
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of TsDPEN based imine-containing ligands for the copper-catalysed asymmetric Kinugasa reaction†
Chuanlong Xu‡
,
Yuchen Yang‡,
Yue Wu,
Feilong He ,
Huakang He,
Ping Deng* and
Hui Zhou*
,
Huakang He,
Ping Deng* and
Hui Zhou*
Chongqing Research Center for Pharmaceutical Engineering, Research Center for Innovative Pharmaceutical and Excipient Analysis Technology, College of Pharmacy, Chongqing Medical University, Chongqing 400016, China. E-mail: hzhou@cqmu.edu.cn; orangedp@cqmu.edu.cn
First published on 12th May 2020
Abstract
A novel class of chiral N,N,N imine-containing ligands derived from TsDPEN (N-(p-tosyl)-1,2-diphenylethylene-1,2-diamine) has been developed and applied to the copper-catalyzed asymmetric Kinugasa reaction. The copper(II) salt proved to be an efficient catalyst precursor, and it provides an efficient way to synthesize enantioenriched cis-β-lactam. The pathway is air-tolerant and easily manipulated, and the ligands are easy to synthesize. A working model is proposed in which the stereocontrolling step is the [2 + 2] cycloaddition between ketene and imine to explain the observed stereoselectivities.
Introduction
Since the discovery of penicillin in 1928, a number of β-lactam antibiotics have been developed.1 In addition to antibacterial activity, β-lactam containing molecules also exhibit special biological activities,2 such as Ezetimibe,2a dual PPARα/γ agonists,2b a TRPV1 antagonist,2c β-lactamic hFAAH inhibitors2d and neuronal protective effect against brain ischemia.2e At the same time, β-lactam compounds are also widely employed as useful intermediates in organic synthesis.3 Therefore, considerable efforts have been made in developing an efficient synthesis method for the preparation of chiral β-lactams.4The Kinugasa reaction, developed in 1972,5 is a simple and direct reaction between alkynes and nitrones for the synthesis of β-lactams in the presence of copper and a base.6 The group of Miura reported the first catalytic asymmetric version by using CuI (10 mol%) and bis(oxazoline) (20 mol%) with 57% ee and 35% de in 1995.7 Fu et al. achieved the first diastereoselective and enantioselective catalytic Kinugasa reaction by employing 1–2.5 mol% CuCl and bis(azaferrocenes) (up to 93% ee and cis/trans up to 95/5) in 2002.8 Tang and co-workers demonstrated for the first time that a copper(II) catalyst could replace the previously performed air-sensitive copper(I) salts by using a C3-symmetric tris(oxazoline) ligand in 2003.9 Since then, several ligands were developed for the catalytic asymmetric Kinugasa reaction, such as phosphaferrocene-oxazoline,8b HETPHOX,10 Inda-bis(oxazoline),11 diamine,12 N-PINAP,13 and Prolinol-Phosphine.14 Enders et al. developed a method to synthesis spirocyclic β-lactams through Kinugasa/Michael domino reactions by the utilizing of copper-BOX.15 In most cases the main products are cis-β-lactams, except for the use of Cu-diamine by Feng et al.12 Although progress have been achieved, the scope of chiral ligands employed in the Kinugasa reaction is limited and the catalytic highly enantioselective Kinugasa reaction remains a challenge and less developed.
Chiral imino-containing ligands have emerged as a powerful tool in asymmetric catalysis.16 Despite the great success that has been achieved, there is still a need to develop more powerful novel chiral ligands with imino group for asymmetric catalysis.16 Chan & Li have designed two types of chiral tridentate N-tosylated aminoimine ligands containing phenol and pyridine, respectively (Scheme 1a, L1 and L2)17 for the addition of alkynes to imines. Recently, we have developed a new class of chiral polydentate N,O ligands derived from N-(p-tosyl)-1,2-diphenylethylene-1,2-diamine (TsDPEN).18,19 Herein, we successfully developed a novel class of chiral N,N,N imine-containing ligands derived from TsDPEN (Scheme 1b, L3) and applied them in Cu(II)-catalysed asymmetric Kinugasa reaction.
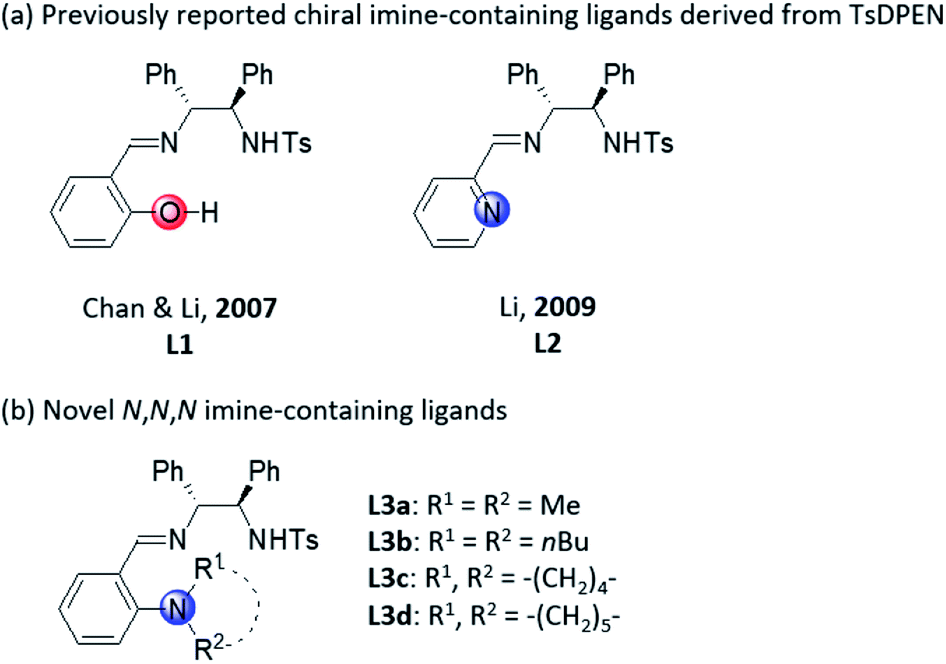 | ||
| Scheme 1 Chiral imine-containing ligands derived from TsDPEN. | ||
Results and discussion
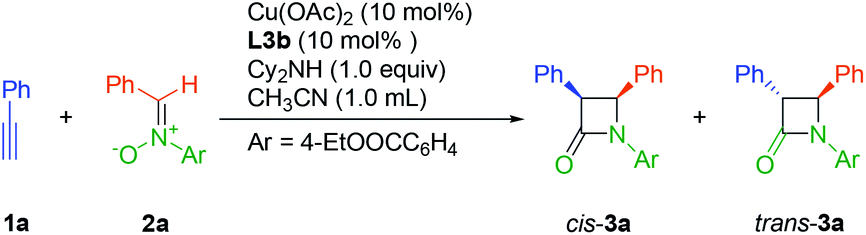
At the outset, we investigated the Kinugasa reaction of alkyne 1a and nitrone 2a in the presence of dicyclohexylamine as the base in CH3CN at 0 °C, evaluating various copper catalysts prepared in situ from copper salts and chiral tridentate ligands L1–L3 (Fig. 1).20 Among them, Cu(OTf)2/L1 (26% yield, 87/13 cis/trans, 39% cis ee),21 CuOTf/L2 (31% yield, 95/5 cis/trans, 36% cis ee),22 Cu(OAc)2/L3b (97% yield, 85/15 cis/trans, 83% cis ee) turned out to be the efficient combination in terms of yield, dr and enantioselectivity. These results indicated that the dialkylamine substitution in aniline obviously affected the reactivity and the enantioselectivity. Then the substituent effect on the amino group of L3 was examined and similar results were obtained (72–97% yield, ∼85/15 cis/trans, 70–83% cis ee). Considering the reactivity and enantioselectivity, we selected L3b for further optimization.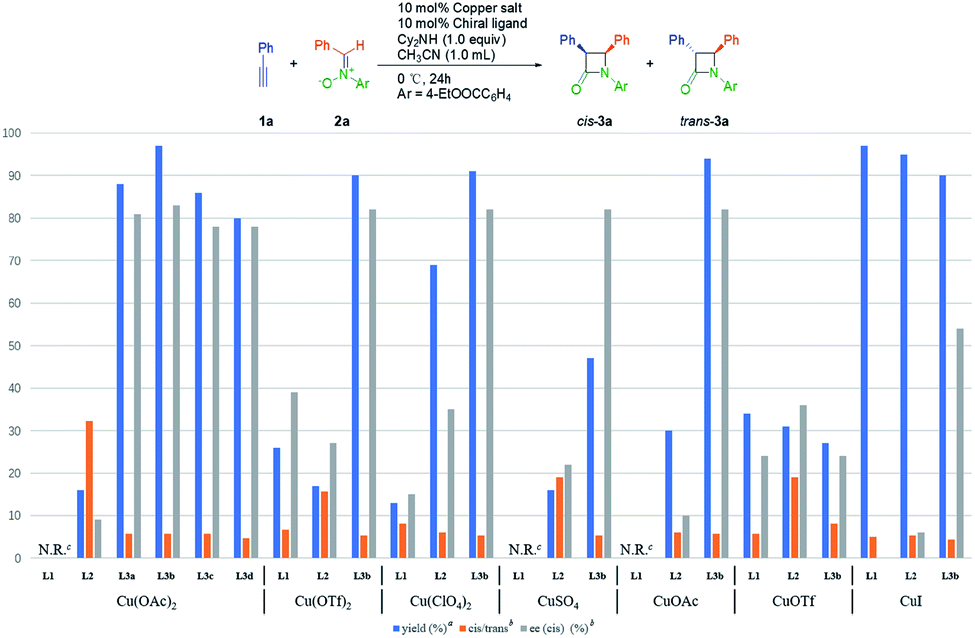 | ||
| Fig. 1 Effect of copper salts and ligands on the asymmetric Kinugasa reaction. Reaction conditions: copper salt (10 mol%), ligand (10 mol%), 1a (0.2 mmol), 2a (0.22 mmol), dicyclohexylamine (1.0 equiv.), CH3CN (1.0 mL), at 0 °C for 24 h. aTotal isolated yield of the cis- and trans-products. bThe ratios of cis/trans (x/1) and the ee of the cis-products were determined by HPLC on a chiral stationary phase. cN.R. = no reaction. | ||
Next, the influences of different bases were studied with the results shown in Table 1. Although the ligand contained a tertiary amine functional group, this transformation hardly proceeded without basic additives (Table 1, entry 1). Tertiary amines and inorganic bases gave poor yield (Table 1, entries 3, 4 and 6). Secondary amines afforded the desired products with better yield and enantioselectivity, and higher sterically hindered secondary amines achieved higher diastereoselectivity (Table 1, entries 2, 5 and 7). Dicyclohexylamine proved to be the best base.
|
||||
|---|---|---|---|---|
| Entry | Base | Yieldb (%) | cis/transc | ee (cis)c (%) |
| a Reaction conditions: Cu(OAc)2 (10 mol%), L3b (10 mol%), 1a (0.2 mmol), 2a (0.22 mmol), base (1.0 equiv.), CH3CN (1.0 mL), at 0 °C for 24 h.b Total isolated yield of the cis- and trans-products.c Determined by HPLC on a chiral stationary phase.d Dicyclohexylamine.e N,N-Dicyclohexylmethylamine. | ||||
| 1 | — | Trace | — | — |
| 2 | Cy2NHd | 97 | 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 :15 |
83 |
| 3 | iPr2NEt | 10 | 87:13 |
44 |
| 4 | K2CO3 | 9 | 61:39 |
71 |
| 5 | iPr2NH | 73 | 80:20 |
79 |
| 6 | Cy2NMee | 24 | 85:15 |
68 |
| 7 | nBu2NH | 90 | 59:41 |
80 |
Solvent effects were studied next, with the results summarized in Table 2. Only moderate enantioselectivity could be obtained in examined solvents other than CH2Cl2 and CH3CN (Table 2, entries 1–7). No diastereoselectivity was observed in DMF (Table 2, entry 5). The best yield and enantioselectivity were obtained by CH3CN (Table 2, entry 1).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Yieldb (%) | cis/transc | ee (cis)c (%) |
| a Reaction conditions: Cu(OAc)2 (10 mol%), L3b (10 mol%), 1a (0.2 mmol), 2a (0.22 mmol), dicyclohexylamine (1.0 equiv.), solvent (1.0 mL), at 0 °C for 24 h.b Total isolated yield of the cis- and trans-products.c Determined by HPLC on a chiral stationary phase. | ||||
| 1 | CH3CN | 97 | 85:15 |
82 |
| 2 | CH2Cl2 | 69 | 84:16 |
81 |
| 3 | THF | 77 | 88:12 |
66 |
| 4 | EtOAc | 70 | 84:16 |
76 |
| 5 | DMF | 68 | 49:51 |
74 |
| 6 | EtOH | 60 | 87:13 |
74 |
| 7 | Toluene | 74 | 81:19 |
73 |
Subsequently, the reaction temperature was examined. As shown in Table 3, both the diastereoslectivity and enantioselectivity were dependent on the reaction temperature. When the temperature was decreased from 0 to −30 °C, the diastereoslectivity and enantioselectivity increased (Table 3, entries 1–3). When the reaction temperature further decreased to −40 °C, the reaction almost stopped by about halfway through (Table 3, entry 4).
|
|||||
|---|---|---|---|---|---|
| Entry | T (°C) | Time (h) | Yieldb (%) | cis/trans | ee (cis)c (%) |
| a Reaction conditions: Cu(OAc)2 (10 mol%), L3b (10 mol%), 1a (0.2 mmol), 2a (0.22 mmol), dicyclohexylamine (1.0 equiv.), CH3CN (1.0 mL).b Total isolated yield of the cis- and trans-products.c Determined by HPLC on a chiral stationary phase.d Determined by 1H NMR. | |||||
| 1 | 0 | 24 | 97 | 85:15c |
82 |
| 2 | −15 | 48 | >99 | 90:10c |
87 |
| 3 | −30 | 48 | >99 | 91:9c (10.5:1)d |
90 |
| 4 | −40 | 48 or 72 | 46 | 92:8c |
90 |
Under the established optimal reaction conditions, the substrate scope of the cycloaddition reaction between various alkynes and nitrones was examined next (Table 4). First, different alkynes participated in the reaction (Table 4, entries 1–6, 12–15, and 21). Weakly electron donating methyl group did not pose a problem for the reaction, and the corresponding cis-β-lactams were isolated in good yields, diastereo- and enantioselectivities (Table 4, entries 2, 3 and 12–15). The reaction conditions were also compatible with electron withdrawing group (Table 4, entries 4 and 21). Phenylacetylene with a strongly electron donating methoxy group had very low reactivity and only trace amount of corresponding product could be observed (Table 4, entry 5). The application of alkyl alkynes in the system is feasible, as exemplified by the formation of product 3e with high diastereoselectivity (99:1), albeit with low enantioselectivity (12% ee). Next, effects of C-substituents of the nitrone were investigated (Table 4, entries 7–15). Methyl group and fused aromatic substituents had no adverse effect on the reaction. While strong decrease in reactivity for electron withdrawing group and heterocyclic substituents was observed (Table 4, entries 10 and 11). The electronic character of the N-bound aromatic groups of nitrones was also investigated (Table 4, entries 16–20). Whether electron-deficient or electron-rich N-aryl nitrones were found to be highly suitable substrates for this asymmetric transformation.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1, R2, R3 | T (°C) | Product | cis/transb | Yield (cis)c (%) | ee (cis)d (%) |
| a Reaction conditions: Cu(OAc)2 (10 mol%), L3b (10 mol%), 1 (0.2 mmol), 2 (0.22 mmol), dicyclohexylamine (1.0 equiv.), CH3CN (1.0 mL), at −30 °C or 0 °C for 48 h.b Determined by 1H NMR of the crude reaction mixture.c Isolated yield of cis-product by chromatography on silica gel.d Determined by HPLC on a chiral stationary phase. | ||||||
| 1 | C6H5, C6H5, Ar | −30 | 3a | 10.5:1 |
92 | 90 |
| 2 | 4-Me-C6H4, C6H5, Ar | −30 | 3b | 13.1:1 |
93 | 88 |
| 3 | 3-Me-C6H4, C6H5, Ar | −30 | 3c | 9.6:1 |
91 | 91 |
| 4 | 4-F-C6H4, C6H5, Ar | 0 | 3d | 6.2:1 |
74 | 88 |
| 5 | 4-MeOC6H4, C6H5, Ar | 0 | Trace | |||
| 6 | Cyclohexyl, C6H5, Ar | 0 | 3e | >99:1 |
68 | 12 |
| 7 | C6H5, 3-Me-C6H4, Ar | −30 | 3f | 14:1 |
80 | 90 |
| 8 | C6H5, 4-Me-C6H4, Ar | 0 | 3g | 6.7:1 |
81 | 83 |
| 9 | C6H5, 2-naphthyl, Ar | 0 | 3h | 5.3:1 |
70 | 77 |
| 10 | C6H5, 4-Cl-C6H4, Ar | 0 | Trace | |||
| 11 | C6H5, 2-furyl, Ar | 0 | Trace | |||
| 12 | 4-Me-C6H4, 3-Me-C6H4, Ar | −30 | 3i | 17.5:1 |
91 | 89 |
| 13 | 3-Me-C6H4, 3-Me-C6H4, Ar | −30 | 3j | 12.5:1 |
90 | 89 |
| 14 | 4-Me-C6H4, 4-Me-C6H4, Ar | 0 | 3k | 11:1 |
80 | 79 |
| 15 | 3-Me-C6H4, 4-Me-C6H4, Ar | 0 | 3l | 6.3:1 |
82 | 79 |
| 16 | C6H5, C6H5, C6H5 | −30 | 3m | 12.9:1 |
93 | 91 |
| 17 | C6H5, C6H5, 4-Me-C6H4 | 0 | 3n | 14:1 |
71 | 85 |
| 18 | C6H5, C6H5, 4-Cl-C6H4 | 0 | 3o | 12.5:1 |
80 | 87 |
| 19 | C6H5, C6H5, 4-Br-C6H4 | 0 | 3p | 11.8:1 |
82 | 87 |
| 20 | C6H5, C6H5, 4-F-C6H4 | 0 | 3q | 16.5:1 |
70 | 86 |
| 21 | 4-F-C6H4, C6H5, C6H5 | 0 | 3r | 16.3:1 |
75 | 84 |
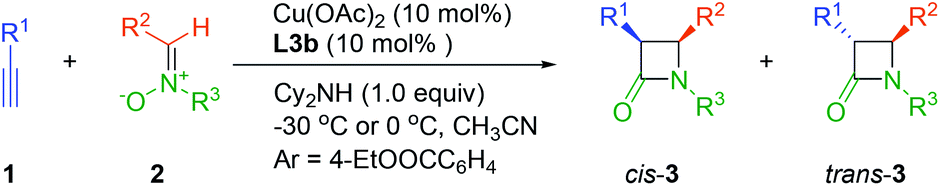
To demonstrate the utility of this method, a mmol scale catalytic asymmetric Kinugasa reaction with 2.0 mmol 1a and 2.2 mmol 2a in the presence of 10 mol% chiral copper complex proceeded smoothly and 3a was isolated with comparably high yield and stereoselectivity (Scheme 2).
 | ||
| Scheme 2 mmol-scale reaction. | ||
The research of Kinugasa reaction mechanism has been an active research area.23 Recently, Hein's group reported an improved mechanism based on the analysis of multiple byproducts and detailed kinetic analysis.23a According to the modified mechanism, the stereocontrolling step is the [2 + 2] cycloaddition between ketene and imine, which are generated by reductive elimination and protonation from the initial [3 + 2] cycloaddition intermediate between nitrone and σ-Cu acetylide. Based on this research and the geometry of Cu complex optimized by DFT,24 a working model to explain the observed stereoselectivities is proposed in Fig. 2. The ketene combined with the tert-amine forming a zwitterionic enolate (phenyl away from N is favourable for the steric repulsion) and the imine coordinated to the Cu in a suitable position by avoiding the steric repulsion of n-butyl group on nitrogen.25
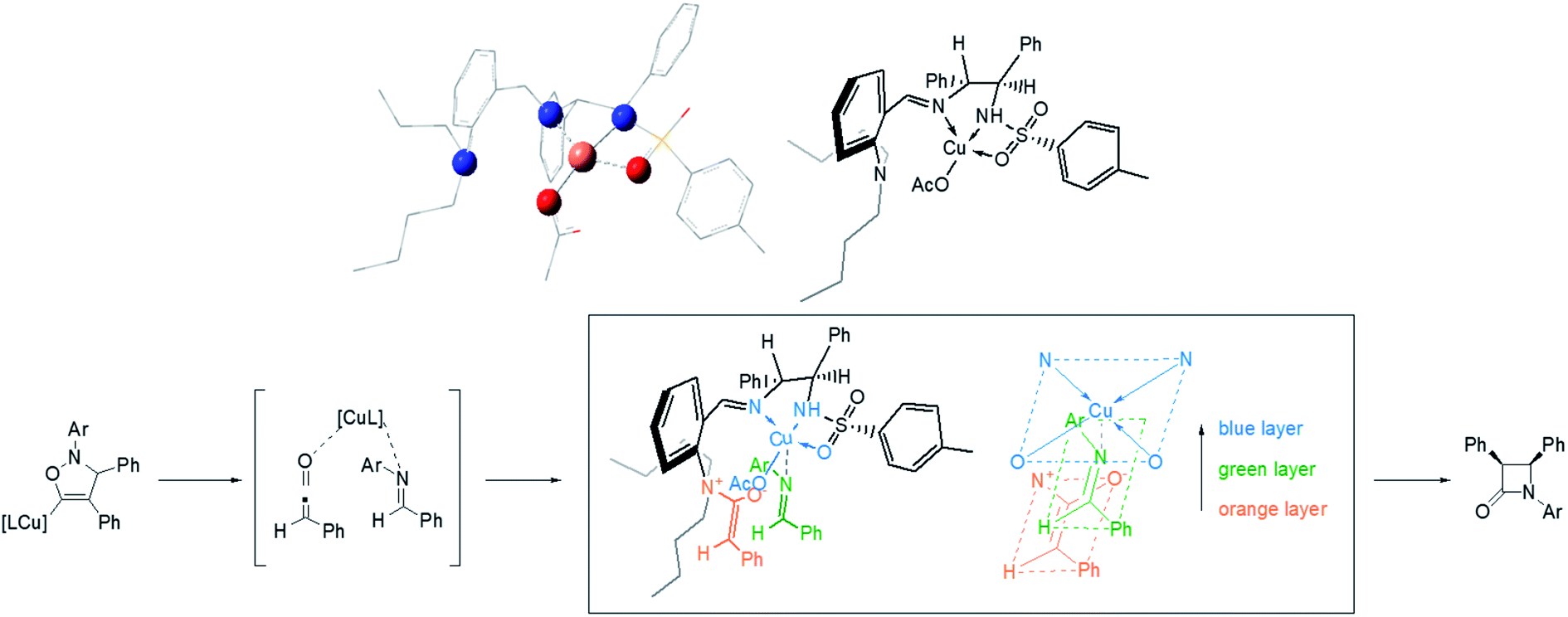 | ||
| Fig. 2 Structure of the Cu complex optimized by DFT and proposed working model for stereoselectivity. | ||
Conclusions
In summary, we have successfully employed the novel chiral N,N,N imine-containing ligands derived from TsDPEN in the catalytic enantioselective Kinugasa reaction. It provides an efficient way to synthesize cis-β-lactam in good to excellent yields (up to 93%) and with good to excellent diastereo- and enantioselectivities (dr up to 17.5:1, ee up to 91%). This new protocol for the Kinugasa reaction is not effective for aliphatic alkynes and phenylacetylenes with a strongly electron donating group. A working model is proposed in which the stereocontrolling step is the [2 + 2] cycloaddition between ketene and imine to explain the observed stereoselectivities. The N,N-dialkylamine substitution is an unprecedented modification on the TsDPEN based imine containing ligand scaffold. The pathway is air-tolerant and easily manipulated, and the ligands are easy to synthesize. Further investigations are underway in our laboratory including further geometry optimization of ligands, the detailed mechanism, and the extending of the substrate scope.
Experimental section
General
The solvents were dried by molecular sieve for more than 72 h before use. Other commercial reagents were used as purchased. NMR (Bruker 600 MHz) spectra were recorded in the deuterated solvents as stated, using residual non-deuterated solvent as internal standard. High resolution mass spectra were recorded with a Bruker Solari XFT-ICR-MS system. The enantiomeric excess (ee) was determined by HPLC (Shimadzu LC-16) analysis using the corresponding commercial chiral column as stated in the experimental procedures at 23 °C with UV detector (254 nm). Optical rotations were measured on a commercial polarimeter (Rudolph Autopol I) and are reported as follows: [α]TD (c = g/100 mL, solvent). The diastereomeric ratios of the β-lactams were determined by integration of 1H NMR spectra (the coupling constant for C3/C4 vicinal protons is 4.5–6 Hz for cis isomers and 2–2.5 Hz for trans isomers).8a The absolute configuration of 3a, 3e and 3m was determined by the comparison of the HPLC retention time and optical rotations with the literature data.8a,12 The absolute configuration of 3b–3d, 3f–3l and 3n–3r was determined by analogy.General preparation procedure of ligands
L1. Yellow solid, 1H NMR (600 MHz, CDCl3) δ 12.66 (s, 1H), 8.16 (s, 1H), 7.41 (d, J = 8.0 Hz, 2H), 7.34–7.31 (m, 1H), 7.19–7.07 (m, 9H), 7.00–6.95 (m, 3H), 6.90–6.83 (m, 3H), 5.32 (d, J = 7.4 Hz, 1H), 4.76 (t, J = 6.7 Hz, 1H), 4.56 (d, J = 5.9 Hz, 1H), 2.30 (s, 3H).
L2. Yellow solid, 1H NMR (600 MHz, CDCl3) δ 8.60 (d, J = 4.5 Hz, 1H), 8.00 (d, J = 7.7 Hz, 1H), 7.85–7.83 (m, 1H), 7.62 (d, J = 8.2 Hz, 2H), 7.32–7.30 (m, 1H), 7.22–7.16 (m, 10H), 6.99 (d, J = 7.1 Hz, 1H), 5.94 (s, 1H), 4.64 (d, J = 6.6 Hz, 1H), 4.32 (d, J = 6.6 Hz, 1H), 2.43 (s, 3H).
:1) on silica gel to obtain a crude product. The crude product was dissolved in the mixture of petroleum ether/ether (v/v 10:1) and acidified to pH 2 with 1 M HCl. The resulting mixture was extracted twice with water, and the aqueous layer was basified to pH 9 with 1 M NaOH and extracted with CH2Cl2. The dichloromethane layers were dried and the solvents were removed in vacuo to afford 4. To a solution of 4 (1.2 mmol) in 30 mL ethanol was added 5 (1.2 mmol) and the mixture was stirred at 75 °C under a N2 atmosphere until the reaction proceed completely detected by TLC. The EtOH was removed in vacuo to give L3.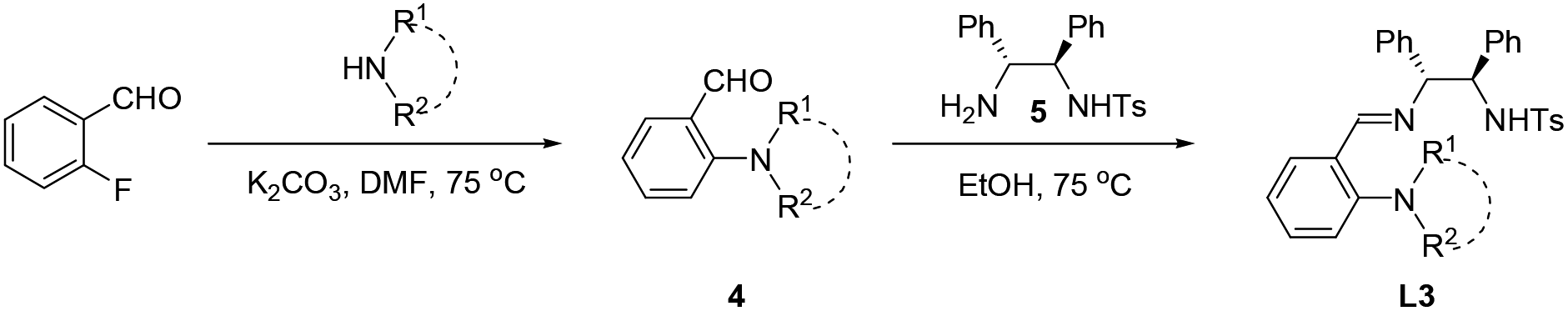
L3a. White solid, (mixture of E- and Z-, E/Z = 5.6/1), 1H NMR (600 MHz, CDCl3) δ 8.14 (E, s, 1H), 7.97 (E, d, J = 7.4 Hz, 1H), 7.91 (Z, d, J = 7.5 Hz, 0.18H), 7.56 (Z, d, J = 8.0 Hz, 0.37H), 7.51 (Z, d, J = 7.5 Hz, 0.38H), 7.32–7.29 (E + Z, m, 3.96H), 7.19–7.00 (E + Z, m, 11.37H), 6.91–6.89 (E + Z, m, 3.12H), 6.19 (E, d, J = 7.0 Hz, 1H) 4.84 (E, q, J = 3.3 Hz, 1H), 4.77 (Z, d, J = 5.2 Hz, 0.18H), 4.51 (E, d, J = 3.4 Hz, 1H), 4.26 (Z, d, J = 5.3 Hz, 0.18H), 2.35 (E, s, 6H), 2.32 (Z, s, 0.71H), 2.25 (E, s, 3H), 2.23 (Z, s, 0.20H). 13C NMR (150 MHz, CDCl3) δ E: 162.1, 154.1, 142.2, 140.9, 140.0, 137.2, 131.5, 129.0, 121.9, 117.8, 78.5, 63.6, 45.0, 21.3; Z: 162.1, 152.5, 143.4, 140.02, 139.4, 135.5, 133.4, 129.2, 124.0, 120.2, 75.2, 71.6, 68.5, 45.4, 21.4.; E + Z: 128.27, 128.22, 127.98, 127.95, 127.84, 127.5, 127.4, 127.03, 127.02, 126.90, 126.89, 126.7, 126.5. ESI-HRMS calcd for C30H32N3O2S+ [M + H]+: 498.2210, found 498.2200.
L3b. White solid, (mixture of E- and Z-, E/Z = 9.1/1), 1H NMR (600 MHz, CDCl3) δ 8.27 (E, s, 1H), 8.03 (E, d, J = 7.3 Hz, 1H), 7.65 (Z, d, J = 7.9 Hz, 0.23H), 7.42 (Z, d, J = 7.6 Hz, 0.25H), 7.34–7.33 (E + Z, m, 3H), 7.17–7.00 (E + Z, m, 12H), 6.93 (E, d, J = 7.9 Hz, 2H), 6.28, (Z, s, 0.11H), 6.20 (E, d, J = 6.5 Hz, 1H), 4.82 (E, q, J = 3.5 Hz, 1H), 4.64 (Z, d, J = 6.4 Hz, 0.11H), 4.51 (E, d, J = 3.7 Hz, 1H), 4.28 (Z, d, J = 6.4 Hz, 0.11H), 3.00–2.95 (Z, m, 0.49H), 2.73–2.64 (E, m, 4H), 2.28 (E, s, 3H), 2.27 (Z, s, 0.16H), 1.20–1.15 (E, m, 4H), 1.03–0.96 (E, m, 4H), 0.71 (E, t, J = 7.4 Hz, 6H). 13C NMR (150 MHz, CDCl3) δ E: 162.5, 152.5, 142.3, 141.1, 140.0, 137.4, 131.2, 78.0, 63.7, 54.2, 29.0, 21.4, 20.3, 13.9; Z: 162.5, 150.0, 143.5, 139.6, 139.2, 137.3, 130.7, 75.8, 71.9, 68.5, 54.6, 31.6, 29.4, 22.6, 21.5, 20.6, 20.3, 14.1, 14.0; E + Z: 129.3, 129.1, 128.3, 128.25, 128.20, 128.1, 128.0, 127.9, 127.7, 127.6, 127.3, 127.2, 127.08, 127.06, 126.9, 126.8, 124.2, 122.9, 122.8, 121.7. ESI-HRMS calcd for C36H44N3O2S+ [M + H]+: 582.3149, found 582.3140.
L3c. White solid, (mixture of E- and Z-, E/Z = 14.3/1), 1H NMR (600 MHz, CDCl3) δ 8.10 (E, s, 1H), 7.88 (E, d, J = 7.7 Hz, 1H), 7.60 (Z, d, J = 7.8 Hz, 0.15H), 7.46 (Z, d, J = 7.5 Hz, 0.17H), 7.31–7.25 (E + Z, m, 3H), 7.23–7.09 (E + Z, m, 10H), 6.9–6.8 (E + Z, m, 3.1H), 6.73 (E + Z, d, J = 8.3 Hz, 1H), 6.19 (E + Z, s, 1.06H), 4.83 (E, q, J = 3.2 Hz, 1H), 4.69 (Z, d, J = 5.8 Hz, 0.07H), 4.47 (E, d, J = 3.2 Hz, 1H), 4.32 (Z, d, J = 5.8 Hz, 0.07H), 2.80–2.76 (E + Z, m, 4H), 2.37 (Z, s, 0.24H), 2.29 (E, s, 3H), 1.69–1.64 (E + Z, m, 4.19H). 13C NMR (150 MHz, CDCl3) δ E: 163.2, 150.4, 142.3, 141.0, 140.2, 137.3, 131.3, 129.2, 129.1, 128.3, 128.0, 127.06, 127.04, 126.98, 126.90, 126.7, 124.1, 118.9, 114.9, 78.7, 63.6, 52.6, 25.3, 21.4. ESI-HRMS calcd for C32H34N3O2S+ [M + H]+: 524.2366, found 524.2358.
L3d. White solid, (mixture of E- and Z-, E/Z = 3.7/1), 1H NMR (600 MHz, CDCl3) δ 8.13 (E, s, 1H), 8.02 (E, d, J = 7.7 Hz, 1H), 7.66 (Z, d, J = 7.6 Hz, 0.57H), 7.58 (Z, d, J = 7.8 Hz, 0.58H), 7.42 (E + Z, t, J = 7.6 Hz, 0.61H), 7.32–7.31 (E + Z, m, 3.49H), 7.25–7.23 (E + Z, m, 1.87H), 7.18–7.05 (E + Z, m, 12H), 6.92–6.90 (E + Z, m, 3.14H), 6.20–6.19 (E + Z, m, 1.24H), 4.91 (Z, d, J = 3.4, 0.27H), 4.84–4.83 (E, m, 1H), 4.53 (Z, s, 0.27H), 4.48 (E, d, J = 3.5 Hz, 1H), 2.59–2.57 (E + Z, m, 2H), 2.49–2.46 (E + Z, m, 2H), 2.29 (Z, s, 0.9H), 2.26 (E, s, 3H), 1.37–1.30 (E + Z, m, 6H). 13C NMR (150 MHz, CDCl3) δ E: 162.0, 154.5, 142.2, 140.9, 140.0, 131.6, 78.5, 63.6, 54.5, 26.0, 23.9, 21.3; Z: 162.0, 152.9, 143.3, 141.1, 139.8, 137.7, 135.6, 134.8, 133.2, 75.0, 70.7, 68.4, 26.7, 26.1, 24.3, 21.4; E + Z: 129.1, 129.02, 128.98, 128.95, 128.5, 128.45, 128.43, 128.29, 128.26, 128.15, 128.07, 128.04, 127.95, 127.7, 127.5, 127.2, 127.1, 127.0, 127.95, 126.7, 126.6, 126.2, 125.2, 124.1, 122.3, 120.3, 118.6. ESI-HRMS calcd for C33H36N3O2S+ [M + H]+: 538.2523, found 538.2518.
General procedure for the asymmetric Kinugasa reaction
The mixture of Cu(OAc)2 (0.02 mmol, 10 mol%) and L3b (0.02 mmol, 10 mol%) was stirring in CH3CN (0.5 mL) under air atmosphere at 35 °C for 1 h. Then, dicyclohexylamine (0.2 mmol) and alkyne 1 (0.2 mmol) were sequentially added, and the test tube was rinsed with CH3CN (0.2 mL), after stirring at room temperature for 10 minutes, the mixture was cooled to −30 °C or 0 °C. After stirring for 10 min at −30 °C or 0 °C, nitrone 2 (0.22 mmol) and CH3CN (0.3 mL) were added. The mixture was further stirred at −30 °C or 0 °C for the time indicated in Table 4. The resulting solution was purified by column chromatography (petroleum ether/AcOEt) on silica gel to afford the products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21672031), Chongqing Natural Science Foundation (cstc2019jcyj-msxmX0034 and cstc2017jcyjAX0352), and Scientific and Technological Research Program of Chongqing Municipal Education Commission (KJQN201900437) for financial support.Notes and references
- (a) G. I. Georg and V. T. Ravikumar, in The Organic Chemistry of β-lactams, ed. G. I.Georg, VCH Publishers, New York, 1992, pp. 295–368 Search PubMed; (b) Heterocyclic Scaffolds I: β-Lactams, ed. B. K.Bank, Springer, 2010 Search PubMed; (c) β-Lactams: Unique Structures of Distinction for Novel Molecules, ed. B. K.Bank, Springer, 2013 Search PubMed; (d) J. D. Fisher and S. Mobashery, The β-Lactam (Azetidin-2-one) as a privileged Ring in Medicinal Chemistry, in Privileged Scaffolds in Medical Chemistry: Design, Synthesis, Evaluation, ed. S.Bräae, The Royal Society of Chemistry, UK, 2015, ch. 3, pp. 64–97 Search PubMed.
- For selected examples of drugs or potent drugs containing β-lactams, see: (a) T. Kosoglou, P. Statkevich, A. O. Johnson-Levonas, J. F. Paolini, A. J. Bergman and K. B. Alton, Clin. Pharmacokinet., 2005, 44, 467–494 CrossRef CAS PubMed; (b) W. Wang, P. Devasthale, D. Farrelly, L. Gu, T. Harrity, M. Cap, C. Chu, L. Kunselman, N. Morgan, R. Ponticiello, R. Zebo, L. Zhang, K. Locke, J. Lippy, K. O’ Malley, V. Hosagrahara, L. Zhang, P. Kadiyala, C. Chang, J. Muckelbauer, A. M. Doweko, R. Zahler, D. Ryono, N. Hariharan and P. T. W. Cheng, Bioorg. Med. Chem. Lett., 2008, 18, 1939–1944 CrossRef CAS PubMed; (c) D. Xiao, A. Palani, R. Aslanian, B. Mckittrick, A. T. McPhail, C. C. Correll, P. T. Phelps, J. C. Anthes and D. Rindgen, Bioorg. Med. Chem. Lett., 2009, 19, 783–787 CrossRef CAS PubMed; (d) M. Feledziaka, C. Michaux, D. M. Lambert and J. Marchand-Brynaert, Eur. J. Med. Chem., 2013, 60, 101–111 CrossRef PubMed; (e) X. Cui, L. Li, Y. Y. Hu, S. Ren, M. Zhang and W. B. Li, Mol. Neurobiol., 2015, 51, 1322–1333 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437–4492 CrossRef CAS PubMed.
- (a) S. Hosseyni and A. Jarrahpour, Org. Biomol. Chem., 2018, 16, 6840–6852 RSC; (b) C. R. Pitts and T. Lectka, Chem. Rev., 2014, 114, 7930–7953 CrossRef CAS PubMed; (c) P. A. Magriotis, Eur. J. Org. Chem., 2014, 2647–2657 CrossRef CAS; (d) S. France, A. Weatherwax, A. E. Taggi and T. Lectka, Acc. Chem. Res., 2004, 37, 592–600 CrossRef CAS PubMed; (e) G. S. Singh, Tetrahedron, 2003, 59, 7631–7649 CrossRef CAS.
- M. Kinugasa and S. J. Hashimoto, J. Chem. Soc., Chem. Commun., 1972, 466–467 RSC.
- For recent reviews, see: (a) S. Murahashi and Y. Imada, Chem. Rev., 2019, 119, 4684–4716 CrossRef CAS PubMed; (b) J. Comas-Barceló and J. P. A. Harrity, Synthesis, 2017, 49, 1168–1181 Search PubMed; (c) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed; (d) M. Chigrinova, D. A. MacKenzie, A. R. Sherratt, L. L. W. Cheung and J. P. Pezacki, Molecules, 2015, 20, 6959–6969 CrossRef PubMed; (e) S. Stecko, B. Furman and M. Chmielewski, Tetrahedron, 2014, 70, 7817–7844 CrossRef CAS; (f) R. H. Khangarot and K. P. Kaliappan, Eur. J. Org. Chem., 2013, 7664–7677 CrossRef CAS; (g) M. Maciejko, S. Stecko, O. Staszewska-Krajewska, M. Jurczak, B. Furman and M. Chmielewski, Synthesis, 2012, 44, 2825–2839 CrossRef CAS.
- M. Miura, M. Enna, K. Okuro and M. Nomira, J. Org. Chem., 1995, 60, 4999–5004 CrossRef CAS.
- (a) M. M. C. Lo and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 4572–4573 CrossRef CAS PubMed; (b) R. Shintani and G. C. Fu, Angew. Chem., Int. Ed., 2003, 42, 4082–4085 CrossRef CAS PubMed.
- (a) M. C. Ye, J. Zhou, Z. Z. Huang and Y. Tang, Chem. Commun., 2003, 2554–2555 RSC; (b) M. C. Ye, J. Zhou and Y. Tang, J. Org. Chem., 2006, 71, 3576–3582 CrossRef CAS PubMed; (c) J. H. Chen, S. H. Liao, X. L. Sun, Q. Shen and Y. Tang, Tetrahedron, 2012, 68, 5042–5045 CrossRef CAS.
- A. G. Coyne, H. Müller-Bunz and P. J. Guiry, Tetrahedron: Asymmetry, 2007, 18, 199–207 CrossRef CAS.
- (a) D. A. Evans, F. Kleinbeck and M. Rüping, in Asymmetric Synthesis – The Essentials, ed. M.Christmann and S.Bräse, Wiley-VCH, Weinheim, Germany, 2007, pp. 72–77 Search PubMed; (b) T. Saito, T. Kikichi, H. Tanabe, J. Yahiro and T. Otani, Tetrahedron Lett., 2009, 50, 4969–4972 CrossRef CAS; (c) B. Baeza, L. Casarrubios and M. A. Sierra, Chem.–Eur. J., 2013, 19, 11536–11540 CrossRef CAS PubMed.
- Z. Chen, L. Lin, M. Wang, X. Liu and X. Feng, Chem.–Eur. J., 2013, 19, 7561–7567 CrossRef CAS PubMed.
- K. Wolosewicz, M. Michalak, J. Adamek and B. Furman, Eur. J. Org. Chem., 2016, 2212–2219 CrossRef CAS.
- (a) Y. Takayama, T. Ishii, H. Ohmiya, T. Iwai, M. C. Schwarzer, S. Mori, T. Taniguchi, K. Monde and M. Sawamura, Chem.–Eur. J., 2017, 23, 8400–8404 CrossRef CAS PubMed; (b) K. Imai, Y. Takayama, H. Murayama, H. Ohmiya, Y. Shimizu and M. Sawamura, Org. Lett., 2019, 21, 1717–1721 CrossRef CAS PubMed.
- T. Shu, L. Zhao, S. Li, X.-Y. Chen, C. von Essen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 10985–10988 CrossRef CAS PubMed.
- For recent reviews about imine-containing ligands, see: X. Chen and Z. Lu, Org. Biomol. Chem., 2017, 15, 2280–2306 RSC.
- (a) B. Liu, J. Liu, X. Jia, L. Huang, X. Li and A. S. C. Chan, Tetrahedron: Asymmetry, 2007, 18, 1124–1128 CrossRef CAS; (b) B. Liu, L. Huang, Y. Zhong, X. Li and A. S. C. Chan, Tetrahedron: Asymmetry, 2007, 18, 2901–2904 CrossRef CAS; (c) B. Liu, Y. Zhong and X. Li, Chirality, 2009, 21, 595–599 CrossRef CAS.
- (a) S. Zhang, P. Deng, J. Zhou, M. Liu, G. Liang, Y. Xiong and H. Zhou, Chem. Commun., 2017, 53, 12914–12917 RSC; (b) G. Chen, G. Liang, Y. Wang, P. Deng and H. Zhou, Org. Biomol. Chem., 2018, 16, 3841–3850 RSC.
- For reviews, see: J. Barrios-Rivera, Y. Xu and M. Wills, Org. Biomol. Chem., 2019, 17, 1301–1321 RSC.
- For detailed data, see ESI.†.
- The results of other solvents in the presence of Cu(OTf)2/L1: CH2Cl2 (no reaction), THF (no reaction), toluene (no reaction).
- The results of other solvents in the presence of CuOTf/L2: CH2Cl2 (44% yield, 98/2 cis/trans, 18% cis ee), THF (70% yield, 96/4 cis/trans, 6% cis ee), toluene (no reaction).
- For recent reports: (a) T. C. Malig, D. Yu and J. E. Hein, J. Am. Chem. Soc., 2018, 140, 9167–9173 CrossRef CAS PubMed; (b) S. Santoro, R. Liao, T. Marcelli, P. Hammar and F. Himo, J. Org. Chem., 2015, 80, 2649–2660 CrossRef CAS PubMed.
- DFT calculations were carried out using the Gaussian 09 software package. For more detailed results about the optimized structure using DFT, see ESI.†.
- For a related mechanistic research in asymmetric Staudinger reaction, see: S. France, M. H. Shah, A. Weatherwax, H. Wack, J. P. Roth and T. Lectka, J. Am. Chem. Soc., 2005, 127, 1206–1215 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03276j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |