DOI:
10.1039/D0RA03192E
(Paper)
RSC Adv., 2020,
10, 18200-18221
From cyclic amines and acetonitrile to amidine zinc(II) complexes†
Received
9th April 2020
, Accepted 29th April 2020
First published on 13th May 2020
Abstract
A seemingly simple combination of [Zn(quin)2(H2O)] (quin− = quinaldinate) and a selected secondary cyclic amine, piperidine (pipe), pyrrolidine (pyro) or morpholine (morph), afforded in acetonitrile a number of products: anionic homoleptic quinaldinate, neutral heteroleptic quinaldinate/amine and quinaldinate/amidine complexes. The piperidine and pyrrolidine systems underwent reaction with acetonitrile to give amidines. The in situ formed piperidinoacetamidine (pipeam) or pyrrolidinoacetamidine (pyroam) coordinated to zinc(II). Reactions with piperidine led to trans-[Zn(quin)2(pipe)2]·2CH3CN (1), [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (2), pipeH[Zn(quin)3]·CH3CN (3), [Zn(quin)2(pipeam)]·CH3CN (4a), [Zn(quin)2(pipeam)]·2CHCl3 (4b), pipeamH[Zn(quin)3] (5) and pipeamH[Zn(quin)2(CH3COO)]·acetamide (6) (pipeH+ and pipeamH+ denote protonated amine or amidine). By analogy, [Zn(quin)2(pyro)2] (7), pyroH[Zn(quin)3]·CH3CN (8), pyroH[Zn(quin)2Cl] (9), [Zn(quin)2(pyroam)]·CH3CN·0.5pyroam·0.5H2O (10a), [Zn(quin)2(pyroam)]·2CHCl3 (10b), [Zn(quin)2(pyroam)]·CH2Cl2 (10c) and pyroamH[Zn(quin)3] (11) were obtained in the pyrrolidine reactions. The morpholine system allowed isolation of only two novel products, trans-[Zn(quin)2(morph)2] (12) and morphH[Zn(quin)3]·CH3CN (13). Importantly, no amidine could be isolated. Instead, in autoclaves at 105 °C morpholine degraded to ammonia, as confirmed by mass spectrometry of the gas phase. pyroamH[Zn(quin)3] exists in two polymorphs which differ in the binding modes of quinaldinate ligands. In 11triclinic, the metal ion of [Zn(quin)3]− features a five-coordinate environment, whereas that in 11monoclinic is surrounded by six donors. Stabilities of the [Zn(quin)3]− isomers were assessed with DFT calculations. The one with a six-coordinate zinc(II) ion was found to be more stable than its five-coordinate counterpart. Favorable intermolecular interactions in the solid state stabilize both and reduce the energy difference between them. The calculations show the conversion of the five-coordinate [Zn(quin)3]− into its coordinatively saturated isomer to be an almost barrierless process.
Introduction
Zinc is an essential trace element that is vitally important for all living beings. The fundamental motive for extensive interest in zinc coordination chemistry is a need for a deeper understanding of its biological role. Its significance was first discovered in 1869 by Raulin, who proved that zinc is necessary for the growth of the fungus Aspergillus niger.1 Another milestone is represented by the discovery of zinc in the enzyme carbonic anhydrase.2 It was not until the 1960s that zinc was established to be essential for humans and that its deficiency causes serious health problems.3,4 Since then, its presence in thousands of human proteins was revealed5 and hundreds of zinc enzymes were discovered,6 belonging to all six classes defined by the International Union of Biochemistry.7 Zinc prevalence in biological systems is due to its unique chemical properties, most of which are ascribed to a fully occupied 3d set of orbitals. The ion Zn2+ has an electron configuration of d10 and therefore no ligand field stabilization effects which would lead to stereochemical preferences. As a result, zinc(II) exists in various coordination environments, from linear to octahedral.8 This coordination flexibility allows the metal ion to meet the needs of a wide spectrum of proteins and enzymes. The ion Zn2+ represents a redox inactive center which cannot participate in the processes of electron exchange. In catalytic sites, it mainly functions as a Lewis acid.9 According to Pearson, Zn2+ is classified as neither ‘soft’ nor ‘hard’,10 and as such, is almost equally fond of oxygen donor atoms from aspartate and glutamate, nitrogen atoms from histidine and sulfur atoms from cysteine.9 Importantly, zinc is considered to be an element with low toxicity. It is not just harmless in moderate amounts to the host organisms, on the contrary, its intake is even desirable.11,12
Because of the inherent coordination flexibility of zinc(II), we introduced into our reaction system quinaldinate, throughout the text abbreviated as quin−, a ligand that is prone to bind in a bidentate chelating manner. This was achieved by using [Zn(quin)2(H2O)] as a starting material.13 Its main structural fragment, {Zn(quin)2} with two tightly bound bidentate chelating ligands, should be preserved even at fairly robust reaction conditions. Herewith, a limit on the number of available binding sites on zinc(II) ion and the reaction outcome was imposed. Quinaldinate is an anion of quinoline-2-carboxylic acid, which is commonly known as quinaldinic acid. Surprisingly, a search of the Cambridge Structural Database (CSD) has revealed that its zinc(II) complexes are fairly scarce.14 There are reports of zinc(II) quinaldinate complexes with water,13 methanol,15 dimethylsulphoxide,16 imidazole,17 1-methylimidazole,18 a tridentate N,N,S ligand,19 pyridine-based ligands20 and recently also amino alcohols.21 The latter two systems are results of our previous work. In continuation of our research, reactions with selected secondary cyclic amines, piperidine, pyrrolidine and morpholine, were investigated (Scheme 1).
 |
| | Scheme 1 Structural formulae of cyclic amines, used in this work: (a) piperidine (pipe), (b) pyrrolidine (pyro), (c) morpholine (morph). | |
The amine NH group complements, in terms of a hydrogen bonding ability, the carboxylate of the quinaldinate ligand. The connectivity in the solid state of thus designed complexes was expected to be governed by the N–H⋯COO− hydrogen bonds. The desired amine complexes should form on a straightforward substitution of water in [Zn(quin)2(H2O)]. Each of the amines would bind to a metal ion via a lone pair on nitrogen atom. Instead, two of the amines, piperidine and pyrrolidine, reacted with acetonitrile, used as a solvent. The product of this reaction was amidine, found as a ligand in novel zinc(II) complexes. Scheme 2 shows the structural formulae of amidines in general and of piperidinoacetamidine and pyrrolidinoacetamidine, prepared in our study.
 |
| | Scheme 2 (a) Amidines in general, Rs denote substituents, (b) piperidinoacetamidine (pipeam), and (c) pyrrolidinoacetamidine (pyroam). | |
Metal complexes with piperidinoacetamidine22,23 or pyrrolidinoacetamidine,24 although very rare, were known prior to this work. However, their zinc(II) complexes were not prepared before. The amidine formation in our reaction system has implications in the synthesis of amidines in general.25 Owing to their reactivity, amidines are valuable precursors in the synthesis of heterocyclic compounds and as catalysts.26 Our study aimed to uncover the role of zinc(II) in the formation of amidines.
Herein, a series of zinc(II) compounds with quinaldinate and amines or amidines is presented. The series encompasses neutral mono-amine, bis-amine, amidine complexes and ionic compounds with protonated amines or amidines. The compounds were characterized by IR and 1H NMR spectroscopy, mass spectrometry and X-ray structure analysis. The ionic compound with protonated pyrrolidinoacetamidine, pyroamH[Zn(quin)3] (11), displayed polymorphism with the most distinctive difference between the polymorphs being in the binding mode of quinaldinate ligands in the [Zn(quin)3]− ions. Stabilities of the two forms in solution and solid state were thus evaluated by molecular modeling methods.
Experimental
General
All reagents were purchased from commercial sources and were used as received with the exception of acetonitrile, which was dried over molecular sieves.27 1H NMR spectra were obtained on a Bruker Avance III 500 at 500 MHz using (CD3)2SO (DMSO-d6) with 0.03% of tetramethylsilane (TMS) as a solvent. All proton spectra were referenced to the central peak of the residual resonance for DMSO-d6 at 2.50 ppm.28 Chemical shifts (δ) are given in ppm and coupling constants (J) in Hz. Multiplicities are reported as follows: s = singlet, d = dublet, t = triplet, m = multiplet and br = broad signal. Data processing was performed using MestReNova program (version 11.0.4).29 High-resolution mass spectra (HRMS) were recorded on an Agilent 6224 Accurate Mass TOF LC Mass Spectrometer. Infrared spectra were recorded on a Bruker Alpha II FT-IR spectrophotometer with ATR module between 4000 cm−1 and 400 cm−1. IR spectra are presented as they were obtained without any corrections made. Microanalyses (C, H, N) were performed on a PerkinElmer 2400 II analyzer. Starting compound [Zn(quin)2(H2O)] was synthesized as previously described.21 Almost all compounds crystalize with solvent molecules of crystallization. No solvate is stable outside of the mother liquor. In each case, a partial or a complete loss of solvent molecules of crystallization was observed. The yields are thus given as masses of air-dried solids. For some compounds, specifically 1, 2, 3, 8 and 13, the elemental analysis data are not given. Although compounds were available in sufficient amounts, numerous attempts at obtaining acceptable results were foiled by the loss of solvent and/or high affinity for water.
Synthesis
[Zn(quin)2(pipe)2]·2CH3CN (1). Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), piperidine (1 mL) and acetonitrile (10 mL) was left in a closed vessel at 4 °C overnight. Mixture of white solid and single crystals of [Zn(quin)2(pipe)2]·2CH3CN (1) was obtained. IR (ATR, cm−1): 3221m, 3054w, 3042w, 2937s, 2869m, 2854w, 1632vvs, 1597s, 1565s, 1509m, 1461s, 1452s, 1432m, 1365vs, 1349s, 1318m, 1297m, 1277m, 1262m, 1219w, 1190m, 1177s, 1157s, 1114w, 1090m, 1070s, 1051m, 1023s, 1000s, 961m, 942m, 893s, 871s, 851s, 801vs, 783vvs, 747m, 635s, 598s, 553w, 523m, 503s, 456m, 428w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.94 (2H, d, J = 8.7 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.41 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 8.04–8.01 (2H, m, quin−), 7.84–7.81 (2H, m, quin−), 2.62–2.60 (8H, m, pipe), 2.08 (3H, s, CH3CN), 1.45–1.40 (4H, m, pipe), 1.38–1.34 (8H, m, pipe). ESI-HRMS m/z calcd for [C25H24N3O4Zn]+ = [Zn(quin)2(pipe) + H]+: 494.1058, found: 494.1051.
[Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (2). [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), piperidine (0.5 mL) and acetonitrile (10 mL) were added to a flask. The contents were stirred at room temperature for 3 days. White solid, [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (2), was filtered off (58 mg) and the filtrate was stored at 4 °C. After a few days single crystals of 2 were obtained. Yield: 58 mg, 46%. Note. The filtrate can contain another crystalline phase, pipeH[Zn(quin)3]·CH3CN (3). IR (ATR, cm−1): 3215w, 3122w, 3061w, 2968w, 2931m, 2853w, 1663s, 1646vs, 1634vvs, 1596s, 1564s, 1507m, 1460s, 1446m, 1368vs, 1359vvs, 1343vs, 1299w, 1263m, 1207w, 1177s, 1149m, 1110m, 1086m, 1073w, 1047m, 1022m, 998m, 963w, 944w, 896s, 881s, 872s, 864m, 855s, 800vvs, 788vs, 777vvs, 749m, 742s, 637s, 601s, 559w, 522m, 498s, 466m, 440w, 429w, 404s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.94 (2H, d, J = 8.7 Hz, quin−), 8.80 (2H, d, J = 8.4 Hz, quin−), 8.42 (2H, d, J = 8.4 Hz, quin−), 8.21 (2H, d, J = 8.1 Hz, quin−), 8.04–8.01 (2H, m, quin−), 7.84–7.81 (2H, m, quin−), 2.63–2.61 (6H, m, pipe), 1.43–1.40 (3H, m, pipe), 1.39–1.34 (6H, m, pipe). ESI-HRMS m/z calcd for [C25H24N3O4Zn]+ = [Zn(quin)2(pipe) + H]+: 494.1058, found: 494.1061.
pipeH[Zn(quin)3]·CH3CN (3). Reaction mixture containing [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (50 mg, 0.05 mmol), quinaldinic acid (32 mg, 0.18 mmol) and acetonitrile (10 mL) was stirred for 3 days at room temperature in a closed flask. Precipitate, compound 3, was filtered off. Yield: 31 mg. IR (ATR, cm−1): 3056w, 3003w, 2943w, 2857w, 2739w, 2534w, 1631vs, 1595s, 1561s, 1506m, 1460s, 1429m, 1389vs, 1375s, 1355vs, 1339vs, 1257m, 1217m, 1204m, 1173s, 1149s, 1110m, 1083w, 1032w, 1021w, 995w, 964w, 945w, 897s, 880m, 858s, 805vvs, 774vvs, 750s, 741s, 635s, 602vs, 558s, 522s, 496s, 441s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.59 (3H, d, J = 8.6 Hz, quin−), 8.55 (3H, d, J = 8.4 Hz, quin−), 8.20 (3H, d, J = 8.4 Hz, quin−), 8.05 (3H, d, J = 8.2 Hz, quin−), 7.75–7.72 (3H, m, quin−), 7.67–7.64 (3H, m, quin−), 2.98–2.96 (4H, m, pipeH+), 1.62–1.57 (4H, m, pipeH+), 1.52–1.48 (2H, m, pipeH+). ESI-HRMS m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0493.
[Zn(quin)2(pipeam)]·CH3CN (4a). Procedure A. A Teflon container was loaded with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), piperidine (0.5 mL) and acetonitrile (7 mL). The container was closed and inserted into a steel autoclave which was heated for 24 hours at 105 °C. The reaction mixture was cooled to room temperature and crystalline solid, [Zn(quin)2(pipeam)]·CH3CN (4a), was filtered off (93 mg). The filtrate was stored at 4 °C overnight. Single crystals of 4a were obtained. Note 1. 4a also forms if the reaction time is longer (3 days) or the temperature is higher (120 °C). Note 2. The filtrate can contain another crystalline phase, pipeamH[Zn(quin)3] (5). Procedure B. Mixture of [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), piperidine (0.5 mL) and acetonitrile (10 mL) was heated under reflux for 8 hours. The yellow solution was first left to cool down to room temperature and was then stored at 4 °C. Single crystals of 4a were obtained. Procedure C. [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), piperidine (1 mL), acetonitrile (5 mL) and ethanol (5 mL) were added to a flask and stirred at room temperature for 3 days. The filtrate was stored at 4 °C and single crystals of 4a were obtained. IR (ATR, cm−1): 3387w, 3282m, 3064w, 3021w, 2934m, 2856w, 2249w, 1635vs, 1584vs, 1569vs, 1562vs, 1506m, 1477s, 1459s, 1434s, 1352vs, 1335vs, 1280m, 1258m, 1217s, 1180s, 1138m, 1113m, 1044m, 1022s, 990m, 968m, 950w, 896s, 853m, 832m, 801vvs, 771vvs, 737s, 637s, 602s, 560m, 522s, 497s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.78 (2H, d, J = 8.4 Hz, quin−), 8.50 (2H, d, J = 8.6 Hz, quin−), 8.35 (2H, d, J = 8.4 Hz, quin−), 8.21 (2H, d, J = 8.2 Hz, quin−), 8.00–7.97 (2H, m, quin−), 7.83–7.80 (2H, m, quin−), 6.98 (1H, br s, pipeam), 3.25–3.23 (4H, m, pipeam), 2.07 (residual CH3CN), 1.90 (3H, s, pipeam), 1.46–1.42 (2H, m, pipeam), 1.23 (4H, br m, pipeam). ESI-HRMS m/z calcd for [C27H27N4O4Zn]+ = [Zn(quin)2(pipeam) + H]+: 535.1324, found: 535.1305. Elemental analysis calcd for C27H26N4O4Zn (%): C, 60.51; H, 4.89; N, 10.45. Found (%): C, 60.18; H, 5.06; N, 10.90.
[Zn(quin)2(pipeam)]·2CHCl3 (4b). A small amount of 4a was dissolved in chloroform and afterwards diethyl ether was carefully layered on top. Single crystals of 4b were thus obtained. IR (ATR, cm−1): 3300m, 3065w, 2971m, 2864w, 1661m, 1644vs, 1584vs, 1570vs, 1509m, 1492m, 1460s, 1432m, 1365s, 1355vs, 1343vs, 1337vs, 1285m, 1254m, 1239s, 1217m, 1178s, 1150m, 1114m, 1049w, 1025m, 992m, 967m, 953w, 897m, 876m, 854m, 801vs, 774vs, 752vvs, 736vvs, 660s, 638s, 603s, 560m, 523m, 498s, 481m.
pipeamH[Zn(quin)3] (5). Dried 4a (50 mg, 0.09 mmol), quinaldinic acid (32 mg, 0.18 mmol) and acetonitrile (10 mL) were added to a flask and stirred at room temperature for 3 days. White crystalline solid, pipeamH[Zn(quin)3] (5), was filtered off. Yield: 44 mg, 67%. IR (ATR, cm−1): 3052w, 3002w, 2935w, 2360w, 1703w, 1658s, 1627vs, 1561s, 1509m, 1463m, 1441w, 1428w, 1369s, 1345vs, 1308m, 1267w, 1214m, 1176m, 1167m, 1150m, 1110w, 1053w, 1022w, 995w, 958w, 894s, 878w, 855s, 806s, 797vs, 776vvs, 742s, 638s, 629s, 604s, 567w, 523m, 500m, 422w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 9.01 (2H, br s, pipeamH+), 8.58 (3H, d, J = 8.6 Hz, quin−), 8.53 (3H, d, J = 8.4 Hz, quin−), 8.18 (3H, d, J = 8.4 Hz, quin−), 8.04 (3H, d, J = 8.1 Hz, quin−), 7.74–7.71 (3H, m, quin−), 7.66–7.63 (3H, m, quin−), 3.51–3.49 (4H, m, pipeamH+), 2.23 (3H, s, pipeamH+), 2.07 (residual CH3CN), 1.62–1.55 (6H, m, pipeamH+). Note. Broad signal around 9.01 ppm belongs to exchangeable protons of amidine ligand, therefore the integral deviates from its true value. ESI-HRMS m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0499. Elemental analysis calcd for C37H33N5O6Zn (%): C, 62.67; H, 4.69; N, 9.88. Found (%): C, 62.23; H, 4.60; N, 9.89.
pipeamH[Zn(quin)2(CH3COO)]·acetamide (6). A Teflon container was loaded with zinc(II) acetate dihydrate (50 mg, 0.23 mmol), piperidine (0.5 mL) and acetonitrile (7 mL). The container was closed and inserted into a steel autoclave which was heated for 24 hours at 105 °C. The reaction mixture was cooled to room temperature and afterwards quinaldinic acid (80 mg, 0.46 mmol) was added. The resulting yellow-orange solution was stored in a closed vessel at 4 °C. Obtained crystals were a mixture of pipeamH[Zn(quin)2(CH3COO)]·acetamide (6) and 4a. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.72–8.71 (4H, m, quin−), 8.31 (2H, d, J = 8.4 Hz, quin−), 8.16 (2H, d, J = 8.2 Hz, quin−), 7.93–7.90 (2H, m, quin−), 7.79–7.76 (2H, m, quin−), 7.28 (1H, br s, acetamide), 6.68 (1H, br s, acetamide), 3.49–3.47 (4H, m, pipeamH+), 2.22 (3H, s, pipeamH+), 2.07 (residual CH3CN), 1.75 (3H, s, acetamide), 1.62–1.55 (6H, m, pipeamH+), 1.53 (3H, s, acetate).
[Zn(quin)2(pyro)2] (7). Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), pyrrolidine (0.5 mL) and acetonitrile (10 mL) was stirred for 3 days in a closed flask. White solid, [Zn(quin)2(pyro)2] (7), was filtered off and air-dried. The filtrate was stored at 4 °C overnight. Crystals of 7, not suitable for X-ray diffraction analysis on single crystal, were obtained. Yield: 78 mg, 60%. Note. The same product, compound 7, forms also at 4 °C. IR (ATR, cm−1): 3188w, 3051w, 2953w, 2873w, 1633vs, 1597m, 1567s, 1507m, 1462m, 1430w, 1362vs, 1349s, 1301w, 1270w, 1208w, 1177m, 1156m, 1105w, 1063m, 1024w, 964w, 911s, 892s, 855s, 809s, 797s, 782s, 775vvs, 743m, 634s, 599s, 522m, 502s, 484m, 407m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.93 (2H, d, J = 8.2 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.40 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 8.03–8.00 (2H, m, quin−), 7.84–7.81 (2H, m, quin−), 2.64–2.62 (8H, m, pyro), 1.52–1.47 (8H, m, pyro). ESI-HRMS m/z calcd for [C24H22N3O4Zn]+ = [Zn(quin)2(pyro) + H]+: 480.0902, found: 480.0897. Elemental analysis calcd for C28H30N4O4Zn (%): C, 60.93; H, 5.48; N, 10.15. Found (%): C, 60.28; H, 5.55; N, 9.86.
pyroH[Zn(quin)3]·CH3CN (8). Reaction mixture containing [Zn(quin)2(pyro)2] (50 mg, 0.09 mmol), quinaldinic acid (31 mg, 0.18 mmol) and acetonitrile (10 mL) was stirred for 3 days at room temperature in a closed flask. Microcrystalline solid, pyroH[Zn(quin)3]·CH3CN (8), was filtered off (43 mg) and the filtrate was stored at 4 °C. Two polymorphic modifications of pyroH[Zn(quin)3]·CH3CN crystalized from the solution, a triclinic (8triclinic) and monoclinic one (8monoclinic). IR (ATR, cm−1): 3470w, 2967w, 2762w, 2470w, 1612vs, 1595s, 1559s, 1507m, 1461s, 1431m, 1367vs, 1342vs, 1298m, 1259m, 1217m, 1176s, 1150s, 1113w, 1022w, 994w, 963w, 897s, 856s, 804vvs, 775vvs, 743s, 636s, 602s, 572m, 552m, 522s, 496s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.58 (3H, d, J = 8.5 Hz, quin−), 8.55 (3H, d, J = 8.5 Hz, quin−), 8.19 (3H, d, J = 8.5 Hz, quin−), 8.05 (3H, d, J = 7.9 Hz, quin−), 7.76–7.73 (3H, m, quin−), 7.68–7.65 (3H, m, quin−), 3.09–3.06 (4H, m, pyroH+), 1.83–1.78 (4H, m, pyroH+). ESI-HRMS m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0494.
pyroH[Zn(quin)2Cl] (9). Procedure A. Zinc(II) chloride (25 mg, 0.18 mmol), quinaldinic acid (64 mg, 0.37 mmol), pyrrolidine (0.25 mL) and acetonitrile (10 mL) were mixed together and left in a closed vessel at room temperature overnight. Single crystals of 9 were filtered off. Yield: 32 mg, 34%. Procedure B. Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), pyrrolidine (1 mL), acetonitrile (5 mL) and dichloromethane (5 mL) was stirred for 3 days at room temperature in a closed flask. The resulting yellowish solution was stored at 4 °C and single crystals of 9 were obtained. IR (ATR, cm−1): 3079w, 2968w, 2718m, 2630w, 2580w, 2465m, 1615vs, 1599s, 1566s, 1509m, 1467s, 1435m, 1388vs, 1376vs, 1351s, 1274m, 1219m, 1206m, 1182m, 1156s, 1116m, 1027m, 1004w, 988m, 965m, 926w, 899s, 857s, 811vs, 803vs, 774vvs, 743s, 634s, 607s, 562m, 522s, 501s, 403s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.78–8.74 (4H, m, quin−), 8.51 (2H, br s, pyroH+), 8.32 (2H, d, J = 8.3 Hz, quin−), 8.19 (2H, d, J = 8.1 Hz, quin−), 7.99–7.96 (2H, m, quin−), 7.82–7.79 (2H, m, quin−), 3.10–3.06 (4H, m, pyroH+), 2.08 (residual CH3CN), 1.83–1.78 (4H, m, pyroH+). ESI-HRMS m/z calcd for [C20H12ClN2O4Zn]− = [Zn(quin)2Cl]−: 442.9777, found: 442.9796. Elemental analysis calcd for C24H22ClN3O4Zn (%): C, 55.73; H, 4.29; N, 8.12. Found (%): C, 55.77; H, 4.40; N, 8.14.
[Zn(quin)2(pyroam)]·CH3CN·0.5pyroam·0.5H2O (10a). Procedure A. A Teflon container was loaded with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), pyrrolidine (0.5 mL) and acetonitrile (7 mL). The container was closed and inserted into a steel autoclave which was heated for 24 hours at 105 °C. The reaction mixture was cooled to room temperature and crystalline solid, [Zn(quin)2(pyroam)]·CH3CN·0.5pyroam·0.5H2O (10a), was filtered off (79 mg). The filtrate was stored at 4 °C for four days. Single crystals of 10a were obtained. Note. 10a also forms if the reaction time is longer (3 days) or the temperature is higher (120 °C). Procedure B. Mixture of [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), pyrrolidine (0.5 mL) and acetonitrile (10 mL) was heated under reflux for 8 hours. The solution was left to cool down to room temperature overnight. Crystalline solid, compound 10a, was filtered off (73 mg) and the filtrate was stored at 4 °C. Single crystals of 10a were obtained. Note. The filtrate can contain another two crystalline phases, either 11monoclinic or 11triclinic. Procedure C. [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), pyrrolidine (1 mL), acetonitrile (5 mL) and ethanol (5 mL) were added to a flask and stirred at room temperature for 3 days. Filtrate was stored at 4 °C and single crystals of 10a were obtained. IR (ATR, cm−1): 3262m, 3063w, 2987w, 2929w, 2873w, 2249w, 1637s, 1591vs, 1570s, 1505m, 1478m, 1460s, 1429s, 1354vs, 1339vs, 1324s, 1255m, 1234m, 1218m, 1182s, 1173s, 1154m, 1137m, 1116m, 1069m, 1025m, 991w, 965m, 922w, 897s, 873m, 853m, 835m, 803vs, 781vs, 772vvs, 739m, 648m, 638s, 602s, 566m, 522s, 497s, 448w, 405s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.77 (2H, d, J = 7.8 Hz, quin−), 8.54 (2H, d, J = 8.7 Hz, quin−), 8.34 (2H, d, J = 7.8 Hz, quin−), 8.20 (2H, d, J = 8.0 Hz, quin−), 7.98 (2H, br m, quin−), 7.81 (2H, br m, quin−), 3.28 (2H, br m, pyroam), 3.10 (2H, br m, pyroam), 1.94 (3H, s, pyroam), 1.76–1.73 (4H, m, pyroam). ESI-HRMS m/z calcd for [C26H25N4O4Zn]+ = [Zn(quin)2(pyroam) + H]+: 521.1167, found: 521.1165. Elemental analysis calcd for C26H24N4O4Zn (%): C, 59.84; H, 4.64; N, 10.74. Found (%): C, 59.54; H, 4.45; N, 10.72.
[Zn(quin)2(pyroam)]·2CHCl3 (10b). A small amount of 10a was dissolved in chloroform and afterwards hexane was carefully layered on top. Single crystals of 10b were thus obtained. IR (ATR, cm−1): 3284w, 3069w, 2965m, 2871w, 1663m, 1645s, 1590vs, 1571s, 1510m, 1492m, 1481m, 1461s, 1430m, 1365s, 1355s, 1344s, 1333s, 1256m, 1237m, 1217m, 1178s, 1150m, 1113m, 1068m, 1024m, 967m, 922w, 898m, 875m, 855m, 801s, 781s, 774s, 751vs, 735vvs, 659s, 638s, 629s, 603s, 590m, 573m, 560m, 523m, 498s, 482m, 445w.
[Zn(quin)2(pyroam)]·CH2Cl2 (10c). A small amount of 10a was dissolved in dichloromethane and afterwards hexane was carefully layered on top. Single crystals of 10c were thus obtained.
pyroamH[Zn(quin)3] (11). Dried 10a (50 mg, 0.10 mmol), quinaldinic acid (33 mg, 0.19 mmol) and acetonitrile (10 mL) were added to a flask and stirred at room temperature for 3 days. White solid, pyroamH[Zn(quin)3] (11), was filtered off. Yield: 50 mg, 75%. IR (ATR, cm−1): 3233w, 3050m, 1695m, 1658s, 1613vs, 1559s, 1509m, 1462s, 1429w, 1387s, 1373vs, 1359s, 1347s, 1307w, 1262w, 1216m, 1184m, 1176m, 1168m, 1151s, 1115w, 1035w, 1023w, 966w, 953w, 895s, 878m, 854m, 808s, 801vs, 777vvs, 745m, 700m, 638s, 630s, 599s, 523s, 501s, 478m, 421m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.93 (2H, br s, pipeamH+), 8.58 (3H, d, J = 8.6 Hz, quin−), 8.53 (3H, d, J = 8.4 Hz, quin−), 8.18 (3H, d, J = 8.4 Hz, quin−), 8.04 (3H, d, J = 7.9 Hz, quin−), 7.74–7.71 (3H, m, quin−), 7.66–7.63 (3H, m, quin−), 3.54 (2H, t, J = 6.8 Hz, pipeamH+), 3.32 (2H, overlapped t, J = 6.8 Hz, pipeamH+), 2.21 (3H, s, pipeamH+), 1.97–1.86 (4H, m, pipeamH+). Note. Broad signal around 8.93 ppm belongs to exchangeable protons of amidine ligand, therefore the integral deviates from its true value. ESI-HRMS m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0501. Elemental analysis calcd for C36H31N5O6Zn (%): C, 62.21; H, 4.50; N, 10.08. Found (%): C, 61.73; H, 4.39; N, 9.92.
trans-[Zn(quin)2(morph)2] (12). Procedure A. Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), morpholine (0.5 mL) and acetonitrile (10 mL) was stirred for 3 days in a closed flask. White precipitate, [Zn(quin)2(morph)2] (12), was filtered off and air-dried (101 mg). The filtrate was stored at 4 °C overnight and single crystals of 12 were obtained. Yield: 101 mg, 74%. Note. The same product, compound 12, forms also at 4 °C. Procedure B. Mixture of [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), morpholine (0.5 mL), acetonitrile (9 mL) and methanol (1 mL) was heated under reflux for 8 hours. White solid, compound 12, was filtered off (68 mg). The resulting filtrate was stored at 4 °C overnight. Single crystals of 12 were obtained. Yield: 68 mg, 50%. Procedure C. A Teflon container was loaded with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), morpholine (0.5 mL) and acetonitrile (7 mL). The container was closed and inserted into a steel autoclave which was heated for 24 hours at 105 °C. The reaction mixture was cooled to room temperature. A mixture of crystalline [Zn(quin)2(NH3)] and [Zn(quin)2(morph)2] (12) was obtained. IR (ATR, cm−1): 3180m, 3060w, 2964w, 2944m, 2925m, 2850w, 1631vs, 1598m, 1568s, 1553m, 1509m, 1461m, 1438s, 1365vs, 1332m, 1322m, 1299w, 1271w, 1258m, 1221m, 1208m, 1198m, 1175m, 1160s, 1114s, 1100s, 1057m, 1049m, 1032s, 996w, 960w, 894m, 876vs, 855s, 829w, 808s, 794s, 775vvs, 742m, 636s, 627s, 600m, 554w, 523w, 499s, 490w, 482w, 431w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.86 (2H, d, J = 8.6 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.41 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.0 Hz, quin−), 8.03–8.00 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 3.49–3.47 (8H, m, morph), 2.65–2.63 (8H, m, morph). ESI-HRMS m/z calcd for [C24H22N3O5Zn]+ = [Zn(quin)2(morph) + H]+: 496.0851, found: 496.0845. Elemental analysis calcd for C28H30N4O6Zn (%): C, 57.59; H, 5.18; N, 9.59. Found (%): C, 57.58; H, 5.00; N, 9.37.
morphH[Zn(quin)3]·CH3CN (13). Reaction mixture containing [Zn(quin)2(morph)2] (50 mg, 0.09 mmol), quinaldinic acid (30 mg, 0.17 mmol) and acetonitrile (10 mL) was stirred for 3 days at room temperature in a closed flask. White solid, compound 13, was filtered off. Yield: 46 mg. Preparation of single crystals of 13. Zinc oxide (814 mg, 10 mmol) was mixed with methanoic acid (5 mL, 0.13 mol). The mixture was refluxed in dichloromethane for 6 hours and afterwards cooled to room temperature. White solid was filtered off and air-dried. A Teflon container was loaded with a 50 mg sample of the solid, morpholine (0.5 mL) and acetonitrile (7 mL). The container was closed and inserted into a steel autoclave which was heated for 24 hours at 105 °C. The reaction mixture was cooled to room temperature and afterwards quinaldinic acid (100 mg, 0.58 mmol) was added. The resulting yellow solution was stored in a closed vessel at 4 °C. Obtained crystals were a mixture of morphH[Zn(quin)3]·CH3CN (13), trans-[Zn(quin)2(morph)2] (12) and morpholinium quinaldinate. IR (ATR, cm−1): 3499m, 3422m, 2970w, 2859w, 2742w, 2660w, 2503w, 1738w, 1623vs, 1613vs, 1562s, 1507m, 1462s, 1432m, 1394s, 1371vs, 1342s, 1303m, 1261w, 1238w, 1218m, 1178m, 1152m, 1107s, 1044w, 1022w, 963w, 897s, 882m, 874m, 858s, 805vvs, 775vvs, 747s, 636s, 602vs, 522s, 497s, 447vs, 424s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.60–8.57 (6H, m, quin−), 8.21 (3H, d, J = 8.4 Hz, quin−), 8.07 (3H, d, J = 7.9 Hz, quin−), 7.79–7.76 (3H, m, quin−), 7.70–7.67 (3H, m, quin−), 3.71–3.69 (4H, m, morphH+), 3.03–3.01 (4H, m, morphH+). ESI-HRMS m/z calcd for [C30H18N3O6Zn]− = [Zn(quin)3]−: 580.0487, found: 580.0500.
X-ray structure analysis
Single crystal X-ray diffraction data were collected on an Agilent SuperNova diffractometer with molybdenum (Mo-Kα, λ = 0.71073 Å) or copper (Cu-Kα, λ = 1.54184 Å) micro-focus sealed X-ray source at 125 or 150 K. Each crystal was placed on a tip of a glass fiber using silicone grease and then mounted on the goniometer head. Data processing was performed with CrysAlis PRO.30 The structures were solved with Olex software31 using ShelXT32 and refined using the least squares methods in ShelXL.33 Anisotropic displacement parameters were determined for all non-hydrogen atoms. The modest quality of the crystal of 11triclinic resulted in large R1 and wR2 residuals. In spite of the latter, its composition, determined to be pyroamH[Zn(quin)3], is not questionable. Repeated attempts to obtain crystals with better diffraction data were not successful. The majority of compounds crystallized as solvates. For many, the interstitial solvent molecules were disordered. Positional disorder was observed for solvent molecules of 4b, 10b and 10c which are either chloroform or dichloromethane solvates. In each case, the disorder was successfully modelled using PART instruction. For 10a, a symmetry-imposed disorder of a non-coordinated pyroam molecule with a hydrogen bonded water molecule was resolved using PART-1 instruction. The disorder of acetonitrile in 1 could not be modelled. The contribution of the disordered solvent to the scattering factors was therefore accounted for by the SQUEEZE program.34 The same procedure was applied for 4a where one of the two solvent molecules appeared in a severe disorder. With a few exceptions, the NH or NH2+ hydrogen atoms of amines, protonated amines, amidines or protonated amidines, located in the final stages of refinement from difference Fourier maps, were refined with isotropic displacement parameters. The remaining hydrogen atoms were added in calculated positions. Programs Platon,35 Ortep36 and Mercury37 were used for crystal structure analysis and preparation of figures. Crystallographic data are collected in Tables 1 and 2. All crystal structures were deposited to the CCDC and were assigned deposition numbers 1973015 (1), 1973016 (2), 1973017 (3), 1973018 (4a), 1973019 (4b), 1973020 (5), 1973021 (6), 1973022 (8monoclinic), 1973023 (8triclinic), 1973024 (9), 1973025 (10a), 1973026 (10b), 1973027 (10c), 1973028 (11monoclinic), 1973029 (11triclinic), 1973030 (12) and 1973031 (13).
Table 1 Crystallographic data for 1 to 8triclinic
| |
1 |
2 |
3 |
4a |
4b |
5 |
6 |
8monoclinic |
8triclinic |
| Empirical formula |
C32H37N5O4Zn |
C55H57N7O8Zn2 |
C37H33N5O6Zn |
C29H29N5O4Zn |
C29H28Cl6N4O4Zn |
C37H33N5O6Zn |
C31H35N5O7Zn |
C36H31N5O6Zn |
C36H31N5O6Zn |
| Formula weight |
621.03 |
1074.81 |
709.05 |
576.94 |
774.62 |
709.05 |
655.01 |
695.03 |
695.03 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/n |
P21/c |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P |
P21/n |
P21/n |
P |
| T [K] |
150.00(10) |
150.00(10) |
150.00(10) |
125.00(10) |
150.00(10) |
150.00(10) |
150.00(10) |
150.00(10) |
150.00(10) |
| λ [Å] |
0.71073 |
0.71073 |
1.54184 |
0.71073 |
1.54184 |
1.54184 |
0.71073 |
0.71073 |
0.71073 |
| a [Å] |
11.8913(6) |
10.2457(4) |
13.1291(2) |
7.6353(2) |
10.23980(10) |
9.5111(2) |
14.3370(4) |
13.2473(7) |
8.8023(3) |
| b [Å] |
7.6503(3) |
35.0251(13) |
13.1804(2) |
15.1456(4) |
20.2129(2) |
12.6993(3) |
8.5075(2) |
14.3337(9) |
12.7903(6) |
| c [Å] |
16.1369(8) |
14.6211(5) |
19.8037(3) |
15.5015(4) |
16.02080(10) |
13.6361(3) |
25.4808(8) |
17.9869(12) |
15.2980(6) |
| α [°] |
90 |
90 |
90 |
110.850(3) |
90 |
88.709(2) |
90 |
90 |
79.309(4) |
| β [°] |
91.748(4) |
109.855(4) |
93.1260(10) |
101.862(2) |
97.6380(10) |
88.368(2) |
102.665(3) |
91.334(5) |
84.720(3) |
| γ [°] |
90 |
90 |
90 |
101.479(2) |
90 |
82.441(2) |
90 |
90 |
70.296(4) |
| V [Å3] |
1467.32(12) |
4935.0(3) |
3421.87(9) |
1565.17(8) |
3286.50(5) |
1631.76(6) |
3032.32(15) |
3414.5(4) |
1592.51(12) |
| Z |
2 |
4 |
4 |
2 |
4 |
2 |
4 |
4 |
2 |
| Dcalc [g cm−3] |
1.313 |
1.447 |
1.376 |
1.224 |
1.566 |
1.443 |
1.435 |
1.352 |
1.449 |
| μ [mm−1] |
0.877 |
1.036 |
1.441 |
0.823 |
5.874 |
1.511 |
0.866 |
0.772 |
0.827 |
| Collected reflections |
7093 |
29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 571 571 |
16434 |
26129 |
33472 |
29230 |
28251 |
17656 |
13888 |
| Unique reflections |
3365 |
13067 |
6980 |
7177 |
6741 |
6194 |
6946 |
7838 |
7320 |
| Observed reflections |
3014 |
9183 |
5830 |
6496 |
6008 |
5975 |
5925 |
4598 |
5614 |
| Rint |
0.0192 |
0.0404 |
0.0258 |
0.0308 |
0.0399 |
0.0455 |
0.0287 |
0.0551 |
0.0337 |
| R1 (I > 2σ(I)) |
0.0304 |
0.0427 |
0.0351 |
0.0321 |
0.0315 |
0.0325 |
0.0314 |
0.0545 |
0.0437 |
| wR2 (all data) |
0.0852 |
0.0909 |
0.0981 |
0.0883 |
0.0859 |
0.0890 |
0.0828 |
0.1331 |
0.1089 |
Table 2 Crystallographic data for 9 to 13
| |
9 |
10a |
10b |
10c |
11monoclinic |
11triclinic |
12 |
13 |
| Empirical formula |
C24H22ClN3O4Zn |
C31H34N6O4.5Zn |
C28H26Cl6N4O4Zn |
C27H26Cl2N4O4Zn |
C36H31N5O6Zn |
C36H31N5O6Zn |
C28H30N4O6Zn |
C36H31N5O7Zn |
| Formula weight |
517.26 |
628.01 |
760.60 |
606.79 |
695.03 |
695.03 |
583.93 |
711.03 |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P2/c |
P |
P21/n |
P |
P21/c |
P |
P21/n |
P21/n |
| T [K] |
150.00(10) |
150.00(10) |
150.00(10) |
125.00(10) |
150.00(10) |
150.00(10) |
150.00(10) |
150.00(10) |
| λ [Å] |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| a [Å] |
9.5603(6) |
7.9798(2) |
10.2026(3) |
7.9088(2) |
10.1666(6) |
9.3854(9) |
10.6517(5) |
13.0755(4) |
| b [Å] |
6.7219(8) |
12.1068(3) |
20.3546(5) |
12.5987(4) |
17.0493(12) |
12.6764(12) |
17.8533(8) |
12.9618(5) |
| c [Å] |
17.1475(12) |
15.2595(4) |
15.8428(5) |
14.7809(5) |
18.2066(9) |
13.5914(12) |
13.5909(5) |
20.0571(7) |
| α [°] |
90 |
81.018(2) |
90 |
114.123(3) |
90 |
93.234(8) |
90 |
90 |
| β [°] |
91.675(7) |
78.984(2) |
97.780(2) |
99.089(2) |
101.886(5) |
90.556(8) |
91.999(4) |
93.157(3) |
| γ [°] |
90 |
86.388(2) |
90 |
93.237(2) |
90 |
99.664(9) |
90 |
90 |
| V [Å3] |
1101.49(17) |
1428.47(6) |
3259.79(16) |
1315.26(8) |
3088.1(3) |
1591.2(3) |
2582.98(19) |
3394.2(2) |
| Z |
2 |
2 |
4 |
2 |
4 |
2 |
4 |
4 |
| Dcalc [g cm−3] |
1.560 |
1.460 |
1.550 |
1.532 |
1.495 |
1.451 |
1.502 |
1.391 |
| μ [mm−1] |
1.274 |
0.910 |
1.285 |
1.179 |
0.853 |
0.828 |
1.003 |
0.780 |
| Collected reflections |
5444 |
24625 |
38010 |
20933 |
17097 |
11616 |
12761 |
16839 |
| Unique reflections |
2536 |
6572 |
7490 |
6040 |
7095 |
7130 |
5925 |
7775 |
| Observed reflections |
2178 |
6213 |
6163 |
5319 |
5059 |
2271 |
4521 |
5844 |
| Rint |
0.0325 |
0.0273 |
0.0340 |
0.0326 |
0.0431 |
0.3299 |
0.0242 |
0.0286 |
| R1 (I > 2σ(I)) |
0.0350 |
0.0374 |
0.0430 |
0.0382 |
0.0648 |
0.1638 |
0.0344 |
0.0397 |
| wR2 (all data) |
0.0867 |
0.0874 |
0.1082 |
0.1030 |
0.1616 |
0.4640 |
0.0888 |
0.1033 |
Detection of ammonia by mass spectrometry
Four samples were subjected to analysis. First sample consisted of neat acetonitrile, second and third were mixtures of acetonitrile and morpholine in the 7:1 and 1:1 volume ratios, respectively, and the fourth was pure morpholine. The Teflon containers with liquid samples were closed and inserted into steel autoclaves which were heated for 24 hours at 105 °C. After an overnight cooling to room temperature, each container was quickly sealed with a foil. A thin capillary with an inner diameter of 220 μm was inserted through the foil and placed above the liquid phase. The gases were then pumped though a 75 cm long capillary, heated to 150 °C, into Pfeiffer Vacuum ThermoStar mass spectrometer where electron impact ionization took place. The operation pressure was approximately 2.0 × 10−6 mbar. The m/z = 17, 41 and 87 ion currents were monitored with time. The m/z = 17 mass spectrum peak belongs to ammonia, 41 to acetonitrile and 87 to morpholine.38 Measurement of each sample lasted 5 minutes and was followed with a 5 minute purging by air.
DFT calculations
Geometry optimization of minima and transition states was carried out with the Gaussian software package.39 Optimizations were carried out without symmetry restrictions using the ωB97xD40 functional that includes empirical dispersion corrections.41 The all electron basis set 6-31+g(d,p)42–45 was used for all atoms. Bulk solvent effects (acetonitrile) were included during optimization with the SMD continuum model.46 The nature of the stationary points as minima of the potential energy surface was confirmed with vibrational analysis, which was also used to calculate the thermal corrections to free energy. Analysis of the electron density was performed within the Atoms In Molecules (AIM) theory of R. F. W. Bader47,48 using the Multiwfn program.49 The CYLview50 and VMD51 visualization software have been used to create some of the figures.
Results and discussion
Syntheses and reactivity
The following discussion is divided into two parts. The first one pertains to reactions which were carried out at ambient or lower temperature and whose outcome were the amine complexes. The second part is focused on the reactions which afforded amidines and were typically carried out under forcing conditions, at reflux or in the autoclave at 105 °C. These were put in the context of known syntheses of amidines. The medium, acetonitrile, was chosen on the account of its usual inertness at mild conditions and has been for that reason even exploited as a solvent in the conversions of amines and activated nitriles into amidines.52 A substitution chemistry on [Zn(quinal)2(H2O)], our starting material, was expected to proceed smoothly, with no interfering reactions. The replacement of water with amine could produce three complexes, a mono-substituted amine complex with the [Zn(quinal)2(amine)] composition and a di-substituted [Zn(quinal)2(amine)2] which could exist as cis and trans geometric isomers. However, only the piperidine system allowed isolation of all three. When the reaction temperature was 4 °C or lower, trans-[Zn(quinal)2(pipe)2]·2CH3CN (1) was obtained. Interestingly, at ambient conditions the other two complexes co-crystallized in [Zn(quinal)2(pipe)]·cis-[Zn(quinal)2(pipe)2] (2). The isolation of the co-crystal 2 speaks of a transient nature of the mono-piperidine complex which under given conditions could not be isolated in pure form. Instead, it reacted further with piperidine to [Zn(quinal)2(pipe)2] until the concentration of the two species reached a critical point and 2 started to precipitate. The remaining filtrate contained yet another complex species, a [Zn(quinal)3]− ion, which crystallized as pipeH[Zn(quinal)3]·CH3CN (3). Its formation raises two questions. One concerns a proton source and the other a fate of a quinaldinate source. Namely, the composition of the [Zn(quinal)3]− ion entails a presence of the quinaldinate-depleted zinc(II) species in the remaining solution. Unable to identify either, a rational synthesis of pipeH[Zn(quinal)3]·CH3CN (3) was carried out by reacting [Zn(quinal)2(pipe)]·cis-[Zn(quinal)2(pipe)2] (2) with quinaldinic acid. The acid combines both required reagents for this reaction in one compound. With the piperidine system, the well-known coordination flexibility of zinc(II) came to light, since the reaction mixtures often contained several species. This arises from the ability of zinc ion to easily alter its coordination geometry, either through contraction or expansion of its coordination sphere or through the ligand exchange.53 On the contrary, pyrrolidine produced at mild conditions only one product, a bis-amine complex [Zn(quinal)2(pyro)2] (7). Currently, its stereochemistry remains unknown, as we did not succeed in growing single crystals of acceptable quality. Based on the NMR and IR spectra,54 we are confident of its composition to be [Zn(quinal)2(pyro)2]. Among several attempts to prepare single crystals of [Zn(quinal)2(pyro)2] (7), two stand out. When the zinc(II) starting material was replaced with zinc(II) chloride, an ionic compound with the pyroH[Zn(quinal)2Cl] (9) composition formed. Same product resulted from the reaction of [Zn(quinal)2(H2O)] with pyrrolidine in the 1:1 volume mixture of dichloromethane and acetonitrile. Chloride is a product of the known dichloromethane reaction with amine which typically results in 1,1′-methylenebis(pyrrolidine), known as aminal, and pyrrolidinum hydrochloride.55,56 Similarly to piperidine complexes, addition of quinaldinic acid to the acetonitrile suspension of [Zn(quinal)2(pyro)2] (7) afforded pyroH[Zn(quinal)3]·CH3CN (8) which, interestingly, crystallized in two polymorphic forms. By analogy, a bis-morpholine complex trans-[Zn(quinal)2(morph)2] (12) reacted with the acid to morphH[Zn(quinal)3]·CH3CN (13). Nevertheless, the morpholine system differs radically from its predecessors. First, trans-[Zn(quinal)2(morph)2] (12), was obtained both at mild and at harsh conditions. An eight-hour reaction at refluxing conditions or a 24 hour autoclave reaction at 105 °C produced an amine complex instead of the expected amidine complex. However, the autoclave reaction yielded another product, [Zn(quinal)2(NH3)], a known zinc(II) complex with ammonia.21 The formation of ammonia was further confirmed by the MS analysis of the gaseous phase obtained when pure morpholine was heated in the autoclave under the same conditions (Fig. S21†). Ammonia was identified by its m/z = 17 peak.38 MS analysis of the acetonitrile/morpholine mixtures in a 7:1 and in a 1:1 volume ratios showed the increase in the m/z = 17 ion currents to be proportional to the morpholine content. In the control sample, e.g. neat acetonitrile treated in the same way, no ammonia was detected. The morpholine degradation at 105 °C, although not complete within 24 h, could obstruct reactions which eventually lead to amidine. More importantly, a smaller nucleophilic character of morpholine, when compared to piperidine, could account for different reactivity. The oxygen atom in the heterocyclic ring, due to its electronegativity, causes nitrogen to be less basic and less reactive towards acetonitrile.57 Conversely, the piperidine or pyrrolidine reaction mixtures that were heated in autoclaves produced zinc(II) complexes with amidines which are products of the amine addition across the triple C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond of acetonitrile. The composition of complexes was [Zn(quinal)2(amidine)]. Frustratingly, they crystallized with weakly-bound solvent molecules, i.e., [Zn(quinal)2(pipeam)]·CH3CN (4a) and [Zn(quinal)2(pyroam)]·CH3CN·0.5pyroam·0.5H2O (10a). Their generous solubility in dichloromethane or chloroform allowed their re-crystallization and highly unstable crystals of [Zn(quinal)2(pipeam)]·2CHCl3 (4b), [Zn(quinal)2(pyroam)]·2CHCl3 (10b) and [Zn(quinal)2(pyroam)]·CH2Cl2 (10c) were obtained. Neither the outcome nor the yields of modified reactions, when the reaction temperature was raised to 120 °C or the reaction time was extended from 1 to 3 days, changed. Amidines formed also when the reactions were carried out at refluxing conditions for 8 hours. The initial assumption that their formation requires forcing conditions was challenged when the acetonitrile/ethanol solvent mixtures at ambient condition and a three-day reaction time produced amidine complexes 4a and 10a. The use of 2-propanol had the same effect, whereas methanol coordinated to metal and trans-[Zn(quin)2(CH3OH)2] was obtained.15 The beneficial effect of alcohols over the amidine synthesis has already been recognized.58 In some instances, the filtrates afforded another product, a salt of the [Zn(quinal)3]− ion with a protonated amidine as a countercation. Salts of both amidines, pipeamH[Zn(quinal)3] (5) and pyroamH[Zn(quinal)3] (11), were isolated. The pyroamH+ compound crystallized in two polymorphic forms, denoted as 11triclinic and 11monoclinic. As will be shown presently, the polymorphs markedly differ in their [Zn(quinal)3]− ions, thereby giving evidence to the structural variety of the [Zn(quinal)3]− ion and its labile nature in the solution. The protonation of amidines has occurred at the imine nitrogen. Again, similarly to systems which afforded protonated amines, an imminent question pertains to the source of protons. A rational synthesis of
pipeamH[Zn(quinal)3] (5) and pyroamH[Zn(quinal)3] (11) was accomplished at ambient conditions by the addition of quinaldinic acid to a corresponding amidine compound. In order to gain information about the significance of the zinc(II) starting material in the amidine formation, an alternative, zinc(II) acetate dihydrate, was used. After a work-up of the autoclave-treated mixture, which included the addition of quinaldinic acid, a resulting solution produced a mixture of the amidine compound 4a and pipeamH[Zn(quinal)2(CH3COO)]·acetamide (6). The pair unequivocally confirms the amidine formation. Compound 6 possesses two constituents that merit comment. It retained the acetate, the ligand introduced through the parent material. More interestingly, it contained acetamide, most likely a result of the amidine hydrolysis. The isolation of 6 could not be reproduced and remains, unfortunately, a one-time event.
N bond of acetonitrile. The composition of complexes was [Zn(quinal)2(amidine)]. Frustratingly, they crystallized with weakly-bound solvent molecules, i.e., [Zn(quinal)2(pipeam)]·CH3CN (4a) and [Zn(quinal)2(pyroam)]·CH3CN·0.5pyroam·0.5H2O (10a). Their generous solubility in dichloromethane or chloroform allowed their re-crystallization and highly unstable crystals of [Zn(quinal)2(pipeam)]·2CHCl3 (4b), [Zn(quinal)2(pyroam)]·2CHCl3 (10b) and [Zn(quinal)2(pyroam)]·CH2Cl2 (10c) were obtained. Neither the outcome nor the yields of modified reactions, when the reaction temperature was raised to 120 °C or the reaction time was extended from 1 to 3 days, changed. Amidines formed also when the reactions were carried out at refluxing conditions for 8 hours. The initial assumption that their formation requires forcing conditions was challenged when the acetonitrile/ethanol solvent mixtures at ambient condition and a three-day reaction time produced amidine complexes 4a and 10a. The use of 2-propanol had the same effect, whereas methanol coordinated to metal and trans-[Zn(quin)2(CH3OH)2] was obtained.15 The beneficial effect of alcohols over the amidine synthesis has already been recognized.58 In some instances, the filtrates afforded another product, a salt of the [Zn(quinal)3]− ion with a protonated amidine as a countercation. Salts of both amidines, pipeamH[Zn(quinal)3] (5) and pyroamH[Zn(quinal)3] (11), were isolated. The pyroamH+ compound crystallized in two polymorphic forms, denoted as 11triclinic and 11monoclinic. As will be shown presently, the polymorphs markedly differ in their [Zn(quinal)3]− ions, thereby giving evidence to the structural variety of the [Zn(quinal)3]− ion and its labile nature in the solution. The protonation of amidines has occurred at the imine nitrogen. Again, similarly to systems which afforded protonated amines, an imminent question pertains to the source of protons. A rational synthesis of
pipeamH[Zn(quinal)3] (5) and pyroamH[Zn(quinal)3] (11) was accomplished at ambient conditions by the addition of quinaldinic acid to a corresponding amidine compound. In order to gain information about the significance of the zinc(II) starting material in the amidine formation, an alternative, zinc(II) acetate dihydrate, was used. After a work-up of the autoclave-treated mixture, which included the addition of quinaldinic acid, a resulting solution produced a mixture of the amidine compound 4a and pipeamH[Zn(quinal)2(CH3COO)]·acetamide (6). The pair unequivocally confirms the amidine formation. Compound 6 possesses two constituents that merit comment. It retained the acetate, the ligand introduced through the parent material. More interestingly, it contained acetamide, most likely a result of the amidine hydrolysis. The isolation of 6 could not be reproduced and remains, unfortunately, a one-time event.
The formation of amidines in our system is a result of a nucleophilic addition of piperidine or pyrrolidine to acetonitrile in the presence of zinc(II). Our knowledge of the amidine formation remains limited. With morpholine, no amidine in any form could be isolated. In a control experiment without zinc(II), neither piperidine nor pyrrolidine produced amidine. Furthermore, in autoclave reactions with catalytic amounts of zinc(II), i.e., one fifth of the amount used in the prototypic reaction, no amidine could be detected by 1H NMR spectroscopy. The formation of amidines in the presence of transition metals is hardly without precedence, as shown by a recent review.59 Namely, a direct addition of amine, a nucleophile, to nitrile, is only possible when the CN carbon atom is made electron-deficient.52 Ordinary nitriles that lack electron-withdrawing substituents, such as acetonitrile, require a presence of a Lewis acid such as AlCl3, ZnCl260 or other metal-based activating agents such as lanthanide(iii) triflates,61 CuI,24 CuCl,58 SmI262 or ytterbium amides,63 combined with lengthy reaction times and harsh conditions. Another strategy exploits the fact that the coordination of nitrile to a metal ion considerably enhances electrophilicity of its carbon atom and thereby facilitates its reaction with amines.64,65 Thus formed amidines acted as ligands to W(II),66 Ir(III),67 Pt(II),68 Ni(II),23 Re(I)22 and Pt(IV).69 Interestingly, acetonitrile in fac-[Re(CO)3(5,5′- or 6,6′-Me2bipyridine)(CH3CN)]BF4 reacted also with morpholine to a corresponding acetamidine complex.22 Only recently, a diamine, N,N-diethyl-1,2-diaminoethane, combined at ambient conditions in the methanol solution of zinc(II) acetate with 2-cyanopyridine.70 Notable differences of the latter system, when compared to ours, are that nitrile group in 2-cyanopyridine is activated and, secondly, since a primary amine was used, the in situ formed amidine underwent a 1,3-H shift.63
Crystal structures
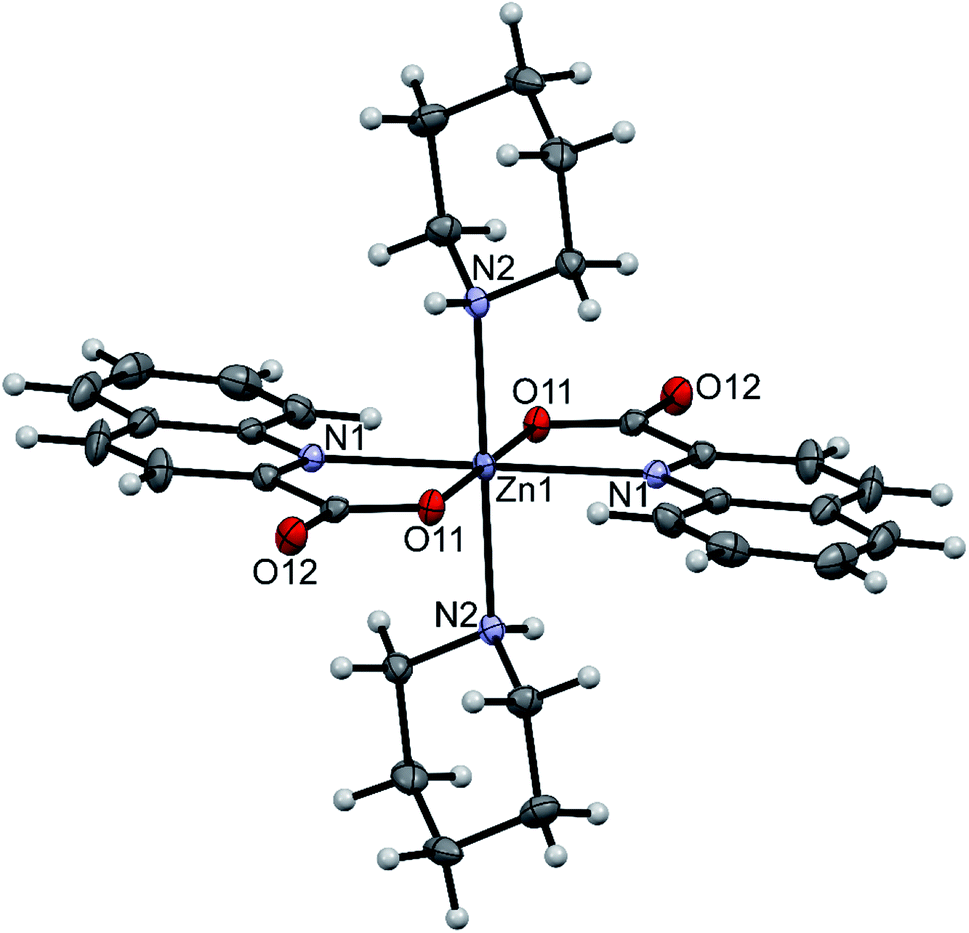
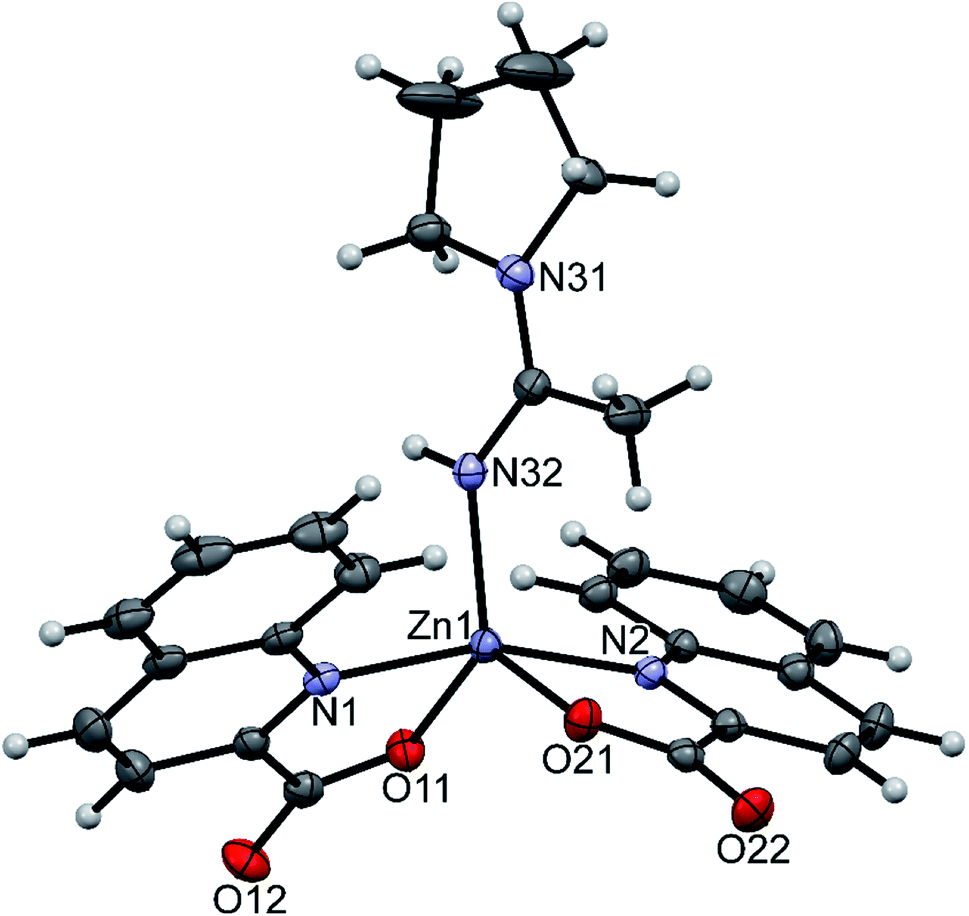
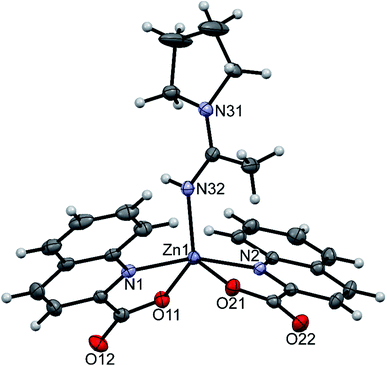
Three compounds that contain coordinated amines were structurally characterized: trans-[Zn(quin)2(pipe)2]·2CH3CN (1), [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (2) and trans-[Zn(quin)2(morph)2] (12). In all, the quinaldinate binds in the usual N,O-bidentate chelating mode and the amine ligands coordinate via their nitrogen donor atom. Compounds 1 and 2 contain all three possible piperidine complexes, a mono-amine complex [Zn(quin)2(pipe)], cis and trans geometric isomers of a bis-amine complex [Zn(quin)2(pipe)2]. Their ORTEP drawings are shown in Fig. 1–3, and their most relevant geometric parameters are summed up in Table 3. Trans disposition of ligands was observed also for the morpholine complex, [Zn(quin)2(morph)2] (12). Both trans complexes are centrosymmetric with the N4O2 donor set occupying vertices of a distorted octahedron. Their geometric parameters are very similar and are different from those of [Zn(quin)2(pipe)] and cis-[Zn(quin)2(pipe)2], the complex species of 2. The amine-to-zinc bond lengths span a wide range, from 2.0670(18) Å to 2.2168(15) Å. The shortest bond is, as expected, in a five-coordinate [Zn(quin)2(pipe)]. As all literature examples are either four- or five-coordinate zinc(II) species, a relevant comparison exists for [Zn(quin)2(pipe)] only. Very similar distances, i.e., 2.0743(6) and 2.0793(8) Å, were displayed in five-coordinate dialkyldithiocarbamate complexes with the [Zn(pipe)(S2CNR2)2] (R = CH3 or C2H5) composition.71 Somewhat longer bonds, 2.094(4) and 2.102(4) Å, are in a four-coordinate [Zn(pipe)2(C6F5)2].72 The N3O2 donor set of the mono-piperidine complex occupies vertices of a distorted square pyramid, as shown by the τ parameter which amounts to 0.35.73 It is to be noted that cis-[Zn(quin)2(pipe)2] features the longest bonds between zinc(II) ion and quinaldinate nitrogen, i.e., 2.3241(19) and 2.3453(18) Å. In another zinc(II) quinaldinate compound with cis arrangements of ligands, cis-[Zn(quin)2(Him)2] (Him = imidazole), the Zn–N distance is even longer, 2.418(4) Å.17 The lengthening is presumably due to the larger trans influence of piperidine in 2 or imidazole in the literature example vs. that of quinaldinate.74 Namely, in 1 and 12 where the corresponding bonds are shorter, 2.2520(14)–2.2696(16) Å, the quinaldinate nitrogens are trans to each other. The quinaldinate ligands in the series of our amine complexes are not strictly planar. Their deviation from planarity can be given by the dihedral angle between the carboxylate and the bicyclic system, which occupies a 3.7(2)–10.6(3)° range.
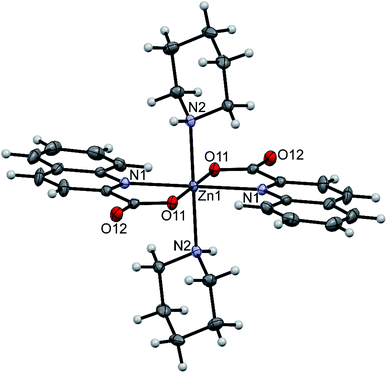 |
| | Fig. 1 ORTEP drawing of trans-[Zn(quin)2(pipe)2], a complex molecule of 1. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
 |
| | Fig. 2 ORTEP drawing of [Zn(quin)2(pipe)], one of the two complex species in 2. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
 |
| | Fig. 3 ORTEP drawing of cis-[Zn(quin)2(pipe)2], one of the two complex species in 2. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
Table 3 Relevant bond lengths [Å] and angles [°] in complexes with amines
| Angle between a pair of quinaldinates. Calculated as an angle between the carboxylate and the quinaldinate bicyclic system. |
| Compound |
1 |
2 |
2 |
12 |
| Complex |
trans-[Zn(quin)2(pipe)2] |
[Zn(quin)2(pipe)] |
cis-[Zn(quin)2(pipe)2] |
trans-[Zn(quin)2(morph)2] |
| Donor set |
N4O2 |
N3O2 |
N4O2 |
N4O2 |
| τ parameter |
— |
0.35 |
— |
— |
| Zn–O(quin−) |
2.0526(12) |
1.9785(15), 1.9888(14) |
2.0080(14), 2.0449(13) |
2.0703(13), 2.0589(13) |
| Zn–N(quin−) |
2.2520(14) |
2.1906(18), 2.1962(17) |
2.3241(19), 2.3453(18) |
2.2582(15), 2.2696(16) |
| Dihedral anglea |
0.00(7) |
35.09(6) |
63.17(4) |
0.00(7), 0.00(6) |
| Non-planarity of quin−b |
6.1(3) |
6.90(18), 10.6(3) |
10.6(2), 10.02(17) |
3.7(2), 4.9(3) |
| Zn–L |
2.2168(15) |
2.0670(18) |
2.1799(18), 2.1755(19) |
2.1959(16), 2.1918(16) |
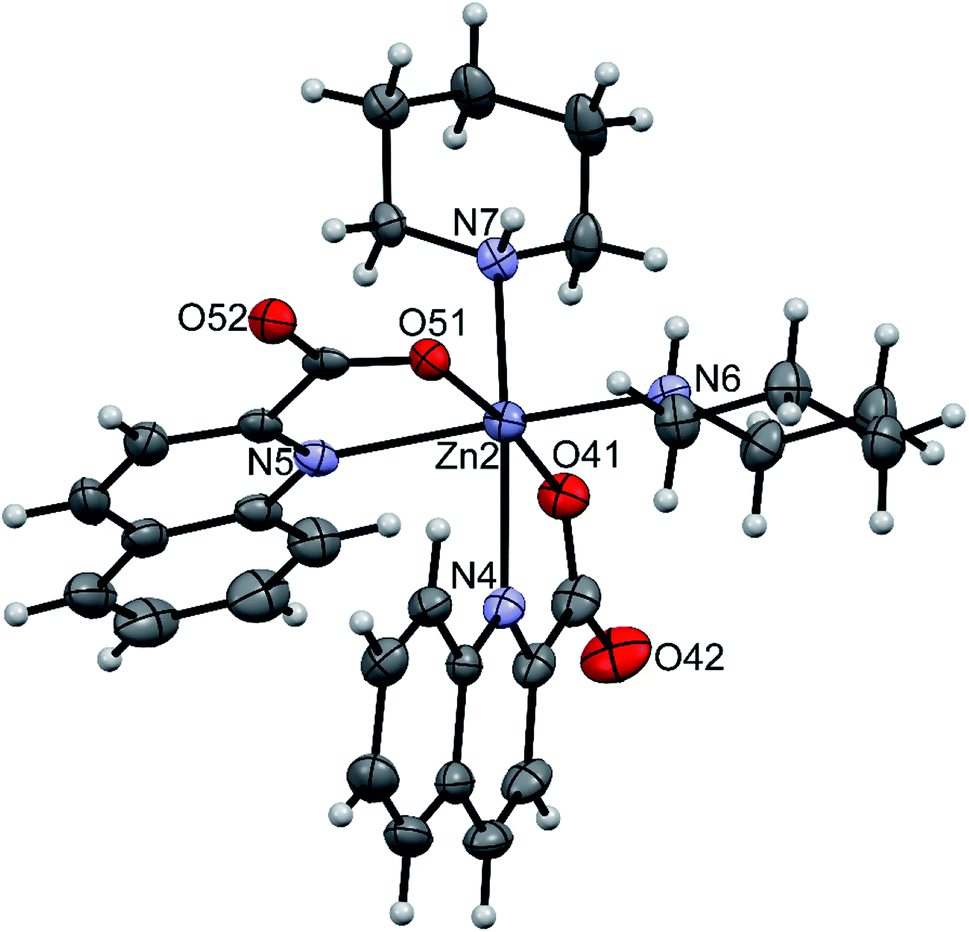
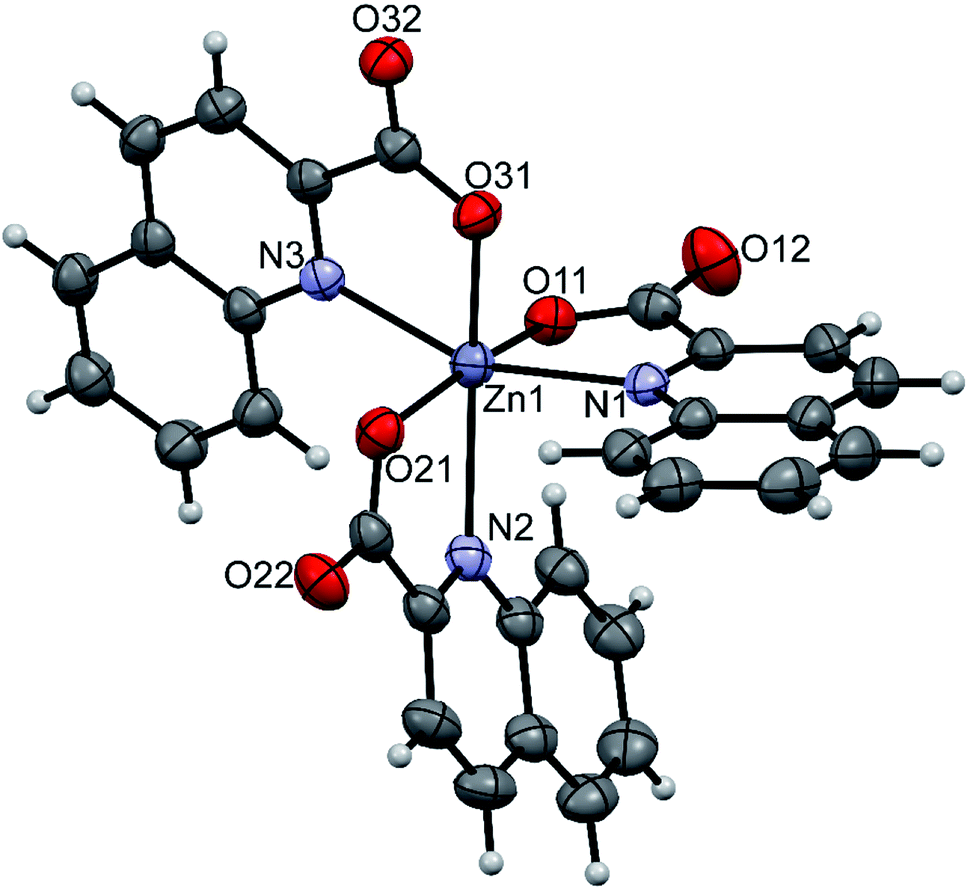
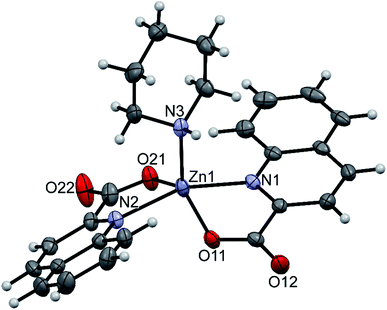
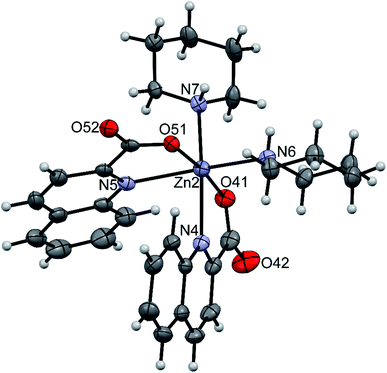
The amidine complexes, [Zn(quin)2(pipeam)] and [Zn(quin)2(pyroam)], were found to crystallize with solvent molecules of crystallization. The dimensions of the [Zn(quin)2(pipeam)] molecules of 4a and 4b are almost the same. The same observation pertains to the pyroam complex of 10a, 10b and 10c as is evident from the Table 4. Fig. 4 shows the ORTEP drawing of [Zn(quin)2(pyroam)] (10a). The five-numbered coordination of zinc(II) consists of two bidentate chelating quinaldinates and a monodentate amidine, bound via its imine nitrogen. The τ parameters in the 0.48–0.66 range suggest that the N3O2 donors describe a polyhedron that may be considered as an intermediate between a square pyramid and a trigonal bipyramid. The quinaldinate-to-zinc bonds are comparable to those in other five-coordinate species. A dihedral angle of 58.46(5)–71.43(3)° is formed between the quinaldinates. Similarly to the amine complexes, quinaldinates in amidine complexes are not strictly planar. The amidine-to-zinc bond, which occupies a 1.974(2)–1.9991(19) Å range, is shorter than the piperidine-to-zinc bond in a five-coordinate [Zn(quin)2(pipe)] of 2, 2.0670(18) Å. Bonds in the piperidinoacetamidine and pyrrolidinoacetamidine ligands are very much alike. The sp2 hybridized carbon is bonded to the imine with bonds in the 1.289(2)–1.304(3) Å range and to the amine with slightly longer bonds, i.e., 1.334(3)–1.352(2) Å. Their lengths are very similar to the ones observed for [Al(pipeam)4]Cl3·CH3CN.75 Owing to the double-bond character of the C–imine bond, the amidine can exist in two, E or Z, configurations, with E isomers being the kinetically favoured products.68 Our pipeam and pyroam ligands are all E isomers. Similarly, the E configuration was observed for a series of bipyridine-based rhenium(I) complexes with piperidinoacetamidine and morpholinoacetamidine. The absence of Z isomers was explained with steric interactions between the bulky ring moiety of the axial amidine and bipyridine ligands in the equatorial plane.22 A cationic nickel(II) complex, [Ni(HNP2)(pipeam)]2+ (HNP2 = a tridentate N,P,P-ligand), also featured pipeam in E configuration,23 whereas pyroam ligands of [Cu(pyroam)2]I were in Z configuration.24
Table 4 Relevant bond lengths [Å] and angles [°] in complexes with amidines
| Angle between a pair of quinaldinates. Calculated as an angle between the carboxylate and the quinaldinate bicyclic system. |
| Compound |
4a |
4b |
10a |
10b |
10c |
| Complex |
[Zn(quin)2(pipeam)] |
[Zn(quin)2(pipeam)] |
[Zn(quin)2(pyroam)] |
[Zn(quin)2(pyroam)] |
[Zn(quin)2(pyroam)] |
| Donor set |
N3O2 |
N3O2 |
N3O2 |
N3O2 |
N3O2 |
| τ parameter |
0.54 |
0.59 |
0.66 |
0.56 |
0.48 |
| Zn–O(quin−) |
2.0114(11), 2.0038(12) |
2.0028(12), 2.0118(12) |
2.0088(16), 2.0209(15) |
2.0033(17), 2.0093(17) |
2.0105(16), 2.0191(15) |
| Zn–N(quin−) |
2.1844(13), 2.1486(13) |
2.1917(14), 2.1787(14) |
2.1589(18), 2.1730(18) |
2.175(2), 2.1786(19) |
2.1578(17), 2.1763(17) |
| Dihedral anglea |
71.43(3) |
60.53(3) |
58.46(5) |
63.69(5) |
67.16(4) |
| Non-planarity of quin−b |
11.73(17), 3.34(12) |
9.9(3), 8.3(2) |
8.52(17), 4.86(11) |
12.1(4), 5.7(3) |
8.98(13), 7.1(2) |
| Zn–N(amidine) |
1.9877(14) |
1.9845(15) |
1.9991(19) |
1.974(2) |
1.9952(18) |
 |
| | Fig. 4 ORTEP drawing of [Zn(quin)2(pyroam)], a complex molecule of 10a. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
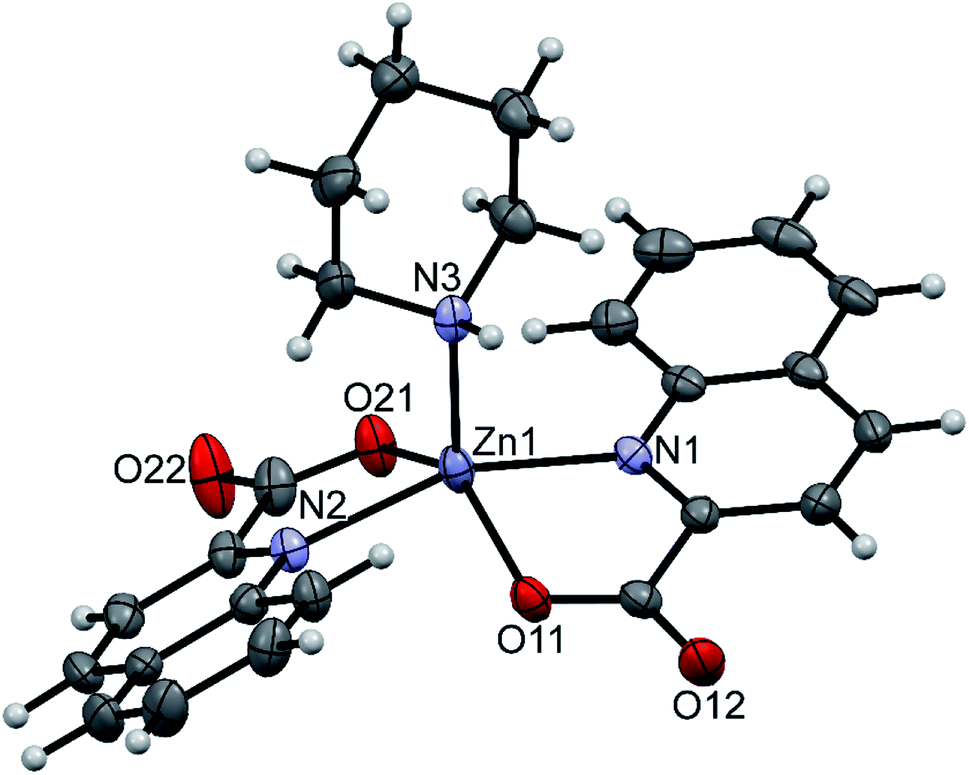
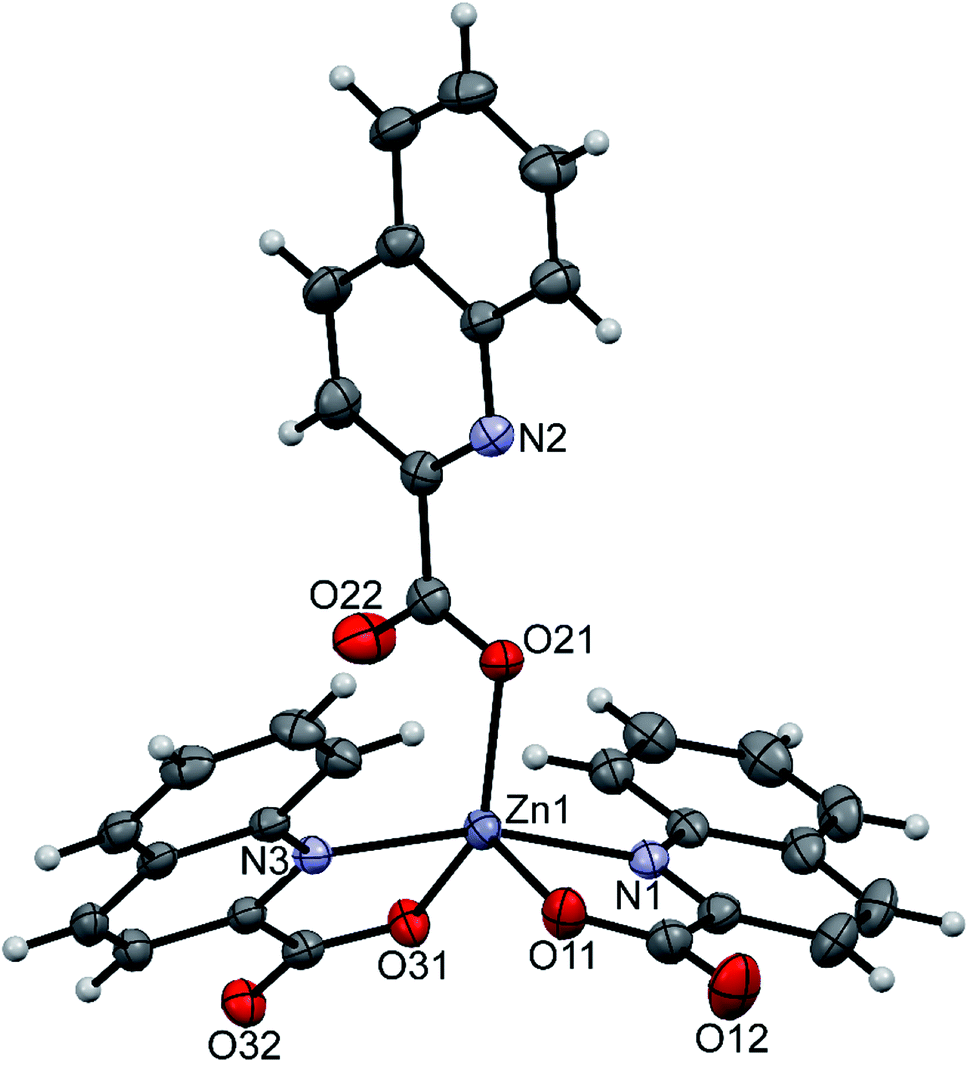
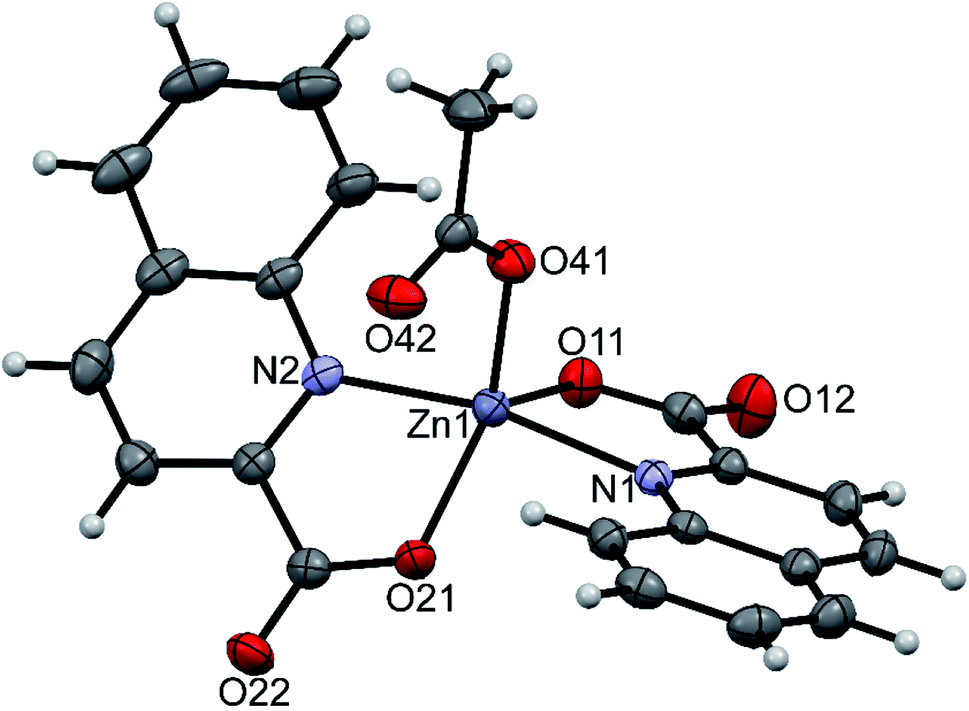
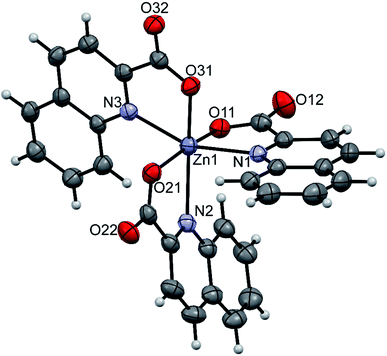
pipeamH[Zn(quin)3] (5) and pyroamH[Zn(quin)3] (11triclinic) consist of the [Zn(quin)3]− ions with a five-coordinate zinc(II) and protonated amidine molecules as countercations. The zinc(II) ion is bound to two bidentate chelating quinaldinates and a monodentate quinaldinate via one carboxylate oxygen. The ORTEP drawing of the [Zn(quin)3]− ion of 5 is shown in Fig. 5. Relevant geometric parameters for five-coordinate zinc(II) complexes are summarized in Table 5. The distribution of N2O3 donor atoms in 5 and 11triclinic is exactly half-way between a square-pyramid and a trigonal bipyramid. The monodentate quinaldinate binds to zinc(II) with a somewhat shorter bond, 1.9780(11) Å (compound 5) or 1.934(9) Å (11triclinic), than the bidentate chelating quinaldinates. The other oxygen of the monodentate quinaldinate does not make a significant bonding interaction as the Zn–O distance exceeds 2.8 Å. The bonding pattern in the complex anion of pipeamH[Zn(quin)2(CH3COO)]·acetamide (6) bears a striking similarity to that in 5: it also features a five-coordinate environment consisting of two bidentate chelating quin− ligands and a monodentate acetate. The τ parameter of 0.52 also shows structural resemblance to complexes of 5 and 11triclinic. The ORTEP drawing of the [Zn(quin)2(CH3COO)]− ion is shown in Fig. 6. The acetate-to-zinc(II) bond length is 1.9766(12) Å, whereas the non-bonding acetate oxygen is at a distance of 3.1652(13) Å. On protonation, the C–N bonds in amidinium cations became more alike: the originally C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond of neutral amidine slightly lengthened, whereas the C–N bond became shorter. Opposite trend was reported for the amidinium salt with nitrate, where the character of the C–N bonds on protonation became even more localized with a difference between them amounting to 0.07 Å. The parent amidine, formed in the presence of platinum(II), was a condensation product of the benzonitrile reaction with 1-methylcytosine.76 For comparison, in our compounds with amidinium cations, the difference between the C–N bonds was not larger than 0.014 Å.
N double bond of neutral amidine slightly lengthened, whereas the C–N bond became shorter. Opposite trend was reported for the amidinium salt with nitrate, where the character of the C–N bonds on protonation became even more localized with a difference between them amounting to 0.07 Å. The parent amidine, formed in the presence of platinum(II), was a condensation product of the benzonitrile reaction with 1-methylcytosine.76 For comparison, in our compounds with amidinium cations, the difference between the C–N bonds was not larger than 0.014 Å.
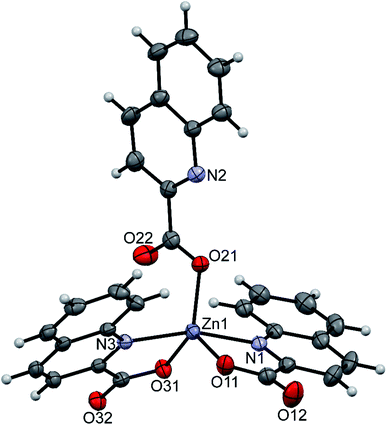 |
| | Fig. 5 ORTEP drawing of the [Zn(quin)3]− ion of 5. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
Table 5 Relevant bond lengths [Å] and angles [°] in five-coordinate [Zn(quin)3]− compounds, 5 and 11triclinic, and in 6 and 9
| Angle between a pair of quinaldinates. For 5 and 11triclinic, the first listed angle is between the bidentate quinaldinates. Calculated as an angle between the carboxylate and the quinaldinate bicyclic system. Not given. Owing to weak data set, the standard deviations of the calculated angles were too large. RCOO− that is bound in a monodentate manner. Distance of a non-coordinated carboxylate oxygen to zinc(II). |
| Compound |
5 |
11triclinic |
6 |
9 |
| |
pipeamH[Zn(quin)3] |
pyroamH[Zn(quin)3] |
pipeamH[Zn(quin)2(CH3COO)]·acetamide |
pyroH[Zn(quin)2Cl] |
| Donor set |
N2O3 |
N2O3 |
N2O3 |
N2ClO2 |
| τ parameter |
0.50 |
0.49 |
0.52 |
0.33 |
| Zn–O(quin−) |
2.0150(11), 2.0179(11) |
2.005(8), 2.011(8) |
1.9965(12), 2.0288(12) |
1.9976(14) |
| Zn–N(quin−) |
2.1478(13), 2.1463(13) |
2.174(10), 2.126(13) |
2.1608(14), 2.1363(15) |
2.2352(15) |
| Dihedral anglesa |
60.16(3) |
58.4(2) |
53.89(3) |
14.303(17) |
| 80.37(3) |
79.5(2) |
| 77.41(3) |
71.4(3) |
| Non-planarity of quin−b |
3.8(3) |
– c |
2.80(16) |
3.66(11) |
| 7.58(15) |
2.5(3) |
| 4.0(2) |
| Zn–O(RCOO−)d |
1.9780(11) |
1.934(9) |
1.9766(12) |
— |
| RCOO− |
quin− |
quin− |
CH3COO− |
— |
| Zn⋯O(RCOO−)e |
2.8838(14) |
2.854(10) |
3.1652(13) |
— |
| Zn–Cl |
— |
— |
— |
2.2670(11) |
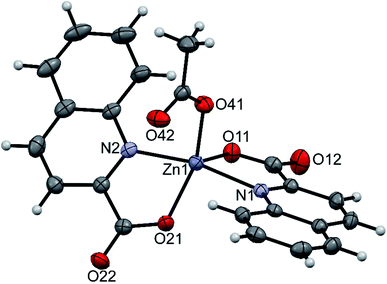 |
| | Fig. 6 ORTEP drawing of the [Zn(quin)2(CH3COO)]− ion of 6. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
The isostructural pipeH[Zn(quin)3]·CH3CN (3), a monoclinic polymorph of pyroH[Zn(quin)3]·CH3CN (8monoclinic) and morphH[Zn(quin)3]·CH3CN (13) contain [Zn(quin)3]− ions with zinc(II) in a six-numbered coordination environment. The same complex anions were found in two more compounds, a triclinic polymorph of pyroH[Zn(quin)3]·CH3CN (8triclinic) and a monoclinic polymorph of pyroamH[Zn(quin)3] (11monoclinic). The dimensions of the [Zn(quin)3]− ions are essentially the same in all compounds. ORTEP drawing of the [Zn(quin)3]− ion of 3 is shown in Fig. 7, and the most relevant geometric parameters are given in Table 6. All three quinaldinate ligands of the [Zn(quin)3]− ion are bound in a bidentate chelate manner. Some of the quinaldinates are nearly planar, for others, the deviation from planarity is more pronounced. The N3O3 donor set occupies vertices of a distorted octahedron. The distribution of the donors is mer: two quinaldinate nitrogens are trans to each other, and the third is trans to quinaldinate oxygen. Of the three Zn–N bonds, the one that is trans to oxygen, is by ca. 0.2 Å longer than the other two. A tightly bound carboxylate apparently weakens the Zn–N bond opposite to itself. The lengthening of this bond has wider consequences. Its cleavage, as the extreme case of the bond labilization, results in a five-coordinate [Zn(quin)3]− species, the one that was actually observed in 5 and in the other polymorph of pyroamH[Zn(quin)3] (11triclinic).
 |
| | Fig. 7 ORTEP drawing of the [Zn(quin)3]− ion of 3. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
Table 6 Relevant bond lengths [Å] and angles [°] in six-coordinate [Zn(quin)3]− compounds
| Angle between a pair of quinaldinates. Calculated as an angle between the carboxylate and the quinaldinate bicyclic system. |
| Compound |
3 |
8monoclinic |
8triclinic |
13 |
11monoclinic |
| |
pipeH[Zn(quin)3]·CH3CN |
pyroH[Zn(quin)3]·CH3CN |
pyroH[Zn(quin)3]·CH3CN |
morphH[Zn(quin)3]·CH3CN |
pyroamH[Zn(quin)3] |
| Donor set |
N3O3 |
N3O3 |
N3O3 |
N3O3 |
N3O3 |
| Distribution of donors |
mer |
mer |
mer |
mer |
mer |
| Zn–O(quin−) |
1.9999(13)–2.0756(13) |
1.996(2)–2.065(2) |
1.9958(17)–2.0859(17) |
2.0016(15)–2.0809(16) |
1.977(3)–2.063(3) |
| Zn–N(quin−) |
2.2141(15)–2.3962(15) |
2.186(3)–2.495(3) |
2.2002(19)–2.407(2) |
2.2108(17)–2.3625(19) |
2.205(3)–2.476(4) |
| Dihedral anglesa |
74.95(4)–81.43(3) |
59.18(6)–70.26(5) |
72.78(5)–77.65(4) |
76.49(4)–84.69(4) |
67.18(9)–84.10(10) |
| Non-planarity of quin−b |
2.5(3)–9.24(18) |
6.0(3)–11.5(3) |
1.0(2)–13.5(4) |
1.5(4)–9.4(3) |
5.6(4)–16.1(5) |
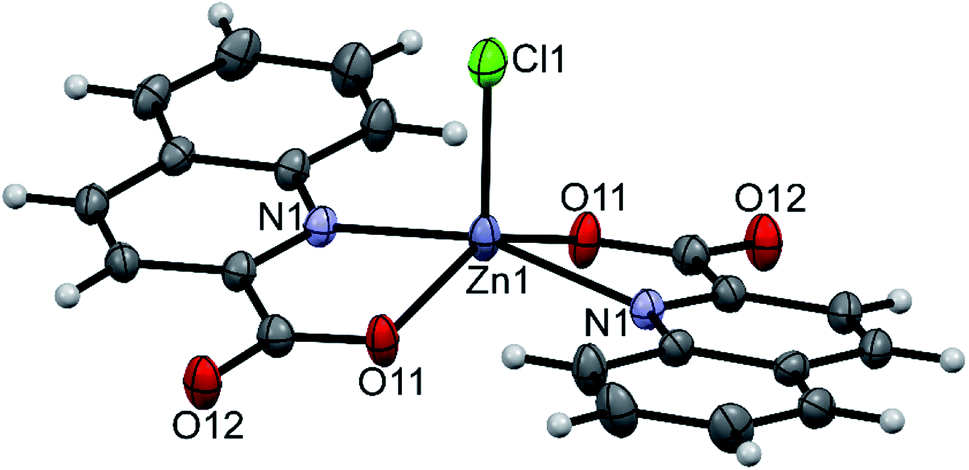
The structure of pyroH[Zn(quin)2Cl] (9) consists of pyrrolidinium cations and the [Zn(quin)2Cl]− complex anions (shown in Fig. 8). The distribution of the N2ClO2 donor set resembles most a distorted square pyramid, as confirmed by the value of 0.33 for τ parameter. The quinaldinate donor atoms define its basal plane, whereas chloride occupies its apical position. The chloride complex features the smallest dihedral angle between the quinaldinate ligands, 14.303(17)°, in all complexes with coordination number five. The chloride-to-zinc bond length of 2.2670(11) Å is very similar to Zn–Cl bonds in other complexes with chloride located at the apex of a square pyramid.77,78
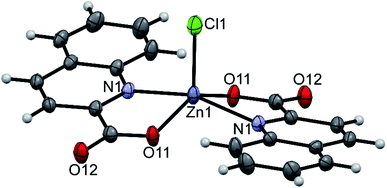 |
| | Fig. 8 ORTEP drawing of the [Zn(quin)2Cl]− ion of 9. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
Intermolecular interactions
The title compounds possess structural elements that are good hydrogen bond donors and/or acceptors. As a result, intricate patterns of intermolecular connectivity are observed in solid state. An exhaustive list of hydrogen bonding interactions in 1 to 13 is given in Tables S2 and S3 (see ESI†). Two types of connectivity patterns prevail: the complex species are either linked into infinite chains or into dimeric clusters. Both motifs are encountered in the structure of [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (2) which contains two different complex species. Piperidine ligands act as hydrogen bond donors via their N–H moieties. On the other hand, the non-coordinated oxygen atoms of quinaldinate carboxylates act as the acceptors. Surprisingly, hydrogen bonds link one type of complex molecules only: [Zn(quin)2(pipe)] molecules are linked via N–H⋯COO− interactions into chains, whereas cis-[Zn(quin)2(pipe)2] molecules are linked into dimers. Both pack with the formation of layers which stack along b-axis. In this stack, a layer of the [Zn(quin)2(pipe)] molecules alternates with the layer of the cis-[Zn(quin)2(pipe)2] molecules. Weak intermolecular interactions occur between the layers. The hydrogen bonding pattern in compound 2 is shown in Fig. 9. The linking patterns, described in graph set notation, are C11(6) and R22(12), respectively.79
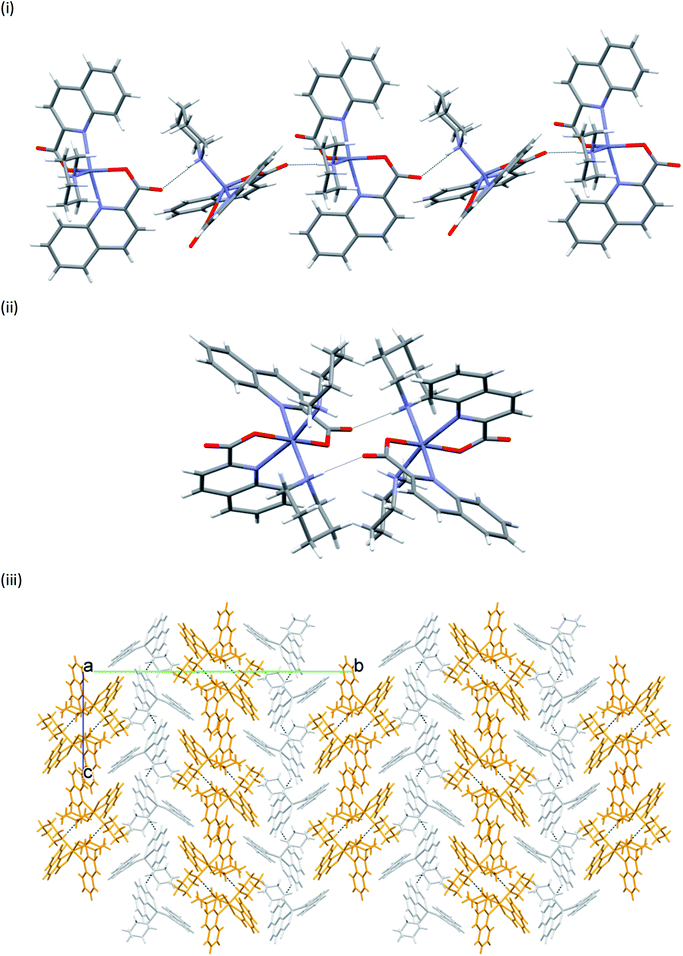 |
| | Fig. 9 Hydrogen bonding in [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (2): (i) section of a chain of the [Zn(quin)2(pipe)] molecules, (ii) a dimer of the cis-[Zn(quin)2(pipe)2] molecules, and (iii) alternating layers of [Zn(quin)2(pipe)] (light grey) and cis-[Zn(quin)2(pipe)2] molecules (gold). | |
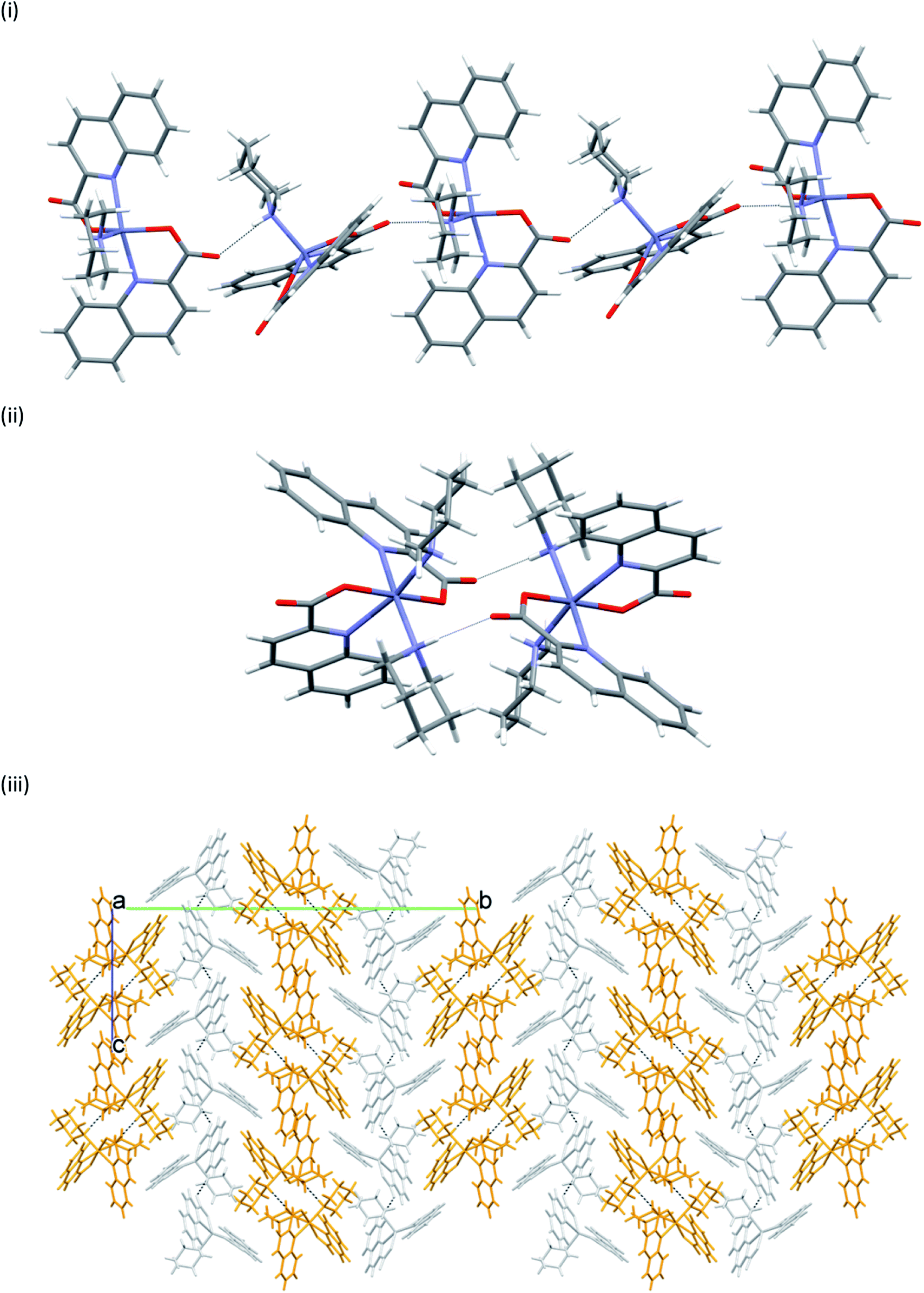
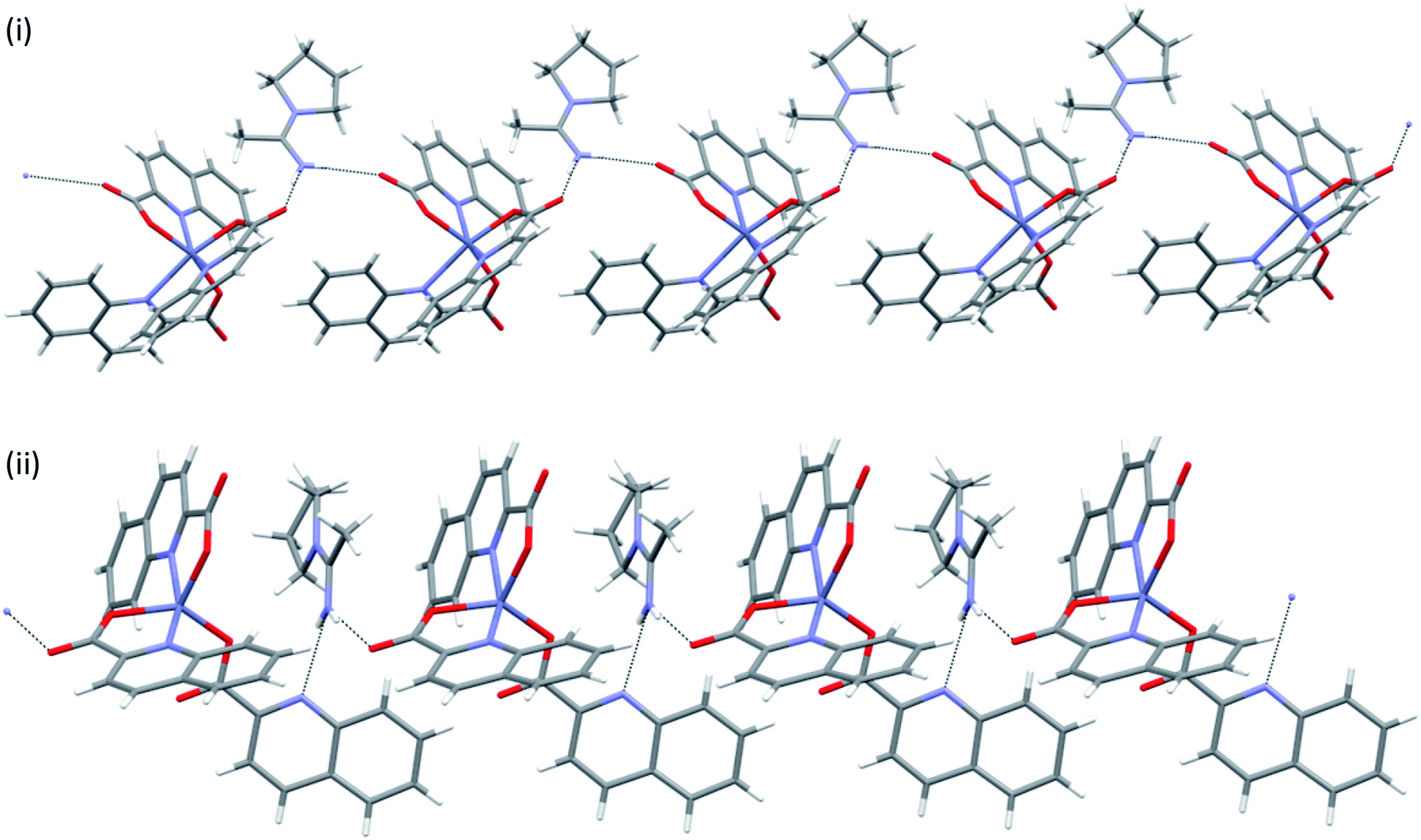
In many structures, the chains pack with the formation of channels that accommodate solvent molecules of crystallization. With their location in channels, facile escape routes are provided. This explains the instability of the crystals of all solvates when taken out from mother liquors. pyroH[Zn(quin)3]·CH3CN merits further comment, as it displays a true polymorphism. It crystallizes in a monoclinic P21/n (8monoclinic) or in a triclinic P (8triclinic) cell. The dimensions of the [Zn(quin)3]− ions in its polymorphs are essentially the same. A major difference between the polymorphs may be observed in intermolecular connectivity, shown in Fig. 10. In the monoclinic polymorph, four N–H(pyroH+)⋯COO− contacts with lengths not longer than 2.776(4) Å link two [Zn(quin)3]− ions with two pyrrolidinium cations into a cyclic motif, denoted as R44(20). In the triclinic polymorph, one N–H⋯COO− contact is longer, it amounts to 2.926(3) Å. The complex anions and the pyroH+ cations are linked into infinite chains, denoted as C22(8). The chains pack in a parallel fashion forming channels that accommodate solvent molecules of acetonitrile.
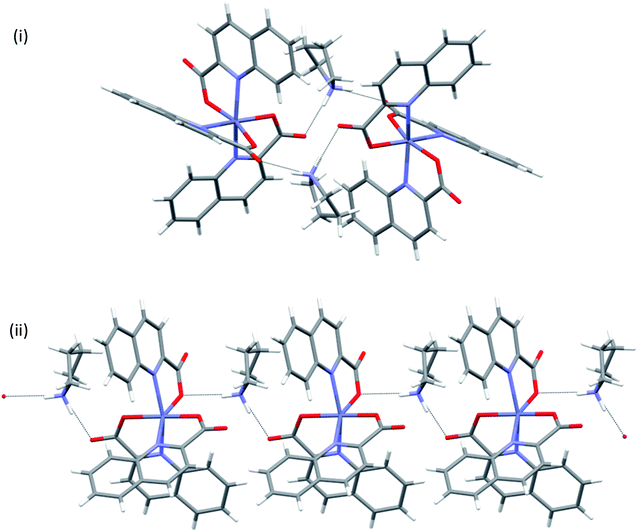 |
| | Fig. 10 Connectivity patterns in polymorphic forms of pyroH[Zn(quin)3]·CH3CN: (i) a cluster of two complex anions and two pyroH+ ions in 8monoclinic, and (ii) a short section of a chain of alternating complex anions and pyroH+ cations in 8triclinic. | |
Drawings of supramolecular motifs in other compounds are given in ESI.†
DFT calculations on the polymorphic forms of pyroamH[Zn(quin)3] (11)
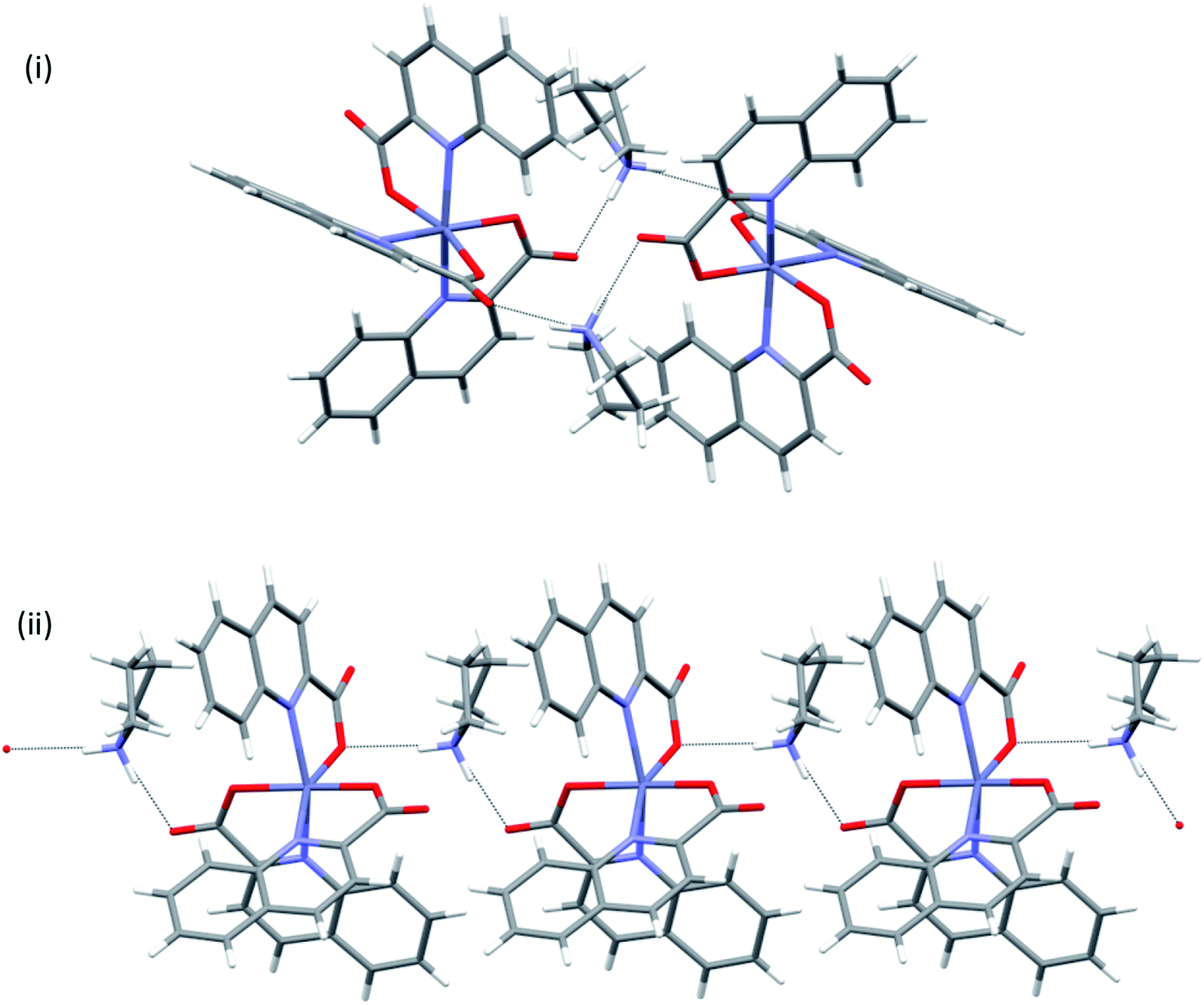
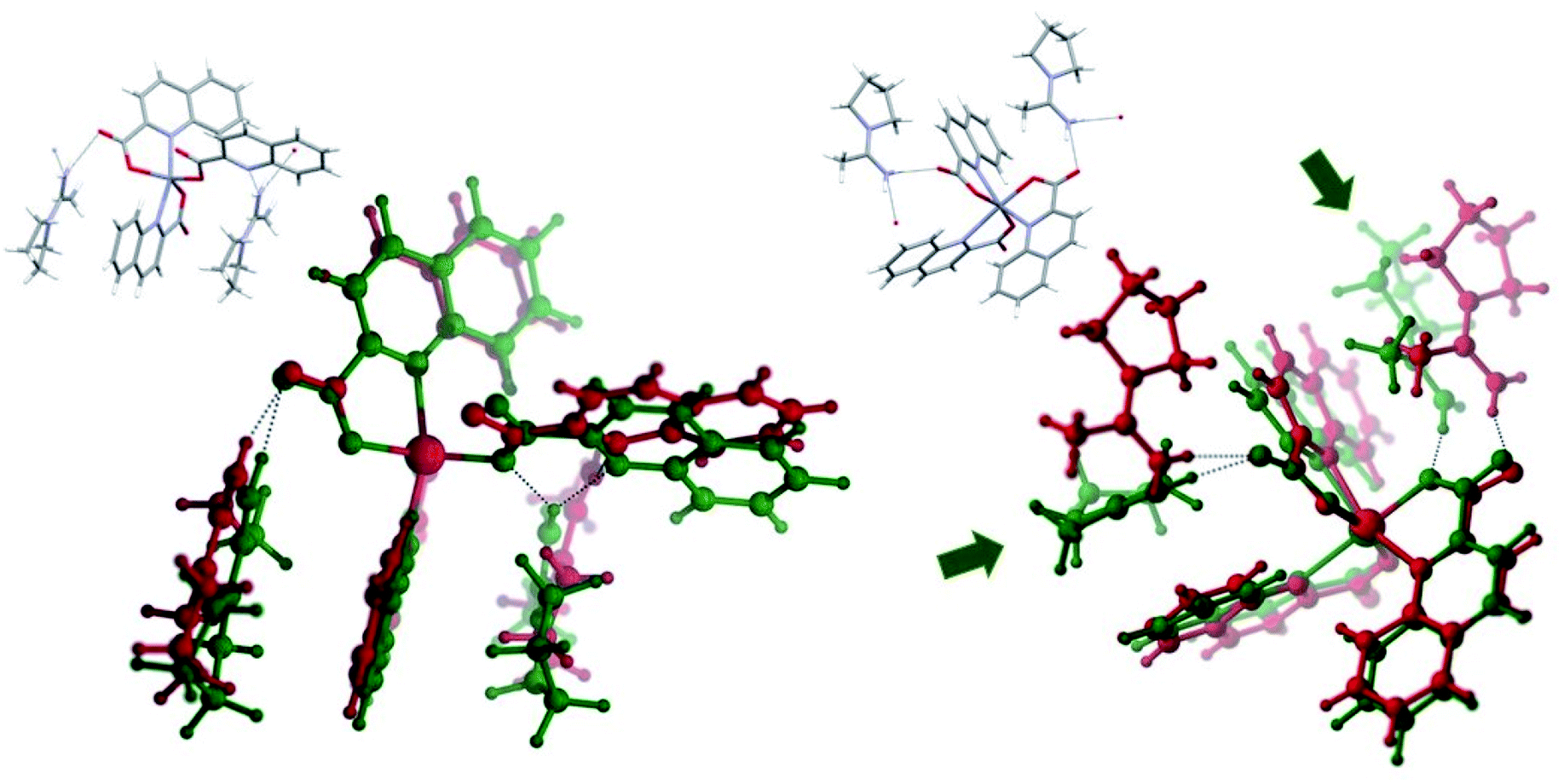
pyroamH[Zn(quin)3] (11) was also found in two polymorphic forms, referred to as 11triclinic and 11monoclinic. A major difference between the polymorphs exists in their [Zn(quin)3]− ions. The anion of the triclinic form features a five-coordinate environment of zinc(II), consisting of two bidentate chelating and a monodentate quinaldinate. The latter is bound through the carboxylate oxygen. For convenience, this isomer was labelled as 5-[Zn(quin)3]−. The same ion, the five-coordinate [Zn(quin)3]−, was found in pipeamH[Zn(quin)3] (5). Conversely, in the [Zn(quin)3]− ion of the monoclinic form, all three quinaldinates are bidentate chelating and the metal ion is in a six-coordinate environment. The complex anion was labelled as 6-[Zn(quin)3]−. The six-coordinate [Zn(quin)3]− ions were also found in salts with protonated amines: 3, 8monoclinic, 8triclinic and 13. The two [Zn(quin)3]− structural isomers, a remarkable demonstration of a flexidentate80 behaviour of the quinaldinate ligand, were subjected to theoretical DFT calculations. Since the calculations were performed also on short hydrogen bonded segments, the X-ray structures of polymorphs were inspected in a more detailed manner. First observation pertains to their densities which do not differ significantly and as such speak of a similar packing efficiency. Differences in the constituent [Zn(quin)3]− ions entail a different intermolecular connectivity. In both structures, hydrogen bonding between the [Zn(quin)3]− ions and the pyroamH+ countercations produces infinite chains of alternating anions and cations, shown in Fig. 11. The six-coordinate [Zn(quin)3]− ions of 11monoclinic form two hydrogen bonds, both of the N–H⋯COO− type, with amidinium cations. The linking motif may be described as C22(10). Within a chain, a methyl hydrogen of the pyroamH+ ion makes a C–H⋯π interaction with the nearest quinaldinate, i.e., H⋯Cg = 2.87 Å, C–H⋯Cg = 120°.81 The chains, packed in a parallel fashion along the a-axis, are held together with π⋯π stacking [parameters of the shortest interaction are Cg⋯Cg = 3.648(2) Å and a dihedral angle of 4.2(2)°] and C–H⋯π interactions [H⋯Cg = 2.69 Å and C–H⋯Cg = 168°].81,82 Hydrogen bonds of the five-coordinate [Zn(quin)3]− ion in 11triclinic are markedly different as they involve a non-coordinated nitrogen of the monodentate quinaldinate. The [Zn(quin)3]− ion forms two hydrogen bonds with the pyroamH+ ions, one is of the N–H⋯COO− type and the other is of the N–H⋯N type. The repeating pattern is described as C22(11). Within a chain, a methylene hydrogen of the pyroamH+ ion makes a short contact to one aromatic ring, i.e., H⋯Cg = 2.88 Å and C–H⋯Cg = 154°. The other two quinaldinates are engaged in the π⋯π stacking with aromatic rings belonging to adjacent chains. In the shortest interaction, the centroid⋯centroid distance amounts to 3.578(7) Å and a dihedral angle is 2.4(6)°.
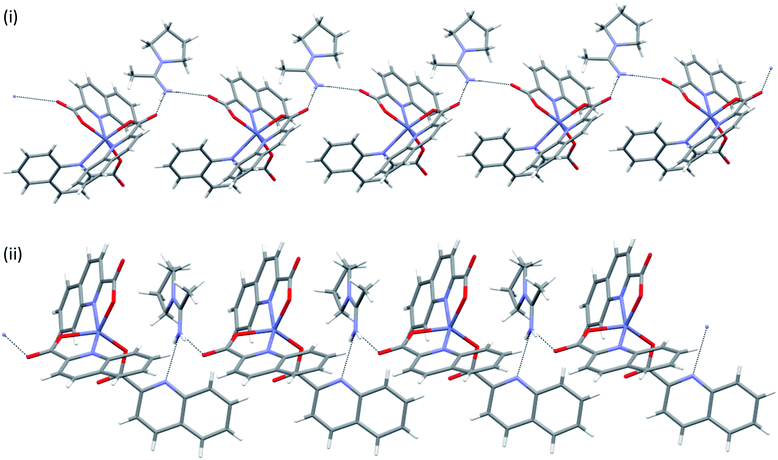 |
| | Fig. 11 Infinite chains in pyroamH[Zn(quin)3] polymorphs: (i) 11monoclinic, (ii) 11triclinic. | |
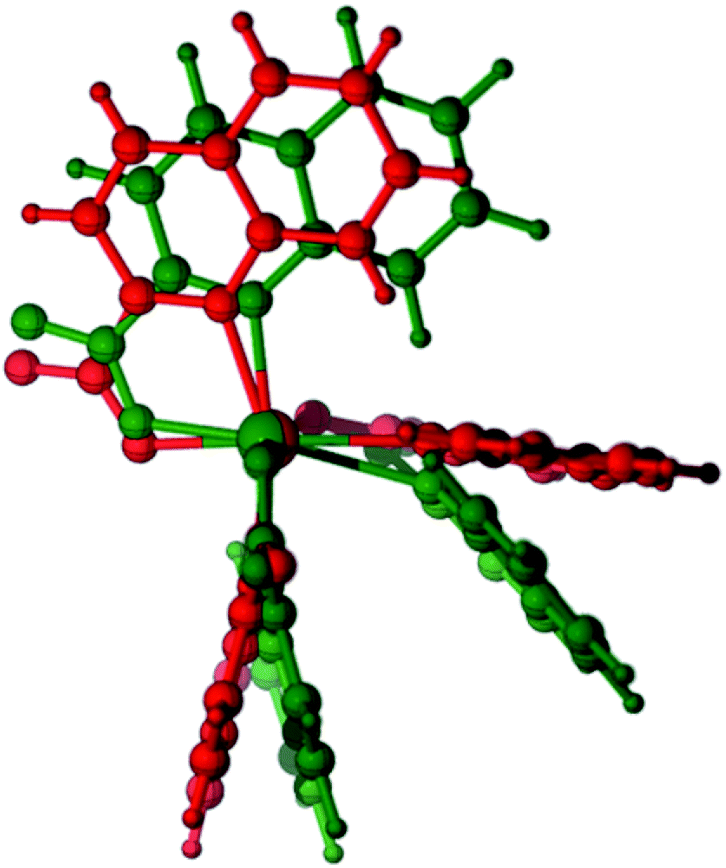
The relative stability of the six- and five-coordinate [Zn(quin)3]− complexes in acetonitrile was assessed with DFT calculations {SMD(acetonitrile)-ωB97XD/6-31+g(d,p)}. Thus, while the optimized geometry of 5-[Zn(quin)3]− does not depart significantly from the solid state geometry of the ion, taken as the starting point for the optimization (RMS deviation = 0.44 Å, excluding hydrogens), optimization of 6-[Zn(quin)3]− results in a severe distortion of the solid state geometry with the orientation of two quinaldinate ligands changing from an almost perpendicular one to a quasi-parallel one, i.e., a dihedral angle between the planes changes from 86.6° to 27.6° (Fig. 12). This distortion hints at, as we shall comment later, an important role of noncovalent π⋯π and C–H⋯π interactions, including intramolecular and anion–cation interactions, in the overall stability of both polymorphs of pyroamH[Zn(quin)3].
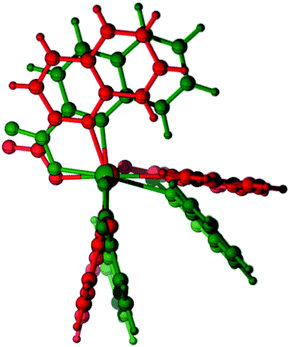 |
| | Fig. 12 Optimized geometry of 6-[Zn(quin)3]− (green) overlaid with the X-ray solid state geometry (red). | |
In bulk acetonitrile, as expected, 6-[Zn(quin)3]− is the most stable by 5.6 kcal mol−1 (ΔZPE). Moreover, 5-[Zn(quin)3]− may convert into its coordinatively saturated isomer through an almost barrierless process, according to the relaxed scan of the Potential Energy Surface (PES) (see the ESI for details†). Interestingly, a new minimum, labelled 5b-[Zn(quin)3]−, was located on the way from the five- to the six-coordinate species. In this new minimum, again, two quinaldinate ligands are found with their planes in a nearly parallel alignment (the corresponding dihedral angle amounts to 9.4°). The stability order (with relative zero-point-energies in kcal mol−1) is as follows: 6-[Zn(quin)3]− (0.00) > 5b-[Zn(quin)3]− (1.79) > 5-[Zn(quin)3]− (5.56). With the 5b-[Zn(quin)3]− intermediate being 1.8 kcal mol−1 above the six-coordinate species, it can be inferred that π⋯π stacking of quinaldinates stabilizes the system by ca. 4 kcal mol−1. Atoms In Molecules (AIM)47,48 and Non Covalent Interactions (NCI)83 analyses clearly reveal this type of contacts in 5b-[Zn(quin)3]−. Fig. 13 shows its optimized geometry, superimposed on its molecular graph and NCI plot, with yellow dots and orange traces representing bond critical points (bcps) and bond paths respectively, green surfaces corresponding to NCIs. Up to four bond paths and bcps connect one of the two κ2-bound quinaldinates with the κ1-quinaldinate, revealing weak, closed shell interactions between the aromatic rings. All four bcps between quinaldinates lie on the green surface, which, in the colour code used for the NCI plot,49 corresponds to weak attractive interactions.
 |
| | Fig. 13 Optimized geometry of 5b-[Zn(quin)3]− with AIM bcps and bond paths, and NCI plot revealing weak attractive interactions. | |
Since the X-ray structures of the pyroamH[Zn(quin)3] polymorphs have revealed hydrogen bonds between the anionic complexes and the pyroamH+ countercations, two short chains with each containing two cations and the anionic complex, either a five- or a six-coordinate one, were freely optimized in acetonitrile. Fig. 14 compares the X-ray determined (red) and DFT-optimized (green) geometries of these chain fragments. In both cases, in the optimized geometry at least one hydrogen bond between each cation and the [Zn(quin)3]− ion is maintained. The most conspicuous difference between the solid state and the calculated geometry is observed for 11monoclinic where one pyroamH+ cation forms a hydrogen bond with the zinc-coordinated carboxylate oxygen (green arrow). Furthermore, both cations shift considerably their relative positions from the solid state geometry and form new C–H⋯π interactions with quinaldinates, as revealed by AIM and NCI analyses (see the ESI for details†). In these contacts, the shortest H⋯C(sp2) distance is 2.85 Å, the H⋯Cg distances occupy a 2.70–2.78 Å range, and the C–H⋯Cg angles are 117.5–125.8°. The latter descriptors are consistent with weak interactions.81,82 The discrepancies between the experimentally observed and the calculated geometries are mostly due to the absence of the extended contacts array in the chain segments. Nevertheless, the calculations inform of the importance of the noncovalent interactions in this system and confirm the anion–cation hydrogen bonding pattern.
 |
| | Fig. 14 Optimized (green) and X-ray solid state (red) chain fragments of 11triclinic (left) and 11monoclinic (right). The insets correspond to the solid state structures. | |
More importantly, the zero-point energy difference between the short chain of 11monoclinic and that of 11triclinic is reduced to 1.6 kcal mol−1, with the six-coordinate species still being the most stable. Although when ΔG is considered, the stability order is reversed and the five-coordinate system appears as the most stable by 0.7 kcal mol−1. We completed the above study with NCI analysis at the promolecular level of frozen fragments of the crystal structures of 11monoclinic and 11triclinic which has confirmed an extended array of weak attractive interactions: (i) intermolecular π⋯π stacking between quinaldinate ligands of neighbouring complex anions, (ii) C–H⋯π interactions, and (iii) N–H⋯O/N hydrogen bonds between the pyroamH+ cations and the complex anions with the latter having an obvious electrostatic component. These results suggest that the isolation of the structural isomers of the [Zn(quin)3]− ion is a likely result of a fine balance between the difference in the stability of the five- and six-coordinate complex and the attractive noncovalent interactions in the crystal lattice.
Infrared spectroscopy
The most intense absorption bands in the spectra of title compounds are due to vibrations of the carboxylate moiety, commonly known as the νas(COO−) and νs(COO−) vibrations.84 Their positions are listed in Table 7. The νas(COO−) vibrations fall between 1646 and 1612 cm−1, whereas the νs(COO−) vibrations may be seen as several bands in the 1394–1324 cm−1 region. Although the quinaldinate binding mode is not the same in all compounds, the binding manner of the carboxylate moiety is. In all, the carboxylate is engaged in coordination through one oxygen atom, whereas the other participates in hydrogen bonding interactions. Monodentate carboxylate coordination is typically reflected in a large difference between the position of the νas(COO−) and νs(COO−) absorption bands, conveniently denoted as a splitting value Δ.84 In the spectra of our compounds, the splitting values occupy a 264–313 cm−1 interval, which agrees with a monodentate binding mode.
Table 7 The νas(COO−) and νs(COO−) absorption bands [cm−1] in the spectra of title compounds
| Compound |
νas(COO−)a |
νs(COO−)a |
| sh = shoulder. |
| 1 |
1632 |
1365, 1349 |
| 2 |
1646, 1634 |
1368sh, 1359, 1343 |
| 3 |
1631 |
1389, 1375sh, 1355, 1339 |
| 4a |
1635 |
1352sh, 1335 |
| 4b |
1644 |
1365sh, 1355, 1343, 1337 |
| 5 |
1627 |
1369, 1345 |
| 7 |
1633 |
1362, 1349sh |
| 8 |
1612 |
1367, 1342 |
| 9 |
1615 |
1388, 1376, 1351 |
| 10a |
1637 |
1354, 1339, 1324 |
| 10b |
1645 |
1365sh, 1355, 1344, 1333 |
| 11 |
1613 |
1387sh, 1373, 1359sh, 1347 |
| 12 |
1631 |
1365 |
| 13 |
1623sh, 1613 |
1394, 1371, 1342 |
A band of weak to medium intensity at ca. 3200 cm−1 in the spectra of 1, 2, 7 and 12 is indicative of the presence of amine ligands (Table 8). The band finds its origin in the N–H stretching vibrations.85 In the spectrum of [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (2) which contains two complex species, two bands are observed in this region. Amine ligands are further corroborated by medium absorptions of the methylene C–H stretching vibrations. The spectra of salts with protonated amines, 3, 8, 9 and 13, display very broad bands in the 3500–2400 cm−1 region. The bands are due to the N–H stretching vibrations of the NH2+ group which is engaged in hydrogen bonding interactions.85 The corresponding bending vibrations of the NH2+ group are hidden by the carboxylate νas(COO−) band. In the spectrum of piperidinium nitrate, the latter absorption occurred at 1619 cm−1.86 The spectra of amidine compounds, 4a, 4b, 10a and 10b, reveal a shift of the ν(N–H) bands to higher frequencies. They are found in the 3300–3262 cm−1 region as compared to 3221–3122 cm−1 for complexes with amines. A series of Pt(IV) complexes with amidines, formed from propionitrile and nitroanilines, revealed ν(N–H) bands at even higher wavenumbers, 3435–3289 cm−1.69 The presence of amidine ligands, pipeam and pyroam, leaves another imprint upon the spectra, a strong and split absorption band at ca. 1580 cm−1 that may be associated with the stretching absorption of the CN amidine bond.87
Table 8 Typical amine and amidine absorption bands [cm−1]
| Compound |
ν(N–H) |
ν(C–H) |
ν(CN) |
| sh = shoulder. |
| Amine complexes |
| 1 |
3221 |
2937, 2869, 2854 |
— |
| 2 |
3215, 3122 |
2931, 2853 |
— |
| 7 |
3188 |
2953, 2873 |
— |
| 12 |
3180 |
2964, 2944, 2925, 2850 |
— |
|
| Amidine complexes |
| 4a |
3282 |
2934, 2856 |
1584, 1569, 1562sha |
| 4b |
3300 |
2971, 2864 |
1584, 1570 |
| 10a |
3262 |
2929, 2873 |
1591, 1570 |
| 10b |
3284 |
2965, 2871 |
1590, 1571 |
The spectra of compounds with protonated amidines, 5 and 11, are different. A very broad band appears at ca. 3000 cm−1 that obscures the ν(N–H) absorptions. A new band at 1658 cm−1 may be observed in the spectra of both compounds. The literature reports that in amidine salts with the CNH2+ structural fragment an interaction occurs between the stretching of the CN bond and the NH2+ deformation vibration.85 As a result, the spectra of amidinium salts lack the ν(CN) band that was observed for amidine complexes at 1580 cm−1.
Many of the title compounds contain solvent molecules of crystallization. Because of the instability of the acetonitrile solvates outside of the mother liquors, the solvent bands are usually absent from the spectra. Surprisingly, for 4b and 10b, traces of chloroform could be seen as a band of moderate intensity at ca. 660 cm−1. Its position agrees well with the literature data.88 Remaining chloroform bands cannot be unambiguously assigned because of the overlap with other absorption bands.
1H NMR spectroscopy
The 1H NMR spectra confirm the composition of the complex species. The integrals of the quinaldinate and amine or amidine signals are in the correct ratio. The chemical shifts for both the quinaldinate and piperidine ligands in the spectra of trans-[Zn(quin)2(pipe)2]·2CH3CN (1) and [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2] (2) are the same suggesting that their DMSO-d6 solutions contain the same complex, most likely an already known DMSO complex.16 The signals of the quinaldinate ligands for the [Zn(quin)3]− compounds, where zinc(II) features in the solid state a six-numbered coordination environment, are slightly upfield when compared to the spectra of the [Zn(quin)2(amine)2] complexes, i.e., 8.60–7.64 ppm vs. 8.94–7.81 ppm, respectively. It is to be noted that the spectrum of pipeamH[Zn(quin)3] (5) with a five-numbered zinc(II) coordination has quinaldinate signals at almost the same position. The latter agrees either with the presence of one form of the [Zn(quin)3]− ion in the solution or the DMSO reaction of both [Zn(quin)3]− forms yielding the same product. The spectra of pipeamH[Zn(quin)2(CH3COO)]·acetamide (6) and pyroH[Zn(quin)2Cl] (9) compounds, which contain Zn(II) complexes with two quinaldinate ligands and a third negatively charged ligand, are ostensibly similar: the quinaldinate signals, five of them, are for both in the 8.78–7.76 ppm range. Same range, 8.78–7.80 ppm, is observed for quinaldinates in the amidine compounds 4a and 10a. The amidine ligands leave a characteristic fingerprint in the spectra of both. Pipeam is characterized by a set of five resonances. The signals at 3.25–3.23, 1.46–1.42 and 1.23 ppm are assigned to methylene groups with the first listed multiplet belonging to hydrogen atoms of the methylene groups that are closest to the ring nitrogen. The resonance at 6.98 ppm belongs to the azomethine hydrogen and the one at 1.90 ppm to the methyl group. Similar chemical shift is observed for the pyroam methyl group in the spectrum of 10a, i.e., at 1.94 ppm. The resonances at 3.28, 3.10 and 1.76–1.73 ppm may be ascribed to the pyroam methylene groups. It is of interest to note that two signals may be observed for the methylene groups that are closest to nitrogen. Upon protonation, the amidine signals shift downfield, i.e., as exemplified by the spectrum of pipeamH[Zn(quin)3] (5) with resonances at 3.51–3.49, 2.23 and 1.62–1.55 ppm. A broad resonance at 9.01 ppm is probably due to the NH2+ moiety.
Conclusions
A series of compounds obtained from the reaction of [Zn(quin)2(H2O)], a selected secondary cyclic amine and acetonitrile has demonstrated two important properties of the zinc(II) coordination chemistry: the ability of zinc(II) to adopt different coordination environments and to activate normally inert organic substrates, in our case, acetonitrile towards the nucleophilic attack by the amine. The products of the straightforward substitution of water by amine include all possible species, from the mono-amine complex with the composition [Zn(quin)2(amine)] to the two geometric isomers of the bis-amine complex, cis- and trans-[Zn(quin)2(amine)2]. In the presence of zinc(II), piperidine and pyrrolidine reacted with acetonitrile to corresponding amidines. The resulting piperidinoacetamidine and pyrrolidinoacetamidine were found as ligands in the neutral [Zn(quin)2(amidine)] complexes, which were unknown prior to this study. The presence of zinc(II) compounds was confirmed as a prerequisite for the acetonitrile reaction with amine. In addition, the reactions had to be carried out either under forcing conditions or in the presence of ethanol or 2-propanol. Morpholine, a homologous amine, did not produce amidine. In autoclaves at 105 °C, its decomposition to ammonia and coordination of the latter to zinc(II) took place. The homoleptic anionic complex [Zn(quin)3]− was found in the solid state in two isomeric forms which differ in the quinaldinate binding manner. The DFT study has shown the isomer with a six-coordinate zinc(II) and all quinaldinates bidentate chelating to be by 5.6 kcal mol−1 more stable than the one with a five-coordinate zinc(II) and one of the three ligands bound in a monodentate manner. The conversion between the two in the solution seems to be almost barrier-free. Intermolecular interactions in the crystal structures of both have a stabilizing effect which reduces the energy difference between the pair.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We would like to express gratitude for financial support from the Junior Researcher Grant for N. P. and the Program Grant P1-0134 of the Slovenian Research Agency. We thank MICIU (Grant CTQ2016-75193-P) for financial support. M. M. A. thanks the MICIU for a research fellowship. The Supercomputing Centre of Galicia (CESGA) and the Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada are acknowledged for providing the computing time. Dr Romana Cerc Korošec is gratefully acknowledged for carrying out the MS analyses of the morpholine mixtures.
References
- J. Raulin, Ann. Sci. Nat., 1869, 11, 93–99 Search PubMed.
- D. Keilin and T. Mann, Nature, 1939, 144, 442–443 CrossRef CAS.
- A. S. Prasad, J. A. Halsted and M. Nadimi, Am. J. Med., 1961, 31, 532–546 CrossRef CAS PubMed.
- A. S. Prasad, A. Miale, Z. Farid, H. H. Sandstead and A. R. Schulert, J. Lab. Clin. Med., 1963, 61, 537–549 CAS.
- C. Andreini, L. Banci, I. Bertini and A. Rosato, J. Proteome Res., 2006, 5, 196–201 CrossRef CAS PubMed.
- B. L. Vallee and D. S. Auld, Biochemistry, 1990, 29, 5647–5659 CrossRef CAS PubMed.
- B. L. Vallee and K. H. Falchuk, Physiol. Rev., 1993, 73, 79–118 CrossRef CAS PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced inorganic chemistry, Wiley, New York, 6th edn, 1999 Search PubMed.
- K. A. McCall, C.-C. Huang and C. A. Fierke, J. Nutr., 2000, 130, 1437S–1446S CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- G. J. Fosmire, Am. J. Clin. Nutr., 1990, 51, 225–227 CrossRef CAS PubMed.
- L. M. Plum, L. Rink and H. Haase, Int. J. Environ. Res. Public Health, 2010, 7, 1342–1365 CrossRef CAS PubMed.
- N. Okabe and Y. Muranishi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m244–m246 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- Z.-Y. Yue, C. Cheng, P. Gao and P.-F. Yan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m82–m84 CrossRef CAS.
- W. Z. Zhang, M. Shuang, M. C. Zhu, L. Lei and E. J. Gao, Russ. J. Coord. Chem., 2009, 35, 874–878 CrossRef CAS.
- D. Dobrzyńska, T. Lis and L. B. Jerzykiewicz, Inorg. Chem. Commun., 2005, 8, 1090–1093 CrossRef.
- T. A. Zevaco, H. Görls and E. Dinjus, Inorg. Chim. Acta, 1998, 269, 283–286 CrossRef CAS.
- U. Brand and H. Vahrenkamp, Inorg. Chem., 1995, 34, 3285–3293 CrossRef CAS.
- B. Modec, Crystals, 2018, 8, 52 CrossRef.
- N. Podjed, P. Stare, R. Cerc Korošec, M. M. Alcaide, J. López-Serrano and B. Modec, New J. Chem., 2020, 44, 387–400 RSC.
- P. Abhayawardhana, P. A. Marzilli, T. Perera, F. R. Fronczek and L. G. Marzilli, Inorg. Chem., 2012, 51, 7271–7283 CrossRef CAS PubMed.
- S. S. Rozenel, J. B. Kerr and J. Arnold, Dalton Trans., 2011, 40, 10397–10405 RSC.
- X. Bao and E. M. Holt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1655–1657 CrossRef.
- G. V. Boyd, in The Chemistry of Amidines and Imidates, ed. S. Patai and Z. Rappoport, Wiley, Chichester, UK, 1991, vol. 2 Search PubMed.
- A. A. Aly, S. Bräse and M. A.-M. Gomaa, Arkivoc, 2018, 6, 85–138 Search PubMed.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- M. R. Willcott, J. Am. Chem. Soc., 2009, 131, 13180 CrossRef CAS.
- Agilent, CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- NIST Mass Spectrometry Data Center, W. E. Wallace, director, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G.Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revisions B.01 and E.01. Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. F. W. Bader, Atom in Molecules: A Quantum Theory, Clarendon Press, Oxford, UK, 1994 Search PubMed.
-
(a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed;
(b) Multiwfn 3.6, http://sobereva.com/multiwfn/ Search PubMed.
- C. Y. Legault, CYLview, 1.0b, http://www.cylview.org Search PubMed.
-
(a) W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS;
(b) VMD 1.9.3, https://www.ks.uiuc.edu/Research/vmd/ Search PubMed.
- J. C. Grivas and A. Taurins, Can. J. Chem., 1961, 39, 761–764 CrossRef CAS.
- J. E. Coleman, Curr. Opin. Chem. Biol., 1998, 2, 222–234 CrossRef CAS PubMed.
- B. Modec, N. Podjed and N. Lah, Molecules, 2020, 25, 1573 CrossRef PubMed . The IR spectrum of 7 is identical to that of homologous copper(II) compound, trans-[Cu(quinal)2(pyro)2], whose true composition was identified by X-ray structure analysis on a single crystal.
- J. E. Mills, C. A. Maryanoff, D. F. McComsey, R. C. Stanzione and L. Scott, J. Org. Chem., 1987, 52, 1857–1859 CrossRef CAS.
- V. S. Rawat, T. Bathini, S. Govardan and B. Sreedhar, Org. Biomol. Chem., 2014, 12, 6725–6729 RSC.
- A. P. Kourounakis, D. Xanthopoulos and A. Tzara, Med. Res. Rev., 2020, 40, 709–752 CrossRef CAS PubMed.
- G. Rousselet, P. Capdevielle and M. Maumy, Tetrahedron Lett., 1993, 34, 6395–6398 CrossRef CAS.
- A. A. Aly and A. M. Nour-El-Din, Arkivoc, 2008, 2008, 153–194 Search PubMed.
- P. Oxley, M. W. Partridge and W. F. Short, J. Chem. Soc., 1947, 1110–1116 RSC.
- J. H. Forsberg, V. T. Spaziano, T. M. Balasubramanian, G. K. Liu, S. A. Kinsley, C. A. Duckworth, J. J. Poteruca, P. S. Brown and J. L. Miller, J. Org. Chem., 1987, 52, 1017–1021 CrossRef CAS.
- F. Xu, J. Sun and Q. Shen, Tetrahedron Lett., 2002, 43, 1867–1869 CrossRef CAS.
- J. Wang, F. Xu, T. Cai and Q. Shen, Org. Lett., 2008, 10, 445–448 CrossRef CAS PubMed.
- R. A. Michelin, M. Mozzon and R. Bertani, Coord. Chem. Rev., 1996, 147, 299–338 CrossRef CAS.
- V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef CAS PubMed.
- S. G. Feng, P. S. White and J. L. Templeton, Organometallics, 1993, 12, 1765–1774 CrossRef CAS.
- C. S. Chin, D. Chong, B. Lee, H. Jeong, G. Won, Y. Do and Y. J. Park, Organometallics, 2000, 19, 638–648 CrossRef CAS.
- U. Belluco, F. Benetollo, R. Bertani, G. Bombieri, R. A. Michelin, M. Mozzon, A. J. L. Pombeiro and F. C. Guedes da Silva, Inorg. Chim. Acta, 2002, 330, 229–239 CrossRef CAS.
- A. N. Chernyshev, N. A. Bokach, P. V. Gushchin, M. Haukka and V. Y. Kukushkin, Dalton Trans., 2012, 41, 12857–12864 RSC.
- S. Roy, S. Bhattacharya and S. Chattopadhyay, J. Coord. Chem., 2016, 69, 112–122 CrossRef CAS.
- A. V. Ivanov, A. S. Zaeva, A. V. Gerasimenko and T. A. Rodina, Russ. J. Coord. Chem., 2008, 34, 688–698 CrossRef CAS.
- A. J. Mountford, S. J. Lancaster, S. J. Coles, P. N. Horton, D. L. Hughes, M. B. Hursthouse and M. E. Light, Organometallics, 2006, 25, 3837–3847 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- A. Pidcock, R. E. Richards and L. M. Venanzi, J. Chem. Soc. A, 1966, 1707–1710 RSC.
- L. M. Engelhardt, P. C. Junk, C. L. Raston, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1996, 3297–3301 RSC.
- D. Montagner, E. Zangrando, G. Borsato, V. Lucchini and B. Longato, Dalton Trans., 2011, 40, 8664–8674 RSC.
- A. B. Kremer, K. M. Osten, I. Yu, T. Ebrahimi, D. C. Aluthge and P. Mehrkhodavandi, Inorg. Chem., 2016, 55, 5365–5374 CrossRef CAS PubMed.
- S. Majumder, L. Mandal and S. Mohanta, Inorg. Chem., 2012, 51, 8739–8749 CrossRef CAS.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef PubMed.
- D. H. Busch, J. Chem. Educ., 1964, 41, 77–85 CrossRef CAS.
- H. Suezawa, T. Yoshida, Y. Umezawa, S. Tsuboyama and M. Nishio, Eur. J. Inorg. Chem., 2002, 2002, 3148–3155 CrossRef.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Wiley, Hoboken, NJ, 6th edn, 2009 Search PubMed.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, San Diego, CA, 3rd edn, 1990 Search PubMed.
- I. Němec, Z. Macháčková, P. Vaněk and Z. Mička, Collect. Czech. Chem. Commun., 2006, 71, 207–214 CrossRef.
- L. E. Clougherty, J. A. Sousa and G. M. Wyman, J. Org. Chem., 1957, 22, 462 CrossRef CAS.
- T. G. Gibian and D. S. McKinney, J. Am. Chem. Soc., 1951, 73, 1431–1434 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ORTEP drawings of [Zn(quin)2(pipeam)] and trans-[Zn(quin)2(morph)2] (Fig. S1 and S2); overlay of [Zn(quin)2(pipeam)] molecules and [Zn(quin)3]− ions (Fig. S3 and S4); geometric parameters of amidine ligands and amidinium cations (Table S1); lists of hydrogen bonds (Tables S2 and S3); intermolecular interactions (Fig. S5–S16); additional computational details (Fig. S17–S20); MS of the morpholine mixtures (Fig. S21); IR spectra of 1 to 13 (Fig. S22–S35); NMR spectra and chemical shifts of 1 to 13 (Table S4 and Fig. S36–S48). Files with xyz coordinates of the calculated species. CCDC 1973015–1973031. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra03192e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Barbara Modec
a,
Barbara Modec