
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, characterization and evaluation of β-hairpin peptide hydrogels as a support for osteoblast cell growth and bovine lactoferrin delivery†
Luis M. De Leon-Rodriguez‡
 a,
Young-Eun Park‡b,
Dorit Naotb,
David S. Mussonb,
Jillian Cornish*b and
Margaret A. Brimble*ac
a,
Young-Eun Park‡b,
Dorit Naotb,
David S. Mussonb,
Jillian Cornish*b and
Margaret A. Brimble*ac
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds Street, Auckland 1010, New Zealand. E-mail: m.brimble@auckland.ac.nz
bDepartment of Medicine, University of Auckland, Auckland 1023, New Zealand. E-mail: j.cornish@auckland.ac.nz
cMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand
First published on 13th May 2020
Abstract
The use of peptide hydrogels is of growing interest in bone regeneration. Self-assembling peptides form hydrogels and can be used as injectable drug delivery matrices. Injected into the defect site, they can gel in situ, and release factors that aid bone growth. We report on the design, synthesis and characterization of three β-hairpin peptide hydrogels, and on their osteoblast cytocompatibility as well as delivery of the lactoferrin glycoprotein, a bone anabolic factor. Osteoblasts cultured in hydrogels of the peptide with sequence NH2-Leu-His-Leu-His-Leu-Lys-Leu-Lys-Val-DPro-Pro-Thr-Lys-Leu-Lys-Leu-His-Leu-His-Leu-Arg-Gly-Asp-Ser-CONH2 (H4LMAX-RGDS) increased the osteoblast cell number and the cells appeared healthy after seven days. Furthermore, we showed that H4LMAX-RGDS was capable of releasing up to 60% of lactoferrin (pre-encapsulated in the gel) over five days while retaining the rest of the glycoprotein. Thus, H4LMAX-RGDS hydrogels are cytocompatible with primary osteoblasts and capable of delivering bio-active lactoferrin that increases osteoblast cell number.
Introduction
Bone defects that fail to heal are a critical clinical problem, especially in older people. With an increasingly aged population, the development of novel methods for the enhancement of bone regeneration have become a main priority. The gold standard material used to fill a bone defect and enhance bone regeneration is autologous bone graft. However, this has issues such as tissue morbidity and limited tissue availability.1 Therefore, there is a need for novel biomaterials that can replace autologous bone grafts.2 In this respect, soft biomaterial hydrogels hold much promise as they can fill any given cavity and thus establish tight contacts with neighbouring tissue.Hydrogels are hydrophilic cross-linked porous polymeric networks which entrap large amounts of water. Among the different hydrogel-based scaffolds (e.g. polysaccharides, proteins, organic polymers),3 synthetic peptide hydrogels (PHGs) are particularly attractive for biomedical applications because of their purported biocompatibility, biodegradability, batch-to-batch manufacturing reproducibility, varied functionality, injectability, and their nanofibrillar structure, which mimics that of the extracellular matrix. However, a downside of PHGs is their low stiffness.4 Therefore, PHGs cannot be used for the repair of load-bearing lesions, when used alone, but as bone defect filling materials, acting as carriers of cells and/or bio-factors to directly promote tissue regeneration and/or to stimulate tissue responses to enhance damaged tissue repair.1
There are a number of studies demonstrating the potential application of single component PHGs for bone regeneration. The 16-amino acid peptide Ac-(Arg-Ala-Asp-Ala)4-CONH2 (RADA16), which contains the repeating unit Arg-Ala-Asp (RAD) similar to the ubiquitous integrin binding sequence Arg-Gly-Asp (RGD), was injected in the calvaria bone defect of mice. RADA16 induced functional bone regeneration more effectively than Matrigel™, a natural biomaterial containing extracellular proteins (e.g. laminin, collagen, entactin, heparan sulfate) and growth factors.5 Another study reported that hydrogels of a RADA16 peptide derivative containing all D-amino acids attached to RGD via a three residue Gly linker unit, Ac-(DArg-DAla-DAsp-DAla)4-Gly-Gly-Gly-Arg-Gly-Asp-Ser-CONH2 (D-RADA16-RGD), promoted bone regeneration when applied to condyle defects of female rats.6 Interestingly, there was no significant difference when comparing the treatment with hydrogels of D-RADA16-RGD alone and the hydrogel containing basic fibroblast growth factor (an important factor which influences migration, proliferation, and differentiation of various cells, including osteoblasts). The latter result suggests that the retention/delivery of the growth factor by the hydrogel is inappropriate, which is attributed to the poor structural stability of RADA16 peptides under neutral conditions. Additionally, RADA16 hydrogels tend to break under mechanical stress. These two disadvantages complicate RADA16 hydrogel preparation and application.7
Nagai et al. reported a modified version of RADA16, Ac-Arg-Leu-Asp-Leu-Arg-Leu-Ala-Leu-Arg-Leu-Asp-Leu-Arg-CONH2 (SPG-178). This 13-residue peptide comprised: (1) four cationic Arg and two anionic Asp to improve the solubility of the peptide under neutral conditions, and (2) Leu residues instead of Ala to increase the hydrophobic interactions among peptide chains, and thus improve the mechanical stability of the hydrogels.7 SPG-178 promoted bone formation when injected as a mixture with cell culture media in rat calvarial bone defects.8 Yet, SPG-178 hydrogel formation is slow and to the best of our knowledge the hydrogel's mechanical stability has not been assessed.
In another work, Castillo-Diaz, et al. demonstrated that the octapeptide NH2-Phe-Glu-Phe-Glu-Phe-Lys-Phe-Lys-COOH promoted not only human osteoblast proliferation but also calcium phosphate deposition over time, thus establishing that the hydrogel was a suitable 3D-support for osteoblast function.9 However, the octapeptide hydrogel concentration required to keep the cells suspended was high (3 wt% versus 0.4 wt% used for SPG-178, when injected alone in bone defects), hydrogel formation was slow and its preparation complex, thus limiting its applicability in a clinical setting.
The previous overview, on the utilization of single component PHGs in bone growth, demonstrates the need for developing novel materials with improved properties e.g. easy to handle and with enhanced growth factor retention/delivery properties. In this work, we designed, synthesized and characterized three β-hairpin peptide hydrogels and evaluated their cytocompatibility and their ability to deliver lactoferrin (LF), a bone anabolic factor. LF is a large ∼80 kDa iron-binding glycoprotein which is present in milk and exocrine secretions in mammals. LF is known to have anti-microbial and immunomodulatory functions.10
Previously, bovine LF was found to increase osteoblast proliferation and differentiation, protect osteoblasts from apoptosis, and inhibit osteoclastogenesis.11 In vivo, systemic administered LF enhanced bone formation in calvaria,11,12 and local delivery of LF in hydrogels improved bone regeneration in mouse and rat calvarial defect model.13,14 Thus, with LF's proven anabolic effect on bone, we proceeded further to find a delivery system for LF suitable for use in the clinic.
The aims of this study were to design and synthesize novel peptide hydrogel scaffolds, to carry out a comprehensive in vitro analysis to evaluate the cytocompatibility of these biomaterials, and to determine whether they have the potential to be used as drug delivery systems when injected with the proven anabolic factor LF for bone regeneration.
Results and discussion
Peptide design and synthesis
MAX1 is a well-known 20-residues peptide (NH2-Val-Lys-Val-Lys-Val-Lys-Val-Lys-Val-DPro-Pro-Thr-Lys-Val-Lys-Val-Lys-Val-Lys-Val-CONH2) that adopts a β-hairpin conformation at pH > 9.0 and forms shear thinning hydrogels, a property that makes gel delivery suitable by injection.15 Furthermore, MAX1 is not cytotoxic but presents antibacterial properties attributed to the peptide's cationic nature.16 However, a drawback of MAX1 is its slow kinetics of hydrogelation at neutral pH caused by the peptide high positive charge (+9, given the presence of eight lysines and an amino terminal group), which induces strong intra- and inter-molecular electrostatic repulsions. While hydrogels of MAX1 can be obtained at neutral pH in the presence of salts or at elevated temperature, a peptide capable of forming hydrogels at neutral pH is highly desirable, as indicated before.Inspired by the design of MAX1, we recently reported the synthesis and characterization of peptide NH2-Leu-His-Leu-His-Leu-Lys-Leu-Lys-Val-DPro-Pro-Thr-Lys-Leu-Lys-Leu-His-Leu-His-Leu-CONH2 (H4LMAX), where Val was substituted by Leu and four Lys by His (Fig. 1). The latter substitution decreased repulsions of positively charged residues (+5 overall charge) allowing H4LMAX to form hydrogels at neutral pH without requiring an additional stimulus.17 Therefore, for this work we selected hydrogels of H4LMAX as a suitable system to study osteoblast viability and proliferation.
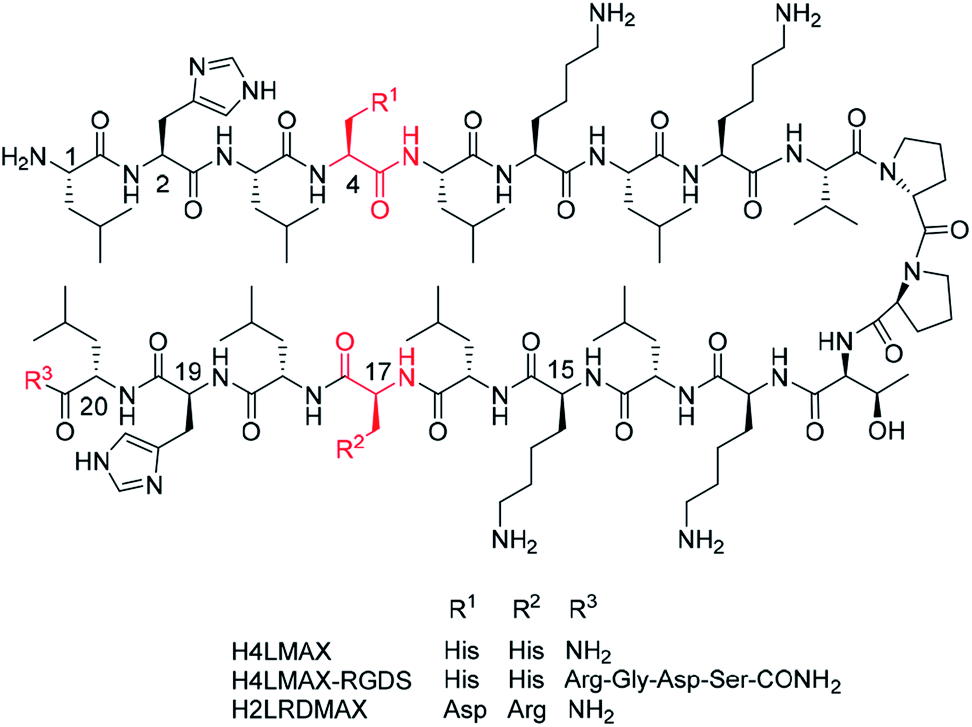 | ||
| Fig. 1 Peptide sequences studied in this work depicted in a β-hairpin conformation. | ||
Peptide H4LMAX was successfully prepared via automated Fmoc solid phase peptide synthesis protocols as reported in the literature17 (Fig. S1†). However, when attempting to culture mouse osteoblasts on H4LMAX hydrogels pre-conditioned with cell culture media, the cells did not attach to the hydrogel but to the plastic cell culture surfaces (not shown). Interestingly, the cells remained viable, suggesting that although the hydrogel did not support cell attachment and growth, it was not cytotoxic.
Derived from the previous result, we deemed necessary to add the RGD subunit to H4LMAX in order to improve the interaction between osteoblasts and the hydrogel. Therefore, two peptides were designed. In one peptide the Arg-Gly-Asp-Ser (RGDS) sequence was directly added to the C-terminus of H4LMAX, resulting in peptide H4LMAX-RGDS (Fig. 1). We envisioned that this design maximized the presence of alternating hydrophobic/hydrophilic amino acids in the sequence, a characteristic of β-sheet peptide hydrogelators,4 which in turn should minimize a detrimental effect on the peptide's mechanical properties. In a second peptide, His4 in H4LMAX was substituted with Asp and His17 with Arg leading to peptide H2LRDMAX (Fig. 1). This arrangement placed Arg and Asp facing each other in opposite arms of the peptide in a β-hairpin conformation, thus resembling a pseudo RGD unit. Substitution of the two His was selected to maintain the overall charge of H2LRDMAX at physiological pH (+5) unchanged in relation to the parent peptide H4LMAX. Substitution of Lys4 with Glu has been previously documented for MAX1. This substitution allowed hydrogelation at neutral pH.18 Additionally, the exchange of Lys15 by Glu was also reported yielding the peptide labelled as MAX8, which shows improved hydrogelation properties near physiological pH. The substitution of Lys with Arg has also been documented without dramatically altering the mechanical properties of the resulting gels.18,19
The synthesis of H4LMAX-RGDS was non-problematic under conventional Fmoc solid phase peptide synthesis protocols (Fig. S2†). In contrast, the initial preparation of H2LRDMAX under similar methods, yielded the desired peptide together with two impurities, identified as the one and two amino acid (His and His–Leu) N-terminally truncated peptide analogues in an approximate 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:10 ratio. Further attempts to drive the peptide synthesis to completion (including double or triple amino acid couplings from the point of truncation and/or microwave assisted synthesis) had limited success, highlighting the strong aggregation propensity of the peptide sequence. The desired peptide was purified by RP-HPLC (Fig. S3†). However, the amount of recovered pure peptide relative to the crude material was low given that the impurities eluted close to the desired product.
:15:10 ratio. Further attempts to drive the peptide synthesis to completion (including double or triple amino acid couplings from the point of truncation and/or microwave assisted synthesis) had limited success, highlighting the strong aggregation propensity of the peptide sequence. The desired peptide was purified by RP-HPLC (Fig. S3†). However, the amount of recovered pure peptide relative to the crude material was low given that the impurities eluted close to the desired product.
Hydrogel formation and characterization
Both H4LMAX-RGDS and H2LRDMAX formed clear hydrogels at room temperature at a 1 wt% concentration (50 mM Tris buffer, pH 7.4) in the absence or presence of salts (e.g. NaCl).Rheology
Rheology was used to determine the viscoelastic characteristics of the hydrogels described herein. Two consecutive time sweep experiments at constant shear strain (γ = 0.2%) and angular frequency (ω = 6 rad s−1) were carried out at 37 °C. A large strain step (γ = 1000%) was applied in between experiments to determine the recoverability of the system. Hydrogel formation was evidenced by an elastic module (G′) significantly larger than the viscous module (G′′) (Fig. 2 and S4†). Importantly, the G′ of H4LMAX-RGDS after two hours was ∼5 times smaller than that of H4LMAX (40 and 210 Pa respectively) and hydrogel formation was slower for the former peptide indicating that the addition of the RGDS unit at the C-terminus has a negative effect in the hydrogelation properties of H4LMAX. The last result can be ascribed to the presence of a negatively charged Asp close to the N-terminus, which has been reported to destabilize β-hairpins.20 Furthermore, a similar detrimental effect on mechanical characteristics was observed when the MAX8 peptide was derivatized with the heptapeptide Met-Leu-Pro-His-His-Gly-Ala, which lacks negatively charged residues.21 H2LRDMAX showed a G′ ∼510 Pa after two hours, which is ∼2.4 times larger than that of H4LMAX. This result agrees with the increase in stiffness when substituting Lys15 by Glu in MAX1.18 It is important to note that the peptides discussed herein are soft, however, their G′ are comparable or higher than the ones for RADA16 and SPG-178 (∼29 and 48 Pa for 1 wt% hydrogels) previously discussed.22 Additionally, one can highlight that all the peptides reported in this work showed sheer-thinning properties.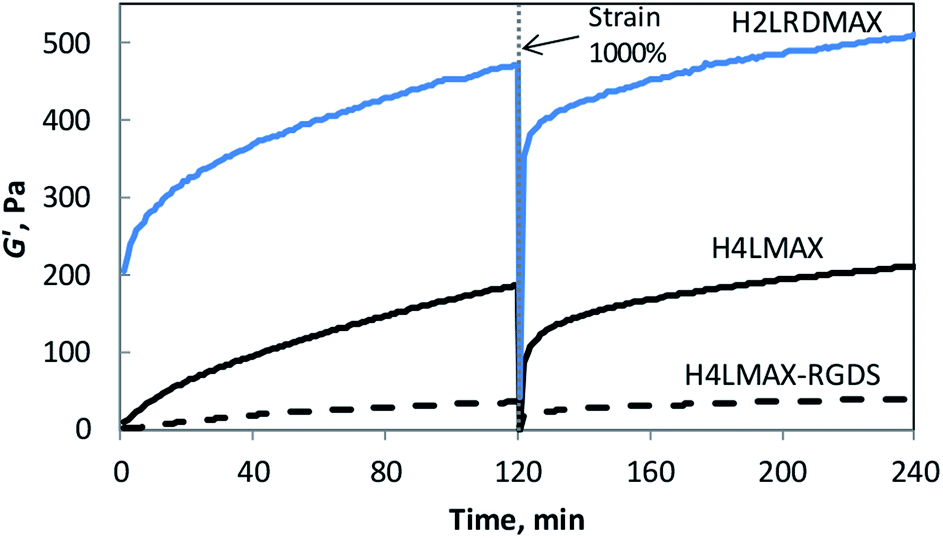 | ||
| Fig. 2 Storage modulus (G′) of 1 wt% peptide hydrogels in 50 mM Tris buffer pH 7.4 with 30 mM NaCl at 37 °C as a function of time (6 rad s−1, 0.2% strain). | ||
Angular frequency sweep experiments at constant shear strain (γ = 0.2%) showed that both G′ and G′′ are independent of the frequency, up to 10 rad s−1 for 2 (Fig. S5†). The linear viscoelastic regime of the system was stablished from shear strain amplitude sweep experiments (ω = 6 rad s−1), which showed a constant G′ and G′′ up to γ = 5% for both peptide hydrogels (Fig. S6†).
FTIR spectroscopy
The secondary structure of peptides H4LMAX-RGDS and H2LRDMAX was determined by ATR-FTIR. The IR spectra showed the typical signals attributed to β-sheet and β-turns at ∼1618 and 1673 cm−1 respectively, which is characteristic of this family of peptides (Fig. S7†). A peak at ∼1683 cm−1 attributed to an antiparallel β-sheet structure, was also observed.17 Furthermore, albeit in lower proportion, the presence of other secondary structures (e.g. random coil at ∼1650 cm−1) was also revealed in the FTIR spectra.Transmission electron microscopy
Transmission electron microscopy (TEM) images of diluted samples of the hydrogels were acquired in order to gain further structural information. The images of both H4LMAX-RGDS and H2LRDMAX showed the typical network of twisted fibrils observed in hydrogel forming peptides (Fig. 3). Furthermore, H4LMAX-RGDS and H2LRDMAX presented cross sections of 4.9 (SD = 0.7, n = 30) and 3.6 nm (SD = 0.5, n = 30) respectively. These values agree with the 3.5 nm cross section corresponding to the length of a folded β-hairpin structure of MAX1, when considering that the RGDS segment in H4LMAX-RGDS will add ∼1.4 nm (ca. 3.5 Å per residue in an extended conformation) to the fibril's width.23 | ||
| Fig. 3 Transmission electron micrographs of a sample of H4LMAX-RGDS (fibril cross section = 4.9 ± 0.7 nm (mean ± SD)) (left) and H2LRDMAX (fibril cross section = 3.6 ± 0.5 nm) (right) 50-fold water diluted from a 1 wt% hydrogel in 50 mM Tris buffer pH 7.4 with 30 mM NaCl. The black scale bars correspond to 50 nm. | ||
Cytocompatibility of H4LMAX-RGDS and H2LRDMAX hydrogels
The cytocompatibility of the hydrogels was determined by culturing primary mouse osteoblasts on H4LMAX-RGDS and H2LRDMAX. The cells attached and grew on both gels (Fig. 4A) and their viability increased during seven days of culture (Fig. 4B). There was no difference in viability between the two groups (p = 0.0978). However, the cells on H2LRDMAX looked morphologically abnormal, whereas the cells on H4LMAX-RGDS appeared more confluent and morphologically closer to native osteoblasts, although aggregate formation was seen on day 7 (Fig. 4A). Therefore, the following studies were carried out with H4LMAX-RGDS peptide. This result can be attributed to Arg and Asp residues in hydrogels of H2LRDMAX not being able to attain the appropriate conformation to properly signal the integrins cell's machinery involved in diverse cell processes. This point is further supported if one considers that the inter/intra strand distance in an assembled β-hairpin of MAX1 (4.5–4.8 Å) is shorter than distance between the alpha carbons of Arg and Asp (6.6 Å) as measured from the crystal of the αVβ1 integrin bound to an RGD peptide.24 However, considering that hydrogels of H2LRDMAX are stiffer than those of H4LMAX-RGDS and that cells remain viable in the former, warrants the need to further study analogues of the H2LRDMAX peptide.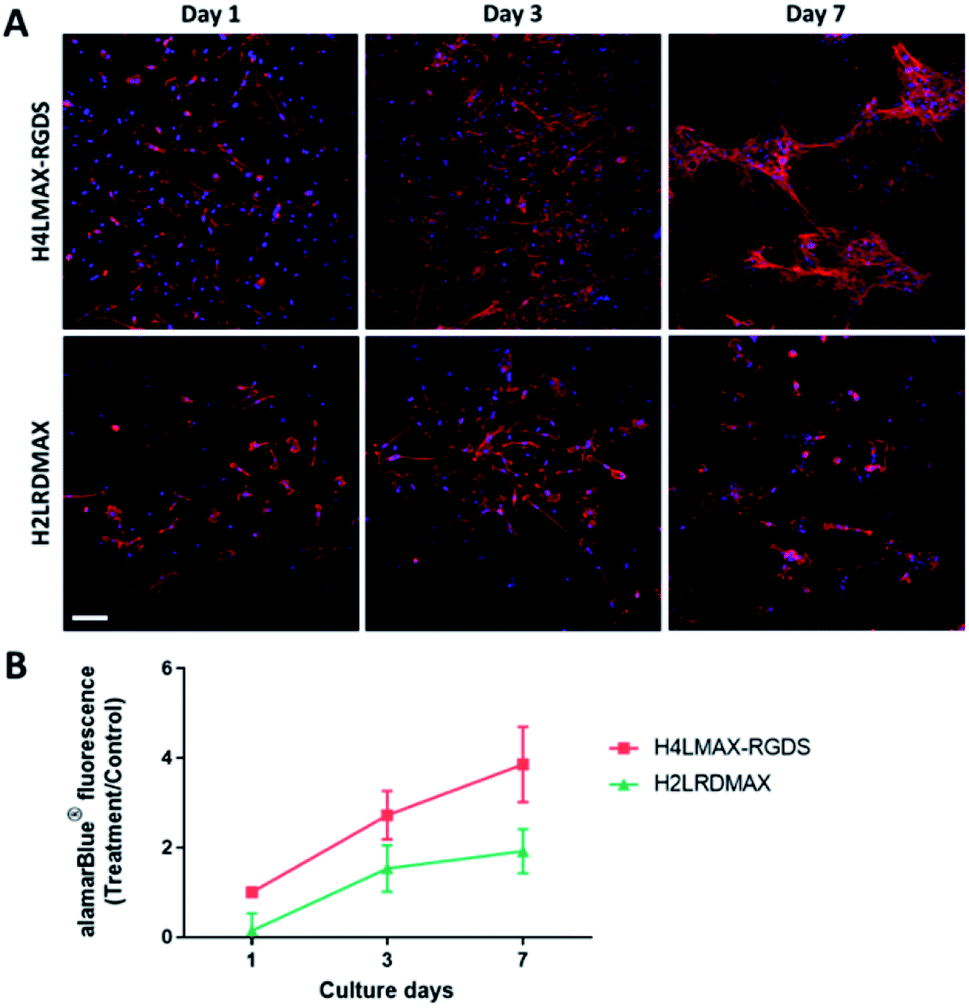 | ||
| Fig. 4 Viability of mouse primary osteoblasts on H4LMAX-RGDS compared to H2LRDMAX. (A) Representative confocal images showing F-actin (red) and nucleic acid (blue). Gels without cells were used as a negative control which did not demonstrate any staining and fluorescence (results not shown). Scale bar = 100 μm. (B) Results from three biological experiments were pooled. | ||
Cumulative release of lactoferrin from hydrogels
Given the previous results, we proceeded to study the effect that encapsulated bovine lactoferrin will have on the viability of osteoblasts cultured in H4LMAX-RGDS hydrogels.Rheology measurements demonstrated hydrogel formation for the samples containing LF (80 μg of LF encapsulated in a 1 wt% H4LMAX-RGDS gel), with a storage modulus slightly higher than the one observed for gels without the protein (Fig. S8†).
Protein hydrogel release studies were performed first by directly encapsulating LF in H4LMAX-RGDS hydrogels. Then, the release of the protein was monitored over a maximum of 12 days. Fig. 5 shows the accumulative amount of LF released from the hydrogel (Mt) relative to the initial amount of encapsulated protein (M0) over time. The data indicates that approximatively 60% of the LF was released from the hydrogel within the first 5 days while the remaining amount (∼40%) was retained within the gel over the time period of the experiments.
 | ||
| Fig. 5 Cumulative LF release profiles from 1 wt% H4LMAX-RGDS hydrogels in 50 mM Tris buffer pH 7.4 with 150 mM NaCl at room temperature (open squares). The data points are connected with a black solid line to guide the eye of the reader. Error bars are smaller than the symbol where not evident. The black dashed line represents a theoretical LF release scenario driven by the mass action law. Dissolution of the hydrogel was not observed during the experiments. | ||
The LF hydrogel release data suggests that there are two distinct pools of proteins encapsulated in the network, a mobile fraction, which is gradually released and an immobile fraction that is retained within the gel. This type of behaviour has been previously observed for hydrogel peptides, which charge favoured electrostatic interactions with proteins of opposite charge.23,25 However, H4LMAX-RGDS (+5 charge) and LF (pI ∼ 8.4–9.0) are both positively charged at physiological pH, hence one would expect electrostatic repulsion interactions between LF and the hydrogel which in turn should lead to complete release of the protein from the gel. In fact, LF encapsulated in hydrogels of MAX1 (with +9 charge at physiological pH) and a related peptide derivative (with +5 charge at physiological pH) was practically fully released from the gels after 10 days.23,26
The next factor to consider is the mesh size (ξ) of the H4LMAX-RGDS hydrogel. If the size of the protein approaches the ξ of the gel, then the mobility of the macromolecule will be constrained by the ξ and physical interactions with the fibril network. The ξ of H4LMAX-RGDS hydrogels is 51.7 nm (SD = 1.8 nm, n = 4) (see Experimental section for calculation details), that is ∼8.5 larger than the hydrodynamic diameter of LF (6.1 nm if present in monomeric form). Therefore, one cannot solely attribute the retention of LF to the ξ of the H4LMAX-RGDS hydrogel. For reference one can cite that a 1 wt% MAX8 hydrogel (+7 charge at physiological pH) with an average ξ of 23 nm showed a 15% LF retention in bulk protein release studies. In this case LF retention in the gel is attributed to the protein physical restraint within the MAX8 fibril network.25
Considering the previous results and the fact that an important difference between the peptides reported herein and the MAX1 family is the presence of His, we hypothesized that the strong LF retention in H4LMAX-RGDS gels is due to non-specific intermolecular interactions within the protein and the His residues of the peptide. To validate this idea, we performed bulk LF release studies in hydrogels of H4LMAX and H2LRDMAX (Fig. S9†). The data shows that while the LF release curve of H2LRDMAX resembled that of H4LMAX-RGDS hydrogels (Fig. 5 with a ca. 40% protein retention), LF was practically fully retained in hydrogels of H4LMAX, thus supporting our hypothesis of the role of His on LF retention. Furthermore, the calculated mesh size values of H4LMAX and H2LRDMAX hydrogels were 36.6 nm (SD = 0.8 nm, n = 4) and 30.3 nm (SD = 0.3 nm, n = 4), respectively, which indicates that physical restraint of LF within the hydrogel network does not command protein retention. Lastly, TEM images of diluted samples of LF encapsulated in the H4LMAX-RGDS hydrogel showed rounded bodies with an average diameter of 12.7 nm (SD = 2.2 nm, n = 30). This diameter corresponds to twice the hydrodynamic diameter of LF (6.1 nm), which indicates that LF is present in a dimeric form.27 Importantly, ∼22% of the protein appears attached to the peptide fibrils, further validating the presence of a strong intermolecular interaction (Fig. S10†).
Activity of lactoferrin encapsulated in H4LMAX-RGDS
To examine whether the addition of lactoferrin to the gel improved cell viability, primary rat osteoblasts were cultured on the H4LMAX-RGDS gel with and without LF for 3 days, the latter group acting as control. The addition of LF to the gel increased the alamarBlue® absorbance which corresponds to an increase in the number of cells, compared to the control gel without LF on day 3 (p < 0.05). Importantly, the cell morphology was similar between the two groups (Fig. 6).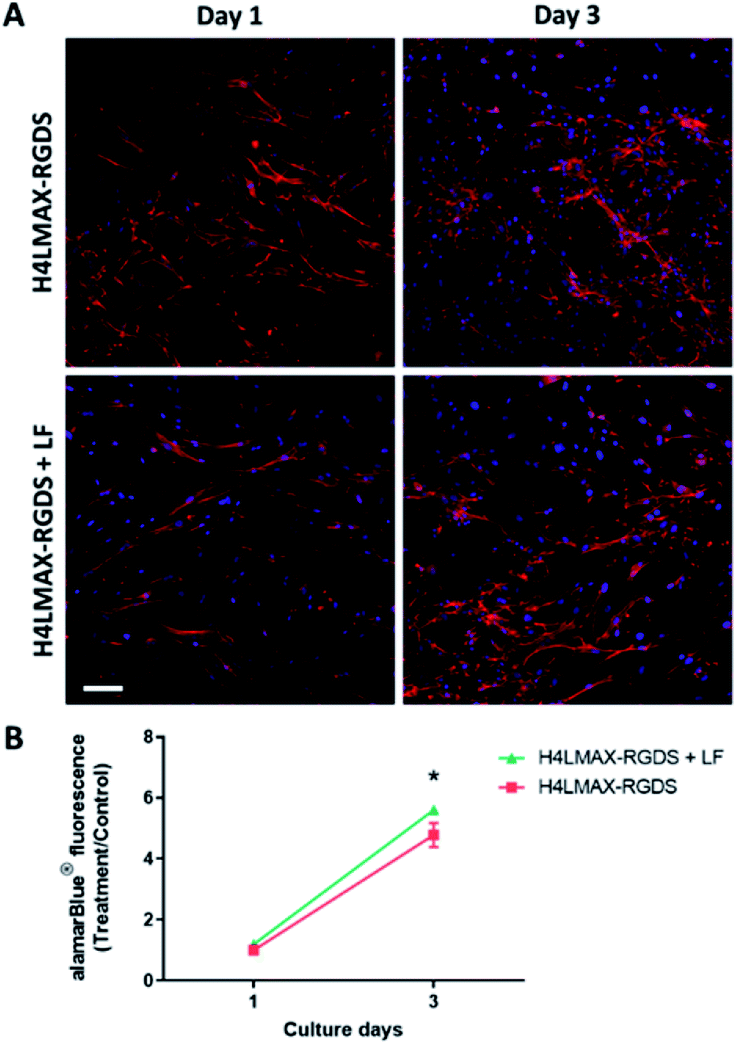 | ||
| Fig. 6 LF increase rat primary osteoblasts number on H4LMAX-RGDS hydrogel. (A) Representative confocal images showing F-actin (red) and nucleic acid (blue). Scale bar = 100 μm. (B) Results from three biological experiments were pooled. | ||
The results show that the peptide hydrogels are cytocompatible with primary osteoblasts and have the potential to be used as a sustainable drug delivery matrix for LF.
The desired features of hydrogels in bone tissue engineering include biocompatibility, non-toxicity, and formation of new bone by stimulating osteoconduction and osteoinduction.28,29 Osteoconduction is the property of biomaterial that promotes bone growth by stimulating cell adhesion to the material from surrounding environment. Osteoinduction is the property of biomaterial that induces osteogenesis by stimulating cell differentiation into the bone-forming cell lineage, often as a result of incorporation of growth factors into the biomaterial.29,30
The peptide hydrogels that we examined in this paper fulfilled these features. The hydrogel was cytocompatible with osteoblasts, and the addition of the RGD motif to the gel allowed osteoconduction, by allowing cell attachment. Incorporation of bone anabolic factor LF in the hydrogel promoted an increase in cell number, and allows potential osteoinduction as LF is proven to promote osteogenic differentiation.31,32
Previously, our group demonstrated that LF promoted calvarial bone regeneration in vivo when it was delivered in soft collagen gel with an initial burst release of LF.14 In our peptide hydrogels, LF was released over a 5 days period and then plateaued. Thus, this showed that the LF was released from the hydrogel in an improved manner than from the collagen gel in the previous in vivo study.
Experimental section
General information
Solvents for RP-HPLC were acquired as HPLC grade and used without further purification. All other reagents were used as supplied. Conventional Fmoc protected amino acids, O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU) and O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) were purchased from GL Biochem (Shanghai, China). N-Methylmorpholine (NMM), N,N′-diisopropylcarbodiimide (DIC), 6-chloro-1-hydroxybenzotriazole (6-Cl-HOBt), piperidine and Tris free base were purchased from Aldrich (St Louis, USA). Dichloromethane (DCM), and sodium chloride (NaCl) were purchased from ECP limited (Auckland, New Zealand). Hydrochloric acid (HCl) and sodium hydroxide (NaOH), dimethylformamide (DMF, AR grade) and acetonitrile (ACN, HPLC grade) were purchased from Scharlau (Barcelona, Spain). Trifluoroacetic acid (TFA) was purchased from Oakwood Chemical (River Edge, USA). Triisopropylsilane (TIPS) was purchased from Alfa Aesar (Wardhill, MA). The aminomethyl polystyrene resin was purchased from Rapp Polymer GmbH (Tübingen, Germany). Bovine lactoferrin (LF) was kindly donated by Fonterra Co-operative Group (New Zealand).TEM images were analyzed with ImageJ 1.50i.33
Peptide synthesis and purification
Peptide synthesis was performed via the Fmoc/tBu strategy on an aminomethyl polystyrene resin. All peptides were synthesised on a 0.1 or a 0.5 mmol scale either at room temperature in a Tribute or PS3 (Gyros Protein Technologies, Tucson, AZ, USA) or in a microwave-assisted Biotage® Initiator+ Fourth Generation alstra (Biotage, Uppsala, Sweden) peptide synthesizers.A Fmoc rink amide linker (for amidated peptides) was attached to the resin first by adding 4 equiv. DIC and 4 equiv. 6Cl-HOBt in DCM and incubating the mixture for 1 h at RT.
For the synthesis of peptides at RT in the Tribute™ synthesizer, HCTU (0.23 M in DMF) and NMM (2 M in NMP) solutions were used. The Fmoc group was removed using 20% piperidine in DMF (2 × 5 min). Protected amino acids were weighed in individual vials and incorporated using FmocAA-OH (5.0 eq.), HCTU (4.6 eq.) and NMM (10 eq.) in DMF, for 10 min per coupling.
For the synthesis of peptides at RT in the PS3™ synthesizer, a solution of NMM (0.4 M in DMF) was used. The Fmoc group was removed using 20% piperidine in DMF (2 × 5 min). Protected amino acids and coupling agent were weighed in individual vials. Protected amino acids were incorporated using Fmoc-AA-OH (5.0 eq.), HATU (4.5 eq.) and NMM (10 eq.) in DMF, for 20 min.
For the microwave assisted synthesis the amino acid couplings were performed using Fmoc-AA-OH (5.0 equiv., 0.2 M), HATU (4.98 equiv., 0.25 M) and 10 equiv. of 4-NMM in DMF, for 5 min at a maximum temperature of 75 °C and at 25 W. The Fmoc group was removed using 20% piperidine in DMF (2 × 3 min at a maximum temperature of 70 °C and at 62 W).
Amino acid couplings were performed as single stage, except for H2LRDMAX where triple couplings were performed from the point of truncation under microwave conditions.
The peptides were released from the resin with concomitant removal of the side chain protecting groups by treatment with a mixture of TFA/TIPS/H2O 95:2.5:2.5 (v/v/v) at room temperature for 1.5 h. The crude peptides were precipitated with cold diethyl ether, isolated by centrifugation, and washed with cold diethyl ether (3×). The washed peptides were dissolved in 1:1 (v/v) ACN/H2O containing 0.1% TFA and lyophilised. Purification of crude peptides was performed by semipreparative RP-HPLC (Dionex Ultimate 3000 (Sunnyvale, CA, USA) equipped with a 4 channel UV detector) at 210, 230, 254, and 280 nm. The solvent system used was A (0.1% TFA in H2O) and B (0.1% TFA in ACN). X-Terra Prep MS C18, 10 μm, 19 × 300 mm, using a 0.5% B per minute linear gradient, a flow of 4 mL per min at 60 °C.
Peptide characterization was done by LC-MS using ESI in positive mode (Agilent 1120 compact LC system equipped with Agilent 6120 Quadrupole MS and a UV detector at 214 nm (Palo Alto, CA.)) using a Zorbax Eclipse XDB-C8 column (5 μm; 4.6 × 150 mm; Agilent), at a flow rate of 0.3 mL min−1 and a linear gradient from 5% B to 80% B over 20 min at room temperature. The solvent system consists of A (0.1% formic acid in H2O) and B (0.1% formic acid in ACN).
Hydrogel preparation
Samples were dissolved in ice chilled MQ grade water to give a 2 wt% peptide concentration, samples were then diluted to 1 wt% by adding chilled Tris buffer (100 mM pH 7.4) with 60 mM NaCl to give a final 50 mM Tris and 30 mM NaCl concentration. The resultant solutions were allowed to stand at room temperature. Hydrogel formation was assessed by inverting the vials.Rheology
The rheological measurements were performed on a stress-controlled rheometer (MCR 302, Anton Paar Austria) fitted with a 25 mm diameter plate geometry, with a gap of 0.2 mm. The plate was pre-equilibrated to 5 °C. The samples were dissolved in ice chilled MQ grade water to give a 2 wt% peptide concentration, samples were then diluted to 1 wt% by adding chilled Tris buffer (100 mM pH 7.4) with 60 mM NaCl to give a final 50 mM Tris and 30 mM NaCl concentration. A sample containing lactoferrin (LF) was prepared as indicated above but an appropriate amount of LF was added to get 80 μg of LF encapsulated in the gel. Samples were carefully placed in the rheometer plate and a thin layer of oil was added in the gap between the plate and sample to prevent evaporation. Dynamic sweep experiments were carried out as follows: (1) temperature ramp from 5 °C to 37 °C over 100 s, (2) time sweep at 37 °C for 120 min at constant strain (0.2%) and frequency (6 rad s−1), (3) frequency sweep from 0.1 to 100 rad s−1 at 37 °C and constant strain (0.2%), (4) strain sweep from 0.1 to 1000% strain at 37 °C and constant frequency (6 rad s−1) and (5) time sweep at 37 °C for 120 min at constant strain (0.2%) and frequency (6 rad s−1).The average mesh size (ξ) is calculated from eqn (1) in accord to the Mackintosh theory, which considers that the viscoelastic behaviour of peptide hydrogels is similar to the one of semi-flexible polymer networks.34
In this equation:
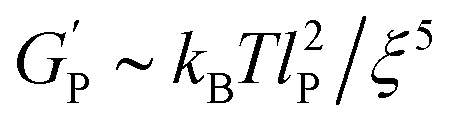 | (1) |
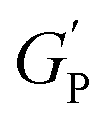 is the hydrogel plateau storage modulus, defined as the storage modulus at the frequency where the loss modulus is a minimum in a frequency sweep experiment and which is almost identical to the maximum value of time sweep measurement (where 1 Pa = 1 kg m−1 s−2); kB is the Boltzmann's constant (1.38 × 10−23 kg m2 s−2 K−1); T is the temperature at which the hydrogel is formed; and lP is the persistence length of the fibrils, which is the length over which they appear straight in the presence of Brownian forces (lP = 55 nm for MAX1 peptide analogues with morphological equivalent fibrils).34,35
is the hydrogel plateau storage modulus, defined as the storage modulus at the frequency where the loss modulus is a minimum in a frequency sweep experiment and which is almost identical to the maximum value of time sweep measurement (where 1 Pa = 1 kg m−1 s−2); kB is the Boltzmann's constant (1.38 × 10−23 kg m2 s−2 K−1); T is the temperature at which the hydrogel is formed; and lP is the persistence length of the fibrils, which is the length over which they appear straight in the presence of Brownian forces (lP = 55 nm for MAX1 peptide analogues with morphological equivalent fibrils).34,35
ATR-FTIR
Spectra were recorded in a PerkinElmer Spectrum 100 FT-IR spectrometer. Peptide samples were dissolved in 25 mM aqueous HCl and freeze dried to remove residual TFA. The process was repeated three times. Peptides were further dissolved in 50 mM Tris buffer containing 30 mM NaCl pH 7.4 to give a 1 wt% final peptide solution and the resultant solutions were freeze dried. The resulting solids were re-dissolved in D2O and freeze dried. This process was repeated three times. The final dried samples were dissolved in an appropriate volume of D2O to give 1 wt% peptide solutions and allowed to gel at room temperature. Each spectrum was an average of 36 scans taken at a resolution of 4 cm−1. The spectrum from 50 mM Tris buffer containing 30 mM NaCl pH 7.4 in D2O was subtracted as a background.Transmission electron microscopy
Sample microstructure was examined using a Tecnai12 electron microscope (FEI) operated at 120 kV equipped with a 2Kx2K GATAN CCD camera. Carbon-coated copper TEM grids (400 mesh from Agar Scientific) were rendered hydrophilic by glow discharging for 30 s. Two microliters of the sample (7.5 × 10−5 (w/v), 2.5 mM Tris buffer pH 7.4) was placed on the grid and allowed to settle for 1 min and excess solution was wicked off by a filter paper. Next, 2 μL of 2 wt% uranyl acetate solution was applied to the grid for 1 min and finally the grid was blotted with a piece of filter paper and allowed to dry overnight at RT.Lactoferrin release studies
Peptide solutions (100 μL prepared by triplicate) were first prepared at 2 wt% peptide concentration in chilled MQ water containing an appropriate amount of lactoferrin (LF) to get 80 μg LF per final gel. One hundred microliters of chilled Tris buffer (100 mM, pH 7.4) containing 300 mM NaCl were added to each vial in order to get a final peptide concentration of 1 wt%, 50 mM Tris buffer and 150 mM NaCl (physiological total salt concentration). Samples were carefully mixed (centrifuged if necessary, to remove bubbles) and allowed to set for 24 h at room temperature. Eight hundred microliters of 50 mM Tris (pH 7.4) buffer containing 150 mM NaCl was carefully added to the top of each gel. At scheduled time points, the entire volume of buffer above the gel was removed and replaced with fresh buffer (800 μL). Each removed aliquot was freeze dried and the solid residue was re-dissolved in an appropriate volume of MQ water for HPLC analysis (120 or 180 μL of water). To determine the amount of LF retained in the hydrogel, the gel was dissolved upon addition of 800 μL of fresh buffer and adjustment of the pH of the mixture to 3 by adding TFA. The amount of LF was determined for an aliquot of each sample as a function of time. Each time point was performed in triplicate. The amount of LF in each aliquot was determined by HPLC-UV (214 nm) from the peak area (calculated areas were converted to the amount of LF via a calibration curve) of the chromatographic signal. The chromatographic analysis was carried out in a Vydac 214MS C4 (5 μm, 50 × 2.1 mm) column at 25 °C at a flow rate of 0.3 mL min−1 and a linear gradient from 5% B to 65% B over 20 min at room temperature. The solvent system is composed of (A) 0.1% TFA in H2O and (B) 0.1% TFA in acetonitrile.To eliminate analyte carryover between samples, a blank was injected after each sample using a flow rate of 0.2 mL min−1 at 65% B for 5 min and then at 10% B for 15 more minutes.
Two calibration curves were acquired: (1) from 0.5 to 2.5 μg LF (peak area = 2142.8 LF – 868.97, R2 = 0.9832) and from 0.1 to 0.25 μg LF (peak area = 1204.2 LF – 49.49, R2 = 0.9723). The detection limit was 0.1 μg LF.
The data is reported as the cumulative amount of protein released from the gels (Mt) normalized to the initial protein loaded (M0) as a function of time.
Ethical approval
Primary rat and mouse osteoblasts were isolated in accordance with the Animal Ethics Committee of The University of Auckland, New Zealand.Osteoblast cell culture
Rat osteoblasts were isolated from 20 days fetal rat calvariae, as previously described.36 Briefly, the frontal and parietal bones of calvariae were collected, free of suture and periosteal tissue. The calvariae were sequentially digested using collagenase and the cells from digests 3 and 4 were pooled.Mouse osteoblasts were isolated from 3 days old mice calvariae, free of sagittal suture. The calvariae were collected in PBS containing collagenase (1 mg mL−1) and dispase (2 mg mL−1). Cells were prepared by four sequential digestions and the cells from digest 2, 3 and 4 were pooled.
Cells were grown in T75 flasks in 10% fetal bovine serum (FBS)/Dulbecco's Modified Eagle's Medium (DMEM) (Gibco) with ascorbate 2-phosphate (A2P) for 2 days and then changed to 10% FBS/Minimum Essential Medium (MEM) (Gibco) with A2P and grown to 90% confluency. Cells were then seeded into 24-well plates in 5% FBS/MEM.
alamarBlue® assay
Primary rat and mouse osteoblasts were seeded onto hydrogels (250 μL per well) pre-soaked in culture media, with or without LF. At each time point of day 1, 3 and 7, 5% alamarBlue® (Invitrogen) (final concentration in well) was added for 4 h at 37 °C. At the end of this incubation, 200 μL of the alamarBlue®-conditioned medium was transferred from each well to a 96-well plate (Greiner Bio-One) and fluorescence (excitation 540 nm; emission 630) read using a Synergy 2 multidetection microplate reader (BioTek Instruments, Inc.). For analysis, the results were normalised to the fluorescence readings from blank gels without cells (negative control).Confocal imaging
At the end of the culture, cells were fixed with 4% PFA for 20 min at room temperature. Then samples were permeabilised with 0.5% Triton-X100 at room temperature overnight. F-actin was stained with 1:40 dilution of Alexa Fluor™ 594 Phalloidin (ThermoFisher Scientific) overnight and cell nuclei were stained with 300 nM 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) (ThermoFisher Scientific) for three hours. Confocal images were obtained using ZEISS LSM 710 Inverted Confocal Microscope (Carl Zeiss, Germany).
Statistics
Data were analysed using two-way analysis of variance (ANOVA) with post hoc Bonferroni's test using the GraphPad Prism Software (GraphPad Software). A 5% significance level is used throughout. Data are presented as mean ± SEM.Conclusions
Overall, our in vitro study showed that the peptide hydrogel H4LMAX-RGDS was cytocompatible with osteoblasts. The presence of LF in the gel improved cell viability and the gel has proven controlled release of LF. Hence, the peptide hydrogel has a potential to be used as a drug delivery system for LF in vivo.Future work will continue to demonstrate applicability of H4LMAX-RGDS in vivo and on varying the position of Arg and Asp within the H2LRDMAX peptide sequence and studying the effect on hydrogelation and cell growth.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Professor Y. Hemar for assistance during the rheology measurements. This work was funded by New Zealand Ministry of Business, Innovation and Employment (Smart Idea grant, no. 9101 370 6100).References
- M. Maisani, D. Pezzoli, O. Chassande and D. Mantovani, J. Tissue Eng., 2017, 8, 1–26 CrossRef CAS PubMed.
- O. B. Betz, V. M. Betz, A. Abdulazim, R. Penzkofer, B. Schmitt, C. Schröder, S. Mayer-Wagner, P. Augat, V. Jansson and P. E. Müller, Tissue Eng., Part A, 2010, 16, 1093–1101 CrossRef CAS PubMed.
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405–414 CrossRef CAS PubMed.
- L. M. De Leon Rodriguez, Y. Hemar, J. Cornish and M. A. Brimble, Chem. Soc. Rev., 2016, 45, 4797–4824 RSC.
- H. Misawa, N. Kobayashi, A. Soto-Gutierrez, Y. Chen, A. Yoshida, J. D. Rivas-Carrillo, N. Navarro-Alvarez, K. Tanaka, A. Miki and J. Takei, Cell Transplant., 2006, 15, 903–910 Search PubMed.
- B. He, Y. Ou, A. Zhou, S. Chen, W. Zhao, J. Zhao, H. Li, Y. Zhu, Z. Zhao and D. Jiang, Drug Des., Dev. Ther., 2016, 10, 1379 CrossRef CAS PubMed.
- Y. Nagai, H. Yokoi, K. Kaihara and K. Naruse, Biomaterials, 2012, 33, 1044–1051 CrossRef CAS PubMed.
- J. Tsukamoto, K. Naruse, Y. Nagai, S. Kan, N. Nakamura, M. Hata, M. Omi, T. Hayashi, T. Kawai and T. Matsubara, Tissue Eng., Part A, 2017, 23, 1394–1402 CrossRef CAS PubMed.
- L. A. Castillo Diaz, A. Saiani, J. E. Gough and A. F. Miller, J. Tissue Eng., 2014, 5, 2041731414539344 Search PubMed.
- D. Naot, A. Grey, I. R. Reid and J. Cornish, Clin. Med. Res., 2005, 3, 93–101 CrossRef PubMed.
- J. Cornish, K. E. Callon, D. Naot, K. P. Palmano, T. Banovic, U. Bava, M. Watson, J.-M. Lin, P. Tong and Q. Chen, Endocrinology, 2004, 145, 4366–4374 CrossRef CAS PubMed.
- T. Yoshimaki, S. Sato, K. Tsunori, H. Shino, S. Iguchi, Y. Arai, K. Ito and B. Ogiso, J. Oral Sci., 2013, 55, 343–348 CrossRef CAS PubMed.
- R. Takaoka, Y. Hikasa, K. Hayashi and Y. Tabata, J. Biomater. Sci., Polym. Ed., 2011, 22, 1581–1589 CrossRef CAS PubMed.
- R. Gao, M. Watson, K. E. Callon, D. Tuari, M. Dray, D. Naot, S. Amirapu, J. T. Munro, J. Cornish and D. S. Musson, J. Tissue Eng. Regener. Med., 2018, 12, e620–e626 CrossRef CAS PubMed.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS PubMed.
- L. M. De Leon-Rodriguez, Y. Hemar, A. K. Mitra and M. A. Brimble, Biomater. Sci., 2017, 5, 1993–1997 RSC.
- K. Rajagopal, M. S. Lamm, L. A. Haines-Butterick, D. J. Pochan and J. P. Schneider, Biomacromolecules, 2009, 10, 2619–2625 CrossRef CAS PubMed.
- A. S. Veiga, C. Sinthuvanich, D. Gaspar, H. G. Franquelim, M. A. Castanho and J. P. Schneider, Biomaterials, 2012, 33, 8907–8916 CrossRef CAS PubMed.
- S. T. Phillips, G. Piersanti and P. A. Bartlett, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13737–13742 CrossRef CAS PubMed.
- M. Gungormus, M. Branco, H. Fong, J. P. Schneider, C. Tamerler and M. Sarikaya, Biomaterials, 2010, 31, 7266–7274 CrossRef CAS PubMed.
- S. Komatsu, Y. Nagai, K. Naruse and Y. Kimata, PLoS One, 2014, 9, e102778 CrossRef PubMed.
- K. Nagy-Smith, Y. Yamada and J. P. Schneider, J. Mater. Chem. A, 2016, 4, 1999–2007 RSC.
- M. Nagae, S. Re, E. Mihara, T. Nogi, Y. Sugita and J. Takagi, J. Cell Biol., 2012, 197, 131–140 CrossRef CAS PubMed.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2010, 31, 9527–9534 CrossRef CAS PubMed.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2009, 30, 1339–1347 CrossRef CAS PubMed.
- B. A. Persson, M. Lund, J. Forsman, D. E. Chatterton and T. Åkesson, Biophys. Chem., 2010, 151, 187–189 CrossRef CAS PubMed.
- M. Liu, X. Zeng, C. Ma, H. Yi, Z. Ali, X. Mou, S. Li, Y. Deng and N. He, Bone Res., 2017, 5, 17014 CrossRef CAS PubMed.
- M. Maisani, D. Pezzoli, O. Chassande and D. Mantovani, J. Tissue Eng., 2017, 8, 2041731417712073 Search PubMed.
- T. Albrektsson and C. Johansson, Eur. Spine J., 2001, 10, S96–S101 CrossRef PubMed.
- J. Cornish, K. E. Callon, D. Naot, K. P. Palmano, T. Banovic, U. Bava, M. Watson, J. M. Lin, P. C. Tong, Q. Chen, V. A. Chan, H. E. Reid, N. Fazzalari, H. M. Baker, E. N. Baker, N. W. Haggarty, A. B. Grey and I. R. Reid, Endocrinology, 2004, 145, 4366–4374 CrossRef CAS PubMed.
- M. Icriverzi, A. Bonciu, L. Rusen, L. E. Sima, S. Brajnicov, A. Cimpean, R. W. Evans, V. Dinca and A. Roseanu, Materials, 2019, 12, 3414 CrossRef CAS PubMed.
- C. Schneider, W. Rasband and K. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- B. Ozbas, K. Rajagopal, J. P. Schneider and D. J. Pochan, Phys. Rev. Lett., 2004, 93, 268106 CrossRef PubMed.
- M. C. Branco, F. Nettesheim, D. J. Pochan, J. P. Schneider and N. J. Wagner, Biomacromolecules, 2009, 10, 1374–1380 CrossRef CAS PubMed.
- J. Cornish, K. E. Callon, C. Q.-X. Lin, C. L. Xiao, T. B. Mulvey, G. J. S. Cooper and I. R. Reid, Am. J. Physiol., 1999, 277, E779–E783 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra03011b |
| ‡ These first authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |