
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDityrosine suppresses the cytoprotective action of thyroid hormone T3 via inhibiting thyroid hormone receptor-mediated transcriptional activation†
Yin-Yi Ding‡
 *ab,
Fang-Fang Wang‡c,
Yu-Ge Jiangde,
Yi-Jing Shenga,
Meng-Qi Jianga,
Xuan Zhua,
Yong-Hui Shide and
Guo-Wei Lede
*ab,
Fang-Fang Wang‡c,
Yu-Ge Jiangde,
Yi-Jing Shenga,
Meng-Qi Jianga,
Xuan Zhua,
Yong-Hui Shide and
Guo-Wei Lede
aCollage of Food Science and Biotechnology, Zhejiang Gongshang University, No.18, Xuezheng Street, Hangzhou, 310018, China. E-mail: dyy-198655@163.com; Fax: +86 571-28877777; Tel: +86 571-28877777
bFood Nutrition Science Centre, Zhejiang Gongshang University, Hangzhou, 310018, China
cSchool of Life Science, Linyi University, Linyi, 276000, China
dThe State Key Laboratory of Food Science and Technology, Jiangnan University, Wuxi, 214122, China
eCenter of Food Nutrition and Functional Food Engineering, School of Food Science and Technology, Jiangnan University, Wuxi, 214122, China
First published on 2nd June 2020
Abstract
Dityrosine (Dityr) is the most common oxidized form of tyrosine. In the previous studies of mice treated with dityrosine, cell death in the pancreas, kidneys, and liver was detected in the presence of enhanced plasma triiodothyronine (T3) content. Due to its structural similarity with the thyroid hormone T3, we hypothesized that dityrosine might disrupt T3-dependent endocrine signaling. The cytotoxic effect of dityrosine was studied in C57BL/6 mice by gavage with a dityrosine dose of 320 μg per kg per day for 10 weeks. Cell death in the liver was detected in the presence of enhanced plasma thyroid hormone content in mice treated with dityrosine. The antagonistic effect of dityrosine on T3 biofunction was studied using HepG2 cells. Dityrosine incubation reduced T3 transport ability and attenuated the T3-mediated cell survival via regulation of the PI3k/Akt/MAPK pathway. Furthermore, dityrosine inhibited T3 binding to thyroid hormone receptors (TRs) and suppressed the TR-mediated transcription. Dityrosine also downregulated the expressions of T3 action-related factors. Taken together, this study demonstrates that dityrosine inhibits T3-dependent cytoprotection by competitive inhibition, resulting in downstream gene suppression. Our findings offer insights into how dityrosine acts as an antagonist of T3. These findings shed new light on cellular processes underlying the energy metabolism disorder caused by dietary oxidized protein, thus contributing to a better understanding of the diet–health axis at a cellular level.
1. Introduction
Some food ingredients, such as oxidized protein in protein-rich processed food, can affect glucose and lipid metabolism by disrupting physiological processes, particularly through direct or indirect interaction with thyroid hormones.1 Recent public and scientific interest has been mostly focused on protein oxidation in food systems during food processing and storage2 and the effects of dietary oxidized protein on human health.3 During food processing and storage, proteins are vulnerable to oxidation by both radical and non-radical oxidant factors,4 such as tyrosine, being readily oxidized to dityrosine (Dityr).5 Dityrosine, which exhibits pro-oxidant behavior, is considered an endocrine disruptor6 that causes oxidative damage in the liver7 and kidneys.8 Exposure to dityrosine changes many systemic metabolic processes, including reduced choline bioavailability as well as cell death in the liver, kidneys, and pancreas.9–12 Dityrosine exposure also induces learning and memory impairments.13,14 The endocrine disrupting effects of dityrosine are seen in other nuclear receptors, such as thyroid hormone receptors (TRs), acting as an antagonist to triiodothyronine (T3).15T3 is widely known for the ability to influence various cellular processes,16 energy expenditure, and metabolic rate.17,18 Numerous of studies have documented that T3 is a survival factor in cells, counteracting both physiological and pharmacological cell death.19 Evidence of protein–protein interactions between cytosolic TRs and phosphatidylinositol 3-kinase (PI3K) as well as activation of PI3K activity by T3 treatment have been reported.20,21 T3 can directly stimulate the activation of Akt, and activate cellular processes strictly related to cell function such as cell proliferation and survival, cell size regulation, protein synthesis, and insulin production.22,23 Thyroid hormone receptor β1 (TRβ1) mediates T3 upregulation of protein synthesis and cell survival playing a crucial role in T3 regulation of the PI3K/Akt pathway.24 TRβ1 seems to be an essential component in mediating T3 action on Akt pathway in the pancreas.24,25 We previously demonstrate that the increase of TRβ1 protein level induced by T3 is significantly inhibited by dityrosine, which subsequently suppress the T3-induced phosphorylation of Akt.15 Taken together, these results suggest that dityrosine may act as an antagonist in T3-regulated cell survival.
The structures of dityrosine and T3 are shown in Fig. 1. The resemblance between dityrosine and T3 is high at 70% similarity according to the database of Chemical Entities of Biological Interest (ChEBI, http://www.ebi.ac.uk/chebi/). Because of its structure homology with T3, the thyroid endocrine system disrupted by dityrosine is studied. Previous studies in our laboratory found that dityrosine administration disrupts regulation of the thyroid hormone T3 by targeting glucose-stimulated insulin synthesis in the pancreas.1 Furthermore, thyroid hormone receptor β1 (TRβ1) and translational factors involved in Akt-mTOR signaling pathway are modulated by dityrosine.15
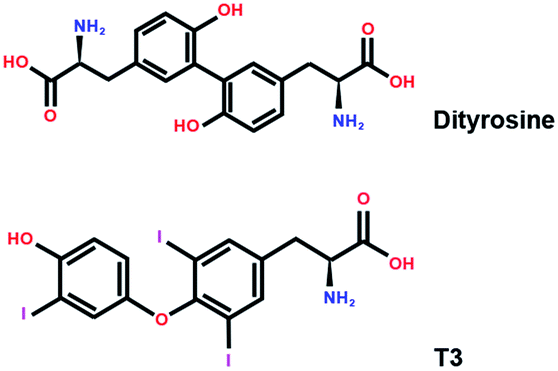 | ||
| Fig. 1 The chemical structure of T3 and dityrosine. | ||
T3 binds to TRs and then binds to a target DNA sequence known as a thyroid hormone response elements (TRE), composed of two half-site core motifs (AGGTCA) with specific nucleotide spacing and orientation.26,27 Besides, RAR-related orphan receptors α (RORα) is a novel member of the nuclear hormone receptor superfamily. Although the ligand of RORα has not been identified and its bio-function is not clear, RORα is widely expressed, including in central nervous system.28 RORα binds to a hormone-response element (ROR-response element [RORE]), which is composed of a 6-bp AT-rich sequence 5′, a half core motif (GGTCA), and an active transcription.28 As TRs and RORα are transcription factors that share a common core motif within the response elements, previous studies have demonstrated that RORα modulates TRs action on TRE by competitively binding to the TRE and forming heterodimers with the TRs.29
Given the findings of the aforementioned researches, examining the relationship between dityrosine and T3 action is at present a major research topic. However, previous studies have not fully explained the antagonistic effects and mechanisms of dityrosine on T3 action. In the present study, we investigate the effects of dityrosine on the cytoprotection action of T3 in liver via a 10 week gavage experiment involving C57BL/6 mice administrated with dityrosine. We used molecular docking simulations to determine the effects of dityrosine on the TRβ1–T3 binding ability. Moreover, we performed T3–TRβ1 binding studies using [125I]T3 to determine the effects of dityrosine on the T3–TRβ1 binding ability. Finally, we conducted a cell experiment exposing HepG2 cells to dityrosine and T3 to evaluate whether dityrosine could suppress T3 action and investigated the effects of dityrosine on the crosstalk between TRs and T3 on TRE via a transient transfection study.
2. Materials and methods
2.1. Animal experiment using C57BL/6 mice
We previously performed a 6 week preliminary study and found that gavage with a dityrosine dose of 320 μg per kg per day caused thyroid hormone resistance and novel object recognition deficits in C57BL/6 mice.15,29 In this study, mice were randomly divided into two groups that received either dityrosine (Ruidong Biotechnology, Shanghai, China; dityrosine > 95%) (Dityr group) or saline solution (control group) (n = 10) after 7 days of acclimatization. After a 10 week period, the mice were anesthetized by pentobarbital sodium (50 mg kg−1, i.p.). Blood was collected from the orbital sinuses and placed into anticoagulant tubes. Plasma was obtained from the blood samples after centrifugation (1570×g for 10 min at 4 °C). All mice were sacrificed by cervical dislocation. The liver was dissected by an animal care technician based on morphological features and fixed in 10% formaldehyde phosphate buffer. The rest of the liver was rinsed with saline and flash frozen in liquid nitrogen for further analysis.
All animal treatments were performed in accordance with the ethical standards set by the National Institutes of Health Guide for the Care and Use of Laboratory Animals (China) and in the 1964 Declaration of Helsinki and its later amendments. All animal studies were approved by the Laboratory Animals Ethics Committee of Zhejiang Gongshang University.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500) (Servicebio, China) at 4 °C overnight. The slices were subsequently incubated with horseradish peroxidase-conjugated secondary antibody (1:200) (Servicebio, China) for 30 min at 37 °C. DAB coloration kit (DAKO, Denmark) was used for color-reaction. Slices were counterstained with hematoxylin. The pictures of the slices were taken by optical microscope (BX41, Olympus, Japan). The immunostaining intensity was analyzed using the Image-pro Plus 5.0 software (Media Cybernetics Inc., USA).
:500) (Servicebio, China) at 4 °C overnight. The slices were subsequently incubated with horseradish peroxidase-conjugated secondary antibody (1:200) (Servicebio, China) for 30 min at 37 °C. DAB coloration kit (DAKO, Denmark) was used for color-reaction. Slices were counterstained with hematoxylin. The pictures of the slices were taken by optical microscope (BX41, Olympus, Japan). The immunostaining intensity was analyzed using the Image-pro Plus 5.0 software (Media Cybernetics Inc., USA).2.2. Cell experiment using HepG2 cells
The cells were seeded in plates and permitted to adhere at 37 °C in a humidified atmosphere containing 95% O2 and 5% CO2 for 24 h. When the cells reached 40% confluence, they were stimulated with dityrosine (final concentration: 0, 0.1, 1, or 10 μM) or T3 (final concentration: 0 or 1 μM for 72 h).
2.3. Protein extracts and western blot analysis
Protein was extracted from HepG2 cells using RIPA Lysis Buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS and protease inhibitors (sodium orthovanadate, sodium fluoride, EDTA and leupeptin). One mM (final concentration) phenylmethanesulfonyl fluoride was added to the lysis buffer before use. Liver (200 mg) and cells were homogenized in 1.5 mL lysis buffer on ice. After lysis, the homogenate was centrifuged for 5 min at 12000×g. The protein concentration in the supernatant was determined by the BCA assay kit (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer's instructions. All extracts were mixed with 1× loading buffer (50 mM Tris–HCl pH 6.8, 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol and 5% β-mercaptoethanol) in the ratio of 4:1 and then were boiled at 95 °C for 10 min to denature the protein.
The proteins were separated by 12% SDS-PAGE gel with 10 μg of total protein per lane and then transferred onto nitrocellulose membrane. The membranes were blocked in Tris-buffered saline (pH 7.4) with 5% bovine serum albumin (BSA) and 0.1% Tween 20. The blocked membrane was incubated with the following primary antibody: rabbit TRβ1, MAPK (ERK1/2), phosphor-MAPK (ERK1/2) (Thr202/Tyr204), caspase 3, Bax, Bcl-2 antibody and rabbit β-actin antibody (1:1000; Cell Signaling Technology; USA) at 4 °C for 10 h. The blots were incubated with secondary antibody (1:4000; Sigma-Aldrich, USA) and fluorescence images were obtained with an automatic chemiluminescence imaging analysis system (LI-COR Biosciences, USA).
2.4. Total mRNA isolation and quantitative RT-PCR (qRT-PCR)
Total RNA was extracted with Trizol reagent according to the manufacturer's protocol (Applied Biosystems; U.S.A.). qRT-PCR was performed using an SYBR green based qRT-PCR kit according to the manufacturer's instructions on a 7900HT instrument (Applied Biosystems, USA). The specificity of the product was assessed from melting curve analysis. Gene expressions were determined using the 2−ΔΔCT method. The primer's sequences for the genes are shown in ESI Table 2.†2.5. Molecular docking simulations
Molecular docking simulations of the ligands (dityrosine and T3) into the TRβ1 (1NAX) binding sites were performed using AutoDock (version 4) in the present work,30 the crystal structures of 1NAX were extracted from the Protein Data Bank (http://www.rscb.org/pdb). The grid-based docking program was used to obtain the binding modes of the compounds. The interaction energy between the ligands and TRβ1 were evaluated using atom affinity potentials calculated on a grid similar to the one described by Goodford.31 During the molecular docking process, the molecule in the crystal structure was used as a standard docking model.For molecular docking, AutoGrid was used for preparing the grid map using a grid box with a docking box of 60 × 60 × 60 Å, the box spacing was set to 0.375 Å. The grid center was designated with the following dimensions (xyz): 9.421 × 16.953 × 26.432. The docking possibilities were calculated by the genetic algorithm with local search (GALS). For each ligand, the simulation was composed of 100 docking runs using the standard AutoDock parameters.
2.6. T3 binding study
2.7. TRs-mediated transcriptional activation
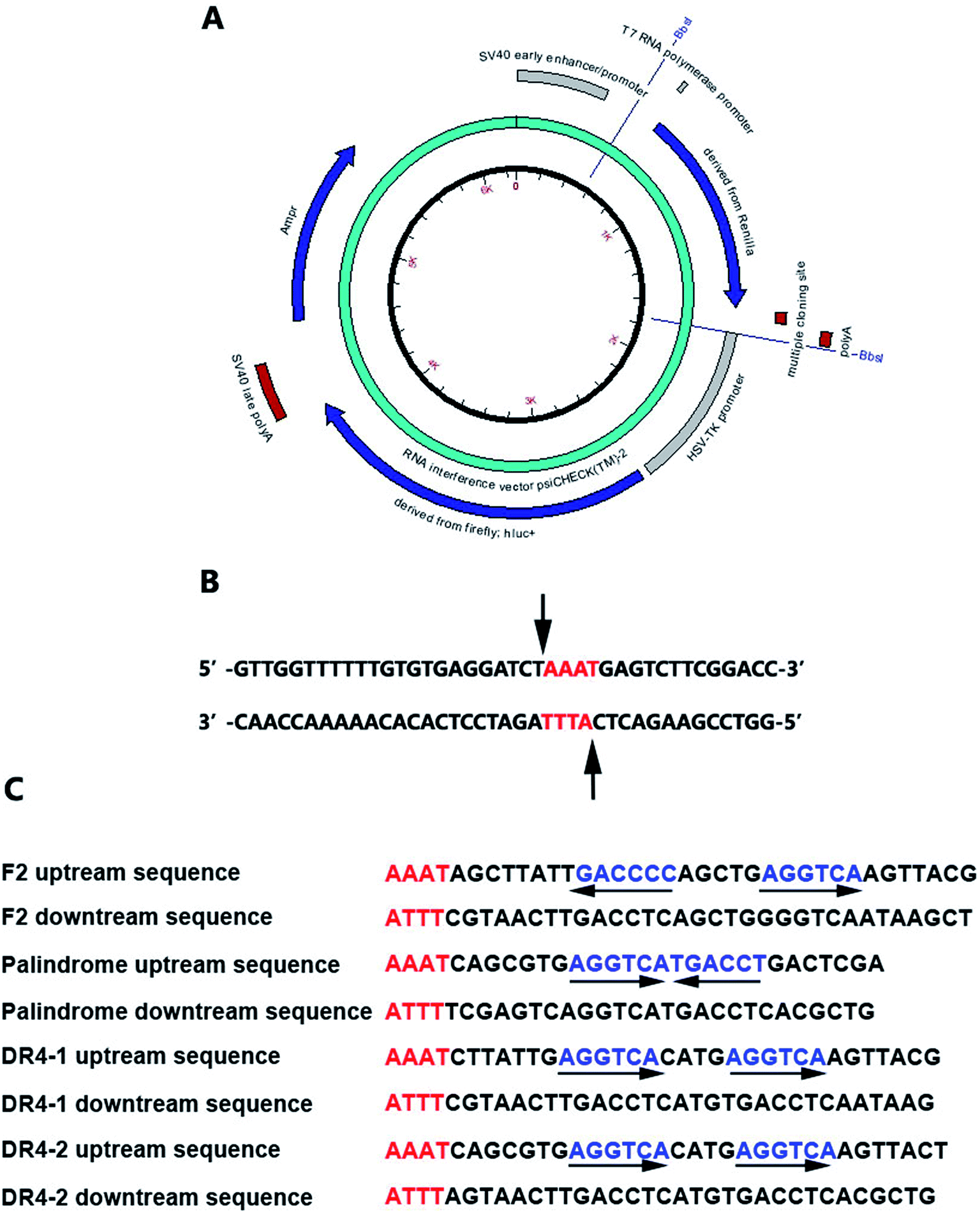 | ||
| Fig. 2 (A) The reporter plasmids; (B) the cutting site; (C) the nucleotide sequences of double-stranded oligonucleotides containing TRE or RORE. | ||
Accelrys DS Gene software (Accelrys Software, Inc., U.S.A.) was adopted to analyze the enzyme-cutting site of the psiCHECK2 promoter between the Poly A and hsv-tk promoter. The Bbs I enzyme was selected and the cutting site is shown in Fig. 2B. The nucleotide sequences of the double-stranded oligonucleotides containing TRE or RORE are shown in Fig. 2C.
2.8. Statistical analysis
All measurement value are expressed as mean ± S.E.M. Significant differences between groups were determined by one-way analysis of variance (ANOVA) using SPSS 20.0 software (IBM, Armonk, NY, USA) for Windows. Differences are considered significant at p < 0.05.3. Results
3.1. Dityrosine causes histopathological changes and apoptosis in liver accompanied by increased THs level
As shown in Fig. 3A and B, at the morphological level, the liver of control mice showed normal hepatocellular architecture with the trabeculae of hepatocytes separated by blood sinusoids radiating from the central vein. The hepatocytes with mostly round or oval nuclei located on the side of the cell membrane were arranged with each other. The shapes of the cells were regular and the cell boundary were clear. The livers of dityrosine-treated mice showed improvement in the hepatocellular damage caused by dityrosine, including loss of architecture, unclear cell boundaries, and area of hepatocyte swelling, vacuolation and inflammation (leukocyte infiltration). In addition, dityrosine treatment resulted in a much higher number of TUNEL-positive cells compared to that in the control mice.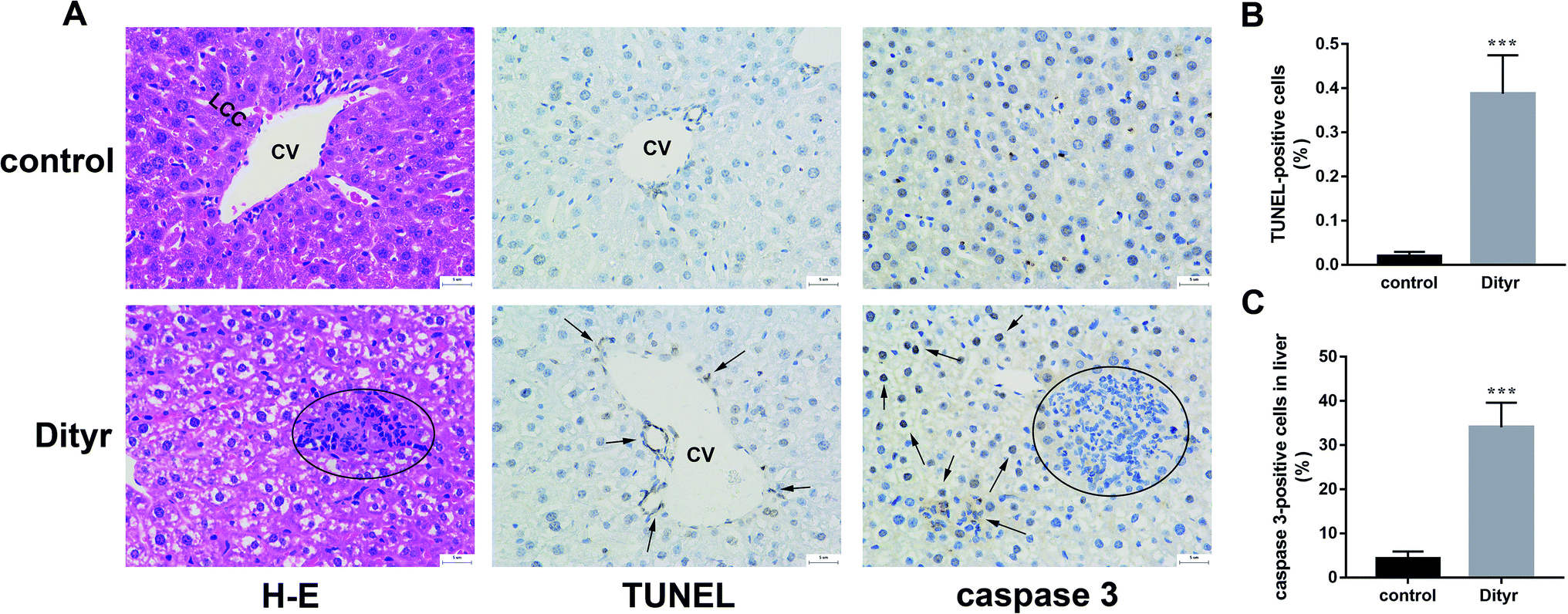 | ||
| Fig. 3 Dityrosine causes apoptosis in liver of mice. (A) H & E staining image (first panel, 400×, space bar: 50 μm, the circle indicates inflammatory cell infiltration), the TUNEL staining image (second panel, 400×, space bar: 50 μm, the arrows indicate TUNEL-positive cells), the caspase 3 immunohistochemistry image (third panel, 400×, space bar: 50 μm, the circle indicates inflammatory cell infiltration, the arrows indicate caspase 3-positive cells); (B) the percentage of TUNEL-positive cells of liver (%); (C) the percentage of caspase 3-positive cells (%).***p < 0.001 when compared with the control group. | ||
To deepen into the molecular changes induced by dityrosine in the apoptosis cascade, two major pro-apoptotic molecules and one anti-apoptotic molecule were analyzed by qRT-PCR. As shown in Fig. 4, dityrosine clearly induced the mRNA expression of Bax and caspase 3, and significantly decreased the expression of Bcl-2. However, TT3, FT4, and FT3 in plasma increased in dityrosine-treated mice (Fig. 5). These results showed that the cell death observed in livers of dityrosine-treated mice accompanied by increased T3 level, which suggested that dityrosine administration might suppresses the cytoprotection action of T3.
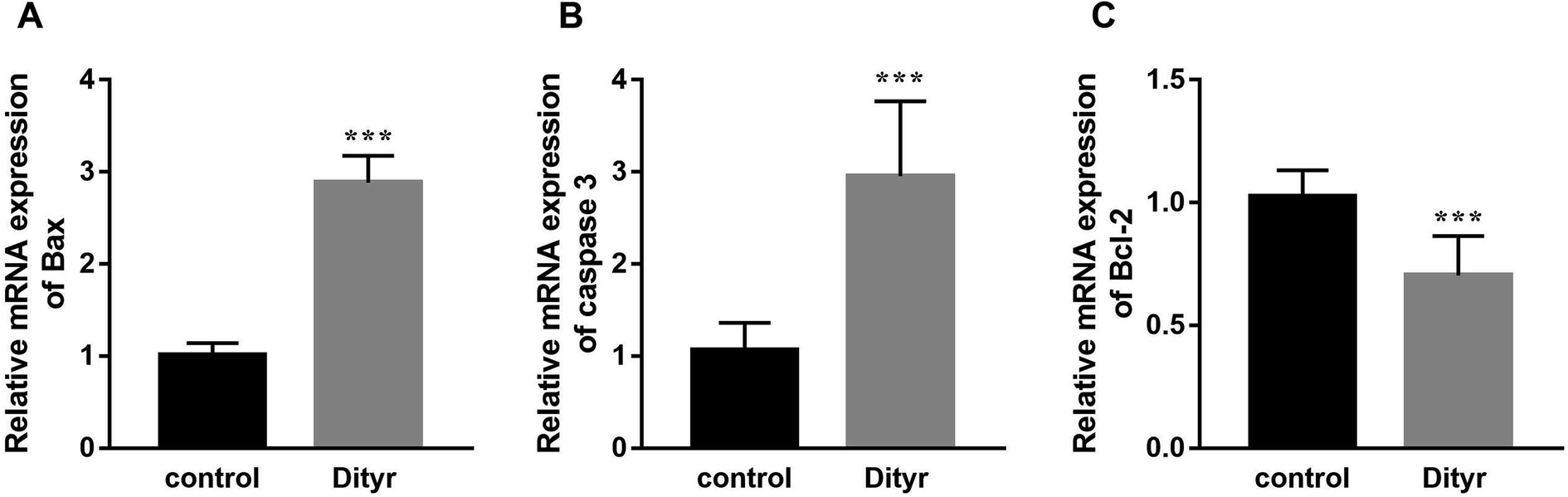 | ||
| Fig. 4 The mRNA expression of Bax, caspase 3, and Bcl-2 of liver. ***p < 0.001 when compared with the control group. | ||
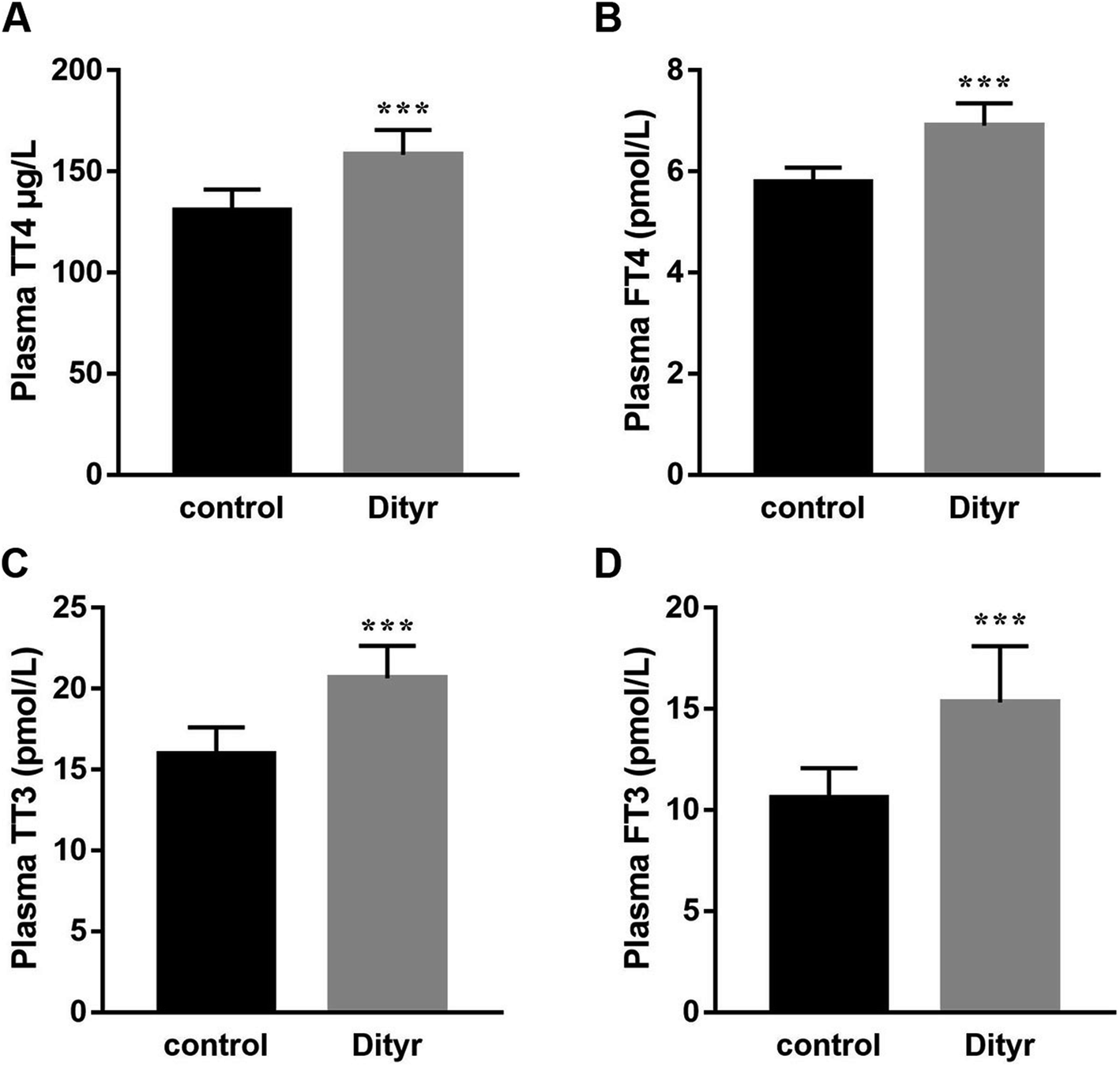 | ||
| Fig. 5 Dityrosine changes the plasma content of THs. (A) Plasma TT4 content of mice; (B) plasma TT3 content of mice; (C) plasma FT4 content of mice; (D) plasma FT3 content of mice. ***p < 0.001 when compared with the control group. | ||
3.2. Dityrosine suppresses T3-dependent survival of HepG2 cells
We used HepG2 cells as model to verify the hypothesis that cytoprotection action of T3 was disrupted by dityrosine as an antagonist. As shown in Fig. 6, dityrosine-only treatment caused a markedly increased degree of apoptosis in HepG2 cells, which was accompanied by alteration of the expression of apoptosis-related genes. Meanwhile, the survival of cells with 1 μM T3 treatment in the presence of dityrosine was notably declined when compared to cells exposed to T3 only.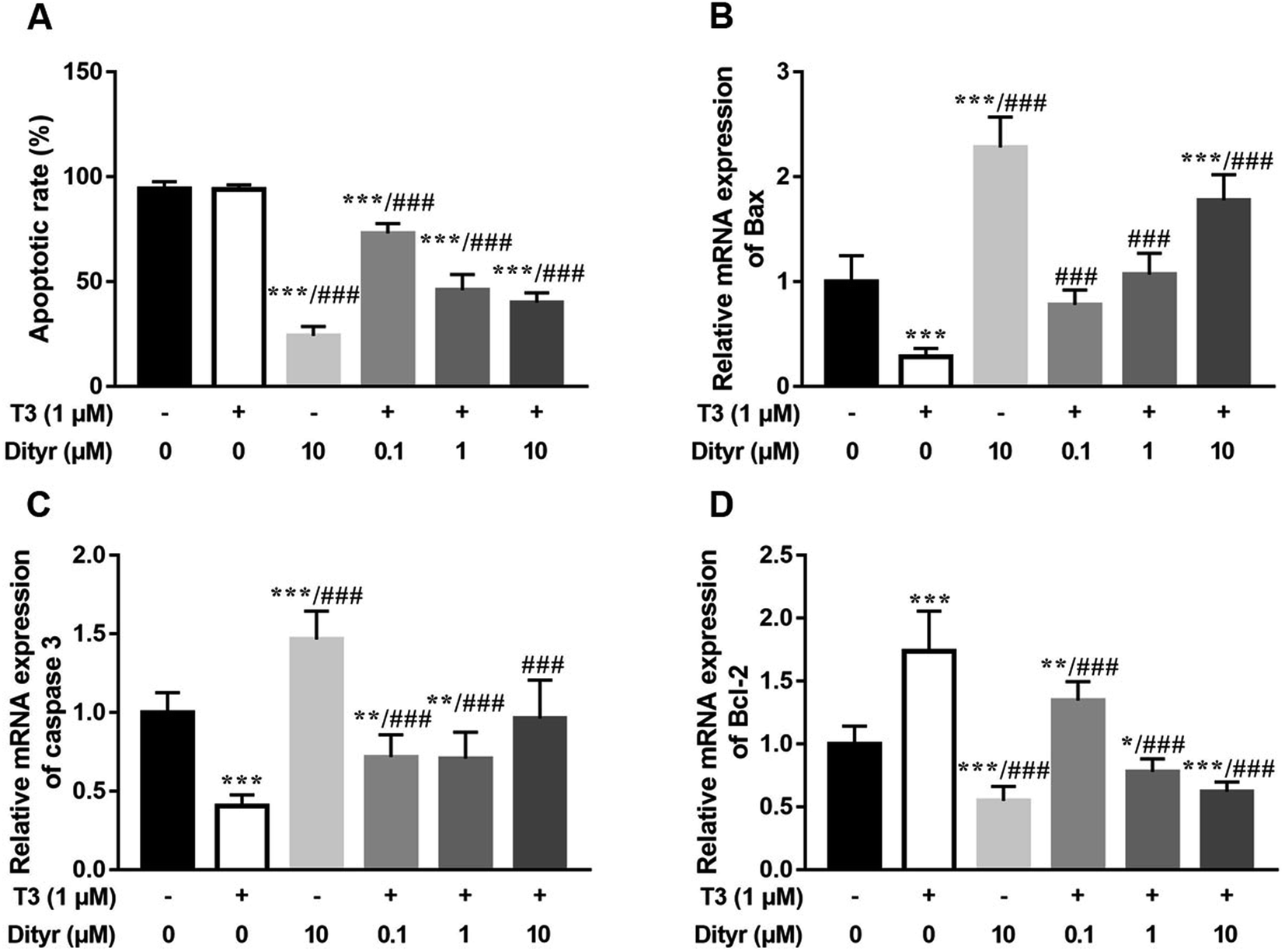 | ||
| Fig. 6 Dityrosine causes apoptosis in HepG2 cells. (A) the proportion of apoptotic cells in each group evaluated by flow cytometry; (B–D) the mRNA expression of Bax, caspase 3, and Bcl-2. *p < 0.05, **p < 0.01, ***p < 0.001 when compared with the control group. ###p < 0.001 when compared with T3-only treated cells. | ||
As shown in Fig. 7, the western blot results confirmed that protein level of PI3k, the phosphorylation of Akt (Ser473/Thr308), and phosphorylation of MAPK (Thr202/Tyr204) increased when the cell were treated with only T3. Dityrosine exposure markedly declined the protein level of PI3k, p-Akt (Ser473/Thr308), and p-MAPK (Thr202/Tyr204). Meanwhile, the protein expressions of PI3k and phosphorylated Akt and MAPK induced by T3 were suppressed with the addition of dityrosine.
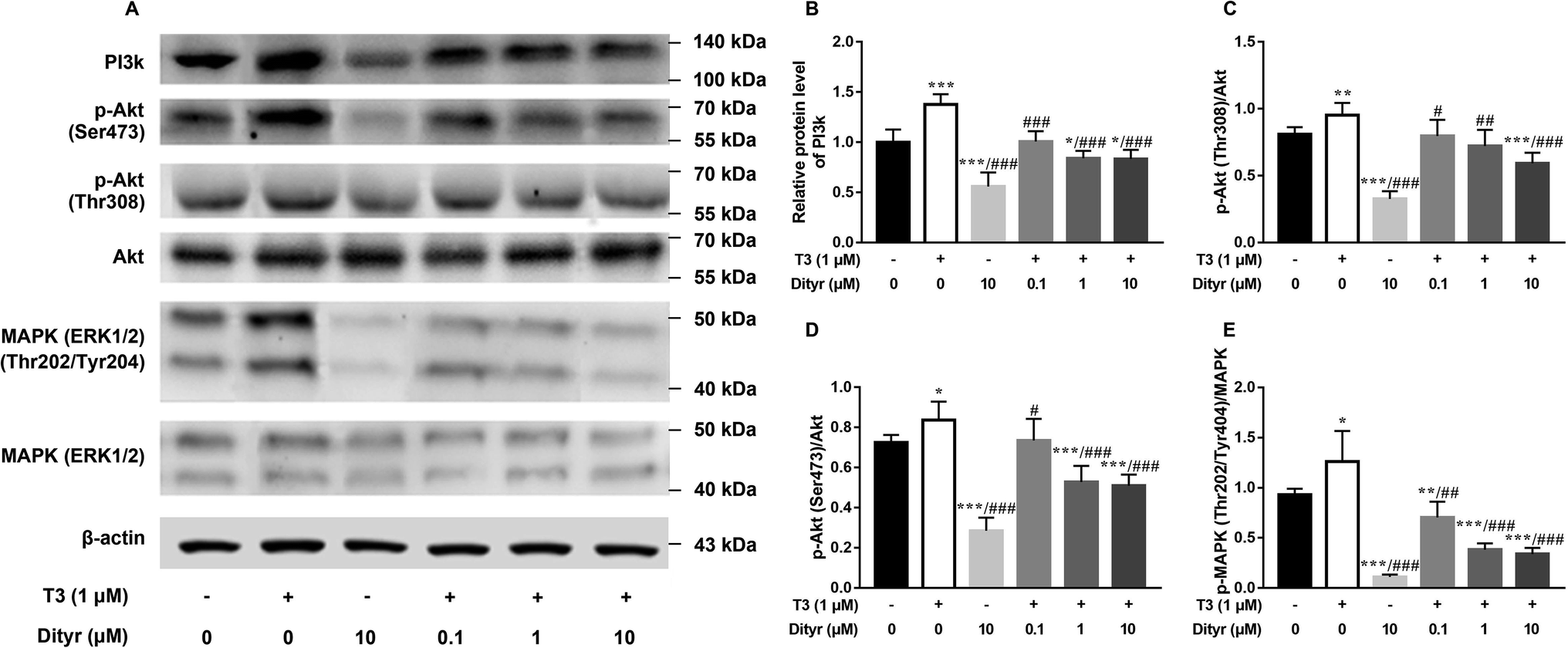 | ||
| Fig. 7 Dityrosine suppresses the activity of PI3k/Akt/MAPK pathway. (A) The western blot image; (B) the quantitative result of the PI3k protein expression; (C and D) ratio of phosphorylated (Thr 308 and Ser 473) Akt and Akt; (E) ratio of phosphorylated (Thr 202/Tyr 404) MAPK and MAPK. *p < 0.05, **p < 0.01, ***p < 0.001 when compared with the control group. #p < 0.05, #p < 0.01, ###p < 0.001 when compared with T3-only treated cells. | ||
Together with our previous evidences in mice model in this paper, these results strongly suggested that dityrosine inhibited the T3-denpendent protective effects on cells at least in part through the PI3k/Akt/MAPK pathway and apoptotic cascade.
3.3. Dityrosine reduces T3 transport and the expression of T3 action-related factors
As shown in Fig. 8A and B, the cellular T3 content and mRNA level of the T3 membrane transporter (MCT8) in dityrosine + T3-treated cells were significantly lower than those of the in cells treated with T3 only. These results suggested that T3 transport ability was suppressed by dityrosine.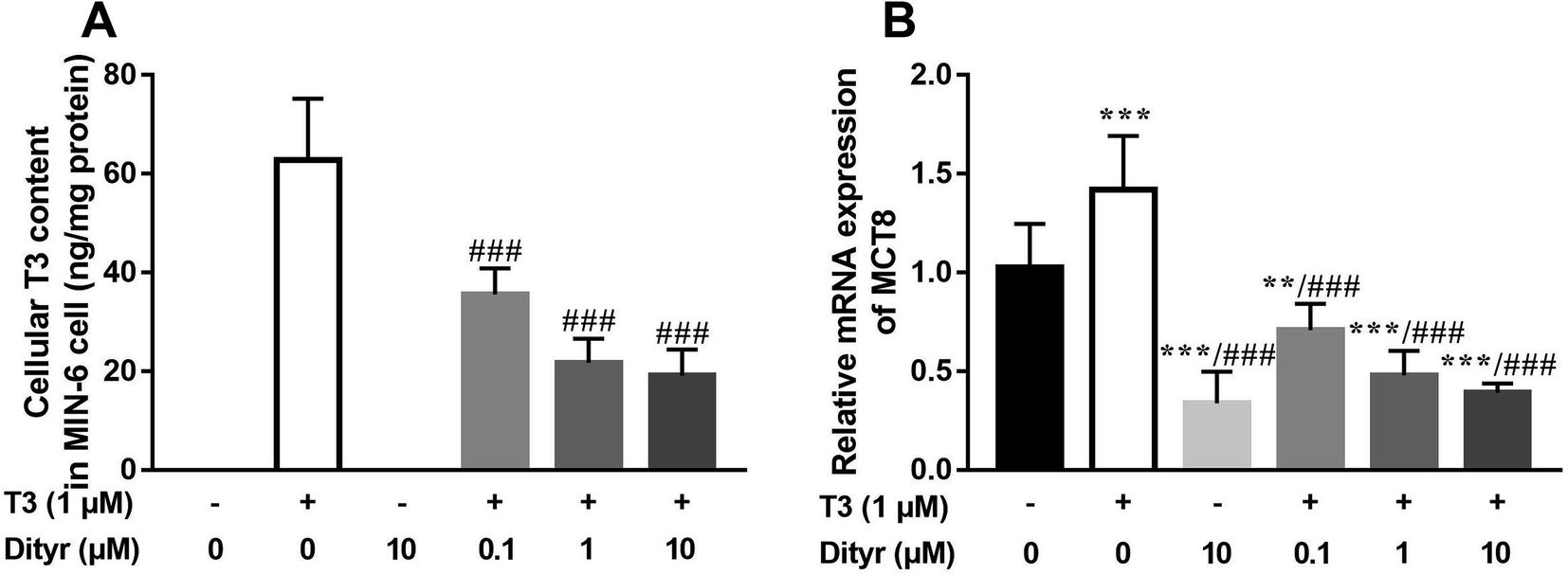 | ||
| Fig. 8 Dityrosine reduces the transport ability of T3. (A) The cellular T3 concentration in MIN-6 cell; (B) the mRNA expression of MCT8. **p < 0.01, ***p < 0.001 when compared with the control group. ###p < 0.001 when compared with T3-only treated cells. | ||
3.4. Dityrosine and T3 competitively bind to TRβ1
To investigate the role of dityrosine in the function of T3, we determined the competitive inhibition of dityrosine on T3 action. A molecular docking study showed the protein–inhibitor interactions and the structural features of active site of the receptor. As with models of 1NAX-DT, the combine pocket of the receptor was exposed in Fig. 9A. Dityrosine was packaged in the pocket, which was composed by the residues Arg282, Leu341, Phe455, Phe269, Ser314, Gly345, Arg320, Phe272, Met442, Arg317, Gly344, Met313, Thr273, Ile275, Ile276, His435, Leu346, Ala279, Met310, and Arg316. Fig. 9B showed dityrosine had three hydrogen bond interactions with the receptor. The –COOH made two hydrogen bonds with the Arg282 (H-1, –O⋯HN, 151.4°) and Arg320 (H-2, –O⋯HN, 155.8°) amino acids at the distances of 2.11 Å and 2.25 Å, respectively, further enhancing the ligand–receptor interactions.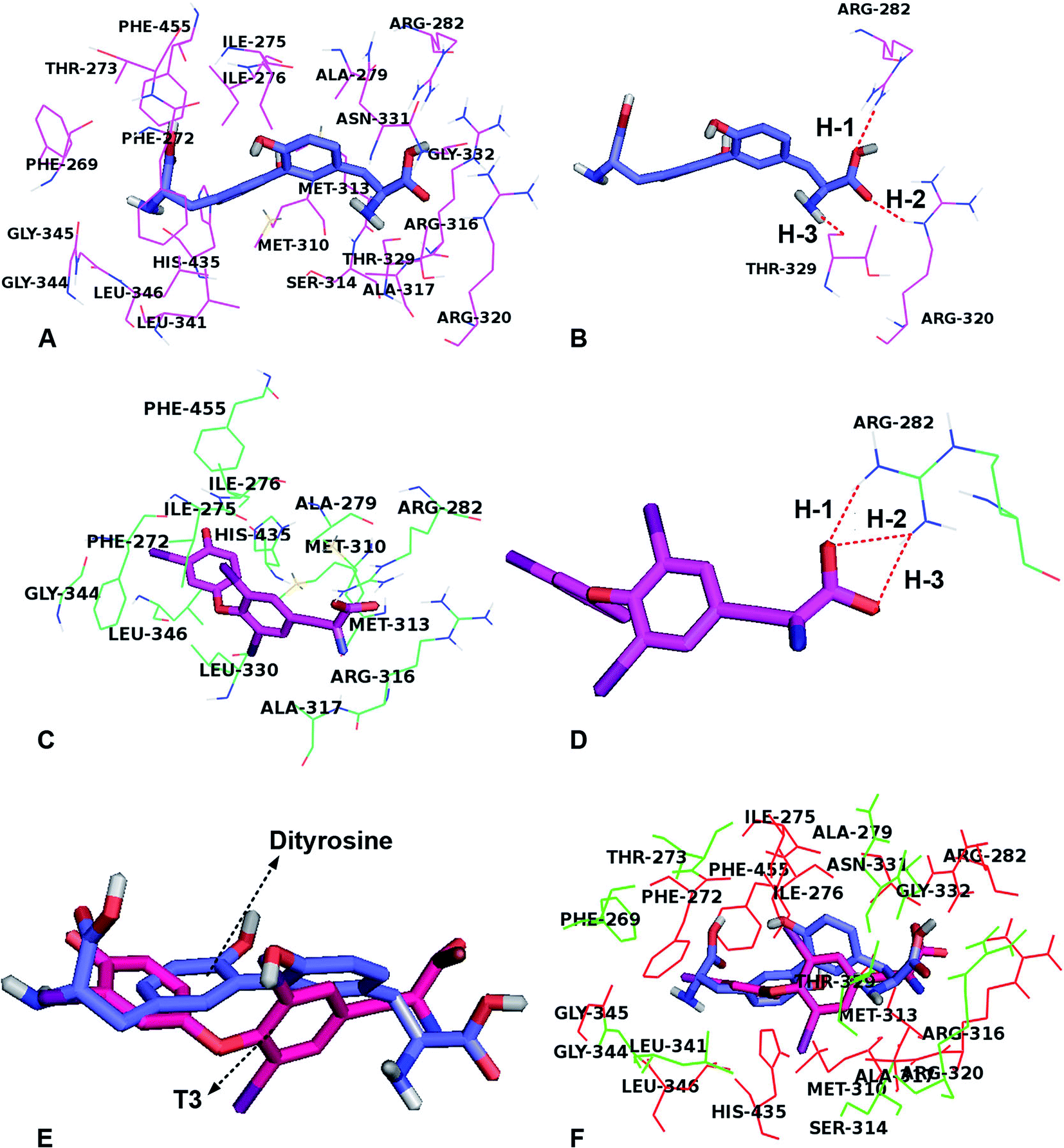 | ||
| Fig. 9 Dityrosine inhibits T3 binding capacity to TRs. (A) The TRβ active site amino acid residues around dityrosine; (B) the enlargement for the ligand in the binding site after molecular docking, which is displayed in stick, hydrogen bonds are shown as dotted red lines, and the nonpolar hydrogens were removed for clarity; (C) the TRβ active site amino acid residues around T3; (D) the enlargement for the ligand in the binding site after molecular docking, which is displayed in stick, hydrogen bonds are shown as dotted red lines, and the nonpolar hydrogens were removed for clarity; (E) structural superposition of TRβ-dityrosine, TRβ-T3; (F) the amino acids located around ligands, the ligands are displayed in stick (dityrosine-blue, T3-magenta), and the related amino acids are displayed in lines (the same residues-red, different residues-green and magenta). | ||
Fig. 9C and D showed the docking-binding mode of T3 in the active site containing the highly conserved residues Phe272, Ile275, Ile276, Ala279, Arg282, Met310, Met313, Arg316, Ala317, Leu330 Gly344, His435 and Phe455 in model 1NAX-T3 (Fig. 9C). Fig. 9D showed the –COOH group acted as the hydrogen bond acceptor, forms hydrogen bond with amino acid Arg282 (–O⋯HN, 1.94 Å, 145.3°) (H-1), (–O⋯HN, 2.10 Å, 139.4°) (H-2) and (–O⋯HN, 2.06 Å, 152.1°) (H-3).
In addition, dityrosine and T3 were buried in the binding pocket of the same receptor (Fig. 9E and F). We noted that the two compounds possessed the same binding site and exhibit a similar orientation, because the two compounds were structurally similar. However, there were differences existed in the structures, leading to the discrepancy of the ligand binding environment. For example, differences were observed for the two compounds in the same receptor derived from the benzene ring, caused a shift in the –COOH groups, leading to the carboxyl group of dityrosine form hydrophilic interactions with amino acid residues Arg320, Thr329, Asn331 and Gly332. Furthermore, the hydrogen bond interactions for dityrosine and T3, influence the receptor binding activity. As shown in Table 1, the two compounds displayed similar binding mode and energies (dityrosine = −10.0 kcal mol−1, T3 = −10.2 kcal mol−1). Therefore, we believe that they competitively bind to the same receptor.
| Ligand–receptor | The biological binding energy |
|---|---|
| TRβ1–T3 | −10.2 kcal mol−1 |
| TRβ1–dityrosine | −10.0 kcal mol−1 |
3.5. Dityrosine inhibits T3 binding to the TRs
Dityrosine displaced [125I]T3 from endogenous TRs with an IC50 of 11.02 nM (Fig. 10A). In Fig. 10B, the Scatchard analysis revealed that addition of dityrosine decreased the association constant (Ka) from 0.0324 to 0.0213, which was expressed as the absolute value of the slope. These results suggested that dityrosine might impair thyroid hormone action by inhibiting T3 binding to the TRs.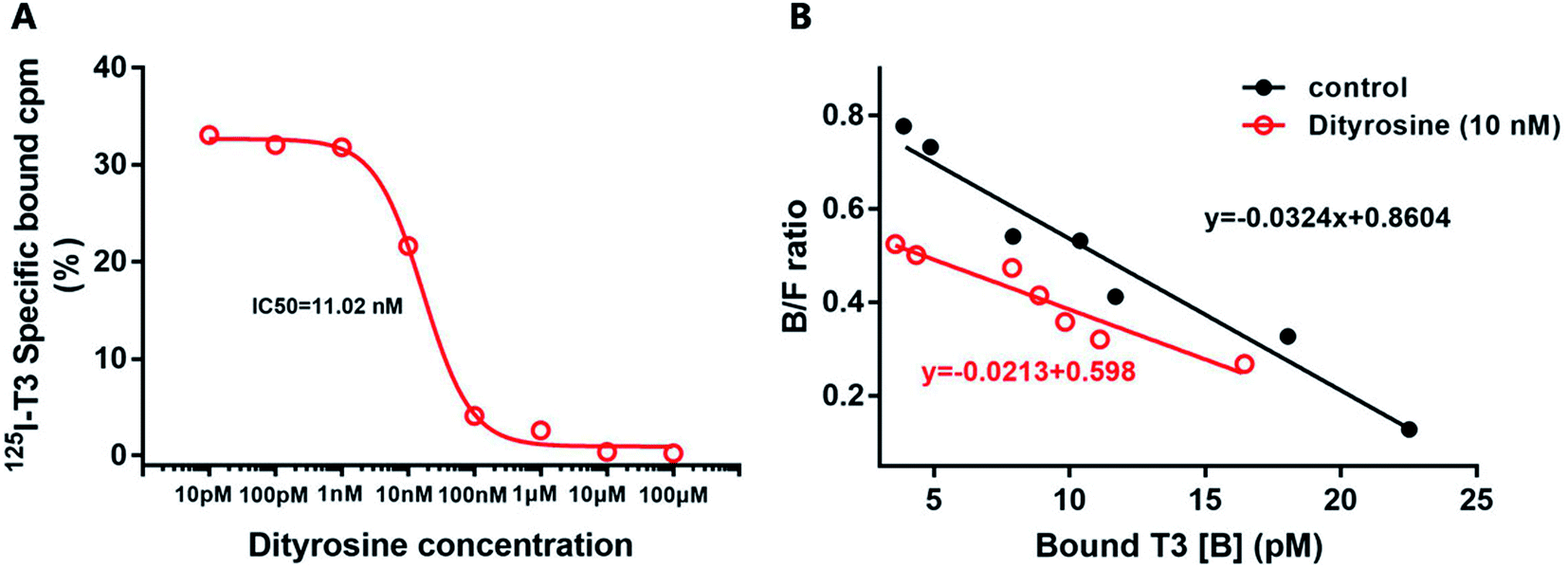 | ||
| Fig. 10 (A) The inhibition binding assay. Nuclear receptor from HepG2 cell is incubated with [125I] T3 and increasing amounts of dityrosine. (B) Scatchard analysis. Nuclear receptor from HepG2 cell is incubated with increasing amounts of T3 and [125I] T3 in the presence or absence of dityrosine (10 nM). B, bound; F, free. | ||
3.6. Dityrosine down-regulates the expression of TRβ1 and its co-activator factors
Fig. 11A and B showed the down-regulation of the mRNA levels of TRs co-activator factors (Rxrα, Src-1) in dityrosine-treated cells, which also suggested that the T3-dependent activation of downstream genes are suppressed by dityrosine. Moreover, T3 treatment increases TRβ1 protein level in cells. However, this action was significantly inhibited by dityrosine (Fig. 7C and D), suggesting that the T3 transport ability and T3-dependent stimulation of downstream genes are suppressed by dityrosine.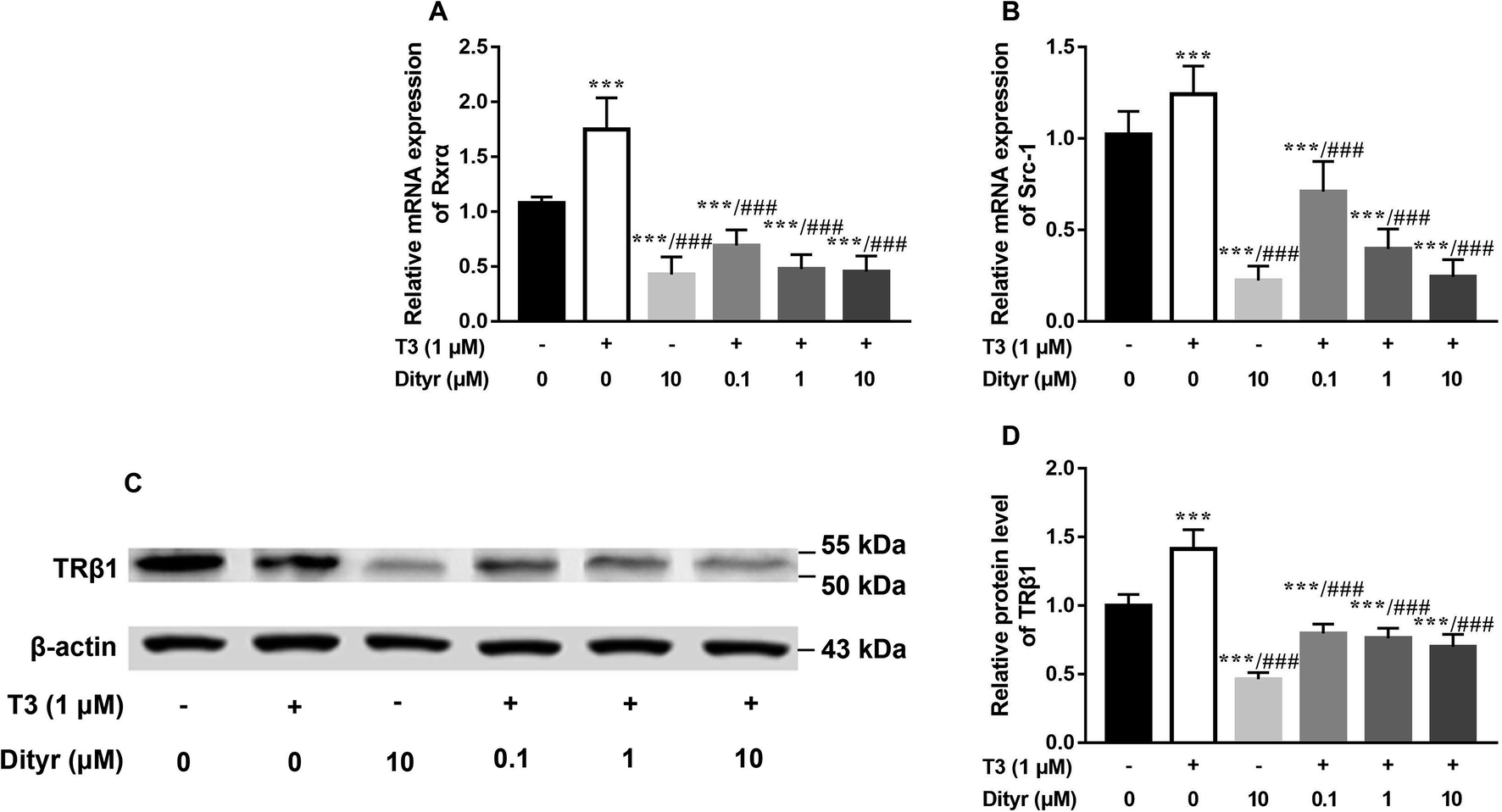 | ||
| Fig. 11 Dityrosine suppresses the expression of T3 function-related factors in HepG2 cells. (A and B) The mRNA expression of Rxrα, and Src-1; (C and D) the western blot image and quantitative results of TRβ1. ***p < 0.001 when compared with the control group. ###p < 0.001 when compared with T3-only treated cells. | ||
3.7. Dityrosine suppresses TR-mediated transcription
Fig. 12 showed the activities of the four luciferases increased when cells were exposed to T3. The highest luciferase activities were observed in cells incubated with 1 μM T3. In the presence of 1 μM T3, an increasing amount of dityrosine was added to the medium, and transcriptional activity was measured. In Fig. 13, dityrosine suppressed the transcriptional activity mediated by TR in a dose-dependent manner. These results suggested that dityrosine might impair thyroid hormone action by suppressing TR-mediated transcription.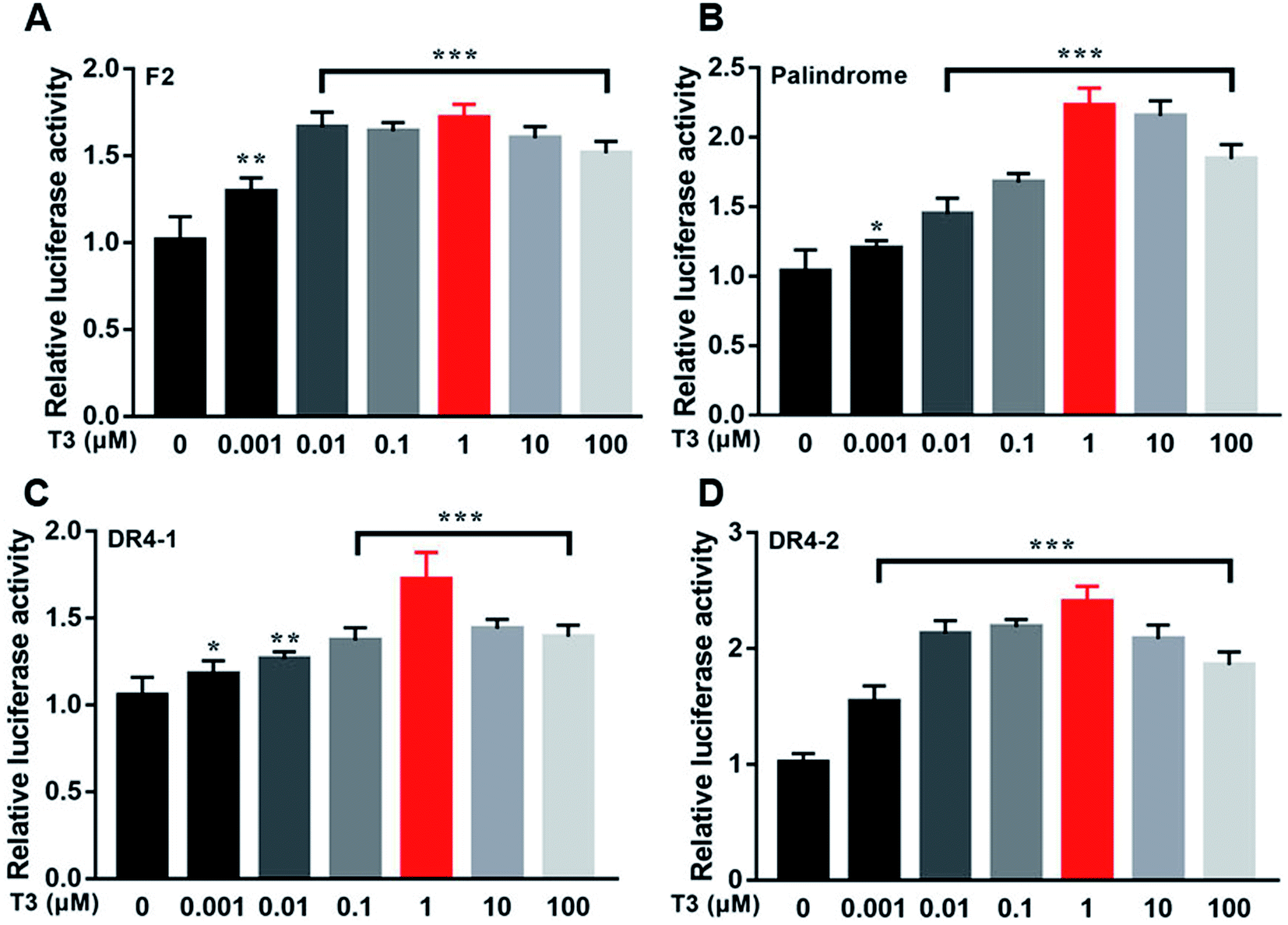 | ||
| Fig. 12 The activity of the four luciferases when cells are incubated with a tracer dose of T3. ***p < 0.001 when compared with the control group. | ||
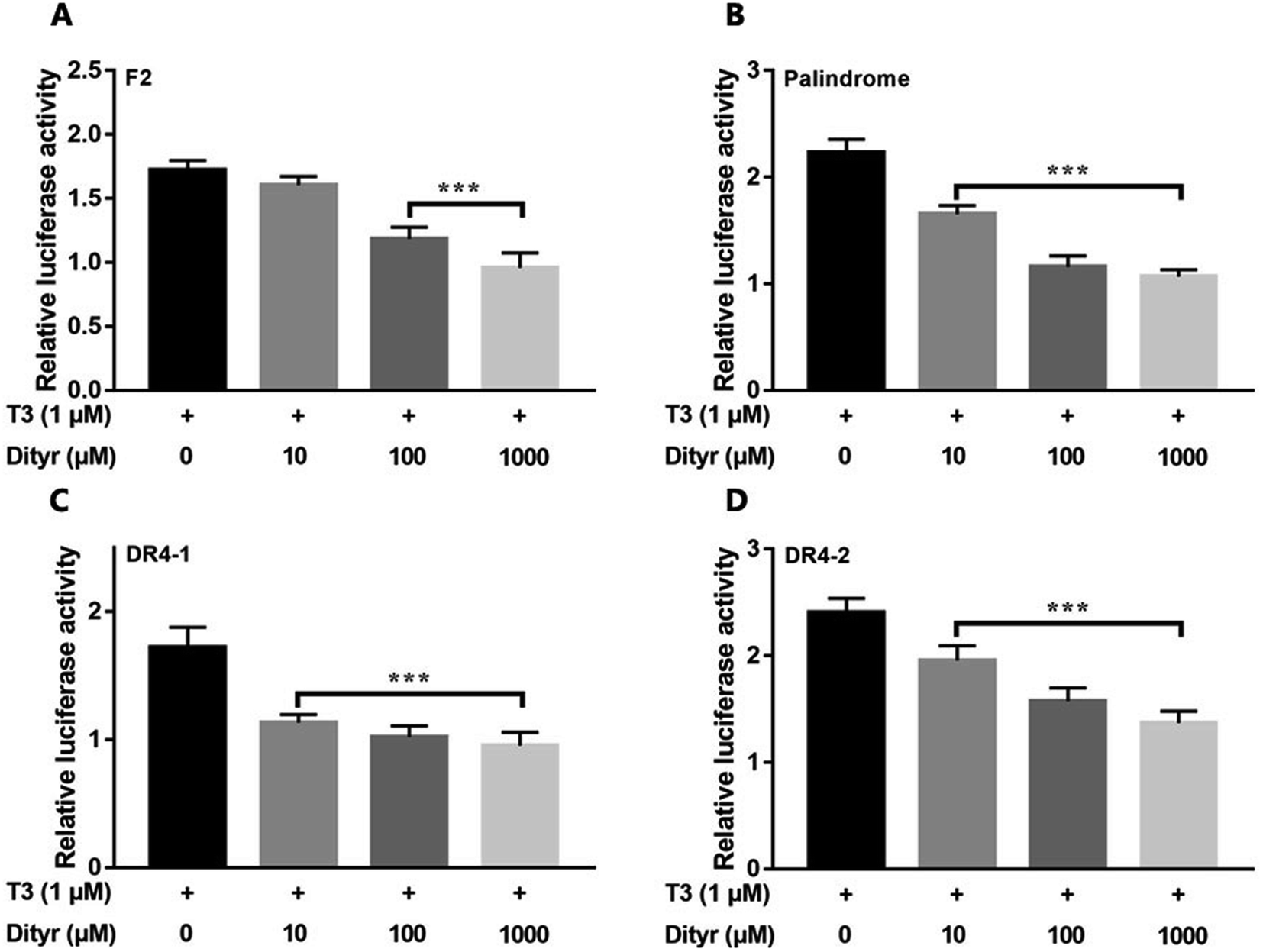 | ||
| Fig. 13 The activity of the four luciferases when cells are incubated with T3 (10−6 M) and increasing amounts of dityrosine. ***p < 0.001 when compared with the control group. | ||
4. Discussion
Structure changes in amino acids induced by oxidation results in loss of the nutritional value and functionality of food proteins. Oxygen radicals can oxidize tyrosine residues leading to the conversion of tyrosine to 3-3′-dityrosine.38 The structure of dityrosine is stable and does not undergo enzymatic hydrolysis easily, therefore, dityrosine is widely detected in the food system including meat and dairy products.39,40 Dityrosine can be transported to important organs by the systemic circulation and produce harmful effects,38,41,42 including impaired protein synthesis and cytotoxicity.9 In this study, we observed that the liver from dityrosine-treated mice showed increased apoptosis in the present of enhanced plasma THs level. It is probable that dityrosine has the potential to interfere with the cytoprotective activity of T3.We subsequently investigated the underlying mechanism involved in the inhibition of T3 activity by dityrosine in liver. Decreased T3 transport is one plausible cause of dityrosine-dependent T3 suppression.43 The transport of T3 into target cells requires the presence of transport proteins on the cell membrane. Monocarboxylate transporter 8 (MCT8) is a highly specific transporter of T3.44 Decreased cellular T3 content and mRNA expression of MCT8 in dityrosine-treated HepG2 cells suggest that dityrosine inhibits T3 transport capacity in cells.
Activation of PI3k/Akt/MAPK pathway leads to events that include the triggering of the Akt kinase activation and expression of its downstream genes.45 Herein, we show that the T3-induced anti-apoptotic effects were depressed by dityrosine, which were associated with decreased Akt activity. In addition, we also observed a key role off phosphorylated MAPK in inducing cytotoxicity during dityrosine exposure. Reports on T3 regulation of cell viability suggest that T3 is a MAPK-dependent cell growth factor.46 In this paper, we observe that dityrosine inhibits T3-stimulated MAPK activity, which may contribute to apoptosis. MAPK is a potential upstream regulator of Bax family and its downstream caspase family members.47 Translocation of Bax can also be regulated by the PI3k/Akt pathway,48 and PI3k/Akt signaling promotes cell survival by up-regulating the expression of the anti-apoptotic factor Bcl-xL.49
The aforementioned observations demonstrate that dityrosine interferes with T3 physiological action in the process of cell survival. It has been suggested that the disruption of some industrial compounds, such as polychlorinated biphenyls (PCBs) and bisphenol A (BPA), on T3 function is thought to be due to their toxicological effects, which may be related to the structural similarities shared by PCBs, BPA and T3.50,51 In this study, the two benzene cores are either linked by an ether bond (T3) or by a carbon-carbon bond (dityrosine). Both dityrosine and T3 molecules contain tyrosine residues. Molecular docking simulations of ligands (dityrosine and T3) binding to TRβ1 binding sites were performed in this study, which demonstrated that the two ligands were embedded into the active site with favorable poses, and the position of dityrosine in TRβ1 bore a close resemblance to that of T3, indicating that the molecular docking domain for dityrosine and T3 might be the same sites. In addition, the simulations showed that dityrosine and T3 display similar binding mode and binding energy. Therefore, we believe that these molecules bind to the same receptor in a competitive way. A novel finding from this study is that dityrosine can impair T3 action by inhibiting T3 binding to TRs and suppressing its transcriptional activity, resulting in inhibition of T3 bio-function. Suppression of T3 function is partly attributed to the inhibited T3 binding to TRs, which further suggests that dityrosine might act as an antagonist to T3.
One confounding factor which induce the disruption of T3 action may be the deficiency of thyroid hormone receptors (TRs).52 Increasing evidence has shown that besides the classical mode of nuclear actions, TRs could act via extra-nuclear signaling. One important extra-nuclear pathway is mediated by PI3k signaling.53 TRs play important roles in the regulation of cell survival via the PI3k pathway is first demonstrated by the finding that T3 protects pancreatic β-cells from pharmacologically induced apoptosis.54 Subsequently, it was shown that T3-induced AKT phosphorylation is via TRβ1 in the rat pancreatic β-cell line (rRINm5F) and human pancreatic insulinoma cells (hCM cells).55 This activation involves binding between the TRβ1 and the p85 submit of PI3k.56 The binding of T3-TRs complex is located in both nuclear and cytosolic compartments to activate signaling pathways, such as MAPK and PI3k-Akt.57–60 In the present research, dityrosine suppressed T3-dependent cytoprotection, which may be partially due to the diminished protein level of TRβ1. This, in turn would cause decreased sensitivity to T3 peripheral tissue cells.
We then quantified the luciferase activities in transgenic cells after T3 exposure in the presence or absence of dityrosine. Our results show that on adding dityrosine, the relative activity of luciferase was decreased in comparison to that of the T3 treated cells. These data explain the ability of dityrosine to modify T3-mediated effects. In addition, TRs regulate the transcriptional activity of T3 to target genes by recruitment of the 9-cis retinoic acid receptor (Rxr). On positive TREs, Rxr favours binding of TRs-T3 heterodimer to DNA and stimulates transcription.61 Conformational changes of TRs induced by T3 binding release the co-repressor complex and recruit co-activators such as steroid receptor co-activator 1 (Src-1) receptors.62,63 In this study, we observed a significant downregulation of mRNA expressions of Rxrα and Src-1 in dityrosine-treated cells. These findings suggest that the dityrosine-dependent decreased in cell sensitivity to T3 is also attributed to the absence of some TRs co-activator factors.
In summary, our findings demonstrate that dityrosine, which is one of the most prevalent form of oxidized tyrosine, suppresses the cytoprotective effects of T3 by inhibiting TRs-mediated transcriptional activation. Fig. 14 shows the mechanism diagram of this work, dityrosine inhibits T3 binding to TRs and downregulates its co-activator factors, which then disrupts T3-dependent PI3k/Akt/MAPK pathway activity. This study improves our understanding of the molecular mechanisms whereby dityrosine affects T3 function and establishes fundamental data for future experimental studies on dietary dityrosine. Our findings offer new light on cellular processes underlying the health threats caused by dietary oxidized protein, thus contributing to a better understanding of the diet–health axis at a cellular level.
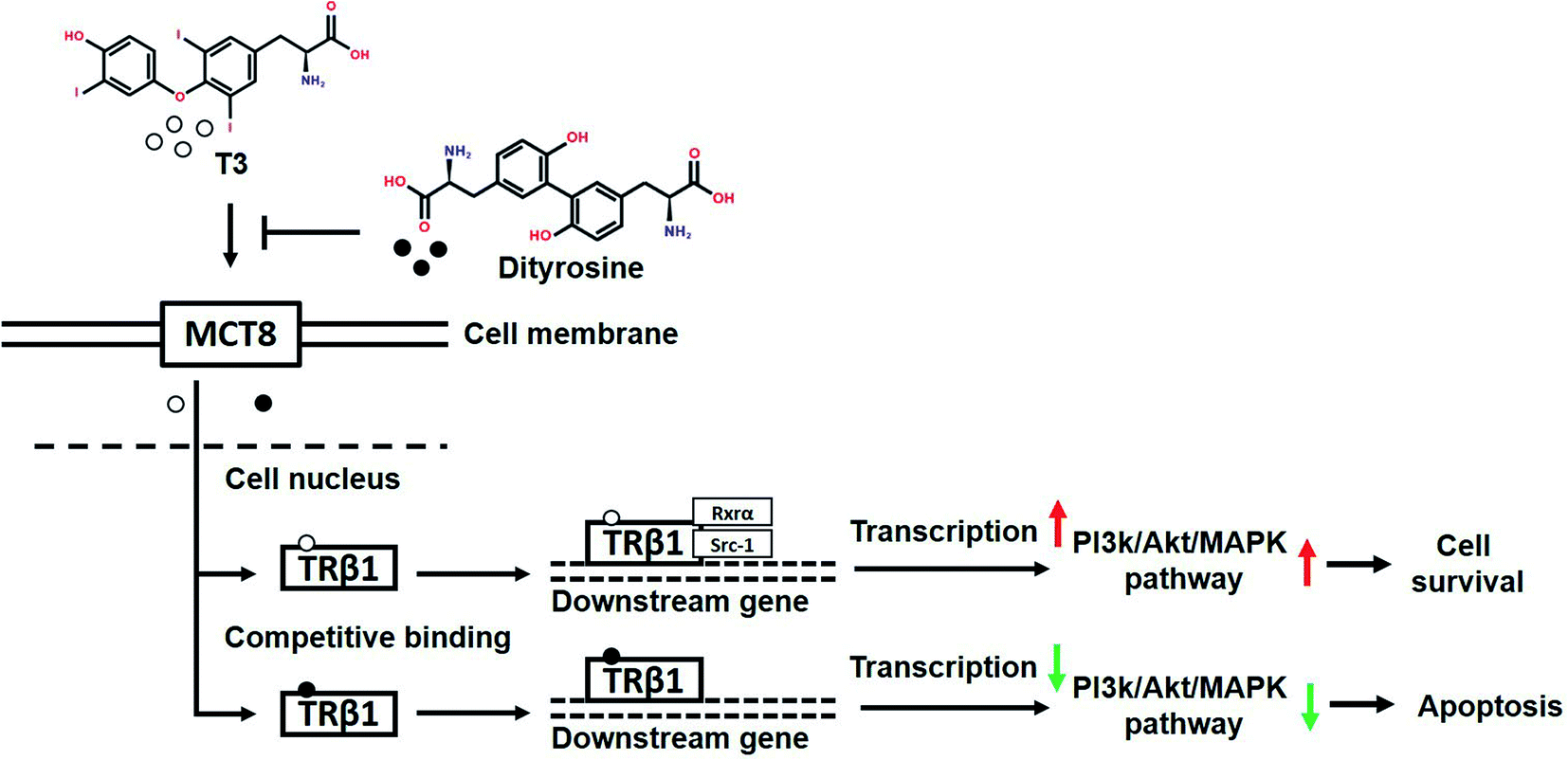 | ||
| Fig. 14 The mechanism diagram of this work. | ||
Conflicts of interest
The authorship declare no conflict of interest.Acknowledgements
T3 binding study using [125I] T3 was performed with the help of Dr Shichen Xu from Jiangsu Institute of Nuclear Medicine, Key Laboratory of Nuclear Medicine, Ministry of Health, China. This work was supported by Talent Research Start-up Foundation of Zhejiang Gongshang University 1110XJ22318010 (to Y. D.), Zhejiang Provincial Nature Science Foundation LQ19C110001 (to Y. D.), and Chinese Nature Science Foundation 31571841 (to G. L.).References
- Y. Y. Ding, Z. Q. Li, X. R. Cheng, Y. M. Ran, S. J. Wu, Y. Shi and G. Le, Amino Acids, 2017, 49, 1401–1414 CrossRef CAS PubMed.
- M. Estévez, Poult. Sci., 2015, 94, 1368–1378 CrossRef PubMed.
- M. Estevez and C. Luna, Crit. Rev. Food Sci. Nutr., 2016, 57, 3781–3793 CrossRef PubMed.
- M. J. Davies, Biochim. Biophys. Acta, 2005, 1703, 93–109 CrossRef CAS PubMed.
- M. A. Malik, S. N. Basahel, A. Y. Obaid and Z. Khan, Colloids Surf., B, 2010, 76, 346–353 CrossRef CAS PubMed.
- Z. L. Li, L. Mo, G. Le and Y. Shi, Food Chem. Toxicol., 2014, 64, 86–93 CrossRef CAS PubMed.
- Z. Li, Y. Shi, G. Le, Y. Ding and Q. Zhao, Oxidative Medicine and Cellular Longevity, 2016, pp. 1–12 Search PubMed.
- Z. L. Li, Y. Shi, Y. Ding, Y. Ran and G. Le, Amino Acids, 2016, 49, 1–20 Search PubMed.
- Y. Yang, H. Zhang, B. Yan, T. Zhang, Y. Gao, Y. Shi and G. Le, J. Agric. Food Chem., 2017, 65, 6957–6971 CrossRef CAS PubMed.
- Y. Yang, B. Yan, X. Cheng, Y. Ding and G. Le, RSC Adv., 2017, 7, 28591–28605 RSC.
- Y. Y. Ding, X. R. Cheng, Z. Q. Li, S. J. Wu, Y. Yang, Y. H. Shi and G. W. Le, RSC Adv., 2017, 7, 26809–26826 RSC.
- M. Estévez and Y. Xiong, J. Food Sci., 2019, 1–10 Search PubMed.
- Y. Ran, B. Yan, Z. Li, Y. Ding, Y. Shi and G. Le, Physiol. Behav., 2016, 164, 292–299 CrossRef CAS PubMed.
- B. Li, Y. Ge, Y. Xu, Y. Lu, Y. Yang, L. Han, Y. Jiang, Y. Shi and G. Le, J. Agric. Food Chem., 2019, 67, 9039–9049 CrossRef CAS PubMed.
- Y. Y. Ding, X. Tang, X. R. Cheng, F. F. Wang, Z. Q. Li, S. J. Wu, X. R. Kou, Y. Shi and G. Le, RSC Adv., 2017, 7, 54610–54625 RSC.
- A. Oetting and P. M. Yen, Best Pract. Res., Clin. Endocrinol. Metab, 2007, 21, 193–208 CrossRef CAS PubMed.
- C. S. Mitchell, D. B. Savage, S. Dufour, N. Schoenmakers, P. Murgatroyd, D. Befroy, D. Halsall, S. Northcott, P. Raymond-Barker, S. Curran, E. Henning, J. Keogh, P. Owen, J. Lazarus, D. L. Rothman, I. S. Farooqi, G. I. Shulman, K. Chatterjee and K. F. Petersen, J. Clin. Invest., 2010, 120, 1345–1354 CrossRef CAS PubMed.
- E. Danforth Jr and A. Burger, Clin. Endocrinol. Metab., 1984, 13, 581–595 CrossRef CAS.
- F. C. Verga, C. Mangialardo, S. Raffa, A. Mancuso, P. Piergrossi, G. Moriggi, S. Piro, A. Stigliano, M. R. Torrisi and E. Brunetti, Islets, 2010, 2, 96–103 CrossRef PubMed.
- X. Cao, F. Kambe, L. C. Moeller, S. Refetoff and H. Seo, Mol. Endocrinol., 2005, 19, 102–112 CrossRef CAS PubMed.
- T. Simoncini, A. Hafezi-Moghadam, D. P. Brazil, K. Ley, W. W. Chin and J. K. Liao, Nature, 2000, 407, 538–541 CrossRef CAS PubMed.
- F. C. Verga, L. Panacchia, B. Bucci, A. Stigliano, M. G. Cavallo, E. Brunetti, V. Toscano and S. Misiti, J. Cell. Physiol., 2010, 206, 309–321 CrossRef PubMed.
- X. Meng, H. Wang, J. Zhao and L. Hu, Frontiers in Oncology, 2020, 10, 1–13 CrossRef PubMed.
- V. F. Cecilia, P. Valentina, B. Barbara, M. Claudia, M. Simona, M. Giulia, S. Antonio, B. Ercole, T. Vincenzo and M. Silvia, J. Cell. Biochem., 2009, 106, 835–848 CrossRef PubMed.
- P. J. Davis, J. L. Leonard and F. B. Davis, Front. Neuroendocrinol., 2008, 29, 211–218 CrossRef CAS PubMed.
- W. M. Wiersinga, Neth. J. Med., 1985, 28, 74–82 CAS.
- W. W. Chin and P. M. Yen, Molecular Mechanisms of Nuclear Thyroid Hormone Action, Humana Press, 1997 Search PubMed.
- V. Giguère, M. Tini, G. Flock, E. Ong, R. M. Evans and G. Otulakowski, Genes Dev., 1994, 8, 538 CrossRef PubMed.
- N. Koibuchi, Y. Liu, H. Fukuda, A. Takeshita, P. M. Yen and W. W. Chin, Endocrinology, 1999, 140, 1356–1364 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 2015, 19, 1639–1662 CrossRef.
- P. J. Goodford, J. Med. Chem., 1985, 16, 849–857 CrossRef PubMed.
- Y. Yang, A. Zuo, Z. Zhu, X. Zhao, G. Guo and J. Zhang, Chinese Journal of Endocrine metabolism, 2011, 15–23 Search PubMed.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CAS.
- S. Shafie and S. C. Brooks, J. Lab. Clin. Med., 1979, 94, 784–798 CAS.
- A. Baniahmad, C. Steiner, A. C. Köhne and R. Renkawitz, Cell, 1990, 61, 505–514 CrossRef CAS PubMed.
- M. Tini, R. A. Fraser and V. Giguère, J. Biol. Chem., 1995, 270, 20156 CrossRef CAS PubMed.
- J. Wang, W. Xu, T. Zhong, Z. Song, Y. Zou, Z. Ding, Q. Guo, X. Dong and W. Zou, Sci. Rep., 2016, 6, 38285–38295 CrossRef CAS PubMed.
- M. E. Refaey, C. P. Watkins, E. J. Kennedy, A. Chang, Q. Zhong, K. H. Ding, X. M. Shi, J. Xu, W. B. Bollag and W. D. Hill, Mol. Cell. Endocrinol., 2015, 410, 87–96 CrossRef PubMed.
- T. K. Dalsgaard, J. H. Nielsen, B. E. Brown, N. Stadler and M. J. Davies, J. Agric. Food Chem., 2011, 59, 7939–7947 CrossRef CAS PubMed.
- E. R. Stadtman, Annual Review Biochemistry, 1993, 62, 797–821 CrossRef CAS PubMed.
- G. O. Hande, E. Nuran, M. Suneetha, P. Subramaniam, O. Hilmi and J. W. Heinecke, Biochem. J., 2006, 395, 277–284 CrossRef PubMed.
- S. W. Chan, R. A. Dunlop, R. Anthony, K. L. Double and K. J. Rodgers, Exp. Neurol., 2012, 238, 29–37 CrossRef CAS PubMed.
- P. M. Yen, Metabolism, 2003, 14, 327–333 CAS.
- J. Bernal, Endocrinologia y nutricion: organo de la Sociedad Espanola de Endocrinologia y Nutricion, 2011, 58, 185–196 CrossRef CAS PubMed.
- J. A. Fresno Vara, E. Casado, C. J. De, P. Cejas, C. Belda-Iniesta and M. González-Barón, Cancer Treatment Reviews, 2004, 30, 193–204 CrossRef PubMed.
- H. Y. Lin, H. Y. Tang, A. Shih, T. Keating, G. Cao, P. J. Davis and F. B. Davis, Steroids, 2007, 72, 180–187 CrossRef CAS PubMed.
- R. R. Pan, C. Y. Zhang, Y. Li, B. B. Zhang, L. Zhao, Y. Ye, Y. N. Song, M. Zhang, H. Y. Tie, H. Zhang and J. Y. Zhu, J. Nat. Prod., 2020, 3, 1–11 Search PubMed.
- F. Tsuruta, N. Masuyama and Y. Gotoh, J. Biol. Chem., 2002, 277, 14040–14047 CrossRef CAS PubMed.
- J. Qian, Y. Zou, J. S. M. Rahman, B. Lu and P. P. Massion, Molecular cancer therapeutics, 2009, 8, 101–109 CrossRef CAS PubMed.
- J. D. Mckinney, Enviromental Health Perspectives, 1989, 82, 323–336 CrossRef CAS PubMed.
- M. Kenji, T. Tetsuya, A. Takashi, U. Takeshi, S. Misa, K. Naotetsu, H. Yuji, S. Akira, K. Hideshi and N. Kazuwa, The Journal of Clinical Endocrinology, 2002, 87, 5185–5190 CrossRef PubMed.
- X. Zhu, J. Hanover, G. Hager and S. Cheng, J. Biol. Chem., 1998, 273, 27058–27063 CrossRef CAS PubMed.
- J. W. Park and S. Y. Cheng. SH2 Domain Structures and Interactions, 2015, pp. 91–110 Search PubMed.
- F. C. Verga, E. Petrucci, V. Patriarca, S. Michienzi, A. Stigliano, E. Brunetti, V. Toscano and S. Misiti, J. Mol. Endocrinol., 2007, 38, 221–233 CrossRef PubMed.
- C. V. Falzacappa, V. Patriarca, B. Bucci, C. Mangialardo, S. Michienzi, G. Moriggi, A. Stigliano, E. Brunetti, V. Toscano and S. Misiti, J. Cell. Biochem., 2009, 106, 835–848 CrossRef CAS PubMed.
- C. Liu, L. Li, H. Mei, S. Qi, P. Duan and K. Yang, Chemosphere, 2015, 118, 229–238 CrossRef CAS PubMed.
- X. Mi, J. Hou, S. Jiang, Z. Liu, S. Tang, X. Liu, Y. Wang, C. Chen, Z. Wang and W. Li, Journal of Agriculture and Food Chemistry, 2019, 67, 1392–1401 CrossRef CAS PubMed.
- A. D. Luca, M. R Maiello, A. D'Alessio, M. Pergameno and N. Normanno, Expert Opinion on Therapeutic Targets, 2012, 16, S17–S27 CrossRef PubMed.
- C. Xia, K. Fukushi, L. C. Moeller, R. Samuel and S. Hisao, Mol. Endocrinol., 2005, 19, 102–112 CrossRef PubMed.
- I. Deb and S. Das, Neurochem. Int., 2011, 58, 861–871 CrossRef CAS PubMed.
- L. Laflamme, G. Hamann, N. Messier and S. Maltais, J. Mol. Endocrinol., 2002, 29, 61–72 CAS.
- S. A. Onate, S. Y. Tsai, M. J. Tsai and B. W. Omalley, Science, 1995, 270, 1354–1357 CrossRef CAS PubMed.
- Y. Hu, A. Y. Wu, C. Xu and L. H. Qiu, Journal of Cancer, 2019, 10, 547–555 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00276c |
| ‡ Yin-Yi Ding and Fang-Fang Wang contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |