, 1989, 458–461, DOI:10.1055/s-1989-27289.
- W. Shen, C. A. Coburn, W. G. Bornmann and S. J. Danishefsky, J. Org. Chem., 1993, 58, 611–617 CrossRef CAS.
- K. Paulvannan and J. R. Stille, J. Org. Chem., 1994, 59, 1613–1620 CrossRef CAS.
- C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232–10236 CrossRef CAS PubMed.
- J.-Y. Merour, L. Chichereau, E. Desarbre and P. Gadonneix, Synthesis, 1996, 519–524, DOI:10.1055/s-1996-4236.
- H. M. Sim, K. Y. Loh, W. K. Yeo, C. Y. Lee and M. L. Go, ChemMedChem, 2011, 6, 713–724 CrossRef CAS PubMed.
- F. Souard, S. Okombi, C. Beney, S. Chevalley, A. Valentin and A. Boumendjel, Bioorg. Med. Chem., 2010, 18, 5724–5731 CrossRef CAS PubMed.
- J. Zhou, B. Wang, X.-H. He, L. Liu, J. Wu, J. Lu, C. Peng, C.-L. Rao and B. Han, J. Org. Chem., 2019, 84, 5450–5459 CrossRef CAS PubMed.
- Z. W. An, M. Catellani and G. P. Chiusoli, J. Organomet. Chem., 1990, 397, C31–C32 CrossRef CAS.
- M. Genelot, A. Bendjeriou, V. Dufaud and L. Djakovitch, Appl. Catal., A, 2009, 369, 125–132 CrossRef CAS.
- M. Genelot, V. Dufaud and L. Djakovitch, Tetrahedron, 2011, 67, 976–981 CrossRef CAS.
- R. Li, X. Qi and X.-F. Wu, Org. Biomol. Chem., 2017, 15, 6905–6908 RSC.
- V. L. Gein, A. V. Demeneva, N. A. Rassudikhina and M. I. Vakhrin, Russ. J. Org. Chem., 2006, 42, 617–618 CrossRef CAS.
- J.-M. Contreras, Y. M. Rival, S. Chayer, J.-J. Bourguignon and C. G. Wermuth, J. Med. Chem., 1999, 42, 730–741 CrossRef CAS PubMed.
- M. Muramatsu, J. Tamaki-Ohashi, C. Usuki, H. Araki, S. Chaki and H. Aihara, Eur. J. Pharmacol., 1988, 153, 89–95 CrossRef CAS PubMed.
- A. Turck, N. Ple, L. Mojovic and G. Queguiner, Bull. Soc. Chim. Fr., 1993, 130, 488–492 CAS.
- C. G. Wermuth, G. Schlewer, J. J. Bourguignon, G. Maghioros, M. J. Bouchet, C. Moire, J. P. Kan, P. Worms and K. Biziere, J. Med. Chem., 1989, 32, 528–537 CrossRef CAS.
- A. Abdel Hamid Deeb, F. Abdel Rahman El-Mariah and H. K. Abd El-Mawgoud, Eur. J. Chem., 2015, 6, 211–218 CrossRef CAS.
- B. K. Albrecht, A. Cote, T. Crawford, M. Duplessis, A. C. Good, Y. Leblanc, S. R. Magnuson, C. G. Nasveschuk, F. A. Romero, Y. Tang and A. M. Taylor, WO2016138114A1, 2016.
- C. F. H. Allen and R. K. Kimball, Org. Synth., 1930, 10, 80–81 CrossRef CAS.
- F. G. Baddar and S. Sherif, J. Chem. Soc., 1960, 2309–2312, 10.1039/JR9600002309.
- K. A. Berryman, A. M. Doherty, J. J. Edmunds, W. C. Patt, M. S. Plummer and J. T. Repine, US Pat., US5691373A, 1997.
- J. A. Ciller, C. Seoane and J. L. Soto, Liebigs Ann. Chem., 1985, 51–57, DOI:10.1002/jlac.198519850106.
- V. S. Velezheva, P. J. Brennan, V. Y. Marshakov, D. V. Gusev, I. N. Lisichkina, A. S. Peregudov, L. N. Tchernousova, T. G. Smirnova, S. N. Andreevskaya and A. E. Medvedev, J. Med. Chem., 2004, 47, 3455–3461 CrossRef CAS PubMed.
- A. V. Butin, S. K. Smirnov, T. A. Stroganova, W. Bender and G. D. Krapivin, Tetrahedron, 2007, 63, 474–491 CrossRef CAS.
- Z. Lin, Z. Hu, X. Zhang, J. Dong, J.-B. Liu, D.-Z. Chen and X. Xu, Org. Lett., 2017, 19, 5284–5287 CrossRef CAS PubMed.
- R. P. Barnes, J. H. Graham and M. A. S. Qureshi, J. Org. Chem., 1963, 28, 2890–2893 CrossRef CAS.
- J. R. Carson, R. J. Carmosin, J. L. Vaught, J. F. Gardocki, M. J. Costanzo, R. B. Raffa and H. R. Almond, Jr, J. Med. Chem., 1992, 35, 2855–2863 CrossRef CAS PubMed.
- A. E. Jungk and G. M. J. Schmidt, J. Chem. Soc. B, 1970, 1427–1434, 10.1039/j29700001427.
- R. A. Bunce and B. Nammalwar, J. Heterocycl. Chem., 2011, 48, 613–619 CrossRef CAS.
- B. C. Mahanta, P. L. Nayak and M. K. Rout, J. Inst. Chem., 1970, 42, 49–52 CAS.
- H. V. Kamath and S. N. Kulkarni, Synthesis, 1978, 931–932, DOI:10.1055/s-1978-24946.
- F. Zhao, Q.-J. Zhao, J.-X. Zhao, D.-Z. Zhang, Q.-Y. Wu and Y.-S. Jin, Chem. Nat. Compd., 2013, 49, 206–214 CrossRef CAS.
- V. Colotta, D. Catarzi, F. Varano, G. Filacchioni, L. Cecchi, A. Galli and C. Costagli, J. Med. Chem., 1996, 39, 2915–2921 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data. CCDC 1992506. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03520c |
| This journal is © The Royal Society of Chemistry 2020 |