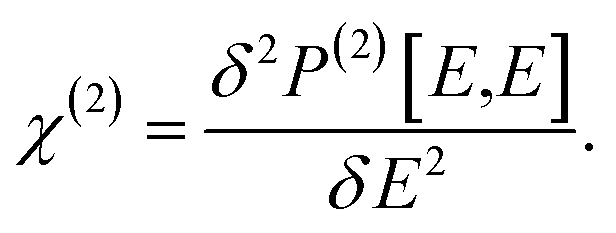DOI:
10.1039/D0RA03136D
(Paper)
RSC Adv., 2020,
10, 26479-26485
Second harmonic generation responses of KH2PO4: importance of K and breaking down of Kleinman symmetry†
Received
7th April 2020
, Accepted 7th July 2020
First published on 16th July 2020
Abstract
The second harmonic generation (SHG) responses of the paraelectric and ferroelectric phases of KH2PO4 (KDP) were calculated by first-principles density functional theory (DFT) calculations, and the individual atom contributions to the SHG responses were analyzed by the atom response theory (ART). We show that the occurrence of static polarization does not enhance the SHG responses of the ferroelectric KDP, and that the Kleinman symmetry is reasonably well obeyed for the paraelectric phase, but not for the ferroelectric phase despite that the latter has a larger bandgap. This is caused most likely by the fact that the ferroelectric phase has lower-symmetry local structures than does the paraelectric phase. The contribution to the SHG response of an individual K+ ion is comparable to that of an individual O2− ion. The contributions of the O2− and K+ ions arise overwhelmingly from the polarizable parts of the electronic structure, namely, from the valence bands of the O-2p nonbonding states and from the conduction bands of the K-3d states.
1 Introduction
NLO crystals play a crucial role in modern laser technology as electro-optical materials for optical modulators, Q-switches, deflectors, and frequency converters because of their nonlinear interactions with incident light.1 Single crystals of potassium dihydrogen phosphate (KH2PO4, KDP) are one of the first NLO materials known. Although KDP is hygroscopic and exhibits relatively low NLO responses, it possesses extraordinary properties such as transparency in a broad range of light frequencies, adequate birefringence for phase matching, and good crystal growth habit.2–4 These properties enable one to produce meter-sized crystals used in laser-driven nuclear fusion reaction.3 KDP crystalizes in a paraelectric phase (P-KDP) with space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d, which transforms into a ferroelectric phase (F-KDP) with space group Fdd2 below the Curie temperature Tc = 123 K. This phase transition has been examined in a number of studies5–9 since the discovery of its unusually large isotope effect; the Tc of the deuterated KDP, ∼220 K, is about 100 K higher than that of the undeuterated KDP, which strongly indicates the involvement of a proton movement in the phase transition.9,10 The space groups I2d and Fdd2 are both noncentrosymmetric, so both phases of KDP exhibit nonzero SHG responses. So far, there have not been many theoretical studies on the origin of its NLO properties. Levine calculated the SHG coefficient of P-KDP using the bond charge model, omitting the structural details and neglecting the contributions from the K+ and H+ cations.11 The SHG coefficients of P-KDP calculated using the CRYSTAL code were reported by Lacivita et al.12 and by Rérat et al.,13 but their work did not probe the atomic origin of the SHG responses. Lin et al. calculated the optical properties of the P-KDP using the CASTEP code, to report that the anionic groups (PO4)3− contribute ∼99% to the SHG coefficients using their “real-space atom cutting” analysis, in which the contributions of the K+ and H+ cations were excluded.14 According to the neutron diffraction measurements9,15 and recent theoretical studies,16,17 the paraelectric to ferroelectric phase transition of KDP involves the zero-point vibration of the H+ ions leading to the anti-phase shift of the K+ and P5+ ions along the c-axis. On F-KDP, there is one theoretical study reported.6 Lacivita et al. calculated the SHG responses of F-KDP using the CRYSTAL code,12 but did not explore the origin of its SHG responses.
2d, which transforms into a ferroelectric phase (F-KDP) with space group Fdd2 below the Curie temperature Tc = 123 K. This phase transition has been examined in a number of studies5–9 since the discovery of its unusually large isotope effect; the Tc of the deuterated KDP, ∼220 K, is about 100 K higher than that of the undeuterated KDP, which strongly indicates the involvement of a proton movement in the phase transition.9,10 The space groups I2d and Fdd2 are both noncentrosymmetric, so both phases of KDP exhibit nonzero SHG responses. So far, there have not been many theoretical studies on the origin of its NLO properties. Levine calculated the SHG coefficient of P-KDP using the bond charge model, omitting the structural details and neglecting the contributions from the K+ and H+ cations.11 The SHG coefficients of P-KDP calculated using the CRYSTAL code were reported by Lacivita et al.12 and by Rérat et al.,13 but their work did not probe the atomic origin of the SHG responses. Lin et al. calculated the optical properties of the P-KDP using the CASTEP code, to report that the anionic groups (PO4)3− contribute ∼99% to the SHG coefficients using their “real-space atom cutting” analysis, in which the contributions of the K+ and H+ cations were excluded.14 According to the neutron diffraction measurements9,15 and recent theoretical studies,16,17 the paraelectric to ferroelectric phase transition of KDP involves the zero-point vibration of the H+ ions leading to the anti-phase shift of the K+ and P5+ ions along the c-axis. On F-KDP, there is one theoretical study reported.6 Lacivita et al. calculated the SHG responses of F-KDP using the CRYSTAL code,12 but did not explore the origin of its SHG responses.
Over the years, several guidelines were influential in explaining the SHG responses of known NLO materials as well as searching for new ones. One was the assumption that the metal cations of an NLO compound can be neglected in describing the SHG response.18–20 However, this assumption has been found to be inappropriate by the atomic response theory (ART)21 based on the first-principles methods. The ART analysis enables one to determine the individual atom contributions to the total SHG of a given NLO compound by employing the partial response functionals. The ART analyses have been carried out for numerous NLO compounds, which include LiCs2PO4,21 Ga2Se3,22 ABC2 chalcopyrites (A = Zn, Cd; Si, Ge, Sn; C = P, As),23 borates KBe2BO3F2, β-BaB2O4, LiB3O5, CsB3O5 and CsLiB6O10,24 as well as Na2MIIMIV2Q6 (MII = Zn, Cd; MIV = Ge, Sn; Q = S, Se).25 Thus, in the case of P-KDP and F-KDP, the metal cations K+ cations are expected to contribute strongly to the SHG responses, as found for those in KBe2BO3F2 (KBBF).24
Another influential guideline was the belief that the SHG response of an NLO compound becomes stronger when its static (i.e., spontaneous) polarization is larger, which was first noted by Jerphagnon26 and has been invoked in explaining a number of NLO materials.27–29 However, the SHG responses of certain NLO compounds do not depend explicitly on the static polarization.24,30 For example, KBBF has a large SHG response although it has no static polarization. Thus, it is doubtful that Jerphagnon's observation is a reliable guideline. As inversion symmetry nullifies the second order NLO effect, non-centrosymmetry of a structure becomes necessary to have a SHG response. However, this condition is not a sufficient one, because it does not guarantee a significant SHG response. The role of acentricity has been well studied by Poeppelmeier et al. by means of the SAMD model.31–33 The paraelectric and ferroelectric phases of KDP differ in their static polarization; P-KDP has no static polarization because it has D2d point group symmetry, whereas F-KDP has a nonzero static polarization. Thus, Jerphagnon's observation suggests that F-KDP should have a greater SHG response than does P-KDP. Experimentally, the SHG coefficients of P-KDP are known (Table 1(a)), but those of F-KDP are not. The SHG coefficients of both P-KDP and F-KDP have been examined in one computational study,12 but it is unclear whether P-KDP or F-KDP has a stronger SHG response (Table 1).
Table 1 The SHG coefficients dij in pm V−1 reported for P-KDP (I2d) and F-KDP (Fdd2) in the literature
| (a) Experimental and computational results on the SHG responses of P-KDP |
| Experimental |
Computational |
| Input beam |
d36 |
Code used |
Method |
d14 = d36 |
| 1.08 eV |
0.62 (ref. 2) |
CASTEP |
LDA |
0.420 (ref. 14) |
| 1.17 eV |
0.41,34 0.39,35 0.38,36 0.39 (ref. 2) |
CRYSTAL |
LDA |
0.487 (ref. 13) |
| 2.33 eV |
0.57 (ref. 2) |
GGA-PBE |
0.467 (ref. 13) |
| (b) Computational results on the SHG responses of F-KDP12 |
| Code used |
Method |
d15 = d31 |
d24 = d32 |
d33 |
| CRYSTAL |
LDA |
0.516 |
−0.510 |
0.001 |
| GGA-PBE |
0.507 |
−0.284 |
0.012 |
To answer the above question, it is necessary to evaluate the effective SHG coefficients deff for P-KDP and F-KDP without using the Kleinman symmetry,37 which was yet another influential guideline. This symmetry is valid only when the applied field frequency (of the input laser) is very much smaller than the resonance frequency of the NLO compound (i.e., the bandgap). If the latter condition is satisfied, the SHG response becomes independent of the applied field frequency and the number of SHG coefficients to evaluate becomes reduced, thereby simplifying both experimental and computational studies of SHG responses. However, it has been reported that the Kleinman symmetry is not rigorously correct in several cases.22–24,38 Thus, it is of interest to probe the validity of the Kleinman symmetry, as well as the factors influencing it, by studying the SHG responses of P-KDP and F-KDP.
In the present work we probe the three issues raised above by examining the electronic structures and the SHG responses of P-KDP and F-KDP based on first principles DFT calculations.
2 Calculations of SHG responses and ART analyses
The crystal structure of KDP at room temperature was studied by Hassel,39 Hendricks40 and West.41 Hassel determined the unit cell parameters and the space group, I2d, in the modern terminology, and Hendricks obtained the positions of the P and K atoms as well as the approximate position of the O atom in this body-centered tetragonal unit cell. West further determined the position of O and suggested the location of H in midway between the two O atoms of two adjacent PO4 groups. The structural model of West has proven to be correct for P-KDP above the Curie temperature. In our work, the structure optimization of P-KDP was performed by using the Vienna Ab initio Simulation Package (VASP)42,43 to find that the optimized structure agrees quite well with the experimental one. The details of the optimized structure are given in Table S1 as well as Fig. S1 and S2 of the ESI.† Throughout this work the optimized structure was used for the structure description, electronic structure and optical calculations at various levels by using the VASP and ABINIT.44 The structure of F-KDP (SG: Fdd2) below the Curie temperature9 was treated similarly (Table S1 as well as Fig. S3 and S4†).
3 Results and discussion
3.1 Crystal structure
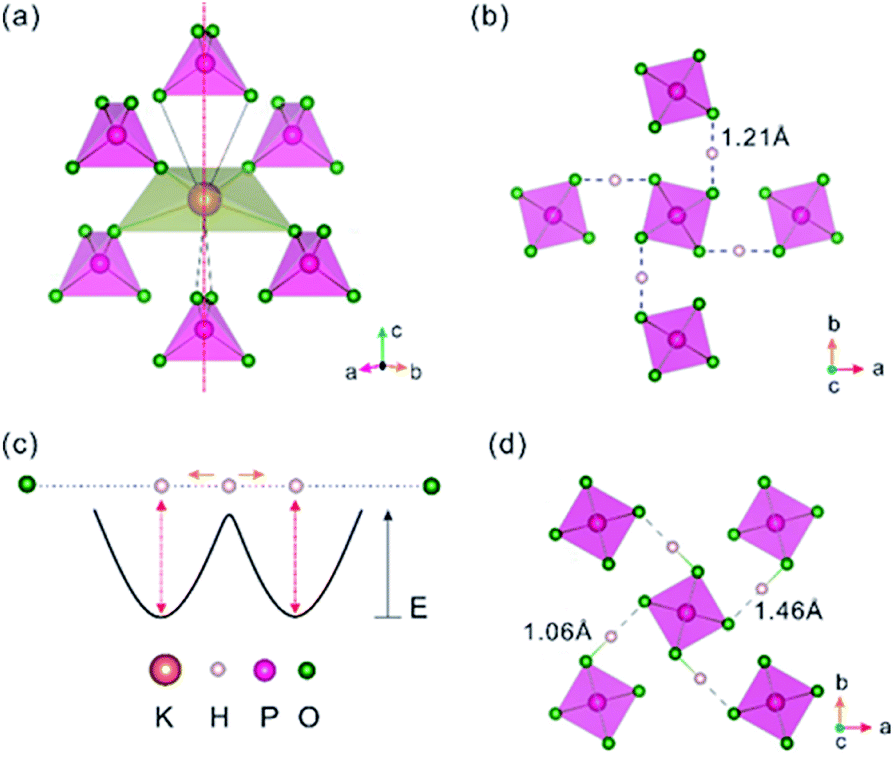
In P-KDP, there is only one unique position for each of P, K, O and H in the unit cell, so each element has a unique coordination polyhedron with its nearest-neighbour elements. For example, each P has a tetrahedral coordination, PO4, of S4 symmetry. Each K forms a KO8 polyhedron with six surrounding PO4 tetrahedra (Fig. 1a), four of which each contributes one O atom to form a flattened KO4 tetrahedron, and the rest two each contributes two O atoms to form an elongated KO4 such that the resulting KO8 polyhedron has a axis (the dashed line in Fig. 1a). The K–O distances of the flattened KO4 tetrahedron are 2.895 Å, ∼0.04 Å shorter than the remaining four K–O distances of KO8. Adjacent PO4 groups are interconnected by a symmetric O–H–O bond with H–O distance of ∼1.21 Å (Fig. 1b). These O–H–O bonds are almost parallel to the a- or b-axis. The symmetric O–H–O bridge is not stable45 against the formation of an asymmetric O–H⋯O bridge made up of a short H–O bond and a long H⋯O hydrogen bond. Slater10 proposed a two-potential-well model to describe the position of the proton, which can hop or tunnel between the two wells thus appearing to be at the midpoint between the two O atoms (Fig. 1c) at high temperature. A trapped proton in either of the two wells can break the tetragonal symmetry hence leading to the paraelectric to ferroelectric phase transition. This proposal has been corroborated by several experimental studies.15,46 A recent neutron diffraction study9 revealed that, below Tc, the proton in each O–H–O bridge becomes trapped in one of the two wells forming an asymmetrical O–H⋯O bridge (Fig. 1d) and induces an anti-phase movement of the P5+ and K+ ions such that a static electric polarization along the c-axis is created. Its influence on the electronic band gap is discussed below.
 |
| | Fig. 1 (a) Six PO4 tetrahedra surrounding a K atom in P-KDP, leading to a KO8 polyhedron. The latter consists of two concentric KO4 tetrahedra, and only the compressed tetrahedron is shown for clarity. The vertical dashed line represents the axis. (b) Projection view along the c-axis of the PO4 tetrahedra in P-KDP, which are interconnected via symmetric O–H–O bridges. (c) Double-well potential model for the movement of H in between two O atoms. (d) Projection view along the c-axis of the PO4 tetrahedra in F-KDP, which are interconnected via asymmetric O–H⋯O bridges. | |
3.2 Electronic structure
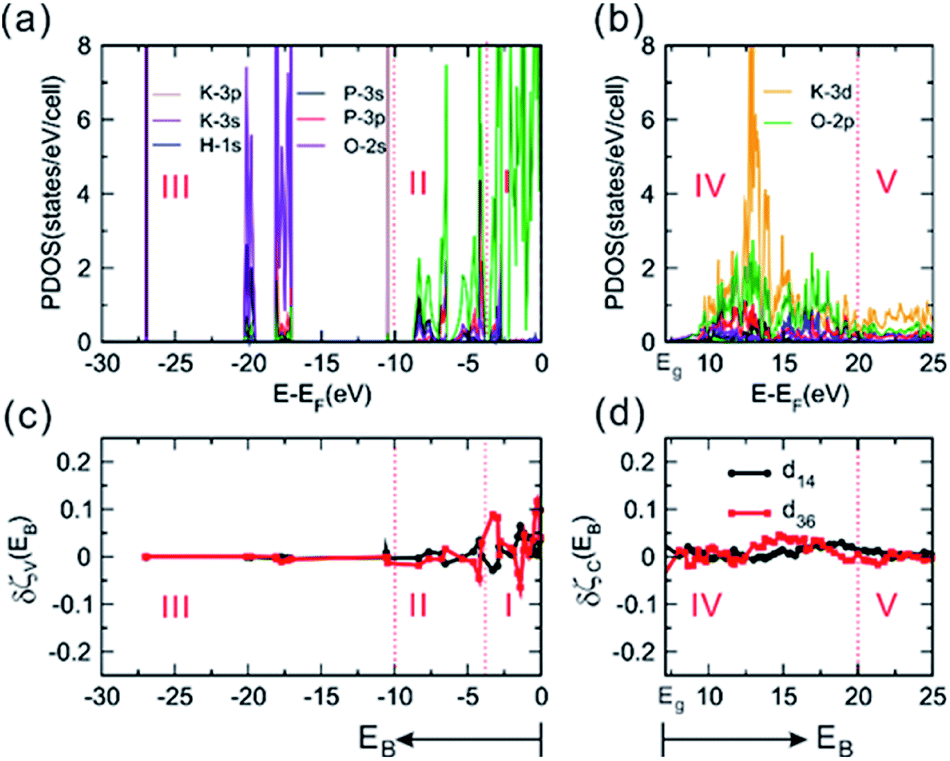
The calculated electronic structures of P-KDP and F-KDP are presented in Fig. S5–S8† in terms of band dispersion relations and projected density of states (PDOS). The calculated band gap of P-KDP, ∼5.61 eV, is smaller than the experimental value of ∼7.12 eV,35 which is a well-known deficiency of the DFT method.47 For F-KDP, the calculated band gap of ∼5.45 eV is again smaller than the experimental value of ∼8.0 eV.1 The measured expansion of the electronic band gap from P-KDP to F-KDP is another consequence of the symmetry breaking of the O–H–O bond, which, however, can only be an indirect effect,48–50 because the electronic states of H atoms contribute negligibly to both the bottom of the conduction band (CBM) and the top of the valence band (VBM) (see Fig. 2a, b and S5–S8†). The analyses on the VBM and CBM states (see Table S2†) indicate that the VBM states of both P-KDP and F-KDP consist predominantly of O-2p orbitals, while the CBM states consist mainly of O-2p, K-3d, P-3p orbitals. These results indicate that the ferroelectric phase transition changes only a small amount of band gap (∼0.9 eV). The mainly ionic nature remains unchanged. The discrepancies between the calculated and measured band gaps can mainly be attributed to the intrinsic deficiency of the DFT method.47 To calculate the optical properties correctly, we employ the scissor operation,51,52 which raises the energies of the unoccupied states from those of the occupied ones to have the experimental band gap without modifying the nature of these states.
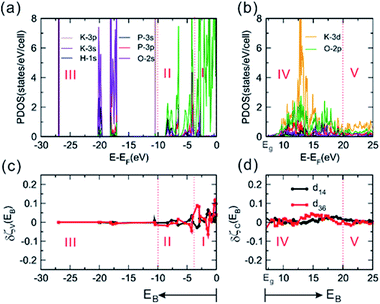 |
| | Fig. 2 The calculated electronic structure and optical properties of P-KDP. (a) The PDOS plots calculated for the valence bands, and (b) those for the conduction bands. (c) The derivative functions δζV(EB)-vs-EB for the valence bands, and (d) δζV(EB)-vs-EB for the conduction bands. | |
As shown in Fig. 2a for P-KDP, the valence bands are divided into three regions I–III, and the conduction bands into two regions IV and V. The region I (−3.75 to 0 eV) is composed mainly of the O-2p nonbonding states. The region II (−3.75 to −10 eV) consists of mainly O-2p, P-3p and H-1s orbitals, representing the P–O bonding and O–H bonding states. The region III (−10 to −30 eV), far below the Fermi level, consists of more strongly bound states (i.e., O-2s, K-3s, 3p and P-3s, 3p and H-1s states). The region IV (7.12 to 20 eV) is composed of largely the K-3d states with substantial amount of the O-2p states, where the two states make antibonding interactions. In the region V (20 to 25 eV), there are not many states. The K-3d states describe the high-energy excited states of KDP, and contribute strongly to the SHG response of F-KDP because they are polarizable. As can be seen from the PDOS plots (Fig. S7 and S8†), the electronic structure of F-KDP is very similar to that of P-KDP in basic features. The electronic polarization of F-KDP from our VASP calculations is ∼2.3 μC cm−2, a value smaller than to the experimental one (i.e., ∼4.5 μC cm−2).9,53 With the Bader charges calculated54,55 for each element of F-KDP (+0.891, +3.610, ∼−1.455 and 0.659 for K, P, O and H, respectively), we used the point charge model to find that all ions contribute only through their z components, with their contributions decreasing in the order K > P > O ≫ H. The static polarization of P-KDP is exactly zero due to the symmetry forbiddance. In fact, among the 21 non-centrosymmetric point groups, only 10 of them allow the occurrence of static polarization.56 Thus, a ferroelectric material satisfies the necessary condition of SHG, but the vice versa need not be true. Except for the creation of the necessary condition for SHG, the static polarization does not influence the SHG phenomenon as shown in the following.
In a strong electric field E of a light, the total polarization P can be written as a functional form of E,57
| | |
P[E] = P(0) + P(1)[E] + P(2)[E,E]+…,
| (1) |
where
P(n)[
E] is the
n-th order term of
E, denoting the
n-th order induced polarization. By definition, the second order NLO coefficient tensor can be written as the functional derivative as follows,
| |
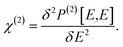 | (2) |
Obviously, the static polarization
P(0) makes no explicit contribution to
χ(2), which is also argued for by Jiang
et al. from a different angle.
30
3.3 SHG coefficients
In our study on the NLO responses of P-KDP and F-KDP, all optical transition matrix elements were calculated using the ABINIT code as well as the VASP code to cross-check. Based on these results, our own code based on the sum of states (SOS) method58,59 was used to calculate the SHG coefficients for P-KDP and F-KDP. The contributions of the individual atomic orbitals and the individual atoms to the SHG responses were calculated by performing the ART analyses.21 The details of the computational methods and chosen parameters are given in ESI.† For P-KDP, only two independent nonzero components of the SHG tensor (i.e., d14 and d36) occur due to the constraints of the crystal symmetry. The Kleinman symmetry was not assumed throughout this work, because it is not rigorously correct in some cases.22–24,38 The calculated results are given in Table 2. Our results agree quite well with those obtained from the latest calculations using the CRYSTAL code,13 and are slightly larger than those from the CASTEP calculations (Table 1(b)).14 The experimental values varying in the range of 0.38–0.62 pm V−1 for the d36 component were measured with lasers of ω = 1.08, 1.17 and 2.33 eV, respectively2,34–36 (Table 1a). The variation of the laser frequency does not significantly affect the measured SHG response. For this reason, the values calculated at ω = 0 are quite often used to compare with the experimental values. To quantify how well the Kleinman symmetry is obeyed, we define the % error, Δ, for the Kleinman pair of (dij, dkl) of SHG coefficients as| | |
Δ = 100|dij − dkl|/[(dij + dkl)/2]
| (3) |
where dij and dkl are two SHG coefficients that are identical if the Kleinman symmetry is obeyed. For the (d14, d36) pair of P-KDP, the Kleinman symmetry is correct within error lower than 3.6% (Table 2a), which agrees very well with the early experimental results.2 The symmetry reduction from P-KDP to the F-KDP introduces more independent nonzero SHG components, i.e., (d15, d31), (d24, d32) and d33 (Table 2b). The values for the two Kleinman pairs of F-KDP are significantly greater than that for the Kleinman pair of P-KDP. This is rather striking because one would have expected the opposite because F-KDP has a greater bandgap than does P-KDP (8.0 vs. 7.12 eV) so that the condition required for the Kleinman symmetry is better satisfied in F-KDP. The calculated bandgaps of F-KDP and P-KDP are much larger than the energies of the pump light, but are smaller than the energy gaps involved in the SOS method of calculating the SHG coefficients. The breaking down of the Kleinman symmetry means that the polarization directions of the mixed pump light and that of the exit light cannot be permuted, indicating the occurrence of dynamic anisotropy in the SHG response of the lower-symmetry structure. For example, d31 (i.e., dzxx) represents the z-polarized SHG response to the x-polarized light fields Ex × Ex, while d15 (i.e., dxzx) represents the x-polarized SHG response to the mixed light fields Ez × Ex. In P-KDP, (d15, d31) is forced to be (0, 0) due to its D2d symmetry, thus the Kleinman symmetry is satisfied trivially. The reduction of the symmetry from D2d of P-KDP to C2v of F-KDP releases the crystal symmetry constraint between d15 and d31, and the breaking down of Kleinman symmetry manifests (Table 2). In the current case, it is tempting to attribute this phenomenon to the static anisotropy of the F-KDP structure, because the occurrence of the static electric polarization in the z-direction makes it inequivalent to those in the x, y-directions. However, the vanishing value of d33 (i.e. dzzz) indicates that the z-polarized SHG response to the pump light fields Ez × Ez is not favored. The breaking down of the Kleinman symmetry may be attributed to the anisotropic lossy nature of F-KDP related to the flexibility of the hydrogen bond network mainly in the xy-plane (i.e., perpendicular to the z axis). In general, the breaking down of the Kleinman symmetry shows up with the lowering of structural symmetry.
Table 2 The SHG coefficients dij and deff (in pm V−1) calculated for P-KDP (I2d) and F-KDP (Fdd2) in the present work
| (a) P-KDP |
| The first row indicates the code used for electronic structure calculations, and the second the code used for optical property calculations. Here the “local code” refers to the one developed in our laboratory. |
| Methods |
GGA-PBE |
GGA-ONCV |
GGA-ONCV |
| Codes useda |
VASP |
ABINIT |
ABINIT |
| Localb |
Localb |
ABINIT |
| (d14, d36) Δ (%) |
(0.505, 0.490) 3.02 |
(0.469, 0.486) 3.56 |
(0.470, 0.486) 3.35 |
| deff |
0.423 |
0.401 |
0.402 |
| (b) F-KDP |
| Methods |
GGA-PBE |
GGA-ONCV |
GGA-ONCV |
| Codes useda |
VASP |
ABINIT |
ABINIT |
| Localb |
Localb |
ABINIT |
| (d15, d31) Δ (%) |
(−0.435, −0.275) 45.07 |
(−0.366, −0.345) 5.91 |
(−0.375, −0.337) 10.67 |
| (d24, d32) Δ (%) |
(0.221, 0.390) 55.32 |
(0.250, 0.281) 11.68 |
(0.243, 0.301) 21.32 |
| d33 |
−0.044 |
−0.059 |
−0.06 |
| deff |
0.266 |
0.245 |
0.246 |
To see the overall effect of the static polarization, we calculate the effective SHG coefficient, deff,60,61 which is a quantity comparable to the measured SHG coefficient on a powder sample. The calculated deff values for P-KDP are all greater than the corresponding ones of F-KDP for all computational methods we tested. These results provide a clear evidence that the presence of a static polarization is not essential for the SHG response. The result that deff of P-KDP is greater than that of F-KDP can be simply understood by means of the quasi-linear relation23,24 between dav and V/Eg, where dav is the calculated average SHG coefficient or the effective SHG response deff, V is the volume of the primitive unit cell, and Eg is the experimental band gap. The calculated values of V/Eg are 28.09, 25.4 Å3 eV−1 for P-KDP and F-KDP, which show the same order as their SHG responses (Table 2).
3.4 Atom contributions to SHG responses
To determine the contributions of the atomic orbital states to the SHG coefficients, we carry out the ART analyses by using the derivative functions of the partial response functions for the valence bands and conduction bands as a function of the energy EB,21,23 as shown in Fig. 2c and d for d36 and d14 of P-KDP. In region I of Fig. 2c where the nonbonding O-2p states dominate, the derivative functions δζV(EB) changes drastically. In region II where the P–O bonding and O–H bonding states occur, δζV(EB) shows much less pronounced peaks indicating smaller contributions from the P–O bonding and O–H bonding states to the SHG responses than the O-2p states in region I. As can be seen from Fig. 2d, the derivative functions δζC(EB) of both d14 and d36 exhibit a sawtooth-like shape in a wide range of energy. This property results from the very dispersive character of the K-3d orbitals. Our analyses reveal that the O-2p nonbonding states contribute dominantly in the valence bands, and so do the K-3d states in the conduction bands where the K-3d and O-2p orbitals make antibonding. Results very similar to those discussed above for P-KDP are found for F-KDP, as summarized in Fig. S9.†
To determine quantitatively the contribution, Aτ, of an individual atom τ to each independent nonzero component of a SHG response tensor, we carry out the ART analyses.21,23 The calculated results for the d14 and d36 components of P-KDP are listed in Table 3. The individual atom contributions in d14 are not the same as those in d36, but in both cases the individual contributions of K and O are comparable and are much greater than those of P and H. The total contribution of an element is equal to the individual atom contribution multiplied by a factor counting the number of its equivalent atoms in the unit cell (see WA in Table 3). The ratio of such factors is equal to the stoichiometry of a compound. These factors make the relative contributions of individual atoms differ from those of elements. The resulting changes are not the same for every element in a compound, we call this phenomenon an uneven stoichiometry effect as suggested in our previous work on borates.24 In KDP (KH2PO4), the stoichiometry is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:4 for K, H, P and O, which quadruples the contribution of O element compared to that of K element, though the individual atom contributions of K and O are almost the same. The uneven stoichiometry effect is determined by the valences of the constituent elements of a compound, thus it is a chemical effect. As already pointed out, the contribution of the O atoms arises mainly from the region I of the valence bands, and that of the K atoms from the region IV of the conduction bands. The detailed quantitative information is provided in Table S3 and S4.† The analogous analyses were also carried out for F-KDP, which results in the individual atom contributions presented in Table S5 and S6.† The main features found for P-KDP hold also true for F-KDP.
:2:1:4 for K, H, P and O, which quadruples the contribution of O element compared to that of K element, though the individual atom contributions of K and O are almost the same. The uneven stoichiometry effect is determined by the valences of the constituent elements of a compound, thus it is a chemical effect. As already pointed out, the contribution of the O atoms arises mainly from the region I of the valence bands, and that of the K atoms from the region IV of the conduction bands. The detailed quantitative information is provided in Table S3 and S4.† The analogous analyses were also carried out for F-KDP, which results in the individual atom contributions presented in Table S5 and S6.† The main features found for P-KDP hold also true for F-KDP.
Table 3 The calculated atomic contributions (in %) to the SHG coefficients d14 and d36 of P-KDP. WA denotes the number of crystallographically equivalent atoms in a unit cell, Aτ the contribution of a single atom τ, and CA the total contribution of equivalent atoms
| Atom |
WA |
d14 |
d36 |
| Aτ |
CA |
Aτ |
CA |
| K |
2 |
8.94 |
17.88 |
5.77 |
11.54 |
| H |
4 |
1.15 |
4.62 |
1.75 |
7.02 |
| P |
2 |
2.92 |
5.84 |
2.05 |
4.10 |
| O |
8 |
8.96 |
71.67 |
9.67 |
77.34 |
4 Conclusions
Our study of P-KDP and F-KDP demonstrates that the presence of static polarization does not influence the SHG response although it guarantees a necessary condition for the occurrence of the SHG phenomenon. The Kleinman symmetry is reasonably well obeyed for P-KDP, but this is not the case for F-KDP although it has a larger bandgap than does P-KDP. Our analysis suggests that the Kleinman symmetry is poorly obeyed for an NLO compound of lower-symmetry. The individual atom contributions of the K and O atoms to the SHG responses are comparable in magnitude. The oxygen atom contributes mainly through the O-2p nonbonding states of the valence bands, and the potassium atom mainly through the K-3d states of the conduction bands. The total contribution of a chemical element depends not only on its individual atom contribution but also on the stoichiometry of the compound. This and our previous works23–25 and the others62–65 suggest a simple approach to enhance the SHG efficiency that is to substitute the cations and anions with larger (i.e., more polarizable) ones by exploiting the valence differences in the constituent elements. NLO compounds of various types are needed to test this approach.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation (NSF) of China (21921001, 21703251, 61874122); the Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) (XDB20000000); the National Key Research and Development Program of China (2016YFB0701001); the NSF of Fujian Province (2019J05151, 2019J01121); Youth Innovation Promotion of CAS (2019302).
Notes and references
- I. N. Ogorodnikov, V. A. Pustovarov, B. V. Shul'gin, V. T. Kuanyshev and M. K. Satybaldieva, Opt. Spectrosc., 2001, 91, 224–231 CrossRef CAS.
- D. Eimerl, Ferroelectrics, 1987, 72, 95–139 CrossRef CAS.
- J. J. De Yoreo, A. K. Burnham and P. K. Whitman, Int. Mater. Rev., 2002, 47, 113–152 CrossRef CAS.
- N. Zaitseva and L. Carman, Prog. Cryst. Growth Charact., 2001, 43, 1–118 CrossRef CAS.
- G. Busch and P. Scherrer, Naturwissenschaften, 1935, 23, 737 CrossRef CAS.
- A. Otani and S. Makishima, J. Phys. Soc. Jpn., 1969, 26, 85–90 CrossRef CAS.
- A. Ioanid, M. Popescu, N. Vlahovici and I. Bunget, Phys. Status Solidi A, 1984, 82, K125–K128 CrossRef CAS.
- T. Matsubara, Jpn. J. Appl. Phys., 1985, 24, 1–5 CrossRef CAS.
- T. Miyoshi, H. Mashiyama, T. Asahi, H. Kimura and Y. Noda, J. Phys. Soc. Jpn., 2011, 80, 044709 CrossRef.
- J. C. Slater, J. Chem. Phys., 1941, 9, 16–33 CrossRef CAS.
- B. F. Levine, Phys. Rev. B: Solid State, 1973, 7, 2600–2626 CrossRef CAS.
- V. Lacivita, M. Rérat, B. Kirtman, M. Ferrero, R. Orlando and R. Dovesi, J. Chem. Phys., 2009, 131, 204509 CrossRef PubMed.
- M. Rérat, L. Maschio, B. Kirtman, B. Civalleri and R. Dovesi, J. Chem. Theory Comput., 2016, 12, 107–113 CrossRef PubMed.
- Z. Lin, Z. Wang, C. Chen and M.-H. Lee, J. Chem. Phys., 2003, 118, 2349–2356 CrossRef CAS.
- G. E. Bacon and R. S. Pease, Proc. R. Soc. London, Ser. A, 1955, 230, 359–381 CAS.
- S. Koval, J. Kohanoff, J. Lasave, G. Colizzi and R. L. Migoni, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 184102 CrossRef.
- S. E. Mkam Tchouobiap and H. Mashiyama, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 014101 CrossRef.
- C. T. Chen, Acta Phys. Sin., 1976, 25, 146–161 CAS.
- C. T. Chen, Sci. Sin., 1979, 22, 756–776 CAS.
- C. T. Chen and G. Z. Liu, Annu. Rev. Mater. Sci., 1989, 16, 203–243 CrossRef.
- X. Cheng, M.-H. Whangbo, G.-C. Guo, M. Hong and S. Deng, Angew. Chem., Int. Ed., 2018, 57, 3933–3937 CrossRef CAS PubMed.
- S.-P. Guo, X. Cheng, Z.-D. Sun, Y. Chi, B.-W. Liu, X.-M. Jiang, S.-F. Li, H.-G. Xue, S. Deng, V. Duppel, K. Jürgen and G.-C. Guo, Angew. Chem., Int. Ed., 2019, 131, 8171–8175 CrossRef.
- X. Cheng, M.-H. Whangbo, M. Hong and S. Deng, Inorg. Chem., 2019, 58, 9572–9575 CrossRef CAS PubMed.
- X. Cheng, Z. Li, X. Wu, M. Hong, M.-H. Whangbo and S. Deng, ACS Appl. Mater. Interfaces, 2020, 12, 9434–9439 CrossRef CAS PubMed.
- R. Ye, X. Cheng, B.-W. Liu, X. M. Jiang, L. Yang, S. Deng and G.-C. Guo, J. Mater. Chem. C, 2020, 8, 1244–1247 RSC.
- J. Jerphagnon, Phys. Rev. B: Solid State, 1970, 2, 1091–1098 CrossRef.
- Y. Shen, Y. Yang, S. Zhao, B. Zhao, Z. Lin, C. Ji, L. Li, P. Fu, M. Hong and J. Luo, Chem. Mater., 2016, 28, 7110–7116 CrossRef CAS.
- S. Zhao, P. Gong, S. Luo, L. Bai, Z. Lin, C. Ji, T. Chen, M. Hong and J. Luo, J. Am. Chem. Soc., 2014, 136, 8560–8563 CrossRef CAS PubMed.
- N. Ye, Q. Chen, B. Wu and C. Chen, J. Appl. Phys., 1998, 84, 555–558 CrossRef CAS.
- X. Jiang, S. Zhao, Z. Lin, J. Luo, P. D. Bristowe, X. Guan and C. Chen, J. Mater. Chem. C, 2014, 2, 530–537 RSC.
- H. Wu, H. Yu, Z. Yang, X. Hou, X. Su, S. Pan, K. R. Poeppelmeier and J. M. Rondinelli, J. Am. Chem. Soc., 2013, 135, 4215–4218 CrossRef CAS PubMed.
- A. Cammarata, W. Zhang, P. S. Halasyamani and J. M. Rondinelli, Chem. Mater., 2014, 26, 5773–5781 CrossRef CAS.
- T. T. Tran, J. Young, J. M. Rondinelli and P. S. Halasyamani, J. Am. Chem. Soc., 2017, 139, 1285–1295 CrossRef CAS PubMed.
- S. Singh, Nonlinear optical materials, in Handbook of Laser Science and Technology, ed. M. J. Weber, CRC Press, Boca Raton, USA, 1995, vol. III, pp. 3–228 Search PubMed.
- V. G. Dmitriev, G. G. Gurzadyan and D. N. Nikogosyan, Handbook of Nonlinear Optical Crystals, Springer: New York, USA, 3rd edn, 1999, vol. 64, pp. 78–85 Search PubMed.
- R. C. Eckardt, H. Masuda, Y. X. Fan and R. L. Byer, IEEE J. Quantum Electron., 1990, 26, 922–933 CrossRef CAS.
- D. A. Kleinman, Phys. Rev., 1962, 126, 1977–1979 CrossRef CAS.
- C. A. Dailey, B. J. Burke and G. J. Simpson, Chem. Phys. Lett., 2004, 390, 8–13 CrossRef CAS.
- H. O. Hassel, Z. Elektrochem., 1925, 31, 523–529 Search PubMed.
- S. B. Hendricks, Am. J. Sci., 1927, 14, 269–287 CrossRef CAS.
- J. West, Z. Kristallogr., 1930, 74, 306–332 CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- X. Gonze, F. Jollet, F. Abreu Araujo, D. Adams, B. Amadon, T. Applencourt, C. Audouze, J.-M. Beuken, J. Bieder, A. Bokhanchuk, E. Bousquet, F. Bruneval, D. Caliste, M. Côté, F. Dahm, F. Da Pieve, M. Delaveau, M. Di Gennaro, B. Dorado, C. Espejo, G. Geneste, L. Genovese, A. Gerossier, M. Giantomassi, Y. Gillet, D. R. Hamann, L. He, G. Jomard, J. Laflamme Janssen, S. Le Roux, A. Levitt, A. Lherbier, F. Liu, I. Lukačević, A. Martin, C. Martins, M. J. T. Oliveira, S. Poncé, Y. Pouillon, T. Rangel, G. M. Rignanese, A. H. Romero, B. Rousseau, O. Rubel, A. A. Shukri, M. Stankovski, M. Torrent, M. J. Van Setten, B. Van Troeye, M. J. Verstraete, D. Waroquiers, J. Wiktor, B. Xu, A. Zhou and J. W. Zwanziger, Comput. Phys. Commun., 2016, 205, 106–131 CrossRef CAS.
- S. Deng and Y. Zhang, Phys. Status Solidi B, 2016, 253, 1688–1696 CrossRef CAS.
- R. Blinc and B. Žekš, Ferroelectrics, 1987, 72, 193–227 CrossRef CAS.
- R. W. Godby, M. Schlüter and L. J. Sham, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 6497–6500 CrossRef CAS PubMed.
- A. Datta and S. K. Pati, Chem. Soc. Rev., 2006, 35, 1305–1323 RSC.
- S. M. Pratik, S. Chakraborty, S. Mandal and A. Datta, J. Phys. Chem. C, 2015, 119, 926–933 CrossRef CAS.
- T. K. Mandal, S. K. Pati and A. Datta, J. Phys. Chem. A, 2009, 113, 8147–8151 CrossRef CAS.
- X. Gonze and C. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10355–10368 CrossRef CAS.
- F. Gygi and A. Baldereschi, Phys. Rev. Lett., 1989, 62, 2160–2163 CrossRef CAS PubMed.
- G. A. Samara, Ferroelectrics, 1973, 5, 25–37 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- R. F. W. Bader, Atoms in Molecules – A quantum theory, Oxford University Press, New York, 1990 Search PubMed.
- R. W. Boyd, Nonlinear Optics, Elsevier, New York, 2003 Search PubMed.
- Y. R. Shen, The principles of nonlinear optics, Wiley-Interscience, New York, 1984 Search PubMed.
- C. Aversa and J. E. Sipe, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 14636–14645 CrossRef CAS PubMed.
- S. N. Rashkeev, W. R. L. Lambrecht and B. Segall, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 3905–3919 CrossRef CAS.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS.
- S. J. Cyvin, J. E. Rauch and J. C. Decius, J. Chem. Phys., 1965, 43, 4083–4095 CrossRef CAS.
- G. Zou, Z. Lin, H. Zeng, H. Jo, S.-J. Lim, T.-S. You and K. M. Ok, Chem. Sci., 2018, 9, 8957–8961 RSC.
- H.-X. Tang, Y.-X. Zhang, C. Zhuo, R.-B. Fu, H. Lin, Z.-J. Ma and X.-T. Wu, Angew. Chem., Int. Ed., 2019, 58, 3824–3828 CrossRef CAS PubMed.
- R.-L. Tang, C.-L. Hu, F.-F. Mao, J.-H. Feng and J.-G. Mao, Chem. Sci., 2019, 10, 837–842 RSC.
- B.-L. Wu, C.-L. Hu, F.-F. Mao, R.-L. Tang and J.-G. Mao, J. Am. Chem. Soc., 2019, 141, 10188–10192 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details; description of the PRF method and ART analysis; optimized crystal structures of P-KDP and F-KDP; calculated electronic structures; Fig. S1–S9; Tables S1–S6 (PDF). See DOI: 10.1039/d0ra03136d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Shuiquan Deng
a and
Shuiquan Deng