
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of symmetric imidazolium ionic liquids N,N-disubstituted with long alkyl chains†
Giacomo Damilano ,
Demian Kalebić,
Koen Binnemans and
Wim Dehaen*
,
Demian Kalebić,
Koen Binnemans and
Wim Dehaen*
KU Leuven, Department of Chemistry, Celestijnenlaan 200F – P.O. Box 2404, B-3001 Leuven, Belgium. E-mail: wim.dehaen@kuleuven.be
First published on 3rd June 2020
Abstract
The Debus–Radziszewski imidazole synthesis was adapted to directly yield long-chain imidazolium ionic liquids. Imidazolium acetate ionic liquids with side-chains up to sixteen carbon atoms were synthesised in excellent yields via an on-water, one-pot reaction. The imidazolium acetate ILs acted as surfactants when dissolved in various solvents. The imidazolium acetate ionic liquids were also derivatised via an acid metathesis to the chloride, nitrate, and hydrogen oxalate derivatives. The thermal behaviour of all the ionic liquids was determined via thermogravimetric and calorimetric analysis.
Introduction
Generally comprised of an organic asymmetrical bulky cation and an inorganic or organic anion, room temperature ionic liquids (ILs) have been defined as salts that are liquid at about or below 100 °C (arbitrary temperature). Being salts, ionic liquids are generally non-volatile and non-flammable. This is in clear contrast to common organic solvents, which are often flammable volatile organic compounds (VOCs). In addition, ionic liquids generally hold a wide liquid range, and a marked chemical, electrochemical, and thermal stability. Because of these features, ionic liquids have fascinated many researchers as alternative solvents with a significant relevance for environmentally sustainable applications.Usually classified by cation, common classes of ionic liquids are imidazolium, ammonium, pyrrolidinium, phosphonium and pyridinium-based. Amongst these, the research on imidazolium ILs advanced swiftly because of their remarkable molecular structure.1,2 The heterocyclic structure is at the origin of the stability of imidazolium compounds and the cause of the C2 proton acidity, which is a central feature of the imidazolium ILs reactivity.3–5
Previous research focused on fundamental studies, which assessed the ILs potential and the suitability to various applications.2,6–9 Imidazolium ILs are of special interest because of the non-innocent nature displayed by the imidazolium cation, which acted not only as a very stable heterocycle but also as an expedient precursor of N-heterocyclic carbenes.7,10–14 This fostered the development of a variety of synthetic procedures.9,15,16 In spite of the importance of green scalable synthetic procedures for ionic liquids production, advanced synthetic procedures are only lately surfacing.17–24
For example, imidazolium ionic liquids synthesis is generally carried out via a Menshutkin reaction between an N-alkyl imidazole and an alkyl halide (Scheme 1). Quarternisation is usually attained via a bimolecular nucleophilic substitution (SN2) between an imidazole and an alkylation agent (often an alkyl halide), which results in the formation of an imidazolium salt. This synthetic pathway is hardly sustainable, both economically and environmentally. Moreover, the quarternisation reaction often involves the use of costly hazardous and harmful chemicals, polar organic solvents, and elevated temperatures for long reaction times, which are conditions not amenable to an industrial scale procedure.
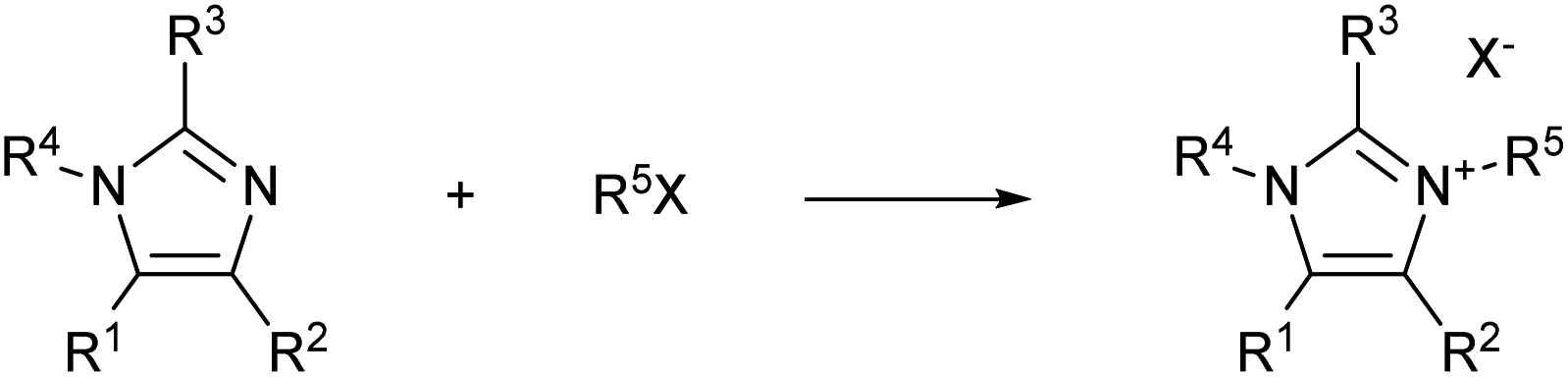 | ||
| Scheme 1 General reaction scheme of the Menshutkin reaction. | ||
Moreover, if the targeted anion is not the leaving group of the alkylation agent, it is impractical to eventually exchange it with another one. A nearly complete exchange can be attained via forcing the reaction equilibrium. A common way to proceed is via an halide precipitation with silver salts (when the anion is a halide).25 However, silver salts are expansive both in purchase and because of the need for appropriate recycling procedures. Even so, salt metathesis rarely results in the material devoid of trace impurities, which are often influential in the final use of the product. This can be detrimental to some applications. For example, trace halides can deactivate an expensive metal, impeding the use of reactive metals and metal complexes in such poisoned media. Hence, this path is not suitable as a generic green synthetic procedure with a wide applicability.
In a recent study, the Debus–Radziszewski imidazole synthesis was modified to yield directly imidazolium ionic liquids in flow.18,26 The Debus–Radziszewski reaction is a four component one-pot cycloaddition directed to the synthesis of imidazoles (Scheme 2). The reaction was developed in the 1880s and its wide scope has been amply studied, but the reaction gives often rather limited yields.26–31 The modification of this reaction for the production of imidazolium ionic liquids was very effective. It improved the product yields and allowed the direct achievement of imidazolium salts in a one-pot reaction from cheap and widely available starting materials. Differently from the Debus–Radziszewski procedure, the scope of the modified imidazolium synthesis was limited to the synthesis of unsubstituted, short-chain, and symmetrical imidazolium ionic liquids (Scheme 3). This limitation was dictated both by the incompatibility of enolisable materials (supposedly because of aldol-type self-condensation) and by the lack of purification and synthetic procedures capable of attaining long-chain N,N-disubstituted imidazolium derivatives.
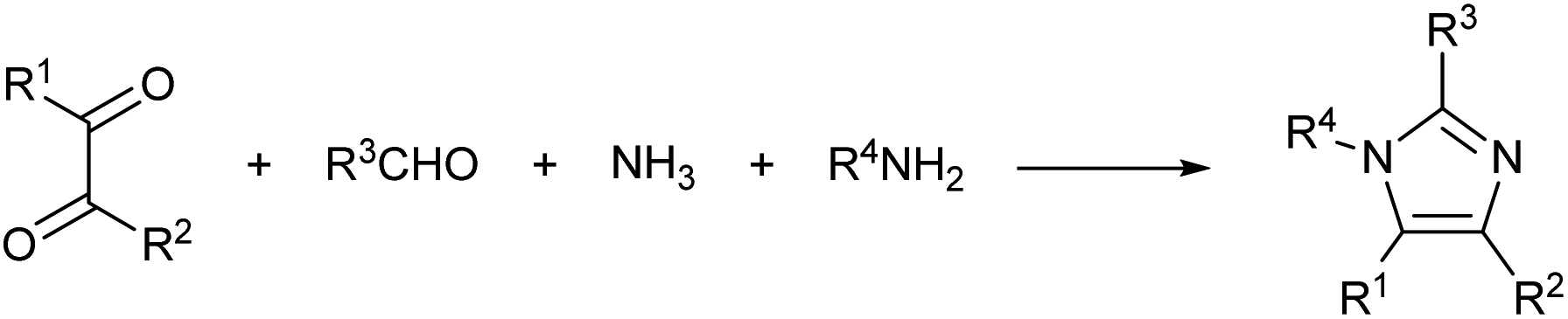 | ||
| Scheme 2 General reaction scheme of the Debus–Radziszewski imidazole synthesis. | ||
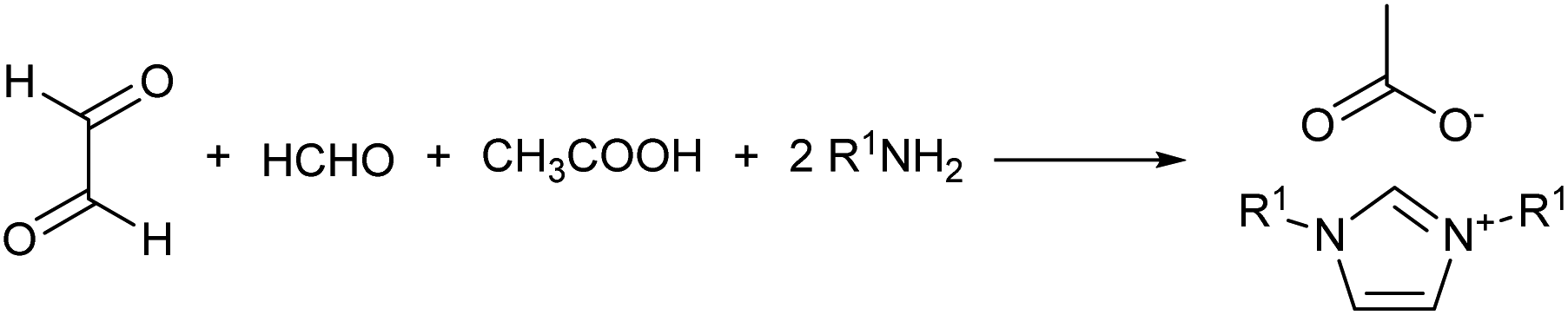 | ||
| Scheme 3 General reaction scheme of the modified Debus–Radziszewski imidazolium synthesis. | ||
Eventually, the procedure was adapted by Depuydt et al. in a solvent-less one-pot synthesis, which was implemented in various studies.17,32 However, none of the studies targeted long chain derivatives, which were a clear limit of the reported procedure.17,32 Besides, some compounds were also reported as a crude mixture because of the difficulties encountered in devising an effective purification procedure capable of isolating the final product.32 This evidenced the need for further studies in order to improve the procedure and scope of the reaction.
Literature reports often have focused on imidazolium ionic liquids comprising N-methyl substituents and with an overall relatively low molecular weight (usually below twenty carbon atoms).33,34 In this study, we adapted the modified Debus–Radziszewski reaction to achieve the synthesis of symmetrical 1,3-dialkylimidazolium ionic liquids comprising linear alkyl chains up to 16 carbon atoms. The procedure is very effective, easy to implement, and avoids the use of hazardous diethyl ether in the purification process. Moreover, this modification gives direct access to a series of high molecular weight imidazoles with a limited water solubility, which is of wide interest for their potential use in several applications (e.g. surfactants, liquid crystals, precursors for lipophilic N-heterocyclic carbenes, extractants, and conductive media for batteries and fuels cells).
We report herein an adaptation of the Debus–Radziszewski reaction to achieve a scalable one-pot procedure for the synthesis of long-chain imidazolium acetate salts. Our analysis showed that the formerly reported modified Debus–Radziszewski reaction is a nearly optimal synthetic procedure and an example of an on-water reaction. In this study however, the reaction procedure was adjusted to favour the homogeneity of the reaction mixture and to isolate the long-chain imidazolium salts from the crude product. Hence, the purification procedure was changed in relation to the increasing lipophilicity of the investigated compounds. Lastly, the synthesised salts were also derivatised to different ion pair couples to investigate the influence of the anion on these N,N-disubstituted long-chain imidazolium salts.
Experimental
Chemicals
Glyoxal (40 wt% in water), oxalic acid (98%, 144-62-7), and 1-octylamine (99+%, 111-86-4) were purchased from Acros Organics (Geel, Belgium). 1-Decylamine (98%, 2016-57-1) was purchased from TCI Europe (Zwijndrecht, Belgium). 1-Hexadecylamine (94%, 143-27-1) and 1-octadecylamine (≥85%, 124-30-1) were obtained from Merck (Heverlee, Belgium). 1-Hexylamine (99%, 111-26-2) and nitric acid (70 wt% in water, 7697-37-2) were bought from Sigma-Aldrich (Diegem, Belgium). 1-Dodecylamine (98%, 124-22-1) and 1-tetradecylamine (98%, 2016-42-4) were purchased from Janssen-Chemica (Beerse, Belgium). Acetic acid, glacial (100%, 64-19-7), hydrochloric acid (37 wt% in water, 7647-01-0) and formaldehyde (36 wt% in water, 50-00-0) were obtained from Fisher Scientific Limited (Loughborough, UK). Petroleum ether (bp 40–65 °C, technical grade) and acetonitrile (≥99.8%) were obtained from VWR (Heverlee, Belgium). Petroleum ether was distilled prior to use to remove high boiling residues. All other chemicals were used as received without further purification.Instrumentation and materials
Nuclear magnetic resonance spectra were performed on a spectrometer (Bruker Avance 400) operating at a 1H frequency of 400 MHz. Fourier transform infrared spectra were recorded with a resolution of 2 cm−1 on a spectrometer (Bruker Vertex 70) in ATR mode (Bruker Platinum ATR module). TGA and DSC analysis were carried out on a TA Instruments TGA Q500 and a TA Instruments DSC Q2000 respectively, in aluminium sample pans with a heating/cooling rate of 10 °C min−1 operating under a nitrogen flow of 60 mL min−1. Ionic liquids were pre-dried for 60 min at 80 °C prior to the TGA measurement. Vacuum measurement were carried out with a digital manometer (Pfeiffer TPG 201). Disposable funnel filters (Chemrus® disposable filter funnel, PP body with PE fritted disk, 18 mL, 25 mm Ø, 10 μm pore Ø) were used for the sample filtration (with exception of the fluoride samples, which melted PP plastics).1,3-Di(octyl)imidazolium acetate ([C8C8IM][OAc])
The synthesis was carried out following the general procedure (ESI Synthetical procedure †) with 1-octylamine (99%, 25.654 g, 20.065 mL, 0.2 mol), formaldehyde (36.5–38%, 8.34 g, 7.65 mL, 0.1 mol), acetic acid (99.9%, 9.01 g, 8.59 mL, 0.15 mol), glyoxal (40%, 14.51 g, 11.15 mL, 0.1 mol). Isolated yield 34.94 g, 92% (≈0.5 eq. of residual acetic acid). Pale to dark orange oil. Decomposition temperature: 223 °C. 1H NMR (300 MHz, CDCl3, Me4Si, δ/ppm): 0.87 (6H, t, J 6.45), 1.29 (24H, dt, J 19.11, 4.48), 1.88 (4H, p, J 7.28, 7.27), 2.02 (3H, s), 4.34 (4H, t, J 7.40), 7.10 (2H, d, J 1.59), 11.67 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 176.7, 139.6, 121.2, 49.9, 31.7, 30.3, 29.0, 29.0, 26.2, 23.7, 22.6, 14.1. IR (Diamond-ATR, neat, cm−1): 2955, 2924, 2856, 1701, 1580, 1466, 1379, 1251, 1165, 1045, 1000, 875, 771, 723, 650, 610, 445.1,3-Di(octyl)imidazolium nitrate ([C8C8IM][NO3])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 5†) with 1,3-di(octyl)imidazolium acetate (0.8061 g, 2.29 mmol). Isolated yield 0.8200 g, 100%. Orange viscous oil. Decomposition temperature: 285 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.83–0.91 (6H, m), 1.23–1.36 (20H, m), 1.89 (4H, dq, J 14.98, 7.85, 7.46), 4.26 (4H, t, J 7.41), 7.26 (2H, d, J 1.68), 10.17 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 137.2, 122.4, 50.1, 31.7, 30.3, 29.0, 28.9, 26.2, 22.6, 14.1. IR (Diamond-ATR, neat, cm−1): 3135, 3089, 3039, 2955, 2924, 2855, 1744, 1671, 1564, 1465, 1334, 1162, 1122, 1070, 1039, 951, 869, 829, 770, 723, 657, 646, 585, 557, 550, 540, 532, 515, 509, 501, 491, 484, 480, 469, 465, 460, 448, 442, 437, 431, 425, 420, 414, 403.1,3-Di(octyl)imidazolium chloride ([C8C8IM][Cl])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 4†) with 1,3-di(octyl)imidazolium acetate (0.4003 g, 1.14 mmol). Isolated yield 0.3051. g, 82%. Pale yellow colloid. Decomposition temperature: 257 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.80–0.94 (6H, m), 1.19–1.38 (20H, m), 1.92 (4H, p, J 7.20, 6.99), 4.28–4.43 (4H, m), 7.21 (2H, d, J 1.64), 11.15 (1H, d, J 1.72). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 137.5, 122.2, 50.0, 31.7, 30.4, 29.1, 29.0, 26.3, 22.6, 14.1. IR (Diamond-ATR, neat, cm−1): 3379, 3132, 3060, 2956, 2923, 2872, 2854, 1637, 1562, 1407, 1377, 1332, 1307, 1246, 1162, 1122, 1023, 773, 723, 658, 607, 569, 502, 438, 410.1,3-Di(octyl)imidazolium hydroxalate ([C8C8IM][Ox])
The synthesis was carried out following the general procedure (ESI. Synthetical procedure 6) with 1,3-di(octyl)imidazolium acetate (0.8149 g, 2.31 mmol). Isolated yield 0.8925 g, 100%. Dark amber solid. Melting point: 74 °C. Decomposition temperature: 227 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.85 (6H, t, J 6.76), 1.26 (24H, dd, J 14.32, 7.52), 1.82 (4H, d, J 7.22), 4.22 (4H, t, J 7.14), 7.33 (2H, s), 9.17 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 161.7, 136.2, 122.2, 50.1, 31.7, 30.0, 29.0, 28.9, 26.2, 22.6, 14.0. IR (Diamond-ATR, neat, cm−1): 3137, 3109, 3080, 2952, 2922, 2871, 2853, 1731, 1679, 1563, 1465, 1421, 1377, 1352, 1320, 1198, 1158, 918, 885, 838, 789, 696, 660, 597, 578, 476.1,3-Di(decyl)imidazolium acetate ([C10C10IM][OAc])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 1†) with 1-decylamine (98%, 31.32 g, 39.80 mL, 0.2 mol), formaldehyde (36.5–38%, 8.34 g, 7.65 mL, 0.1 mol), acetic acid (99.9%, 9.01 g, 8.59 mL, 0.15 mol), glyoxal (40%, 14.51 g, 11.15 mL, 0.1 mol). Isolated yield 40.32 g, 94% (≈0.5 eq. of residual acetic acid). Yellow to orange oil. Melting point: 49 °C. Decomposition temperature: 225 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.81), 1.28 (32H, d, J 30.33), 1.87 (4H, dd, J 9.85, 4.86), 2.02 (3H, s), 4.35 (4H, t, J 7.40), 7.08 (2H, d, J 1.48), 11.94 (1H, d, J 1.61). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 176.8, 139.7, 121.1, 49.9, 31.8, 30.3, 29.5, 29.4, 29.3, 29.0, 26.2, 23.6, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 2923, 2854, 1563, 1466, 1378, 1255, 1166, 1045, 1000, 878, 722, 651, 611, 445, 3135, 3074, 2955, 2923, 2854, 1699, 1644, 1579, 1563, 1466, 1378, 1325, 1255, 1166, 1122, 1045, 1000, 958, 878, 754, 722, 651, 611, 522, 494, 477, 471, 445, 409, 402.1,3-Di(decyl)imidazolium nitrate ([C10C10IM][NO3])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 5†) with 1,3-di(decyl)imidazolium acetate (0.8202 g, 2.01 mmol). Isolated yield 0.8359 g, 101%. Amber solid. Melting point: 27 °C. Decomposition temperature: 296 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.87 (6H, t, J 6.78), 1.28 (29H, d, J 26.69), 1.89 (4H, p, J 7.68, 7.47), 4.25 (4H, t, J 7.39), 7.40 (2H, d, J 1.68), 10.00 (1H, d, J 1.72). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 137.2, 122.4, 50.1, 31.9, 30.3, 29.5, 29.4, 29.3, 29.0, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3136, 3089, 3040, 2955, 2922, 2853, 1744, 1654, 1564, 1466, 1335, 1162, 1077, 1039, 873, 829, 754, 722, 658, 646, 562, 537, 515, 504, 499, 489, 482, 473, 467, 459, 451, 444, 428, 418, 405.1,3-Di(decyl)imidazolium chloride ([C10C10IM][Cl])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 4†) with 1,3-di(decyl)imidazolium acetate (0.8135 g, 1.99 mmol). Isolated yield 0.7132 g, 93%. Viscous red deliquescent solid. Melting point: 57 °C. Decomposition temperature: 256 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.72), 1.29 (32H, d, J 34.20), 1.92 (4H, p, J 7.21, 7.16), 4.36 (4H, t, J 7.43), 7.30 (2H, s), 10.90 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 137.8, 121.9, 50.1, 31.8, 30.4, 29.6, 29.5, 29.4, 29.3, 29.0, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 2956, 2922, 2872, 2853, 1647, 1562, 1466, 1406, 1376, 1333, 1307, 1165, 1123, 1078, 1021, 888, 775, 721, 658, 544, 472, 429.1,3-Di(decyl)imidazolium hydroxalate ([C10C10IM][Ox])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 6†) with 1-decylamine (98%, 31.32 g, 39.80 mL, 0.2 mol), formaldehyde (36.5–38%, 8.34 g, 7.65 mL, 0.1 mol), acetic acid (99.9%, 9.01 g, 8.59 mL, 0.15 mol), glyoxal (40%, 14.51 g, 11.15 mL, 0.1 mol). Isolated yield 40.32 g, 94% (≈0.5 eq. of residual acetic acid). Dark red solid. Melting point: 70 °C. Decomposition temperature: 250 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.87 (6H, t, J 6.81), 1.28 (32H, d, J 26.20), 1.86 (4H, t, J 7.19), 4.34 (4H, t, J 7.35), 7.14 (2H, s), 11.12 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 164.3, 138.3, 122.0, 49.9, 31.8, 30.3, 29.5, 29.4, 29.3, 29.0, 26.2, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3376, 3130, 3067, 2956, 2922, 2872, 2853, 1563, 1529, 1466, 1406, 1376, 1334, 1295, 1215, 1165, 1125, 1042, 721, 659, 533, 502, 434.1,3-Di(dodecyl)imidazolium acetate ([C12C12IM][OAc])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 1†) with 1-dodecylamine (98%, 37.571 g, 47.75 mL, 0.2 mol), formaldehyde (36.5–38.0 wt% in water–methanol, 8.34 g, 7.65 mL, 0.1 mol), acetic acid (99.9%, 9.01 g, 8.59 mL, 0.15 mol), glyoxal (40%, 14.51 g, 11.15 mL, 0.1 mol). Isolated yield 44.292 g, 85% (≈0.5 eq. of residual acetic acid). Orange to beige solid. Melting point: 31 °C. Decomposition temperature: 227 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.81), 1.18–1.39 (40H, m), 1.87 (4H, p, J 7.59, 7.57), 4.34 (4H, t, J 7.40), 7.10 (2H, d, J 1.30), 2.02 (3H, s), 11.78 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 176.7, 139.6, 121.0, 50.0, 31.9, 30.2, 29.6, 29.5, 29.4, 29.3, 29.0, 26.3, 23.4, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3159, 3106, 2952, 2914, 2870, 2849, 1708, 1572, 1560, 1470, 1459, 1449, 1374, 1313, 1301, 1291, 1230, 1171, 1128, 1113, 1038, 1007, 942, 839, 762, 741, 730, 718, 661, 632, 607, 505, 438, 429, 416, 410.1,3-Di(dodecyl)imidazolium chloride ([C12C12IM][Cl])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 4†) with 1,3-di(dodecyl)imidazolium acetate (0.8015 g, 1.0 mL, 1.73 mmol). Isolated yield 0.7194 g, 95%. Dark red solid. Melting point: 42 °C. Decomposition temperature: 265 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.78), 1.20–1.37 (36H, m), 1.91 (4H, q, J 7.39, 7.37), 4.36 (4H, t, J 7.46), 7.17 (2H, d, J 1.46), 11.18 (1H, d, J 1.98). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 137.4, 122.3, 50.0, 31.9, 30.4, 29.6, 29.5, 29.4, 29.3, 29.1, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3390, 3132, 3062, 2953, 2918, 2850, 1643, 1562, 1464, 1404, 1376, 1364, 1305, 1287, 1272, 1246, 1164, 1126, 1108, 1087, 960, 854, 807, 778, 748, 721, 661, 638, 602, 415.1,3-Di(dodecyl)imidazolium nitrate ([C12C12IM][NO3])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 5†) with 1,3-di(dodecyl)imidazolium acetate (0.8018 g, 1.0 mL, 1.73 mmol). Isolated yield 0.7819 g, 97%. Dark red solid. Melting point: 98 °C. Decomposition temperature: 288 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 7.24 (2H, d, J 1.60), 0.88 (7H, t, J 6.80), 1.18–1.36 (46H, m), 1.89 (4H, p, J 8.09, 7.85), 4.25 (4H, t, J 7.41), 10.15 (1H, d, J 1.71). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 138.4, 121.5, 50.3, 31.9, 30.2, 29.6, 29.5, 29.4, 29.3, 29.0, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3419, 3136, 3094, 2954, 2918, 2850, 2019, 2009, 1967, 1949, 1634, 1563, 1466, 1340, 1221, 1159, 1125, 1087, 1040, 993, 947, 856, 828, 775, 720, 658, 603, 571, 518, 491, 485, 477, 469, 455, 446, 430, 426, 419, 411, 404.1,3-Di(dodecyl)imidazolium hydroxalate ([C12C12IM][Ox])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 6†) with 1,3-di(dodecyl)imidazolium acetate (0.8008 g, 1.0 mL, 1.73 mmol). Isolated yield 0.3752 g, 44%. Pale yellow solid. Melting point: 125 °C. Decomposition temperature: 229 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.81), 1.28 (39H, d, J 25.55), 1.86 (4H, p, J 7.64, 7.21), 4.30 (4H, t, J 7.35), 7.16 (2H, d, J 1.12), 10.66 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 163.4, 138.7, 121.5, 50.1, 31.9, 30.3, 29.6, 29.5, 29.4, 29.3, 29.0, 26.2, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3139, 3100, 3083, 2954, 2917, 2871, 2850, 1594, 1563, 1462, 1405, 1370, 1328, 1286, 1256, 1218, 1159, 1086, 963, 925, 841, 813, 789, 711, 656, 637, 601, 543, 458.1,3-Di(tetradecyl)imidazolium acetate ([C14C14IM][OAc])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 2†) with 1-tetradecylamine (98.5%, 4.2688 g, 4.775 mL, 0.02 mol), formaldehyde (36.5–38%, 0.834 g, 0.765 mL, 0.01 mol), acetic acid (99.9%, 0.901 g, 0.859 mL, 0.015 mol), glyoxal (40%, 1.451 g, 1.115 mL, 0.01 mol). Isolated yield 4.451 g, 87% (≈0.5 eq. of residual acetic acid). Yellow solid. Melting point: 43 °C. Decomposition temperature: 229 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.74), 0.97–1.70 (48H, m), 1.87 (4H, p, J 7.50, 7.42), 2.01 (3H, s), 4.33 (4H, t, J 7.39), 7.11 (2H, d, J 1.44), 11.35–11.58 (1H, m). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 176.2, 139.1, 121.1, 50.0, 31.9, 30.2, 29.7, 29.7, 29.6, 29.5, 29.4, 29.4, 29.0, 26.2, 22.8, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3160, 3105, 2952, 2913, 2870, 2849, 1708, 1573, 1562, 1470, 1453, 1376, 1312, 1300, 1170, 1128, 1114, 1050, 1007, 943, 874, 771, 762, 742, 718, 660, 632, 607, 515, 505, 471, 459, 440, 417, 412, 403.1,3-Di(tetradecyl)imidazolium nitrate ([C14C14IM][NO3])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 5†) with 1,3-di(tetradecyl)imidazolium acetate (0.3979 g, 1.0 mL, 0.77 mmol). Isolated yield 0.3938 g, 98%. Yellow solid. Melting point: 111 °C. Decomposition temperature: 285 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.77), 0.96–1.53 (48H, m), 1.88 (4H, q, J 7.13, 6.90), 4.25 (4H, t, J 7.39), 7.33 (2H, d, J 1.57), 10.11 (1H, d, J 1.71). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 138.1, 121.8, 50.2, 31.9, 30.3, 29.7, 29.7, 29.6, 29.5, 29.4, 29.4, 29.0, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3461, 3135, 3092, 3038, 2953, 2916, 2849, 1745, 1651, 1563, 1466, 1339, 1214, 1162, 1128, 1094, 1059, 1041, 1019, 972, 941, 889, 848, 828, 806, 777, 753, 720, 693, 686, 678, 659, 621, 605, 586, 579, 572, 564, 556, 549, 541, 530, 522, 515, 507, 500, 492, 484, 478, 470, 456, 444, 439, 428, 422, 416, 411, 404.1,3-Di(tetradecyl)imidazolium chloride ([C14C14IM][Cl])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 4†) with 1,3-di(tetradecyl)imidazolium acetate (0.4012 g, 1.0 mL, 0.77 mmol). Isolated yield 0.564 g, 91%. Yellow to orange solid. Melting point: 43 °C. Decomposition temperature: 260 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 7.26 (2H, d, J 1.46), 0.88 (6H, t, J 6.74), 1.29 (48H, dd, J 31.53, 4.41), 1.92 (4H, t, J 7.39), 4.36 (4H, t, J 7.43), 11.06 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 138.0, 121.8, 50.1, 31.9, 30.4, 29.7, 29.7, 29.6, 29.5, 29.4, 29.4, 29.0, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3447, 3371, 3256, 3133, 3038, 2954, 2918, 2871, 2850, 1643, 1564, 1464, 1403, 1376, 1365, 1338, 1307, 1258, 1235, 1213, 1191, 1137, 1109, 1094, 1063, 1034, 869, 849, 813, 775, 750, 721, 662, 637, 495, 446, 440, 428, 414.1,3-Di(tetradecyl)imidazolium hydroxalate ([C14C14IM][Ox])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 6†) with 1,3-di(tetradecyl)imidazolium acetate (0.6894 g, 1.69 mmol). Isolated yield 0.6537 g, 88%. Yellow solid. Melting point: 130 °C. Decomposition temperature: 229 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.75), 1.27 (48H, dd, J 20.76, 4.48), 1.86 (4H, t, J 6.98), 4.29 (4H, t, J 7.28), 7.18 (2H, s), 10.53 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 163.6, 138.5, 121.7, 50.0, 31.9, 30.3, 29.7, 29.7, 29.6, 29.5, 29.4, 29.4, 29.0, 26.2, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3139, 3103, 3082, 2954, 2917, 2872, 2849, 1679, 1595, 1563, 1464, 1375, 1328, 1256, 1160, 1094, 1003, 970, 890, 838, 811, 711, 656, 637, 601, 459.1,3-Di(hexadecyl)imidazolium acetate ([C16C16IM][OAc])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 3†) with 1-hexadecylamine (94.0%, 28.67 g, 0.012 mol), formaldehyde (36.5–38%, 4.92 g, 4.43 mL, 0.059 mol), acetic acid (99.9%, 5.32 g, 5.07 mL, 0.0885 mol), glyoxal (40 wt% in water, 8.56 g, 6.58 mL, 0.059 mol). Isolated yield 25.29 g, 67% (≈0.5 eq. of residual acetic acid). Amber to dark brown solid. Melting point: 50 °C. Decomposition temperature: 228 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.64), 1.17–1.37 (56H, m), 1.87 (4H, t, J 7.36), 2.02 (5H, s), 4.32 (4H, t, J 7.41), 7.09 (2H, s), 11.02 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 176.7, 139.8, 121.0, 49.9, 31.9, 30.3, 29.7, 29.7, 29.6, 29.5, 29.4, 29.4, 29.0, 26.3, 23.5, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3157, 3103, 2953, 2913, 2870, 2849, 1705, 1561, 1470, 1456, 1376, 1294, 1172, 1099, 1043, 1007, 962, 878, 767, 739, 717, 672, 661, 633, 607, 507, 484, 468, 442, 414, 403.1,3-Di(hexadecyl)imidazolium nitrate ([C16C16IM][NO3])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 5†) with 1,3-di(hexadecyl)imidazolium acetate (0.7951 g, 1.0 mL, 1.38 mmol). Isolated yield 0.7915 g, 99%. Amber to dark brown solid. Melting point: 127 °C. Decomposition temperature: 290 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 7.25 (2H, d, J 1.58), 4.25 (4H, t, J 7.40), 0.88 (6H, t, J 6.77), 1.19–1.35 (56H, m), 1.89 (4H, p, J 7.75, 7.74), 10.21 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 137.6, 122.1, 50.1, 31.9, 30.3, 29.7, 29.7, 29.6, 29.6, 29.4, 29.4, 29.0, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3460, 3135, 3091, 3044, 2953, 2916, 2849, 1745, 1656, 1563, 1466, 1338, 1214, 1162, 1100, 1067, 1040, 889, 829, 779, 740, 720, 659, 610, 573, 548, 533, 520, 515, 509, 500, 492, 486, 481, 476, 469, 464, 459, 453, 443, 439, 428, 413, 408.1,3-Di(hexadecyl)imidazolium chloride ([C16C16IM][Cl])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 4†) with 1,3-di(hexadecyl)imidazolium acetate (0.7951 g, 1.0 mL, 1.38 mmol). Isolated yield 0.6950 g, 91%. Amber to dark brown solid. Melting point: 128 °C. Melting temperature: 38 °C. Decomposition temperature: 260 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 1.91 (4H, p, J 7.16, 7.01), 0.87 (6H, d, J 7.07), 1.23–1.33 (56H, m), 4.36 (4H, t, J 7.41), 7.37 (2H, s), 10.72 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 138.0, 121.7, 50.2, 31.9, 30.3, 29.7, 29.7, 29.6, 29.5, 29.4, 29.4, 29.1, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 3394, 2954, 2917, 2849, 1671, 1562, 1466, 1407, 1375, 1331, 1248, 1163, 1128, 1100, 887, 862, 842, 779, 739, 721, 658, 575, 523, 500, 493, 484, 461, 453, 445, 430, 418.1,3-Di(hexadecyl)imidazolium hydroxalate ([C16C16IM][Ox])
The synthesis was carried out following the general procedure (ESI Synthetical procedure 6†) with 1,3-di(hexadecyl)imidazolium acetate (0.7943 g, 1.0 mL, 1.38 mmol). Isolated yield 0.4425 g, 53%. Amber to dark brown solid. Melting point: 109 °C. Decomposition temperature: 229 °C. 1H NMR (400 MHz, CDCl3, Me4Si, δ/ppm): 0.88 (6H, t, J 6.77), 1.23–1.34 (56H, m), 1.84 (4H, q, J 7.10, 7.00), 4.37 (4H, t, J 7.39), 7.11 (2H, s), 11.29 (1H, s). 13C NMR (100 MHz, CDCl3, Me4Si, δ/ppm): 168.0, 140.8, 120.9, 49.8, 31.9, 30.4, 29.7, 29.7, 29.6, 29.4, 29.4, 29.1, 29.7, 26.3, 22.7, 14.1. IR (Diamond-ATR, neat, cm−1): 2955, 2917, 2872, 2849, 1554, 1466, 1375, 1334, 1295, 1162, 1128, 1064, 1020, 888, 839, 780, 742, 720, 661, 604, 455, 446, 429, 416.Results and discussion
Procedure optimization
The reaction procedure was optimized for the synthesis of 1,3-di(decyl)imidazolium acetate. Formerly, this derivative was synthesised with only a moderate isolated yield (53%).17 Thus, this was an ideal starting point for the assessment of reaction optimization.17 Depuydt et al. reported losses in the purification procedure of the heaviest compounds synthesised. When the purification of the imidazolium salt was done via several diethyl ether washes, it resulted in significant product losses to the organic layer for the most lipophilic imidazolium salts (determined via 1H NMR analysis). A solvent system capable to isolate these less polar derivatives was therefore investigated. In order to increase the selectivity towards the impurities, diethylether was substituted with petroleum ether (bp 40–65 °C), which has a lower polarity and a lower miscibility with other organic solvents.35 The crude reaction mixture was fully soluble in petroleum ether; therefore, a second solvent was required to selectively partition the product. Unfortunately, there are only a few organic–organic immiscible solvent mixtures.36 Methanol does form a biphasic system with alkanes and it is a fairly green solvent; hence, it was firstly tested to attempt this separation. After a few washing steps, the methanol layer held the product in a relatively higher purity. However, in order to observe a phase separation, methanol had to be used in large excess with respect to petroleum ether and significant product losses were observed to the petroleum ether layer. Using higher alkanes, the product losses to the alkane layer were limited but traces of high weight alkanes impeded the product isolation. To counteract this issue, another purification procedure was tested. Acetonitrile used in combination with petroleum ether allowed for the compound isolation. This procedure was rather effective but traces of acetonitrile and acetic acid were trapped in the isolated product (the impurities were detected via the 1H NMR spectra and collapsed into one signal in the proton spectra at 2.1 ppm in deuterochloroform). Even after a thorough sample drying, impurities were still present. To force acetoniytrile removal, methanol was used to form an azeotrope. Methanol was added and removed via a rotatory evaporator for three times in order to force the acetonitrile entrainal. This procedure was effective for acetonitrile but did not remove the excess of acetic acid.The excess of acetic acid persisted even after drying at high vacuum above the compound melting temperature (1 week, <0.09 mbar). When dried at the Schlenk line, the acetic acid got reduced to about 0.5–1 eq. At this stage the release of acetic acid slowed down to such an extent that further high vacuum drying could not be performed in a reasonable amount of time. During the drying process 1,3-di(decyl)imidazolium acetate solidified and only after heating the mixture it reverted back to a liquid state. This was indicative of the stabilization by the intercalation of the acetic acid in the ionic liquid structure. This hypothesis was compatible with the literature data available on long side-chain imidazolium salts, which reported the formation of an ionic liquid metastable phase, and with the results of Gurau et al., which experienced a strong persistency of acetic acid traces in ionic liquids.37,38
Full removal of the acetic acid excess could only be attained via drying the molten sample under a vigorous flow of air at elevated temperatures (95 °C). The drying of the sample had also an unexpected result, a striking change in the color and increased transparency of the sample (Fig. 1, e.g. 1,3-di(decyl)imidazolium acetate turned to a clear light orange after drying from a dark brown opaque color).
 | ||
| Fig. 1 Comparative picture of the samples prior (above) and after (below) the drying of the sample. From left to right: 1,3-di(octyl)imidazolium acetate, 1,3-di(decyl)imidazolium acetate, 1,3-di(dodecyl)imidazolium acetate, 1,3-di(tetradecyl)imidazolium acetate, and 1,3-di(hexadecyl)imidazolium acetate. | ||
In an attempt to verify if the excess of acetic acid was actually needed, the reaction was repeated with only a slight excess of acetic acid (1.05 eq.). As it was previously observed with shorter chains, this modification did not reduce the acid excess but the overall yield.17 This showed that the acetic acid excess itself favours the reaction completion.
Opposite to what has been previously reported, the solventless synthetic procedure worked with amines up to 1-dodecylamine. 1-Tetradecylamine and the higher homologues were not fluid enough to be used without the addition of a solvent able to improve the mixing at room temperature (Table 2). To check how the solvent would affect the system, the reaction was repeated with 1-dodecylamine in a series of solvents (Table 1). The solventless reaction demonstrated to be the one leading to the best yields.
| Solvent | Volume (mL) | Temperature (°C) | Isolated yield (%) |
|---|---|---|---|
| a The reaction was carried out on a 1 mmol scale. The synthetic procedure can be found within the ESI. | |||
| Methanol | 3 | 22 | 71 |
| Methanol | 4 | 22 | 71 |
| Methanol | 5 | 22 | 69 |
| Ethanol | 3 | 22 | 70 |
| Isopropanol | 3 | 22 | 72 |
| Acetonitrile | 3 | 22 | 75 |
| Neat | — | 22 | 80 |
| Neat | — | 50 | 76 |
| Amine | Solvent | Purification | Isolated yield (%) |
|---|---|---|---|
| a Actually water is the solvent as present in the reagent but there is no additional solvent used and the water volume is minor with respect to the one of the reagents.b Methanol was added after 8 h to ease the runny solid and allow for the reaction to continue.c Amine concentration in toluene is 0.2 M.d Batch extraction with acetonitrile and petroleum ether.e Impurities precipitation via the use of methanol as anti-solvent. | |||
| 1-Octylamine | Neata | Extractiond | 92 |
| 1-Decylamine | Neata | Extractiond | 94 |
| 1-Dodecylamine | Neata | Extractiond | 85 |
| 1-Tetradecylamine | Neata, methanolb | Extractiond | 87 |
| 1-Hexadecylamine | Toluenec | Precipitatione | 67 |
| 1-Octadecylamine | Toluene | — | — |
With amines of at least 14 carbon atoms the reaction had to be modified in order to yield the desired product in satisfactory amounts. Methanol was effective in facilitating the reactivity of 1-tetradecylamine reactivity but not of the 1-hexadecylamine one. Toluene has been reported by Arduengo et al. as a convenient medium for this type of reactions, as it is miscible with both polar and apolar materials.16 Although toluene allowed for the synthesis of the 1,3-di(hexadecyl)imidazolium acetate derivative, it was only marginally effective for 1-octadecylamine and the isolation of the final product was not possible for 1,3-di(octadecyl)imidazolium acetate. This result was ascribed to the reduced solubility of the 1-octadecylamine in toluene, which may have impeded the reaction progression.
Differently from the results of Depuydt et al., there was not a steady reduction in the isolated yield through the series of the amines but a marked reduction between 1-decylamine and 1-dodecylamine (Table 2). This seems to be a result of a poorer mixing, caused by the fact that 1-dodecylamine is a solid (mp 27–29 °C), rather than to a limited applicability of the isolation methodology.17
However, it we found quite surprising that even for clearly heterogeneous systems the reaction performed so well (i.e. 1-dodecylamine and 1-tetradecylamine). To understand why the solventless procedure was favoured with respect to any of the other tested, we investigated the reaction in detail, step by step. It may be pointed out that actually the ‘neat’ reaction conditions are not strictly solventless. Formaldehyde and glyoxal were used as aqueous solutions (36 and 40% w/w, respectively); therefore, the solventless system was a concentrated aqueous system with minor traces of methanol, which was present as formaldehyde stabilizer.
Visual observation of the reaction mixture allowed us to notice a detail which in previous studies got overlooked. The addition of the formaldehyde and acetic acid mixture to the cold amine caused the formation of a biphasic system (Fig. 2). To understand what phased out, the top layer was sampled and analysed via 1H NMR. The analysis showed the top layer to be consisting of the virtually pure 1,3,5-trialkylhexahydro-1,3,5-triazine, formed in situ from the reaction of formaldehyde with the amine. This may explain the reaction effectiveness under neat conditions and give further insights on the mechanism of the reaction (Scheme 4). The segregation of the reagents in two distinct layers impedes potential side reactions, contributing to the reaction selectivity and high yield.
 | ||
| Fig. 2 Biphasic system formed after the addition of acetic acid and formaldehyde during the synthesis of 1,3-di(octyl)imidazolium acetate. The same behavior was observed during the synthesis of 1,3-di(butyl)imidazolium acetate and therefore is expected to happen to all the chains above four carbon atoms if not even to all the linear alkyl amines. The biphasic system remains also after the addition of glyoxal. | ||
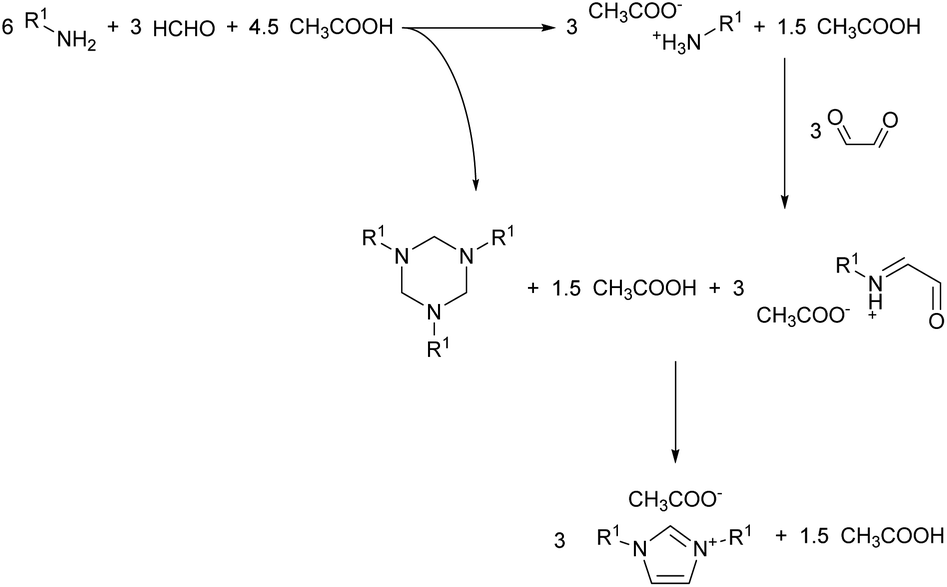 | ||
| Scheme 4 General stepwise reaction scheme of the modified Debus–Radziszewski imidazolium synthesis. | ||
What can be also considered is that the imine self-segregation as a triazine is beneficial to the reaction outcomes only when non-enolisable imines are used, such as formaldehyde.17 If an enolisable imine would be used, it would likely result in self-condensating to a less sterically encoumbered derivative prior to the glyoxal addition (e.g. Mannich condensation to a polyimine). Also, under the current condition, the triazine formation might be a formaldehyde-specific behaviour, explaining why none of the non-enolisable aldehydes tested by Depuydt et al. performed as formaldehyde.17 This gives a further insights in the reaction underlying this modification of the Debus–Radziszewski reaction, which appears to have an analogous reactivity to what has been previously reported for the synthesis of imidazoles from the reaction between an α-hydroximinoketone and 1,3,5-trimethylhexahydro-1,3,5-triazine.39,40
Acid metathesis
Acid metathesis is an effective way to derivatise the imidazolium acetate ionic liquids due to the moderate basicity of the acetate anion. To attain a complete metathesis, the acetate anion has to be fully protonated and the final product has to be successfully partitioned from the acetic acid residue (e.g. solvent extraction, evaporation). To determine a viable acid for the acidic metathesis we applied the Henderson–Hasselbach equation (eqn (1)).
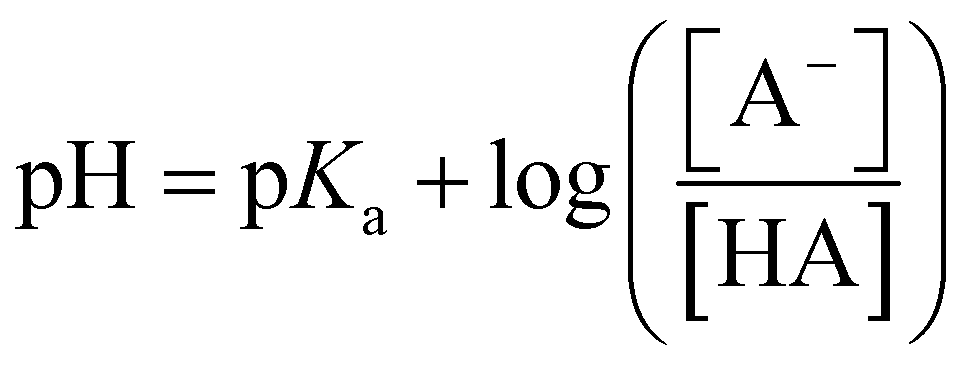 | (1) |
The equation above resolved for the pKa of acetic acid (4.76) allowed us to determine the minimum pH at which the acetic acid is fully protonated, in this case assuming 99.9% (Fig. 3). Resolving the equation with a protonated molar fraction corresponding to 99.9% and the pKa of acetic acid (4.76) it resulted that the acetic acid is fully protonated for solutions with a pH below 1.76. Therefore, any acid with a pKa below 1.76 satisfies the first requirement for achieving complete metathesis.
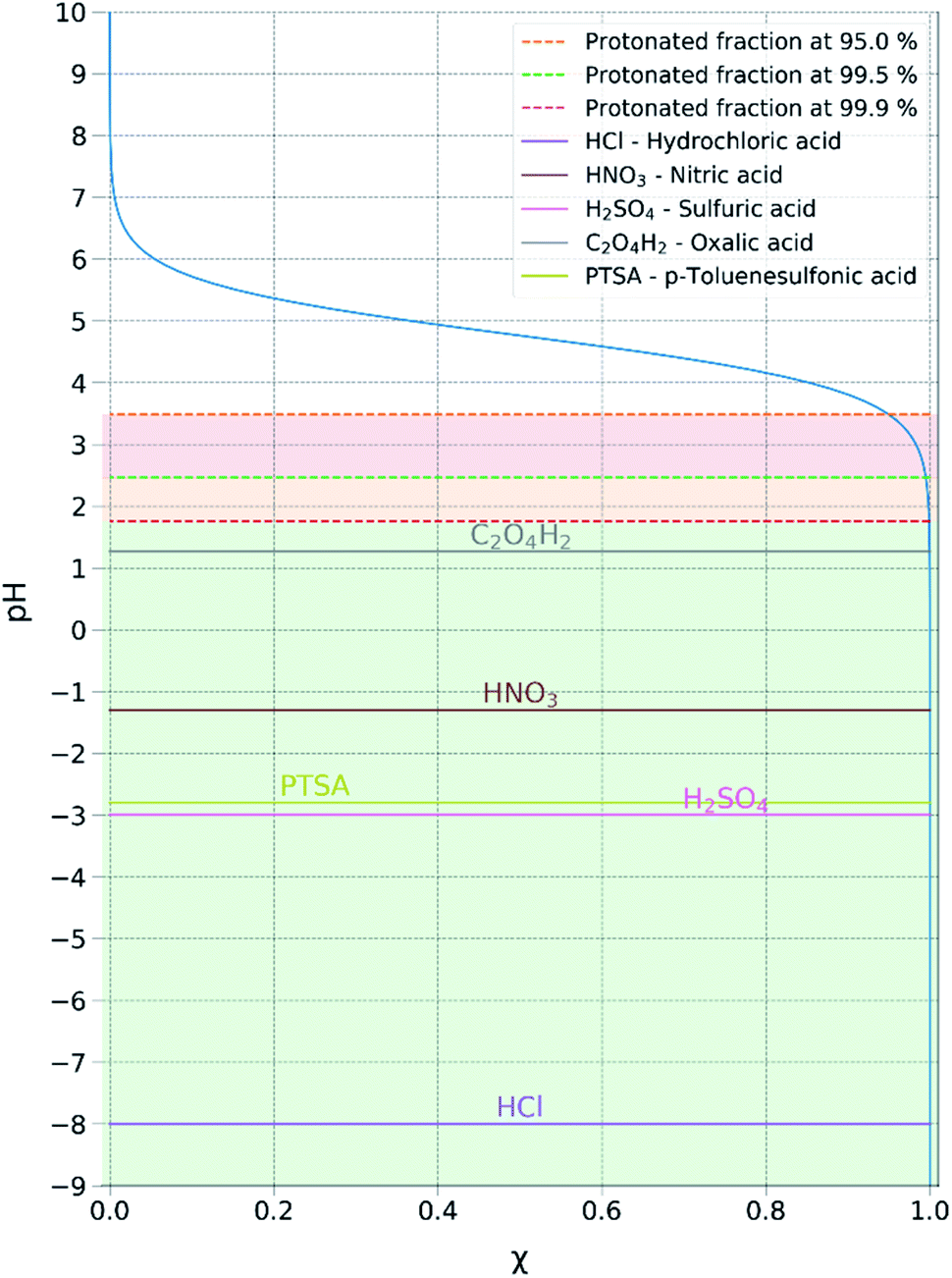 | ||
| Fig. 3 Graphical representation of the molar fraction of protonated acetic acid within an aqueous solution. This can be used to predict the capacity of an acid to displace the acetate anion in the ion-pair couple in an aqueous system. | ||
All acetate salts in this study were soluble in water. In some metathesis, acetic acid removal was problematic, but the hydrophobicity of our class of materials facilitated the isolation of the products.
Imidazolium nitrate salts were partitioned out of the aqueous layer, forming a floating immiscible product (ESI Synthetical procedure 5†). Instead, for the chloride salts partitioning was enforced via the use of concentrated hydrochloric acid, which salted out of the ionic liquid due to the ionic nature of the aqueous layer, caused by the acid self-ionization. The hydrochloric acid excess could be removed either via evaporation under a flow or air or after washing the ionic liquid dissolved in a small amount of an apolar organic solvent with water (ESI Synthetical procedure 4†).
Hydrogen oxalate salts could be isolated in high purity due to the oxalic acid strong acidity (pKa: 1.25, 4.14) via evaporation above the salt melting point under a flow of air.41
Phase characterization and phase interaction
The ILs were studied for their thermal stability and calorimetric phase transition behaviour (Fig. 4, ESI Table S1†). The stability of the ionic liquids appears to be directly correlated to the anion basicity ([NO3]− > [Cl]− > [Ox]− > [OAc]−). This could be correlated to the ability of the anion to extract the front proton from the imidazolium ring, which would imply a decomposition path encompassing a carbene intermediate. However, it is believed that the decomposition pathway goes generally via a reverse Menshutkin reaction.41 Differential scan calorimetry was carried out for each of the salts (ESI Table S1†). Amongst the many ionic liquids tested, 1,3-di(dodecyl)imidazolium nitrate, 1,3-di(tetradecyl)imidazolium nitrate, and 1,3-di(tetradecyl)imidazolium chloride displayed a rather complex transition pattern, with multiple transitions in the range between 20 and 70 °C.42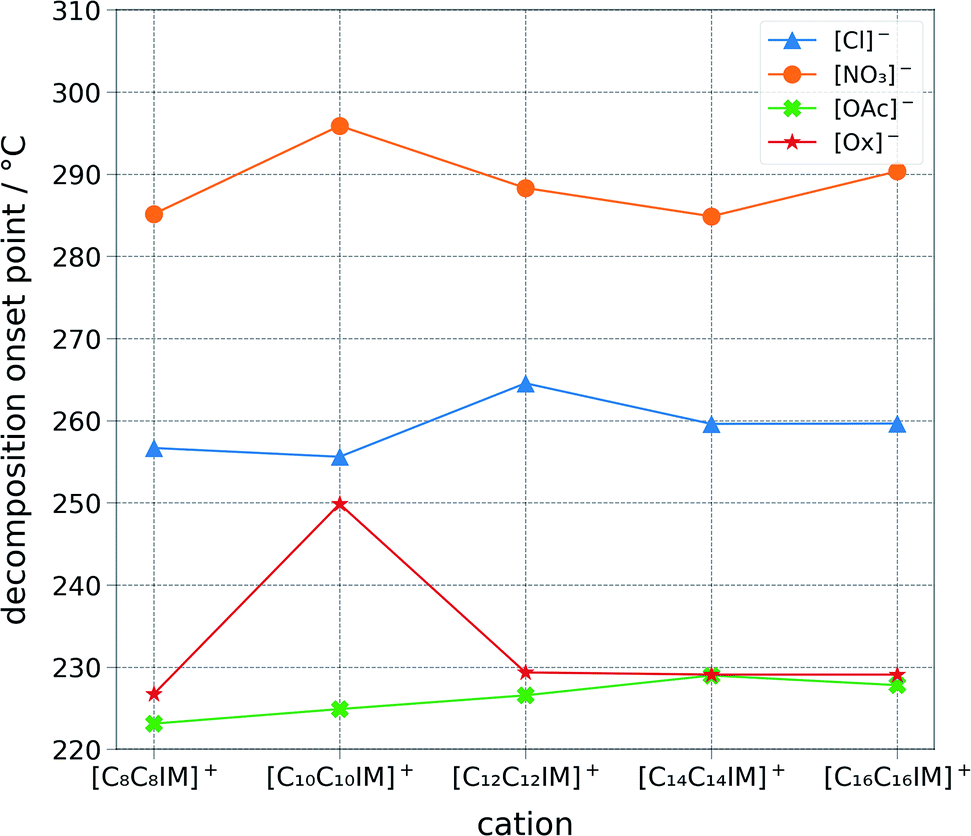 | ||
| Fig. 4 Graphical representation of the one percent decomposition onset point determined via a ramped thermo-gravimetric analysis with a step of 10 degrees per minute scan between 25 and 500 degrees Celsius in an aluminum pan. Decomposition temperatures appears to be chiefly correlated to the anion class and basicity. Ionic liquids stability follows the following order: [NO3]− > [Cl]− > [Ox]− > [OAc]−, which is from the least basic to the most basic anion. The thermo-gravimetric graph of each ionic liquid is available within the ESI.† | ||
The water-soluble imidazolium salts showed a surfactant behaviour during dissolution in water, which was further investigated.43,44 The compounds from 1,3-di(octyl)imidazolium acetate to 1,3-di(tetradecyl)imidazolium acetate developed a marked foam in aqueous solutions. This behaviour was characterised via the use of a capped measuring cylinder (Table 3, ESI Analytical procedure 3†). The analysis showed the 1,3-di(decyl)imidazolium acetate to hold an extremely remarkable foaming behaviour, with a foam height about 3 to 4 times higher than the one obtained with the reference compound (i.e. sodium dodecyl sulfate).
| Compound | Foam height/[cm (pH)] | Notes | Bubble size | |
|---|---|---|---|---|
| Acidic range | Basic range | |||
| a Foamability and foam stability were determined via the use of a 100 mL measuring cylinder (vide ESI Analytical procedure 3). The foam height immediately after shaking and after a minute are herein reported side by side. pH adjustment was done via addition of 5 to 10 drops of either NaOH (1 M) or HCl (1 M). Sodium dodecyl sulfate (SDS) was used as reference compound. Bubble size is registered as small (S), medium (M), and large (L). The test was carried out on 40 mL aqueous solutions with 0.1% w/w of the tested surfactant dissolved within it. | ||||
| SDS | 26–22 (2–3), 36–33 (5) | 14–10 (12) | Readily dissolved, very stable dense foam | S, M |
| [C8C8IM][OAc] | 15–6 (2) | 7–2 (8–9), 9–2 (12–13) | Readily dissolved, pale yellow solution, weak sparse foam | S |
| [C10C10IM][OAc] | 60–56 (1–2) | 47–45 (9), 40–30 (13) | Readily dissolved, pale yellow solution, stable dense foam | S, M, L |
| [C12C12IM][OAc] | 27–24 (1–2) | 27–25 (8), 27–24 (13) | Slowly dissolved, yellow solution, highly stable dense foam | S |
| [C14C14IM][OAc] | 4–4 (1) | 4–4 (8, 4–4 (13) | Partially dissolved, sparse foam with particulate | S |
The foaming ability was especially marked at low pH. The increased foaming capability at low pH could be indicative of a higher foaming strength of the 1,3-di(decyl)imidazolium chloride, which is the main salt present at low pH due to the acidification with hydrochloric acid. Also 1,3-di(dodecyl)imidazolium acetate had a marked foaming ability but, differently from 1,3-di(decyl)imidazolium acetate, it was not influenced by acidification of the solution with hydrochloric acid. This acidification effect was observed up to 1,3-di(decyl)imidazolium acetate, whereas from 1,3-di(dodecyl)imidazolium acetate and above there was no difference with respect to the pH of the environment.
1,3-Di(hexadecyl)imidazolium acetate did not foam in water but under vigorous mixing formed a gel phase. Various concentrations were tested to determine the minimal amount of imidazolium salt needed to have the formation of a gel phase (i.e. 5, 10, 20, 40, 60, 80, and 100 mg mL−1). The samples were homogenised via shaking at 2000 rpm for 2 h at a temperature of 75 °C, then they were allowed to gently cool to room temperature (22 °C). Gelation was tested by turning the sample upside down, in order to observe which was the minimal concentration needed to obtain a firm gel phase. The minimal concentration needed to avoid the collapse of the colloidal phase by turning the vial upside-down was assessed at 100 mg mL−1.
Oxalate precipitation metathesis
Oxalate salts are an extremely expedient category of salts because of the very low solubility of calcium oxalate in aqueous solutions (0.061 g L−1).45 This makes imidazolium hydroxalate salts expedient tools for imidazolium salt metathesis. The salt precipitation metathesis was carried out via the use of a metal salt, embedding the desired anion and a metal cation, which is highly insoluble as its metal oxalate salt (Scheme 5). Calcium salts are optimal because of the low costs and the low aqueous solubility of calcium oxalate. This would allow for the production of imidazolium salts otherwise difficult to synthesise, or use other costly metathesis procedures (e.g. silver halide metathesis). Moreover, the precipitated calcium oxalate can be easily recycled via calcination. Then, the formed calcium oxide can be used to newly form a metal salt via neutralization with the desired acid.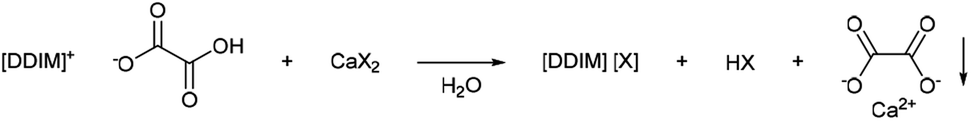 | ||
| Scheme 5 General reaction scheme of the oxalate precipitation metathesis (1,3-di(decyl)imidazolium cation is used here as general example). | ||
A small number of calcium salts were tested with 1,3-di(decyl)imidazolium hydroxalate in order to test the viability of the metathesis procedure. Calcium hydroxide and calcium carbonate were used in the attempt to produce imidazolium hydroxide and imidazolium carbonate, which would be optimal precursors to any imidazolium ionic liquid. None of these salts was isolated, instead complexes of calcium imidazolylidene were obtained with trace signals of the free carbene.46 However, these results demonstrated the procedure to be effective for the production of metal coordinated N-heterocyclic carbene catalysts because being devoid of any halide in the production process, which is a common inhibitor of many metal catalysts.47
Calcium fluoride was also used to attempt the synthesis of imidazolium fluoride salts. Introducing the fluoride anion as counter-ion via reported procedures is particularly cumbersome because of both the extremely reduced acidity of hydrofluoric acid and the high hazard posed by the handling of hydrofluoric acid. The fluoride anion was only partially introduced via the oxalate precipitation metathesis. Calcium fluoride (i.e. 0.016 g L−1) has a lower solubility than calcium oxalate, which partly explains the metathesis outcome.45 The reaction was repeated allowing the reagents to react for a longer time and with an excess of calcium fluoride, resulting only in a partial conversion.
Conclusions
Long-chain imidazolium ionic liquids were synthesised in high yield from readily available chemicals (i.e. formaldehyde, acetic acid, and glyoxal). We optimised the one-pot halide-less synthesis to attain a series of ionic liquids with longer linear alkyl chains with respect to the currently viable ones via the reported procedure. The investigations on the reaction procedure allowed us to identify the 1,3,5-trialkylhexahydro-1,3,5-triazine self-segregation as a key reaction feature, suggestive of an on-water catalytic effect. The formation of a biphasic system explained the high yields achieved by the reaction modification.The stability of the products was found to be chiefly dependent on the anion. Moreover, the longer side-chains induced in the imidazolium salts a complex phase behaviour, which was particularly apparent via the calorimetric analysis.
These ionic liquids showed a surfactant behaviour in aqueous solutions, acting either as a tensioactive or as gelator. With alkyl chains from octyl to tetradecyl, the acetate and chloride imidazolium salts acted as foaming agents, whereas 1,3-di(hexadecyl)imidazolium acetate formed a gel.
Imidazolium hydrogen oxalate salts were studied as precursors for salt precipitation metathesis due to the extreme oxalate salt insolubility in water. In presence of strongly basic counterions (e.g. calcium carbonate, calcium hydroxide) the reaction proceeded to full conversion, but otherwise (e.g. calcium fluoride) the mixture did not reach full conversion, not allowing for a complete metathesis. Although this setback, oxalate precipitation seems a promising methodology for the production of imidazolyldene metal salts via an halide-less procedure, which is of interest for the production of metal catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A particular thanks is due to Daphne Depuydt, whom introduced me to this reaction and stimulated my curiosity in its modification. The research leading to these results received funding from the Horizon 2020 Programmes of the European Community under Grant Agreement no. 721385 (MSCA-ETN SOCRATES) and Grant Agreement no. 812580 (MSCA-ETN SULTAN). This publication reflects only the view of the authors, the European Community is exempted from any liability. Project websites: http://etn-socrates.eu/ and http://etn-sultan.eu/.References
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437–14453 RSC.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2014, 16, 3238–3253 RSC.
- P. A. Hunt, I. R. Gould and B. Kirchner, Aust. J. Chem., 2007, 60, 9–14 CrossRef CAS.
- P. A. Hunt and I. R. Gould, J. Phys. Chem. A, 2006, 110, 2269–2282 CrossRef CAS PubMed.
- P. A. Hunt, B. Kirchner and T. Welton, Chemistry, 2006, 12, 6762–6775 CrossRef CAS PubMed.
- M. De Boeck, G. Damilano, W. Dehaen, J. Tytgat and E. Cuypers, Talanta, 2018, 184, 369–374 CrossRef CAS PubMed.
- M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS.
- M. J. Earle, J. M. S. S. Esperança, M. A. Gilea, J. N. Canongia Lopes, L. P. N. Rebelo, J. W. Magee, K. R. Seddon and J. A. Widegren, Nature, 2006, 439, 831–834 CrossRef CAS PubMed.
- A. K. L. Yuen, A. F. Masters and T. Maschmeyer, Catal. Today, 2013, 200, 9–16 CrossRef CAS.
- H. Kim, S. R. Byeon, M. G. D. Leed and J. Hong, Tetrahedron Lett., 2011, 52, 2468–2470 CrossRef CAS.
- E. Y. Chia, S. Naeem, L. Delaude, A. J. P. White and J. D. E. T. Wilton-Ely, Dalton Trans., 2011, 40, 6645–6658 RSC.
- S. Naeem, L. Delaude, A. J. P. White and J. D. E. T. Wilton-Ely, Inorg. Chem., 2010, 49, 1784–1793 CrossRef CAS PubMed.
- S. P. Nolan, WO2008036084A1, 2008.
- A. J. Arduengo III, US5077414, 1991.
- D. Depuydt, A. Van den Bossche, W. Dehaen and K. Binnemans, RSC Adv., 2016, 6, 8848–8859 RSC.
- J. Zimmermann, B. Ondruschka and A. Stark, Org. Process Res. Dev., 2010, 14, 1102–1109 CrossRef CAS.
- R. S. Kalb, M. Damm and S. P. Verevkin, React. Chem. Eng., 2017, 2, 432–436 RSC.
- R. S. Kalb, E. N. Stepurko, V. N. Emel'yanenko and S. P. Verevkin, Phys. Chem. Chem. Phys., 2016, 18, 31904–31913 RSC.
- S. Kirchhecker, S. Tröger-Müller, S. Bake, M. Antonietti, A. Taubert and D. Esposito, Green Chem., 2015, 17, 4151–4156 RSC.
- N. J. Bridges, C. C. Hines, M. Smiglak and R. D. Rogers, Chem.–Eur. J., 2007, 13, 5207–5212 CrossRef CAS PubMed.
- S. Raiguel, J. Thomas, K. Binnemans and W. Dehaen, Eur. J. Org. Chem., 2018, 2018, 4850–4856 CrossRef CAS.
- G. Damilano, K. Binnemans and W. Dehaen, Org. Biomol. Chem., 2019, 17, 9778–9791 RSC.
- P. Wasserscheid and T. Welton, Ionic liquids in synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- A. J. Arduengo, US6177575, 2007.
- M. R. Grimmett, in Category 2, Hetarenes and Related Ring Systems, ed. R. Neier, Georg Thieme Verlag, Stuttgart, 2002 Search PubMed.
- B. Radziszewski, Ber. Dtsch. Chem. Ges., 1882, 15, 2706–2708 CrossRef.
- B. Radziszewski, Ber. Dtsch. Chem. Ges., 1882, 15, 1493–1496 CrossRef.
- H. Debus, Liebigs Ann., 1858, 107, 199–208 CrossRef.
- E. Gelens, F. J. J. De Kanter, R. F. Schmitz, L. A. J. M. Sliedregt, B. J. Van Steen, C. G. Kruse, R. Leurs, M. B. Groen and R. V. A. Orru, Mol. Divers., 2006, 10, 17–22 CrossRef CAS PubMed.
- S. Tröger-Müller, J. Brandt, M. Antonietti and C. Liedel, Chem.–Eur. J., 2017, 23, 11810–11817 CrossRef PubMed.
- U. Domańska, A. Rȩkawek and A. Marciniak, J. Chem. Eng. Data, 2008, 53, 1126–1132 CrossRef.
- V. Mazan, I. Billard and N. Papaiconomou, RSC Adv., 2014, 4, 13371–13384 RSC.
- C. Reichardt and T. Welton, in Solvents and Solvent Effects in Organic Chemistry, ed. C. Reichardt and T. Welton, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 549–586 Search PubMed.
- W. M. Jackson and J. S. Drury, Ind. Eng. Chem., 1959, 51, 1491–1493 CrossRef CAS.
- R. Rohini, C. K. Lee, J. T. Lu and I. J. B. Lin, J. Chin. Chem. Soc., 2013, 60, 745–754 CrossRef CAS.
- G. Gurau, H. Rodríguez, S. P. Kelley, P. Janiczek, R. S. Kalb and R. D. Rogers, Angew. Chem., Int. Ed., 2011, 50, 12024–12026 CrossRef CAS PubMed.
- G. K. Wagner, D. Kotschenreuther, W. Zimmermann and S. A. Laufer, J. Org. Chem., 2003, 68, 4527–4530 CrossRef CAS PubMed.
- S. A. Laufer, W. Zimmermann and K. J. Ruff, J. Med. Chem., 2004, 47, 6311–6325 CrossRef CAS PubMed.
- L. G. Sillén, A. E. Martell, J. Bjerrum and G. K. Schwarzenbach, Stability constants of metal-ion complexes, London Chemical Society, 2nd edn, 1964 Search PubMed.
- C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963–5977 RSC.
- K. Binnemans, Chem. Rev., 2005, 105, 4148–4204 CrossRef CAS PubMed.
- K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 2005 Search PubMed.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall, M. F. Mahon and P. A. Procopiou, Organometallics, 2008, 27, 3939–3946 CrossRef CAS.
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley-Interscience, New York, NY, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials, equipment specifications, and substances characterization. See DOI: 10.1039/d0ra03358h |
| This journal is © The Royal Society of Chemistry 2020 |