
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalization of pristine graphene for the synthesis of covalent graphene–polyaniline nanocomposite
Jaehyeung Park†
ab,
Xiaojian Yang†a,
Dhanushka Wickramasinghe a,
Madanodaya Sundhoroa,
Nese Orbey*c,
Kwok-Fan Chow*a and
Mingdi Yan*a
a,
Madanodaya Sundhoroa,
Nese Orbey*c,
Kwok-Fan Chow*a and
Mingdi Yan*a
aDepartment of Chemistry, University of Massachusetts Lowell, Lowell, MA 01854, USA. E-mail: mingdi_yan@uml.edu
bDivision of Advanced Materials Engineering, Dong-Eui University, Busan, 47340, Korea
cDepartment of Chemical Engineering, University of Massachusetts Lowell, Lowell, MA 01854, USA
First published on 14th July 2020
Abstract
Polyaniline (PANI) is one of the most studied conducting polymers owing to its high electrical conductivity, straightforward synthesis and stability. Graphene-supported PANI nanocomposite materials combine the superior physical properties of graphene, synergistically enhancing the performance of PANI as well as giving rise to new properties. Covalent nanocomposites have shown to give higher stability and better performance than their non-covalent counterparts, however, the covalent graphene–PANI nanocomposite are primarily prepared from graphene oxide. We report a new method to synthesize covalent graphene–PANI nanocomposites from pristine graphene. Using few-layer graphene (FLG) flakes as the model system, we first conjugated aniline to FLG via a perfluorophenyl azide (PFPA)-mediated coupling chemistry. A subsequent in situ polymerization of aniline gave polyaniline covalently grafted on the FLG surface. Characterization by FTIR, TEM, SEM, XPS, XRD and electrochemistry confirmed the successful conjugation of PANI to FLG. The grafting density of PANI was estimated by thermal analysis to be ∼26%. As the PFPA-mediated coupling chemistry is applicable to other carbon materials including carbon nanotubes and fullerene, the method developed in this work can be readily adapted to grow PANI on these materials.
Introduction
Polyaniline (PANI), one of the most valuable polymers, has a long history, already discovered in the 1850s as a “blue substance” during the electrolysis of aniline sulphate.1 It has gained increasing interest after being found to have intrinsic electrical conductivity comparable to those of semiconductors and metals.2 Its rich chemistry of the polymerization process, the complex redox chemistry and the doping mechanism continue to attract intense investigations from researchers. PANI can be directly synthesized from the inexpensive aniline starting material, resulting in a polymer having higher chemical stability compared to other conducting polymers like polythiophene. Demonstrated applications of PANI include printable electronics, as conductive coating for charge dissipation and in making smart wearables, as chemical and biological sensing material, as energy storage material in supercapacitors, as imaging and drug delivery vehicles, and for tissue engineering.3–7 The high potential of PANI, however, has been hindered by its poor melt and solution processability. To overcome this issue, new polymerization techniques, such as interfacial and electrochemical polymerization, have been developed to fabricate PANI of different morphology, like nanofibers.8 The in situ polymerization is another attract approach where PANI can be directly synthesized on a solid substrate to make composite materials, thus avoiding the processing step altogether.9Graphene is considered as one of the best matrix materials for nanocomposites due to its superior mechanical and electronical properties in addition to its high specific surface area. Blending PANI with graphene is expected to give nanocomposites that not only overcome shortcomings of PANI, but also synergistically enhance the performance of both materials thus giving rise to new properties.10 For example, the relatively high specific capacitance (>950 F g−1) and its ability to undergo multiple redox reactions makes PANI a highly promising pseudo-capacitive materials, however, it suffers from relatively low surface area and poor cycling stability caused by structural degradation during the redox process.11 Blending PANI with graphene is expected to give a nanocomposite that can increase the contact surface area and improve its cycling stability.12 Studies have shown that the covalent binding between graphene and PANI gave rise to higher capacitance and longer cycle life compared to the non-covalent materials by decreasing the interfacial resistance and enhancing the charge transfer stability.13–19 Improved stability, electromagnetic and microwave absorption, and electrochemical performances have also been demonstrated through covalent bonding at graphene–polyaniline interface.20–25
The graphene–PANI nanocomposites have been prepared from graphene oxide (GO) or reduced graphene oxide (rGO), owing to the low chemical reactivity of pristine graphene.10,26,27 GO is synthesized by treating graphite with strong oxidation reagents such as H2SO4, KMnO4 or H3PO4.28 This process produces a number of oxygen-containing functional groups, i.e., hydroxy, carboxy and epoxy,29,30 which serve as functionalization sites for either physisorption or covalent conjugation with PANI. The introduction of these species, on the other hand, also severely disrupts the electronic network of graphene, as such the conjugated sp2 network of graphene and its transport properties cannot be completely restored even after extensive reduction of GO to rGO.31,32 Furthermore, the oxidation/reduction process is difficult to control, and is known to cause permanent structural damages on graphene. Unlike the stable pristine graphene, GO would eventually degrade into humic acid-like materials upon storage,33 which hinders the applications that demand long-term stability of its component materials.
Here, we report a covalent conjugation chemistry to graft PANI onto pristine graphene. The covalent functionalization of graphene is accomplished by perfluorophenyl azide (PFPA)-mediated coupling chemistry previously developed in our laboratory.34–43 Photochemical or thermal activation of PFPA generates the perfluorophenyl nitrene, which undergoes cycloaddition reaction with graphene to form a covalent adduct. In this work, graphene was first functionalized with aniline-derivatized PFPA-ANI to introduce aniline groups on graphene. PANI was subsequently grafted onto the graphene surface by in situ polymerization of aniline (Scheme 1). The resulting nanocomposite, G–PANI, was characterized by spectroscopy, microscopy, X-ray powder diffraction (XRD) and electrochemistry techniques. The grafting density of PANI was estimated from the thermal analysis.
 | ||
| Scheme 1 Synthesis of G–PANI nanocomposite by grafting PANI onto PFPA-ANI-functionalized graphene via in situ polymerization of aniline. | ||
Experimental
Materials
Graphite powders, methyl pentafluorobenzoate (>97%), sodium azide, N-hydroxysuccinimide (NHS), 4-dimethylaminopyridine, 4-(N,N-dimethylamino)pyridine (DMAP), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, phenylenediamine, acetone, ethyl ether, dichloromethane, methanol, N-methyl-2-pyrrolidone (NMP) were purchased from Sigma-Aldrich, and were used as received without further purification. 1H NMR spectra were collected on a Bruker Avance Spectrospin DRX500 spectrometer, referenced to the non-deuterated residual solvent peak in DMSO-d6 (dimethyl sulfoxide) at 2.50 ppm. 19F NMR was done on a JEOL ECZ 400 MHz spectrometer using CF3COOH (−76.55 ppm) as the external standard. The high-resolution mass spectrum (HRMS) was obtained at the University of Illinois at Urbana Champaign Mass Spectroscopy facility.Synthesis of N-(4-aminophenyl)-4-azido-2,3,5,6-tetrafluorobenzamide (PFPA-ANI)
N-Hydroxysuccinimide-4-azido-2,3,5,6-tetrafluorobenzoate (PFPA-NHS) was synthesized from methyl pentafluorobenzoate following the previously reported procedures.44–46 A solution of PFPA-NHS in methanol was added into the solution of 4-(N,N-dimethylamino)pyridine (DMAP) (0.4 equiv.) and excess phenylenediamine (10 equiv.) in methanol, and the mixture was continuously stirred at room temperature overnight. The mixture was then washed with water to remove unreacted phenylenediamine and DMAP. Purification by column chromatography using 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dichloromethane:ethyl acetate as the eluent gave PFPA-ANI as a yellow solid (190 mg, 65%). 1H NMR (CD3CN): δ 8.68 (s, 1H), 7.31 (d, 2H), 6.64 (d, 2H), 4.16 (s, 2H). 19F NMR (CD3CN): δ −141.84 (m, 2F), −150.73 (m, 2F). FTIR: 800.5, 829.6, 991.2, 1259.2, 1331.8, 1335.7, 1430.9, 1485.3, 1511.3, 1560.2, 1610.3, 1652.1, 1670.3, 2130.1, 3037.2, 3247.5, 3379.2 cm−1. HR-MS: calculated for C13H8N5OF4 [M + H]+: 326.06, obtained 326.06.
:1 dichloromethane:ethyl acetate as the eluent gave PFPA-ANI as a yellow solid (190 mg, 65%). 1H NMR (CD3CN): δ 8.68 (s, 1H), 7.31 (d, 2H), 6.64 (d, 2H), 4.16 (s, 2H). 19F NMR (CD3CN): δ −141.84 (m, 2F), −150.73 (m, 2F). FTIR: 800.5, 829.6, 991.2, 1259.2, 1331.8, 1335.7, 1430.9, 1485.3, 1511.3, 1560.2, 1610.3, 1652.1, 1670.3, 2130.1, 3037.2, 3247.5, 3379.2 cm−1. HR-MS: calculated for C13H8N5OF4 [M + H]+: 326.06, obtained 326.06.
Preparation of FLG flakes
Graphite powders were dispersed in NMP at the concentration of 1 mg mL−1, and the mixture was sonicated for 24 h using a sonication probe (SONICS, 20 kHz, 40% Ampl.). The resulting dispersion was then centrifuged for 30 minutes at 500 rpm, and the supernatant was collected. The concentration of FLG flakes in the supernatant was calculated by measuring the absorbance of the dispersion at 660 nm on a UV-vis spectrometer (Perkin Elmer Lambda 45).47Functionalization of graphene with PFPA-ANI
PFPA-ANI was added to a dispersion of FLG flakes in NMP at 1:1 mole ratio of PFPA:C. The mixture was heated at 150 °C for 1 h in a microwave reactor (Mars 6tm, microwave digesting system, CEM corporation). Unreacted PFPA-ANI was removed by repetitive washing and centrifugation in acetone to give the product G–ANI.
Synthesis of PANI-grafted graphene
Graphene–PANI composite (G–PANI) was prepared by way of oxidative polymerization of aniline in the presence of G–ANI using ammonium persulfate (APS) as the oxidant.15,48,49 G–ANI was dispersed in a solution of aniline in 1 M HCl (0.03 M), and the mixture was sonicated for 30 min. The mixture was then cooled in an ice-bath, and APS was slowly added. The mole ratio of aniline:APS was set at 4:1. The polymerization was continued for 4 h in the ice bath, and the resulting dark-green products were filtered and washed with HCl, distilled water and ethanol to remove unreacted monomer and oxidant, and finally dried at 50 °C for 24 h under vacuum.
Electrochemical measurements
Cyclic voltammograms (CVs) were obtained to determine the capacitance of graphite and G–PANI electrodes. In order to carry out an electrochemical measurement, a substrate material was mounted in a cell holder that has a 0.11 cm2 circular opening, which allows the electrolyte solution to interact with the electrode. This cell holder provides a well-defined electrode area for electrochemical measurements. All electrochemical measurements were carried out using a three-electrode cell, where a platinum wire and a silver/silver chloride (Ag/AgCl) were used as the counter and reference electrodes, respectively. The electrolyte solution (0.1 M sulfuric acid) was purged with nitrogen gas for 10 min prior to the measurements.Results and discussions
The covalent graphene–PANI nanocomposites were prepared by in situ polymerization of aniline on aniline-functionalized graphene. The PFPA-mediated coupling chemistry, previously developed in our laboratory, was employed to covalently functionalize pristine graphene, through the cycloaddition reaction of the photochemically- or thermally-generated perfluorophenyl nitrene. Towards this end, an aniline-derivatized PFPA, PFPA-ANI, was synthesized by the amide coupling between PFPA-NHS and phenylenediamine in methanol in the presence of DMAP at room temperature overnight (Scheme 1). The structure of the product was confirmed by FTIR, 1H and 19F NMR, and HRMS. Compared to phenylenediamine, the FTIR spectrum of PFPA-ANI showed strong new absorptions at 2130 and 991 cm−1, originated from the asymmetric stretching of the azido group and the C–F stretching, respectively (Fig. 1). The absorption bands at 3400–3200 cm−1 can be assigned as the asymmetric and symmetric N–H stretching (Fig. 1a). Two peaks at 1610 and 1259 cm−1 were from N–H and C–N vibrations, respectively. The 1H NMR spectrum showed the aniline amine proton at 5.06 ppm and the amide proton at 10.51 ppm in addition to the two sets of aniline aromatic peaks at 6.55 and 7.29 ppm, respectively (Fig. 2). The 19F NMR spectrum contained two sets of F signals at −146.67 and −141.18 ppm, which is consistent with the 2,3,5,6-tetrafluorophenyl structure (Fig. 3).50–52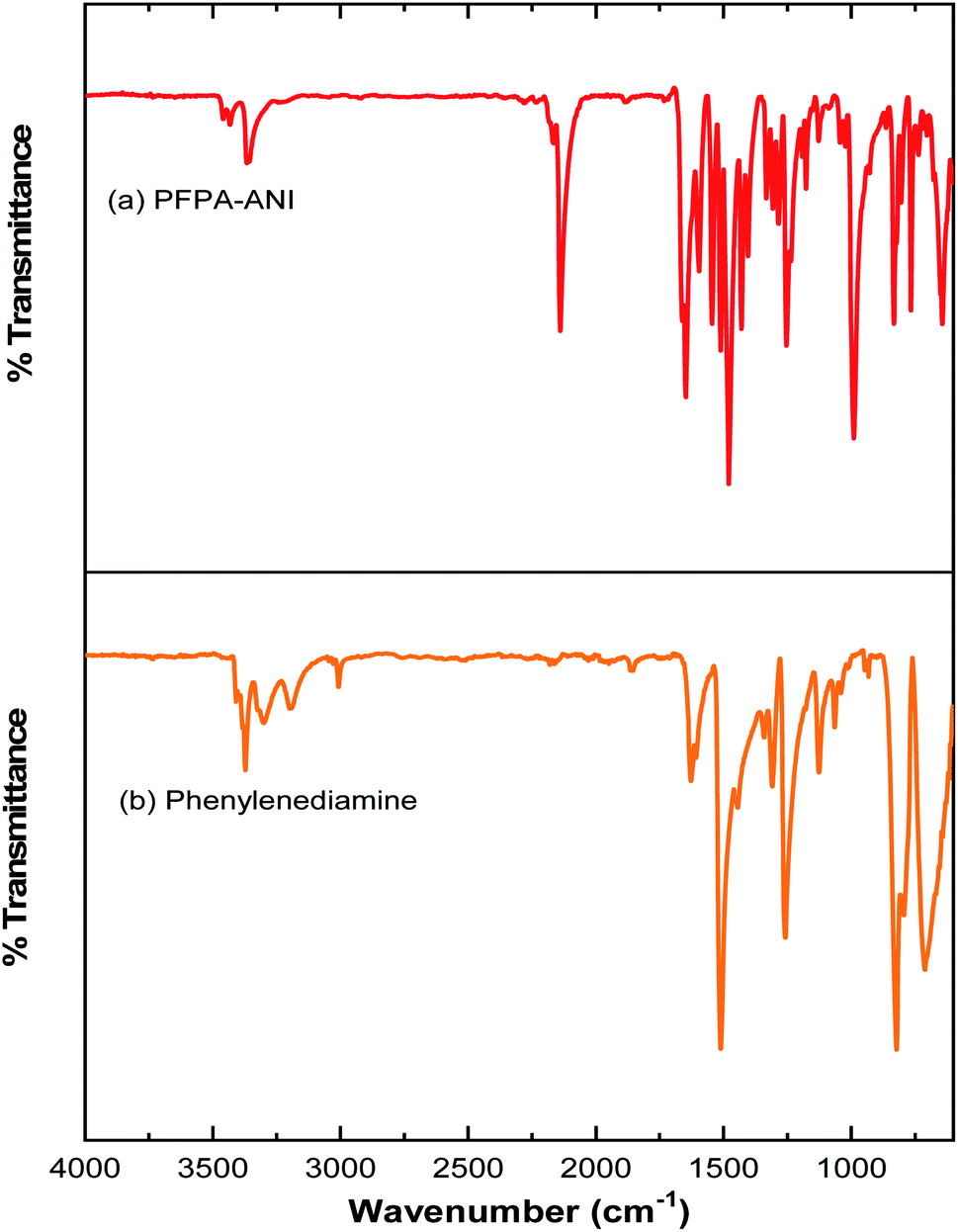 | ||
| Fig. 1 IR spectra of (a) PFPA-ANI and (b) phenylenediamine. | ||
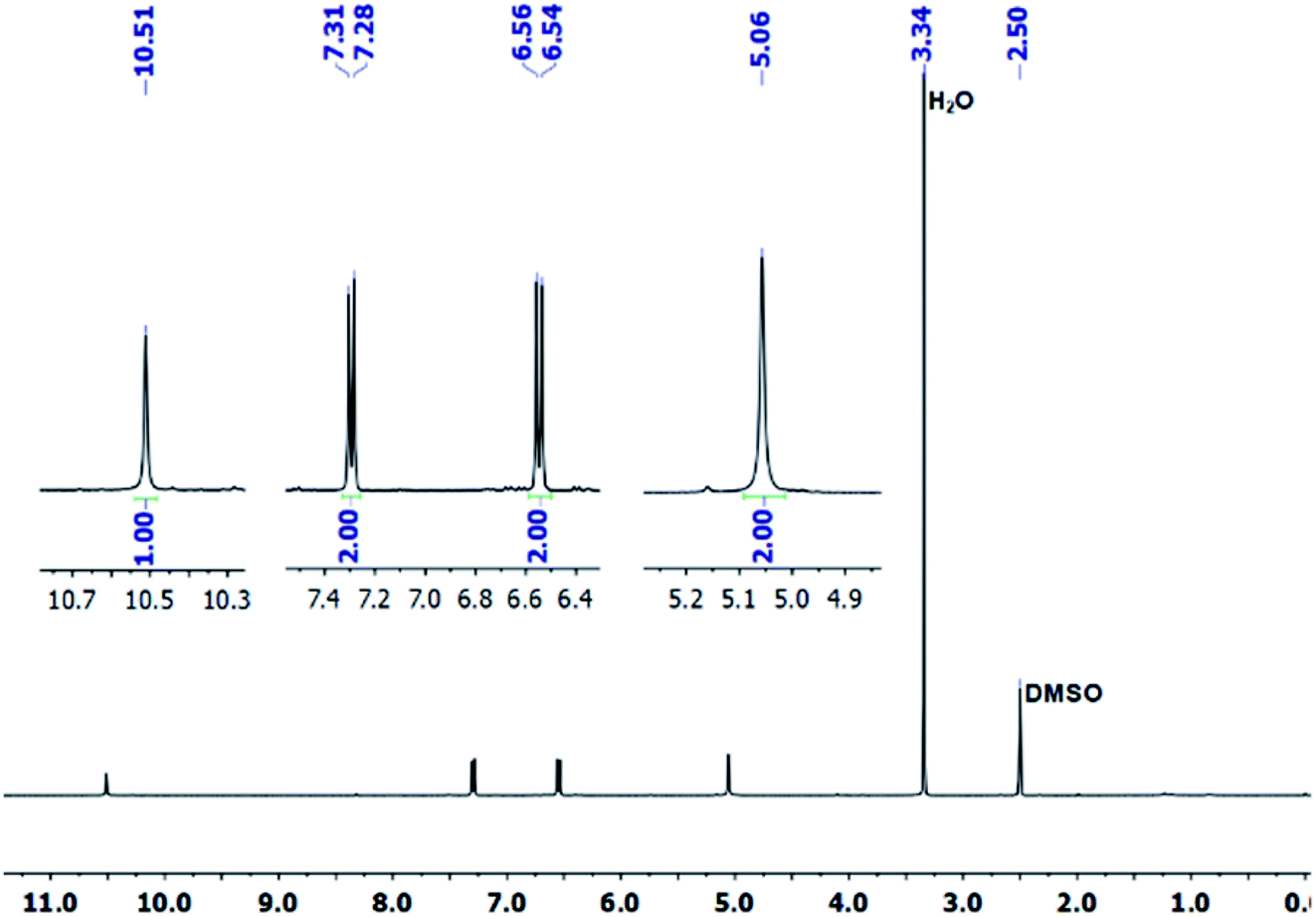 | ||
| Fig. 2 1H NMR spectrum of PFPA-ANI in DMSO-d6. | ||
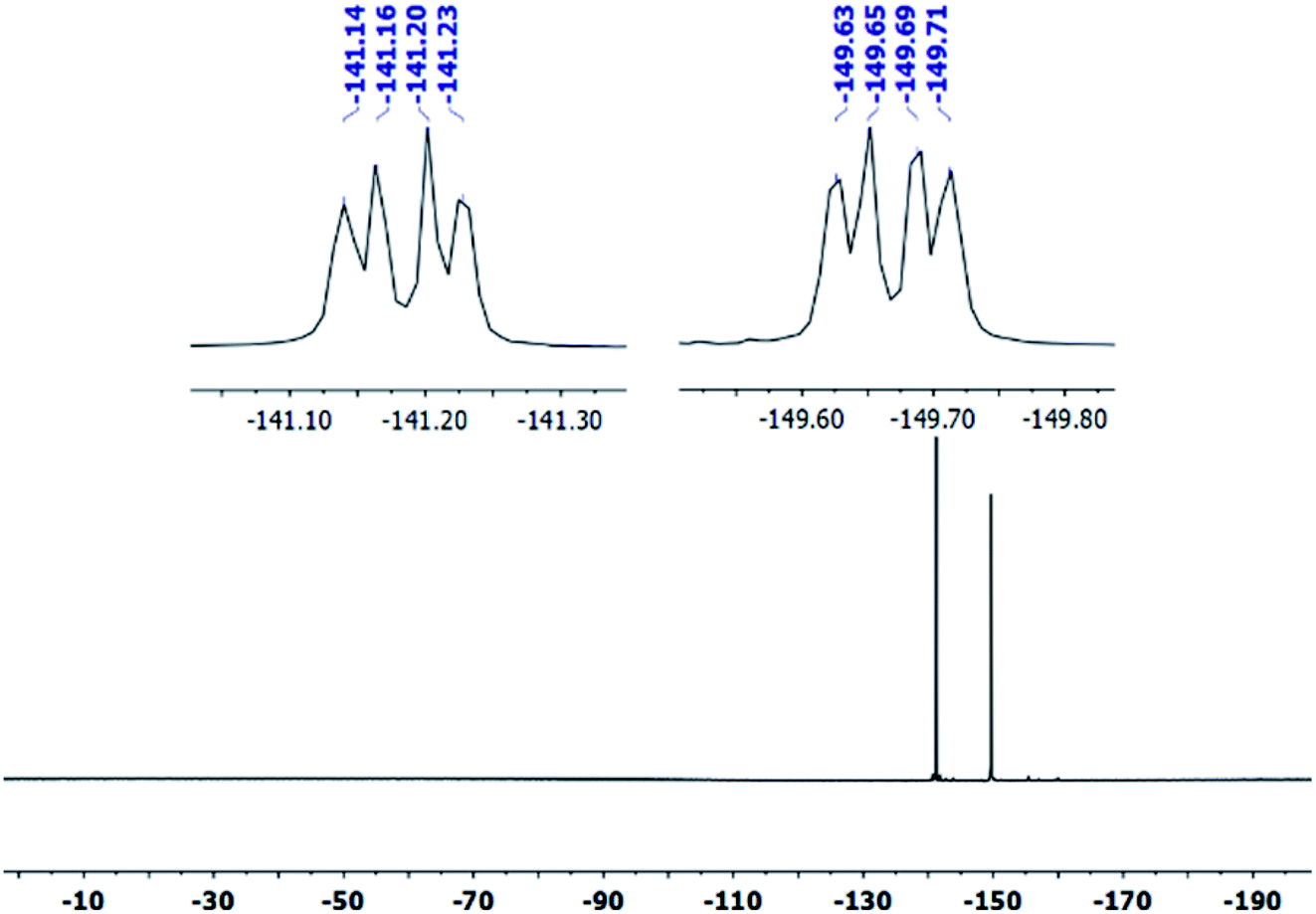 | ||
| Fig. 3 19F NMR spectrum of PFPA-ANI in DMSO-d6. | ||
To facilitate material characterization, liquid-exfoliated few-layer graphene (FLG) flakes were used as the model system to demonstrate the feasibility of the conjugation chemistry. FLG flakes were prepared by subjecting graphite powders to probe sonication and collecting the supernatant.53 The concentration of the FLG suspension was determined by measuring the absorbance at 660 nm.47 FLG was then functionalized with PFPA-ANI by subjecting the reaction mixture to microwave radiation at 150 °C for 1 h to give G–ANI (Scheme 1).43 Under TEM or SEM, no obvious changes in morphology were observed after the FLG flakes were functionalized with PFPA-ANI (Fig. 4b vs. 4a and 5a). Graft polymerization was accomplished from G–ANI in the presence of aniline, APS and HCl, through APS-mediated polymerization that is frequently used to prepare PANI-based composites on materials like graphene oxide.10 After grafting PANI onto G–ANI, densely distributed and vertically grafted needles can be seen on the FLG surface (Fig. 4c and 5b). These are PANI as the polymer synthesized from aniline under the same conditions in solution without G–ANI possessed similar shape and morphology (Fig. 4d). A control experiment was carried out by subjecting FLG flakes under the same polymerization conditions except that the flakes were not functionalized with PFPA-ANI. In this case, the PANI polymer was randomly distributed on the flakes and no vertically grafted PANI was observed (Fig. 5c). The difference in the polymer morphology might be due to the different nucleation mechanism of polymerization.13,54 According to the nucleation theory, both heterogeneous and homogenous nucleation mechanisms are possible. In the control experiment where aniline was polymerized in the presence of un-functionalized FLG flakes, homogenous nucleation in solution and around the FLG surface can be anticipated. The agglomerated PANI observed on the FLG flakes could result from either the deposition of solution-polymerized polymer or polymer grown from the aniline monomer physisorbed on FLG flakes. In the case of aniline polymerized in the presence of aniline-functionalized FLG, G–ANI, the covalently attached aniline on the FLG surface serves as nucleation sites for polymer growth, in which case, aniline polymerization is anticipated to undergo heterogeneous nucleation.13 It has been reported that heterogeneous nucleation on the graphene surface dominates when the aniline concentration is lower than 0.05 M.49 In this case, the polymer would grow directly from the FLG surface, resulting in covalently grafted PANI on FLG.
 | ||
| Fig. 4 TEM images of (a) FLG flake, (b) aniline-functionalized FLG, G–ANI, (c) FLG grafted with PANI prepared by polymerization, G–PANI, and (d) PANI prepared under the same conditions in solution without the addition of G–ANI. | ||
 | ||
| Fig. 5 SEM images of (a) G–ANI, (b) G–PANI, prepared by polymerization of ANI in the presence of G–ANI, and (c) control sample prepared by polymerization of ANI in the presence of un-functionalized FLG. | ||
G–ANI and G–PANI were characterized by X-ray photoelectron spectroscopy (XPS). Both N and F peaks were observed in the XPS survey scan of G–ANI (Fig. 6a), supporting the successful functionalization of the FLG flakes with PFPA-ANI. After grafting of PANI, both N and F peaks were again observed in the survey spectrum of G–PANI but the relative intensity of N 1s increased whereas that of the F 1s decreased. The intensity ratio of N:F changed from 1:1 in G–ANI (Fig. 6a) to 4.2:1 in G–PANI (Fig. 6b). This is anticipated as grafting PANI would drastically decrease the surface concentration of F whereas the concentration of N would increase after PANI was grafted (Scheme 1).
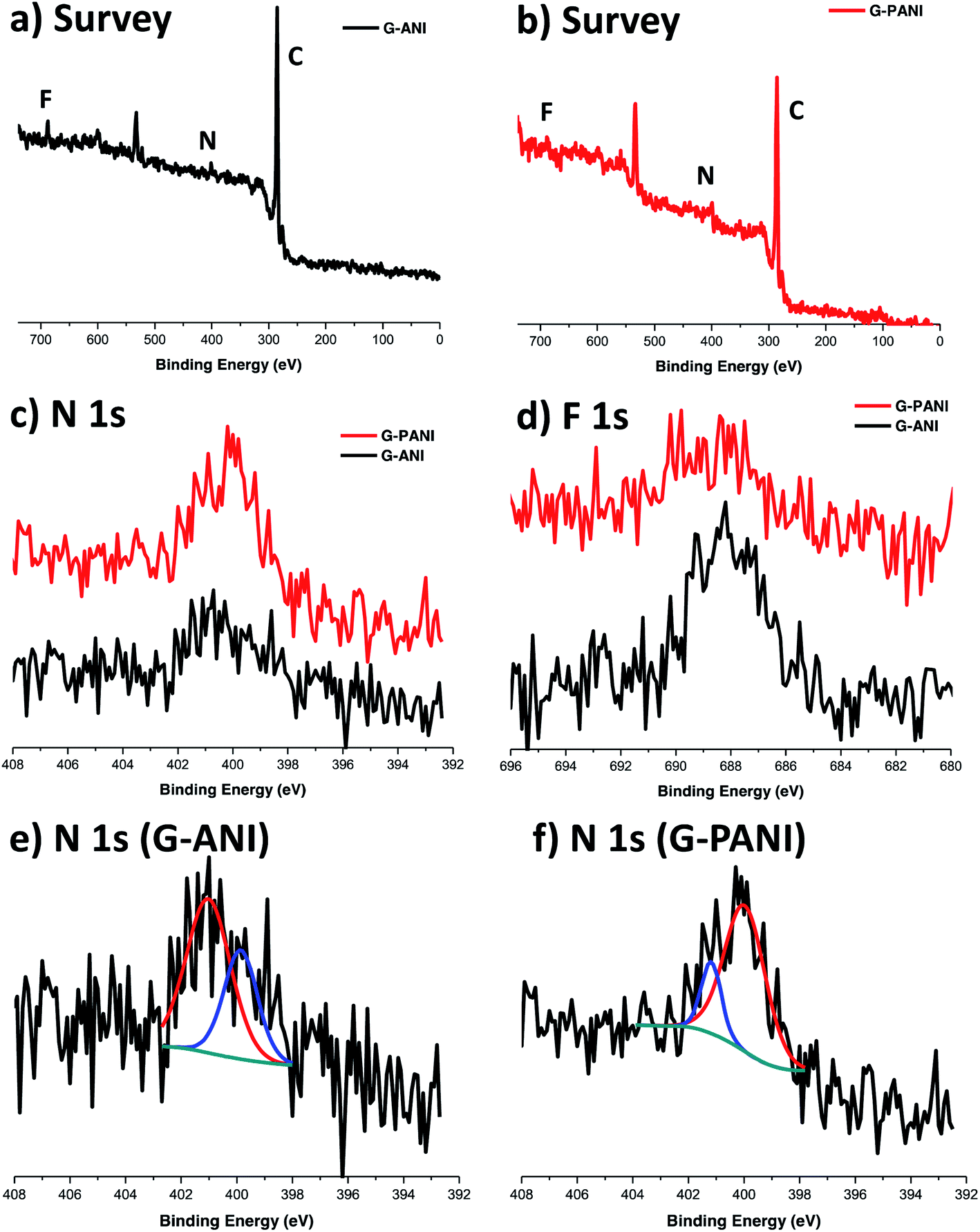 | ||
| Fig. 6 XPS survey spectra of (a) G–ANI and (b) G–PANI. High-resolution (c) N 1s and (d) F 1s spectra of G–ANI and G–PANI. High resolution N 1s XPS spectra of (e) G–ANI and (f) G–PANI. All peaks were referenced to the binding energy of adventitious carbon at 285.0 eV (C 1s). | ||
In the deconvoluted high-resolution N 1s spectrum of G–ANI (Fig. 6e), the peak at ∼401 eV is characteristic of the N in the PFPA-functionalized graphene.40,55,56 The peak at ∼400 eV can be attributed to the aniline N57 and the amide N in PFPA-ANI.55 Polymerization of ANI under the acidic oxidative condition gave the green emeraldine salt of PANI, which consists of un-protonated and protonated aniline structures with the N 1s binding energies at 399.3 eV and 401 eV, respectively.58 Furthermore, the N 1s peak at 399.3 eV dominates in the XPS spectrum of the emeraldine salt regardless of the degree of protonation.58 In our case, after grafting PANI, there should be a net increase in the aniline N concentration and a decrease in the effective concentration of the amide N in PFPA. Indeed, in the high-resolution N 1s spectrum of G–PANI (Fig. 6f), the intensity of N 1s peak at ∼400 eV increased dramatically relative to the peak at ∼401 eV.59 Taken together, these XPS data support the successful grafting of PANI on aniline-functionalized FLG flakes.
The XRD patterns of PANI and G–PANI composite are shown in Fig. 7. For PANI obtained by oxidative polymerization under the same conditions, three peaks at 2θ = 15.3°, 20.7° and 25.2°, corresponding to the (011), (020) and (200) crystal planes of PANI in its emeraldine salt form, were observed.60 The G–PANI composite contained the characteristic peaks of pure PANI. The intense diffraction peak at 2θ = 26.5° corresponds to the (002) hexagonal graphitic carbon which is characteristic to graphene (JCPDS card no. 75-1621). No additional crystalline order was observed in G–PANI composite, supporting the successful formation of PANI on the graphene surface.
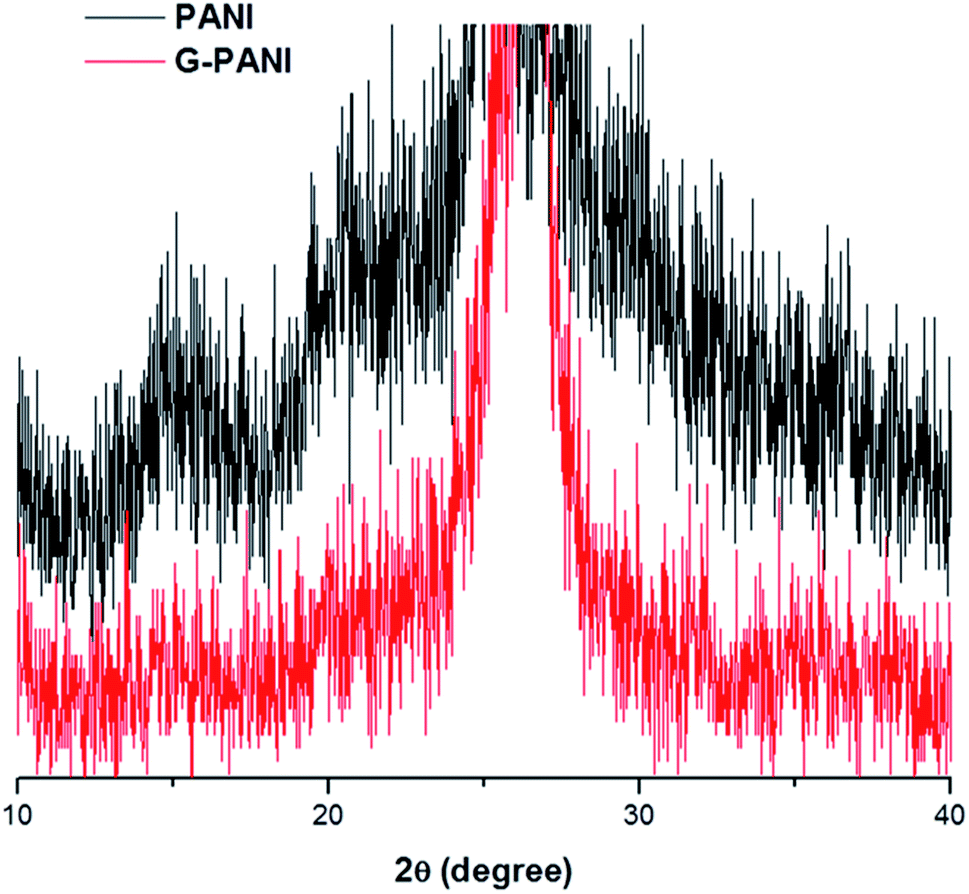 | ||
| Fig. 7 XRD spectra of PANI (top, black) and G–PANI (bottom, red). | ||
The thermal property of G–PANI was characterized by thermal gravimetric analysis (TGA). Fig. 8 show the TGA weight loss curves recorded from room temperature to 800 °C, together with the corresponding differential thermogravimetric analysis (DTGA) curves which identify the onset, maximal and end temperatures of each decomposition event. The FLG flakes had negligible weight loss, ∼0.70% at 800 °C. The TGA and DTGA curves of PANI agreed well with that reported in the literature.61 The initial weight loss before 258 °C is likely due to the loss of moisture and dopant. The major event of PANI decomposition started at above 258 °C,13,62 which was also observed in G–PANI composite, supporting the successful conjugation of PANI to graphene. The additional DTGA peaks observed in G–PANI could be from PFPA-ANI and/or lower molecular weight PANI physisorbed on the FLG surface.
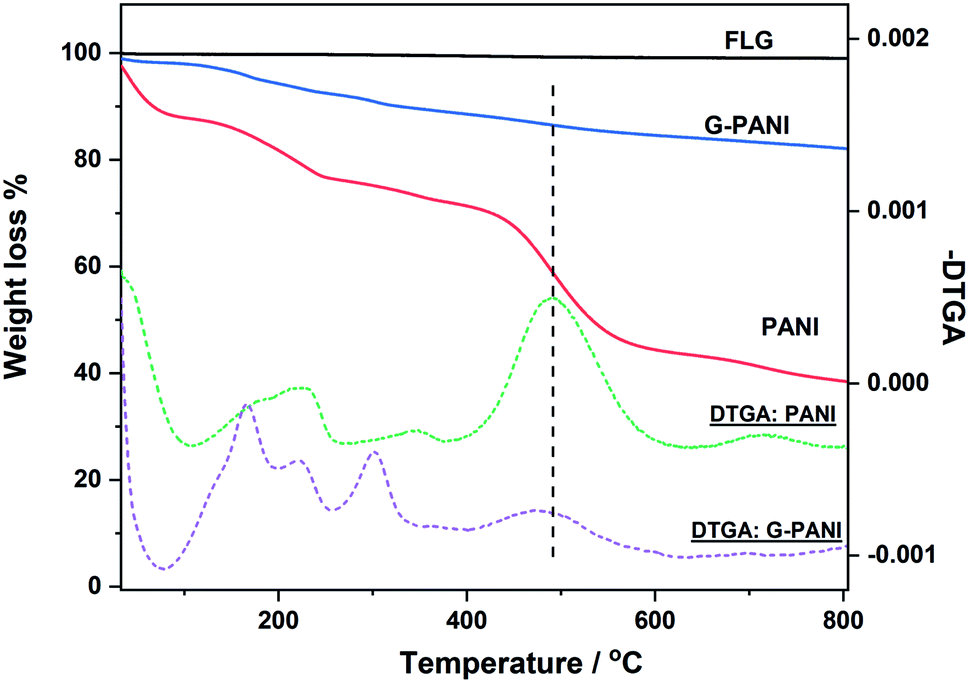 | ||
| Fig. 8 TGA weight loss (solid lines) and differential thermogravimetric (DTGA) curves (dash lines) of FLG flakes, PANI, and G–PANI. Samples were heated at the rate of 10 °C min−1 under N2. | ||
The grafting density of PANI on G–PANI, i.e., the percent of PANI grafted on FLG x, can be estimated from the TGA data in Fig. 8 following eqn (1),
| x × wPANI + (1 − x) × wG = wG–PANI | (1) |
To further confirm the presence of PANI on the FLG surface, the G–PANI electrode was characterized using cyclic voltammetry in a three-electrode cell. Fig. 9 shows the cyclic voltammograms of FLG and G–PANI electrodes at 25 mV s−1. The optimized potential window was from 0.5 to −0.2 V vs. Ag/AgCl, which was determined such that the solution electrochemistry was eliminated at the two extreme ends of each CV. Compared to the FLG electrode, the capacitance of the G–PANI electrode increased from 1.4 F g−1 to 12.3 F g−1. The enhance of specific capacitance in the sample is due to the contribution of both electrical double layer capacitance and pseudocapacitance because PANI can be reduced or oxidized within the potential window under our experimental conditions. The CV results indicate that PANI was successfully grafted onto the FLG surface and remained electrochemical active.
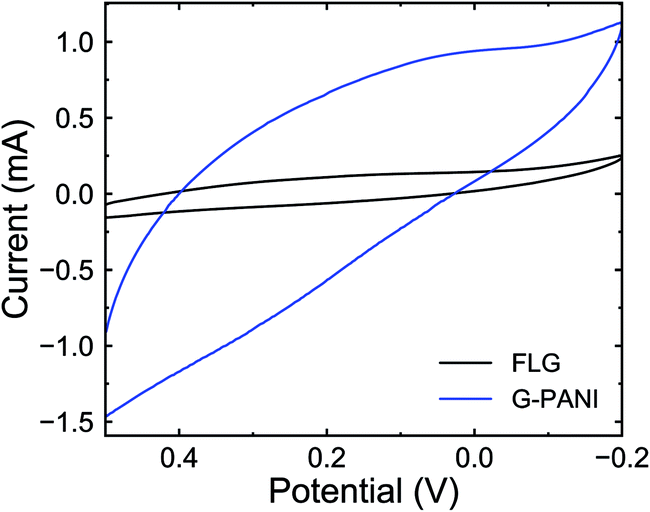 | ||
| Fig. 9 Cyclic voltammograms of FLG (black) and G–PANI (blue) in 0.1 M sulfuric acid at 25 mV s−1 scan rate. | ||
Conclusions
We have successfully synthesized covalently-grafted PANI on FLG flakes using the PFPA coupling chemistry. Functionalization of FLG flakes with PFPA-ANI gave the covalently conjugated aniline that served as the heterogeneous nucleation sites for the in situ polymerization of aniline, leading to polyaniline needles covalently grafted on the FLG flakes. The resulting G–PANI composites were characterized by spectroscopy, microscopy, XRD and electrochemistry, confirming the successful grafting of PANI on FLG. The grafting density of PANI was estimated to be ∼26% from TGA analysis. As the PFPA functionalization chemistry is applicable to other carbon nanomaterials such as carbon nanotubes and fullerene,63,64 the method developed here can be readily adapted to grow polyaniline or other conducting polymers on these technologically-important carbon materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE-1112436 and CHE-1808671 to M. Y.) and an internal seed grant from the University of Massachusetts Lowell (to N. O. and M. Y.). J. P. acknowledges the partial support from the National Research Foundation of Korea (NRF-2019R1G1A1099688).References
- H. Letheby, J. Chem. Soc., 1862, 15, 161–163 RSC.
- E. M. Geniès, A. Boyle, M. Lapkowski and C. Tsintavis, Synth. Met., 1990, 36, 139–182 CrossRef.
- A. M. Grancaric, I. Jerkovic, V. Koncar, C. Cochrane, F. M. Kelly, D. Soulat and X. Legrand, J. Ind. Text., 2018, 48, 612–642 CrossRef CAS.
- B. Guo and P. X. Ma, Biomacromolecules, 2018, 19, 1764–1782 CrossRef CAS PubMed.
- P. Liu, J. Yan, Z. Guang, Y. Huang, X. Li and W. Huang, J. Power Sources, 2019, 424, 108–130 CrossRef CAS.
- B. Wang and A. Facchetti, Adv. Mater., 2019, 31, e1901408 CrossRef PubMed.
- E. N. Zare, P. Makvandi, B. Ashtari, F. Rossi, A. Motahari and G. Perale, J. Med. Chem., 2020, 63, 1–22 CrossRef CAS PubMed.
- C. O. Baker, X. Huang, W. Nelson and R. B. Kaner, Chem. Soc. Rev., 2017, 46, 1510–1525 RSC.
- R. Gangopadhyay and A. De, Chem. Mater., 2000, 12, 608–622 CrossRef CAS.
- L. Wang, X. Lu, S. Lei and Y. Song, J. Mater. Chem. A, 2014, 2, 4491–4509 RSC.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- X. Wang, D. Wu, X. Song, W. Du, X. Zhao and D. Zhang, Molecules, 2019, 24, 2263 CrossRef CAS PubMed.
- Z.-F. Li, H. Zhang, Q. Liu, Y. Liu, L. Stanciu and J. Xie, Carbon, 2014, 71, 257–267 CrossRef CAS.
- L. Lai, H. Yang, L. Wang, B. K. Teh, J. Zhong, H. Chou, L. Chen, W. Chen, Z. Shen, R. S. Ruoff and J. Lin, ACS Nano, 2012, 6, 5941–5951 CrossRef CAS.
- L. Wang, Y. Ye, X. Lu, Z. Wen, Z. Li, H. Hou and Y. Song, Sci. Rep., 2013, 3, 1–9 Search PubMed.
- Z. Gao, F. Wang, J. Chang, D. Wu, X. Wang, X. Wang, F. Xu, S. Gao and K. Jiang, Electrochim. Acta, 2014, 133, 325–334 CrossRef CAS.
- J. Diao, J. Yuan, A. Ding, J. Zheng and Z. Lu, Macromol. Mater. Eng., 2018, 303, 1800092 CrossRef.
- H. Qiu, X. Han, F. Qiu and J. Yang, Appl. Surf. Sci., 2016, 376, 261–268 CrossRef CAS.
- S. Xiong, Y. Shi, J. Chu, M. Gong, B. Wu and X. Wang, Electrochim. Acta, 2014, 127, 139–145 CrossRef CAS.
- Y. Wang, X. Gao, Y. Fu, X. Wu, Q. Wang, W. Zhang and C. Luo, Composites, Part B, 2019, 169, 221–228 CrossRef CAS.
- M. Jamdegni and A. Kaur, J. Electrochem. Soc., 2019, 166, H502–H509 CrossRef CAS.
- S. Kang, S. Qiao, Z. Hu, J. Yu, Y. Wang and J. Zhu, J. Mater. Sci., 2019, 54, 6410–6424 CrossRef CAS.
- J. Yan, Y. Huang, C. Wei, N. Zhang and P. Liu, Composites, Part A, 2017, 99, 121–128 CrossRef CAS.
- V. H. Nguyen, C. Lamiel, D. Kharismadewi, V. C. Tran and J.-J. Shim, J. Electroanal. Chem., 2015, 758, 148–155 CrossRef CAS.
- L. Wang, Y. Huang, C. Li, J. Chen and X. Sun, Compos. Sci. Technol., 2015, 108, 1–8 CrossRef CAS.
- Z. Wang, J.-J. Han, N. Zhang, D.-D. Sun and T. Han, J. Solid State Electrochem., 2019, 23, 3373–3382 CrossRef CAS.
- T. Yu, P. Zhu, Y. Xiong, H. Chen, S. Kang, H. Luo and S. Guan, Electrochim. Acta, 2016, 222, 12–19 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. K. James and J. M. Tour, Acc. Chem. Res., 2013, 46, 2307–2318 CrossRef CAS PubMed.
- C. Gómez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499–3503 CrossRef PubMed.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- A. M. Dimiev, L. B. Alemany and J. M. Tour, ACS Nano, 2013, 7, 576–588 CrossRef CAS PubMed.
- L.-H. Liu and M. Yan, Nano Lett., 2009, 9, 3375–3378 CrossRef CAS PubMed.
- L.-H. Liu, M. M. Lerner and M. Yan, Nano Lett., 2010, 10, 3754–3756 CrossRef CAS.
- L.-H. Liu and M. Yan, Acc. Chem. Res., 2010, 43, 1434–1443 CrossRef CAS.
- L.-H. Liu, G. Nandamuri, R. Solanki and M. Yan, J. Nanosci. Nanotechnol., 2011, 11, 1288–1292 CrossRef CAS PubMed.
- L.-H. Liu and M. Yan, J. Mater. Chem., 2011, 21, 3273–3276 RSC.
- L.-H. Liu, G. Zorn, D. G. Castner, R. Solanki, M. M. Lerner and M. Yan, J. Mater. Chem., 2010, 20, 5041–5046 RSC.
- J. Park, H. S. Jayawardena, X. Chen, K. W. Jayawardana, M. Sundhoro, E. Ada and M. Yan, Chem. Commun., 2015, 51, 2882–2885 RSC.
- J. Park and M. Yan, Acc. Chem. Res., 2013, 46, 181–189 CrossRef.
- J. Park, T. Jin, C. Liu, G. Li and M. Yan, ACS Omega, 2016, 1, 351–356 CrossRef CAS PubMed.
- N. Kong, J. Park, X. Yang, O. Ramstrom and M. Yan, ACS Appl. Bio Mater., 2019, 2, 284–291 CrossRef CAS.
- H. S. N. Jayawardena, K. W. Jayawardana, X. Chen and M. Yan, Chem. Commun., 2013, 49, 3034–3036 RSC.
- Q. Tong, X. Wang, H. Wang, T. Kubo and M. Yan, Anal. Chem., 2012, 84, 3049–3052 CrossRef CAS.
- L. Deng, O. Norberg, S. Uppalapati, M. Yan and O. Ramstroem, Org. Biomol. Chem., 2011, 9, 3188–3198 RSC.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- N. R. Chiou and A. J. Epstein, Adv. Mater., 2005, 17, 1679–1683 CrossRef CAS.
- J. Xu, K. Wang, S.-Z. Zu, B.-H. Han and Z. Wei, ACS Nano, 2010, 4, 5019–5026 CrossRef CAS PubMed.
- S. Xie, O. Ramstroem and M. Yan, Org. Lett., 2015, 17, 636–639 CrossRef CAS PubMed.
- S. Xie, S. Manuguri, G. Proietti, J. Romson, Y. Fu, A. K. Inge, B. Wu, Y. Zhang, D. Haell, O. Ramstroem and M. Yan, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8464–8469 CrossRef CAS PubMed.
- S. Xie, J. Zhou, X. Chen, N. Kong, Y. Fan, Y. Zhang, G. Hammer, D. G. Castner, O. Ramstroem and M. Yan, Mater. Chem. Front., 2019, 3, 251–256 RSC.
- A. Ciesielski and P. Samori, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- N.-R. Chiou, C. Lu, J. Guan, L. J. Lee and A. J. Epstein, Nat. Nanotechnol., 2007, 2, 354–357 CrossRef CAS PubMed.
- G. Zorn, L.-H. Liu, L. Árnadóttir, H. Wang, L. J. Gamble, D. G. Castner and M. Yan, J. Phys. Chem. C, 2013, 118, 376–383 CrossRef PubMed.
- G. Zorn, D. G. Castner, A. Tyagi, X. Wang, H. Wang and M. Yan, J. Vac. Sci. Technol., A, 2015, 33, 021407 CrossRef PubMed.
- L. Lai, G. Huang, X. Wang and J. Weng, Carbon, 2010, 48, 3145–3156 CrossRef CAS.
- K. L. Tan, B. T. Tan, E. T. Kang and K. G. Neoh, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 8070–8073 CrossRef CAS PubMed.
- N. Kohut-Svelko, S. Reynaud, R. Dedryvère, H. Martinez, D. Gonbeau and J. François, Langmuir, 2005, 21, 1575–1583 CrossRef CAS.
- H. K. Chaudhari and D. S. Kelkar, Polym. Int., 1997, 42, 380–384 CrossRef CAS.
- T. L. A. Campos, D. F. Kersting and C. A. Ferreira, Surf. Coat. Technol., 1999, 122, 3–5 CrossRef CAS.
- N. Chandrakanthi and M. A. Careem, Polym. Bull., 2000, 44, 101–108 CrossRef CAS.
- N. Kong, M. R. Shimpi, J. Park, O. Ramstroem and M. Yan, Carbohydr. Res., 2015, 405, 33–38 CrossRef CAS.
- M. Yan, S. X. Cai and J. F. W. Keana, J. Org. Chem., 1994, 59, 5951–5954 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |