DOI:
10.1039/D0RA01962C
(Paper)
RSC Adv., 2020,
10, 18231-18244
In(OTf)3-catalyzed intramolecular hydroarylation of α-phenylallyl β-ketosulfones – synthesis of sulfonyl 1-benzosuberones and 1-tetralones†
Received
1st March 2020
, Accepted 5th May 2020
First published on 13th May 2020
Abstract
In(OTf)3-catalyzed intramolecular hydroarylation of α-phenylallyl β-ketosulfones provides sulfonyl 1-benzosuberones and 1-tetralones in moderate to good yields in refluxing (CH2Cl)2 under open-vessel and easy-operation reaction conditions. A plausible mechanism is proposed and discussed. This highly regioselective protocol provides an atom-economic ring-closure route.
Introduction
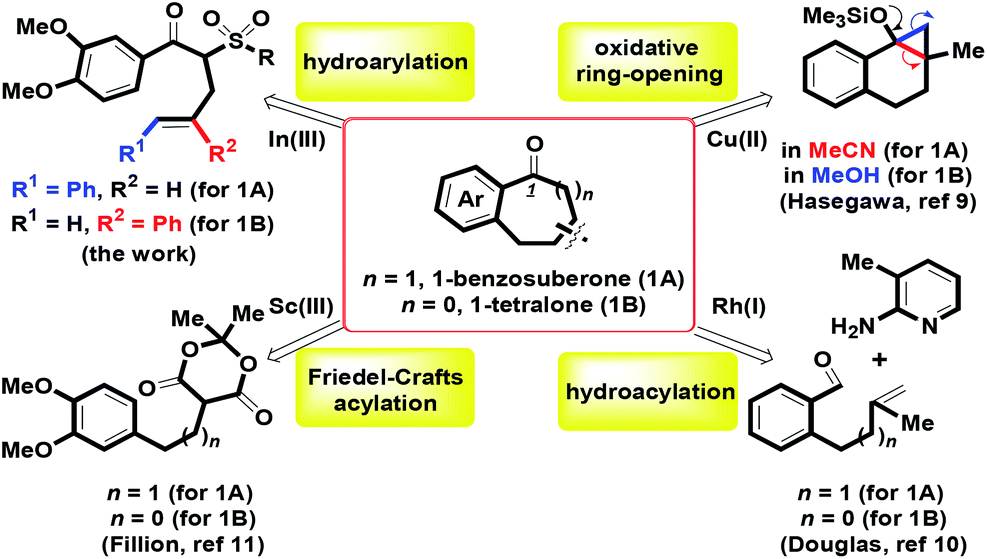
Substituted benzocycles with a medium ring (six and seven-membered rings) commonly serve as core structures in many potential bioactive molecules,1,2 natural products3,4 and versatile synthetic blocks.5,6 According to our recent reports, efficient synthesis of diverse tetralins and benzosuberans has been investigated via BF3·OEt2 mediated stereocontrolled formal (4 + 2) and (5 + 2) cycloaddition of 4-alkenols with veratrol.7 For heterocyclic ring frameworks, we also provided a one-pot route for benzofused six- and seven-membered oxacycles by mCPBA-mediated intramolecular oxidative annulation of ortho-crotyl or cinnamyl arylaldehydes.8 On the basis of previous experience, herein, the major works were focused on the preparation of 1-benzosuberone 1A and 1-tetralone 1B using the same synthetic route. By transition metal-catalyzed and Brønsted acid-promoted intramolecular benzannulation, a considerable number of attempts have been developed to prepare the skeletons of 1-benzosuberone and 1-tetralone via a kind of synthetic method.9–11 For example, by the involvement of different solvents (MeCN and MeOH), Hasegawa and co-workers demonstrated that Cu(BF4)2-catalyzed regioselective oxidative ring-opening of benzofused bicyclic cyclopropyl silyl ethers provided two benzofused ring systems of 1A and 1B (Scheme 1).9 Douglas et al. investigated whether the ortho-side arm length of benzaldehyde could regulate the hydroacylation of homoallyl (n = 1) or o-allyl (n = 0) with 2-aminopyridine to generate 1A and 1B in the presence of rhodium(I) catalysts.10 By controlling the Sc(III) complex as the Lewis acid, the Fillion group explored efficient Friedel–Crafts acylation of α-arylpropyl and α-arylethyl Meldrum's acid derivatives for the formation of 1A and 1B.11
 |
| | Scheme 1 Intramolecular routes of 1-benzosuberone 1A and 1-tetralone 1B. | |
In spite of these attractive advancements, we envisioned that further investigation of a novel and efficient synthetic method for the formation of 1A and 1B was still highly desired. Herein, we present an In(OTf)3-mediated synthesis of 1-tetralone and 1-benzosuberone via one-pot intramolecular hydroarylation of α-phenylallyl β-ketosulfones in refluxing (CH2Cl)2 via one carbon–carbon bond formation. For the hydroarylation of arenes with multiple bonds, different transition metals-catalyzed reactions become one of the most widely used strategies for diversified carbocycles and heterocycles.12 These major metals include iridium,13 cobalt,14 platinum,15 ruthenium,16 bismuth,17 gold,18 rhodium,19 zinc20 and indium.21 Other recent Brønsted and Lewis acid-mediated routes for atom-economic hydroarylation reactions have been thoroughly investigated.22
Results and discussion
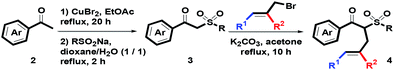
On the basis of our previous reports,23 starting substrates 4 were easily prepared via a three-step route, including (1) CuBr2-mediated α-bromination of oxygenated acetophenones 2 in refluxing EtOAc for 20 h, (2) nucleophilic substitution of the resulting α-bromoacetophenone with RSO2Na in a co-solvent of dioxane and water (v/v = 1/1) at reflux for 2 h and (3) α-phenylallylation of the corresponding β-ketosulfones 3 in the presence of K2CO3 in boiling acetone for 10 h, as shown in Scheme 2.
 |
| | Scheme 2 Synthetic route of starting materials 4. | |
Among our researches on metal triflate-promoted synthetic applications of carbocyclic and heterocyclic skeletons,24,25 especially, the synthesis of substituted pyridazines was accomplished by the In(OTf)3-mediated cyclocondensation of α-propargyl β-ketosulfone with N2H4.24 With the previous synthetic experience in mind, herein, the Lewis acid In(OTf)3 was preferred to examine the hydroarylation of α-phenylallyl β-ketosulfone. The initial study (Table 1) was commenced with the treatment of 3,4-dimethoxyacetophenone (4a, Ar = 3,4-(MeO)2C6H3, R = Tol, R1 = Ph, R2 = H, 1.0 mmol) in (CH2Cl)2 (20 mL) at 25 °C for 20 h using catalytic amounts of In(OTf)3 (10 mol%). However, no desired 5a was detected, and the starting material 1a was recovered in a 90% yield (entry 1). By elevating the reaction temperature to reflux (25 → 84 °C), the yield of 5a was increased to 91% (entry 2).
Table 1 Benzannulation conditionsa

|
| Entry |
Metal triflates |
Temp. |
Solvent |
Time |
5ab (%) |
| The reactions were run on a 1.0 mmol scale with 4a, metal triflate (mol%), temp. (oC), solvent (20 mL), time (h). Isolated yields. No reaction. Complex products. |
| 1 |
In(OTf)3 (10) |
25 |
(CH2Cl)2 |
20 |
—c |
| 2 |
In(OTf)3 (10) |
84 |
(CH2Cl)2 |
20 |
91 |
| 3 |
In(OTf)3 (5) |
84 |
(CH2Cl)2 |
20 |
41 |
| 4 |
In(OTf)3 (15) |
84 |
(CH2Cl)2 |
20 |
80 |
| 5 |
In(OTf)3 (20) |
84 |
(CH2Cl)2 |
20 |
63 |
| 6 |
In(OTf)3 (10) |
101 |
MeNO2 |
20 |
55 |
| 7 |
In(OTf)3 (10) |
77 |
CCl4 |
20 |
61 |
| 8 |
In(OTf)3 (10) |
153 |
DMF |
20 |
—d |
| 9 |
In(OTf)3 (10) |
84 |
(CH2Cl)2 |
15 |
75 |
| 10 |
In(OTf)3 (10) |
84 |
(CH2Cl)2 |
30 |
82 |
| 11 |
Sn(OTf)2 (10) |
84 |
(CH2Cl)2 |
20 |
—c |
| 12 |
Tm(OTf)3 (10) |
84 |
(CH2Cl)2 |
20 |
—c |
| 13 |
Sc(OTf)3 (10) |
84 |
(CH2Cl)2 |
20 |
47 |
| 14 |
Ga(OTf)3 (10) |
84 |
(CH2Cl)2 |
20 |
56 |
| 15 |
Sc(OTf)3 (10) |
84 |
(CH2Cl)2 |
15 |
30 |
| 16 |
AgOTf (10) |
84 |
(CH2Cl)2 |
30 |
72 |
| 17 |
Cu(OTf)2 (10) |
84 |
(CH2Cl)2 |
20 |
78 |
| 18 |
Fe(OTf)3 (10) |
84 |
(CH2Cl)2 |
20 |
86 |
| 19 |
Bi(OTf)3 (10) |
84 |
(CH2Cl)2 |
20 |
84 |
| 20 |
Fe(OTf)3 (10) |
84 |
(CH2Cl)2 |
30 |
70 |
After adjusting the catalytic amounts of In(OTf)3 from 10 to 5, 15 and 20 mol%, however, no better yields of 5a were observed (entries 3–5). This meant that 10 mol% amounts of In(OTf)3 were appropriate to trigger the reaction completely. Solvent screening was performed next. It was found that the reaction had low yields (55% and 61%) in MeNO2 and CCl4, respectively (entries 6 and 7), while no desired product was detected in DMF (entry 8). By changing the reaction time (20 → 15 and 30 h), the yields decreased to 75% and 82% (entries 9 and 10). From these observations, several commercially available metal triflates were examined next including: Sn(OTf)2, Tm(OTf)3, AgOTf, Cu(OTf)2, Sc(OTf)3, Ga(OTf)3, Fe(OTf)3 and Bi(OTf)3. However, no isolation of the desired 5a was observed and only 4a was recovered under Sn(OTf)2 or Tm(OTf)3-mediated reaction (entries 11 and 12). After changing metal triflate to Sc(OTf)3 or Ga(OTf)3, better reactivity (47%, 56%) was observed (entries 13 and 14). To compare with In(OTf)3 and Sc(OTf)3, a diminished time (15 h) showed that Sc(OTf)3 couldn't improve the yield (30%, entries 9 and 15). Subsequently, by replacing metal triflates with AgOTf or Cu(OTf)2, 5a could be isolated in 72% and 78% yields (entries 16 and 17). Furthermore, Fe(OTf)3 and Bi(OTf)3 provided similar yields for In(OTf)3 (entries 18 and 19). In comparison with In(OTf)3 and Fe(OTf)3, an elongated time (30 h) described that Fe(OTf)3 couldn't obtain better yield (70%, entries 10 and 20). From the results, we concluded that In(OTf)3 was a key promoter affecting the benzannulation of α-cinnamyl β-ketosulfone 4a. On the basis of 1H-NMR spectra, the ratios of diastereomer 5a were determined as an approximate ratio of 2/1 (for trans/cis) in entries 1–18. The configuration of major isomer of 5a has to be found out as far as there is a ratio 2/1 of two isomers and one can be isolated from another by crystallization. The stereochemical structure of one isomer 5a with α,δ-trans-configurated centers was determined by single-crystal X-ray analysis.26
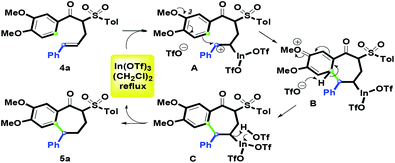
On the basis of the experimental results, a plausible mechanism for the formation of 5a is illustrated in Scheme 3. Initially, 4a reacts with In(OTf)3 to get benzylic carbocation A with an In(OTf)2 arm. Then, the methoxy group on the C-3 position of the aryl ring promotes intramolecular electrophilic annulation to yield B via one carbon–carbon bond (green) formation. Next, the in situ generated triflate anion deprotonates the proton of B to give C under the dehydrogenative aromatization process. Furthermore, 5a is afforded by complexation of In(OTf)2 moiety on C and the resulting TfOH. Subsequently, In(OTf)3 could be regenerated for the next catalytic cycle. For the conversion from C to 5a, Nishizawa et al. reported similar results for the Hg(OTf)2-promoted the synthesis of vinyl-containing tetralins.27
 |
| | Scheme 3 Plausible mechanism. | |
To study the substrate scope and limitation of this route, we applied the optimal conditions established (Table 1, entry 2) to investigate In(OTf)3 mediated intramolecular ring-closure of 4a–4n. As shown in Table 2, 1-benzosuberones 5a–5l were obtained in a yield range of 84–95% except for 5m and 5n. By use of the oxygenated aryl group (Ar = 3,4-(MeO)2C6H3, 3,4-CH2O2C6H3, 3,4,5-(MeO)3C6H2, and 3-MeOC6H4), the R substituents with aliphatic groups, and electron-donating, electron-neutral, electron-withdrawing aromatic groups were well-tolerated (entries 1–12). The diastereomeric ratios of (dr) 5a–5l were determined as an approximate range of 5/1–1/1 based on the 1H-NMR spectrum. On the other hand, when the R group was chosen as Tol and the Ar group was changed from oxygenated arenes to heterocyclic arenes, entries 13 and 14 showed that 2-thienyl and 2-furyl groups could not obtain the desired 5m and 5n and only complex products were observed. These complex cycloadducts with different stereoisomers could result from intramolecular Diels–Alder cycloaddition of 2-thienyl or 2-furyl and the phenylallyl group.
Table 2 Synthesis of 5a–5na
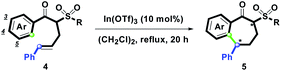
|
| Entry |
4, Ar=, R= |
5b (%, dr) |
| The reactions were run on a 1.0 mmol scale with 4, In(OTf)3 (10 mol%), reflux (84 °C), (CH2Cl)2 (20 mL), 20 h. Isolated yields (ratio: trans/cis). Unknown and unidentified complex products. |
| 1 |
4a, 3,4-(MeO)2C6H3, Tol |
5a, 91, 2/1 |
| 2 |
4b, 3,4-(MeO)2C6H3, Ph |
5b, 90, 4/1 |
| 3 |
4c, 3,4-(MeO)2C6H3, 4-FC6H4 |
5c, 86, 5/1 |
| 4 |
4d, 3,4-(MeO)2C6H3, 4-MeOC6H4 |
5d, 89, 1/1 |
| 5 |
4e, 3,4-(MeO)2C6H3, 4-nBuC6H4 |
5e, 87, 1/1 |
| 6 |
4f, 3,4-(MeO)2C6H3, Me |
5f, 90, 3/1 |
| 7 |
4g, 3,4-(MeO)2C6H3, nBu |
5g, 94, 1/1 |
| 8 |
4h, 3,4-CH2O2C6H3, Tol |
5h, 93, 1/1 |
| 9 |
4i, 3,4,5-(MeO)3C6H2, Tol |
5i, 90, 3/1 |
| 10 |
4j, 3,4,5-(MeO)3C6H2, Ph |
5j, 95, 3/1 |
| 11 |
4k, 3,4,5-(MeO)3C6H2, Me |
5k, 92, 3/1 |
| 12 |
4l, 3-MeOC6H4, Tol |
5l, 84, 1/1 |
| 13 |
4m, 2-Thienyl, Tol |
5m, —c |
| 14 |
4n, 2-Furyl, Tol |
5n, —c |
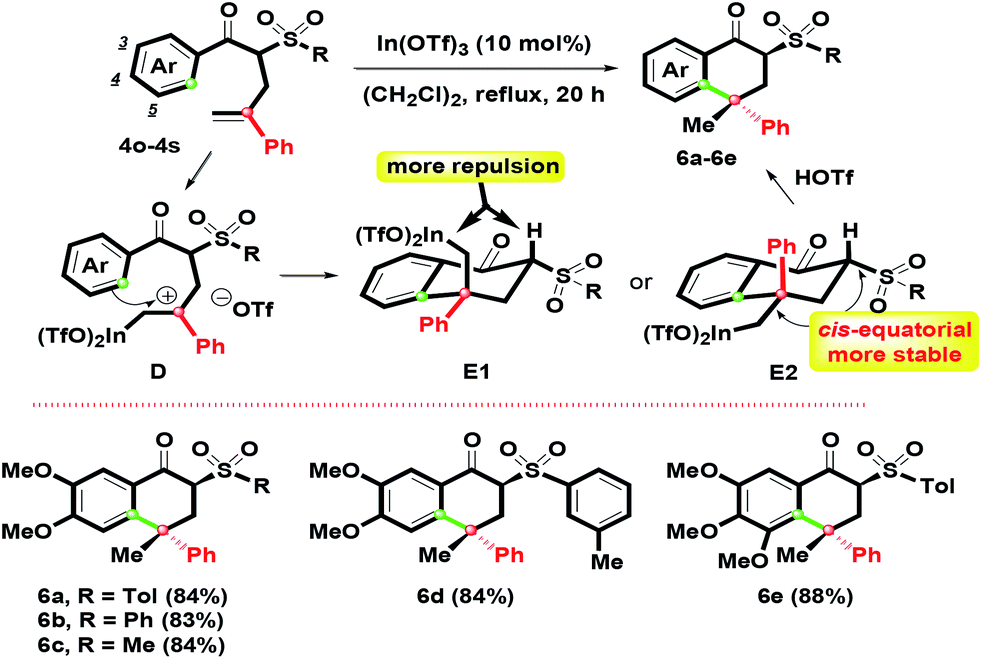
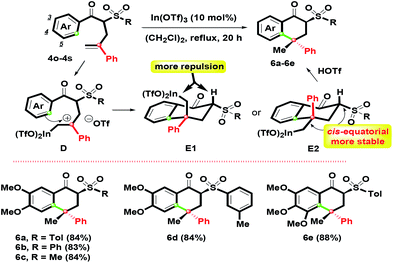
With the results in hand, adjusting the α-substituent of β-ketosulfones 3 from a cinnamyl to 2-phenylallyl group was the next aim, as shown in Scheme 4. By controlling the Ar group as the oxygenated substituents (3,4-(MeO)2C6H3 and 3,4,5-(MeO)3C6H2), 4o–4s with different sulfonyl groups were easily prepared in good yields by the K2CO3-mediated α-phenylallylation of β-ketosulfones with 2-phenylallyl bromide in boiling acetone for 10 h (see ESI†). According to the above conditions, 1-tetralones 6a–6e were obtained in 83–88% yields via the In(OTf)3 mediated intramolecular ring-closure of 4o–4s. Compared with the formation of 5a–5l, interestingly, 6a–6e were generated in the sole isomer.
 |
| | Scheme 4 Synthesis of 6a–6e. | |
By the complexation of 4o–4s and In(OTf)3, the benzylic carbocation D initially formed with a primary In(OTf)2 arm could generate two possible chair-like conformations E1 and E2. For the relative configuration of E1 and E2, the quaternary stereochemical center was the only difference. E1 possessed more repulsion since the hydrogen and the primary In(OTf)2 arm were orientated as a cis-configuration. Compared with E1, the primary In(OTf)2 arm and sulfonyl group in E2 were preferred for arranging as a equatorial position (cis-form) due to its higher stability and lower steric hindrance that was revealed. Then, by the participation of TfOH, 6a–6e were afforded by complexation of In(OTf)2 moiety on E2. This route provided highly effective regio- and stereocontrolled intramolecular hydroarylation to construct two stereochemical centers. The stereochemical structure of 6a with the α,γ-configurated centers was determined by single-crystal X-ray crystallography.26 On the basis of our observations, we found that the chair-like intermediate with a six-membered ring (for 1-tetralone skeleton) could trigger the generation of one isomer easier than the seven-membered ring intermediate (for 1-benzosuberone skeleton) via intramolecular electrophilic annulation.
In the next stage, we switched the α-substituent of β-ketosulfones 3 from aromatic phenylallyl groups into an aliphatic methylallyl (crotyl) group to examine intramolecular annulation. By the K2CO3-mediated α-crotylation of β-ketosulfones 3, 4t–4w with a mixture of E- and Z-form isomers were provided in modest to good (76–80%) yields. In particular, 7a–7d were isolated as the major regioisomers, and the hydrated products displaced the predicted benzofused bicycles (Scheme 5). Firstly, F1 or F2 containing a secondary carbocation was generated via the In(OTf)3-complexation of an α-alkenyl motif on 4t–4w. Then, the lone-pair of oxygen atoms on a para-methoxy group could force the carbonyl group to stabilize the secondary carbocation and provide the oxonium cation G1 or G2 with a five or six-membered ring under intramolecular annulation. Compared with the formation of 1-benzosuberone (derived from A with a stable benzylic carbocation), however, the methoxy group on the arene ring of F1 or F2 could not initiate the intramolecular o-carbon addition process because the seven- or six-membered ring rather than the six (for F1) or five- (for F2) membered ring was undesirable for cyclizing. For the relative configuration of G1, 1,3-repulsion between the sulfonyl group and the indium side arm and 1,2-repulsion between the methyl group and the indium side arm were generated such that an in situ formed triflate anion mediating the reversed pathway may occur. Next, by participation of H2O (from (CH2Cl)2 solvent),28 transformation from G2 to H could be achieved by the intermolecular addition of H2O on the oxocarbenium ion of G2. After ring-opening of tetrahydrofuran ring on H, I was generated. You and coworkers have demonstrated the phenomenon to the formation of 2,2-disubstituted 2,3-dihydrofurans.29a For the In(OTf)3-mediated intermolecular Michael addition of enone with H2O or alcohols, Loh et al. reported similar results for preparing γ-hydroxy or alkoxyketones.29b Following the triflic acid-promoted deindiumation of H, the removal of In(OTf)3 afforded 7a–7d (50–60%). From the above results, we understood that R1 (phenyl or methyl) was a key substituent that controlled intramolecular benzannulation.
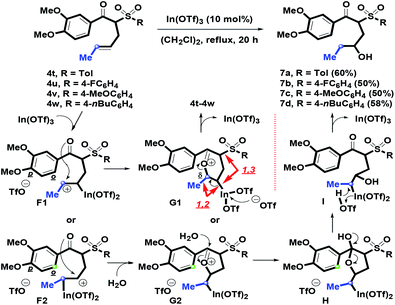 |
| | Scheme 5 Synthesis of 7a–7d. | |
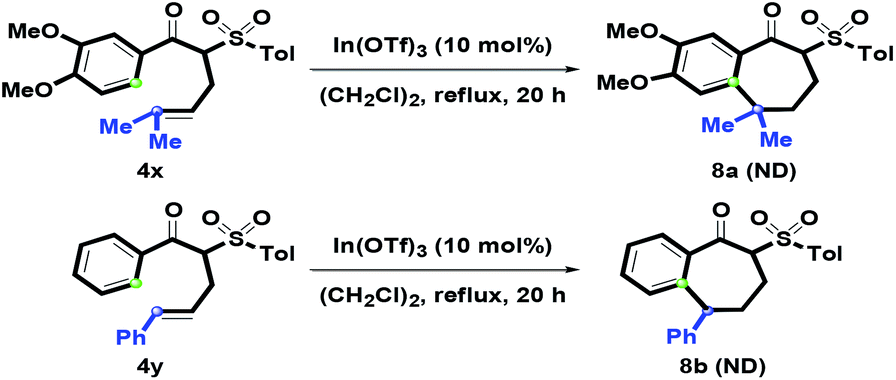
After examining the one-pot synthetic route, 4x with an α-prenyl group was also examined (Scheme 6). In particular, treatment of 4x with In(OTf)3 afforded a complex dehydrated mixture via the formation of tertiary carbocation, and no desired product 8a was detected. Adjusting the oxygenated Ar ring to a simple phenyl ring was also tested, however, no isolation of 8b was observed under the above reaction conditions. After increasing the amount of In(OTf)3 to 50 mol%, there was no generation of 8b. On the basis of this phenomenon, we understood that the oxygenated group on Ar was the key factor in causing the formation of 1-benzosuberone via intramolecular benzannulation.
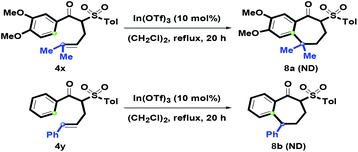 |
| | Scheme 6 Reaction of 4x–4y. | |
For other substituents at alkenyl moiety, we examined three commercially available allyl bromides, including (4-nitrophenyl)allyl bromide, α-bromomethyl methyl acrylate and β-bromomethyl methyl acrylate (Scheme 7). When the alkylation reaction of 3a was treated with the three bromides in the presence of K2CO3, unidentified complex mixture were detected. Many attempts to desired products 4z, 4aa and 4ab failed. Based on the results, we found that alkenyl moiety was not suitable to the two kinds of acrylate groups and the electron-withdrawing 4-nitrophenyl group. Therefore, we had no enough amounts of α-alkenyl β-ketosulfones 4z–4ab to examine the In(OTf)3-catalyzed intramolecular hydroarylation. Although the overall substrate scope is limited, the present route can provide the aromatic group (two kinds of phenylallyl) and the aliphatic group (crotyl) in the alkenyl moiety.
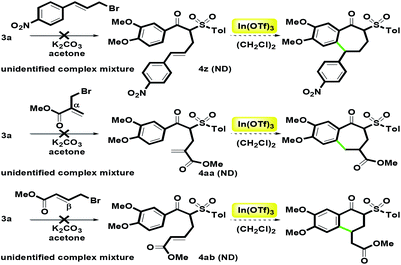 |
| | Scheme 7 Unsuccessful synthesis of 4z–4aa. | |
As the synthetic application of the In(OTf)3-mediated intramolecular ring-closure, 5a was chosen as the starting substrate to investigate synthetic applications (Scheme 8). When 5a was treated with NaBH4 in MeOH, the cyclic vinyl sulfone 9 was generated via a one-pot reduction and the spontaneous dehydration process. The dehydration process of the resulting β-hydroxysulfone could be spontaneous due to the acidic proton alfa to sulfone fragment. Furthermore, conjugated addition of 9 was studied. By choosing the Ar′ group as the aryl substituents (phenyl, 4-fluorophenyl and 2-naphthyl), 10a–10c with the 1,5-syn-diaryl group were afforded in 83%, 76% and 74% yields, respectively, via Michael addition Ar′ copper species to vinyl sulfones with Ar′MgBr and CuI. The stereochemical structure of 10c was determined by single-crystal X-ray crystallography.26 The results showed that the two aryl groups were orientated as syn-face. One possible reason is that the generated sandwich interaction for the aromatic (Ar)–copper–aromatic (Ar′) could control the introduction face of the Ar′ group. After the protonation of α-carbanion, three stereochemical centers were installed as the trans–trans conformation.
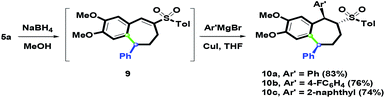 |
| | Scheme 8 Synthesis of 10a–10c. | |
Conclusion
In summary, we have, herein, developed an In(OTf)3-promoted facile and efficient one-pot synthesis of sulfonyl 1-benzosuberones and 1-tetralones via intramolecular hydroarylation of α-phenylallyl β-ketosulfones in refluxing (CH2Cl)2 in moderate to good yields under open-vessel and easy-operational reaction conditions. This highly regioselective protocol provides an atom-economic ring-closure route via one carbon–carbon bond formation. We have also discussed the related plausible reaction mechanisms. Further investigations regarding the synthetic application of metal triflates will be conducted and published in due course.
Experimental
General
All catalysts (metal triflates), reagents and solvents were obtained from commercial sources and used without further purification. Reactions were routinely carried out under an atmosphere of dry nitrogen with magnetic stirring. The heating mantle is used to provide a stable heat source. Products in organic solvents were dried with anhydrous magnesium sulfate (MgSO4) before concentration in vacuo. Melting points were determined with a SMP3 melting apparatus. 1H and 13C NMR spectra were recorded on a Varian INOVA-400 spectrometer operating at 400 and at 100 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) and the coupling constants (J) are given in Hertz. High-resolution mass spectra (HRMS) were measured with a double focusing mass spectrometer by ESI using a hybrid ion-trap. X-ray crystal structures were determined with a diffractometer (CAD4, Kappa CCD).
A representative synthetic procedure of skeleton 3
CuBr2 (450 mg, 2.0 mmol) was added to a solution of commercial available acetophenones (1.0 mmol) in EtOAc (30 mL) at 25 °C. The reaction mixture was stirred at reflux for 10 h. Then, the reaction mixture was cooled to 25 °C, filtered, neutralized with saturated NaHCO3(aq) (30 mL), and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine, dried, filtered, and evaporated to afford crude product under reduced pressure. Without further purification, substituted sodium sulfinate (2.1 mmol) was added to the resulting substituted α-bromoacetocephenones in a cosolvent of dioxane and water (20 mL, v/v = 1/1) at 25 °C. The reaction mixture was stirred at reflux for 3 h. The reaction mixture was cooled to 25 °C and extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with brine, dried, filtered, and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 8/1–4/1) afforded 3. For the starting substrates 3, these materials were known compounds and the analytical data are consistent with those in our previous reports.23a,b
A representative synthetic procedure of skeleton 4
K2CO3 (400 mg, 2.9 mmol) was added to a solution of 3 (1.0 mmol) in acetone (10 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 10 min. Bromides (1.05 mmol) were added to the reaction mixture at 25 °C. The reaction mixture was stirred at reflux for 10 h. The reaction mixture was cooled to 25 °C and the solvent was concentrated. The residue was diluted with water (10 mL) and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 8/1–2/1) afforded 4.
1-(3,4-Dimethoxyphenyl)-5-phenyl-2-(toluene-4-sulfonyl)pent-4-en-1-one (4a). 4a was synthesized according to general synthetic procedure from 3a (334 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 92% (414 mg); colorless solid; mp = 128–129 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O5S 451.1579, found 451.1586; 1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 8.4 Hz, 2H), 7.62 (dd, J = 2.0, 8.4 Hz, 1H), 7.48 (d, J = 2.4 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.24–7.14 (m, 5H), 6.88 (d, J = 8.8 Hz, 1H), 6.40 (d, J = 16.0 Hz, 1H), 5.91 (dt, J = 7.2, 15.6 Hz, 1H), 5.13 (dd, J = 4.0, 10.8 Hz, 1H), 3.94 (s, 3H), 3.91 (s, 3H), 3.03–2.96 (m, 1H), 2.95–2.88 (m, 1H), 2.44 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.0, 154.3, 149.1, 145.4, 136.6, 133.9, 133.3, 130.3, 129.8 (2x), 129.5 (2x), 128.4 (2x), 127.6, 126.2 (2x), 124.6, 123.3, 110.7, 110.0, 69.2, 56.1, 56.0, 31.9, 21.7.
2-Benzenesulfonyl-1-(3,4-dimethoxyphenyl)-5-phenylpent-4-en-1-one (4b). 4b was synthesized according to general synthetic procedure from 3b (320 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 90% (393 mg); colorless solid; mp = 125–127 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25O5S 437.1423, found 437.1429; 1H NMR (400 MHz, CDCl3): δ 7.83–7.80 (m, 2H), 7.67–7.63 (m, 1H), 7.61 (dd, J = 2.0, 8.4 Hz, 1H), 7.55–7.50 (m, 2H), 7.46 (d, J = 2.0 Hz, 1H), 7.23–7.13 (m, 5H), 6.86 (d, J = 8.4 Hz, 1H), 6.41 (d, J = 16.0 Hz, 1H), 5.92 (dt, J = 7.6, 16.0 Hz, 1H), 5.17 (dd, J = 4.0, 10.4 Hz, 1H), 3.92 (s, 3H), 3.89 (s, 3H), 3.05–2.92 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 189.8, 154.2, 149.1, 136.5, 136.4, 134.2, 133.9, 130.1, 129.7 (2x), 128.8 (2x), 128.4 (2x), 127.5, 126.1 (2x), 124.5, 123.2, 110.6, 110.0, 69.1, 56.1, 55.9, 31.7.
1-(3,4-Dimethoxyphenyl)-2-(4-fluorobenzenesulfonyl)-5-phenylpent-4-en-1-one (4c). 4c was synthesized according to general synthetic procedure from 3c (338 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 86% (391 mg); colorless solid; mp = 133–134 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H24FO5S 455.1329, found 455.1324; 1H NMR (400 MHz, CDCl3): δ 7.84–7.79 (m, 2H), 7.60 (dd, J = 2.0, 8.4 Hz, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.22–7.13 (m, 7H), 6.85 (d, J = 8.4 Hz, 1H), 6.41 (d, J = 15.6 Hz, 1H), 5.92 (dt, J = 7.6, 15.6 Hz, 1H), 5.19 (dd, J = 4.0, 10.4 Hz, 1H), 3.91 (s, 3H), 3.88 (s, 3H), 3.04–2.87 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 189.9, 166.1 (d, J = 255.4 Hz), 154.3, 149.1, 136.3, 134.0, 132.7 (d, J = 9.9 Hz, 2C), 132.2, 129.9, 128.4 (2x), 127.5, 126.0 (2x), 124.5, 122.9, 116.1 (d, J = 22.7 Hz, 2x), 110.4, 110.0, 69.0, 56.0, 55.8, 31.8.
1-(3,4-Dimethoxyphenyl)-2-(4-methoxybenzenesulfonyl)-5-phenylpent-4-en-1-one (4d). 4d was synthesized according to general synthetic procedure from 3d (350 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 90% (420 mg); colorless solid; mp = 149–150 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O6S 467.1528, found 467.1525; 1H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 9.2 Hz, 2H), 7.62 (dd, J = 2.0, 8.4 Hz, 1H), 7.48 (d, J = 2.0 Hz, 1H), 7.24–7.14 (m, 5H), 6.98 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.4 Hz, 1H), 6.41 (d, J = 16.0 Hz, 1H), 5.92 (dt, J = 7.2, 15.6 Hz, 1H), 5.13 (dd, J = 4.0, 10.4 Hz, 1H), 3.93 (s, 3H), 3.90 (s, 3H), 3.87 (s, 3H), 3.03–2.87 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.2, 164.2, 154.2, 149.1, 136.6, 133.8, 132.0 (2x), 130.3, 128.4 (2x), 127.7, 127.5, 126.1 (2x), 124.6, 123.4, 114.1 (2x), 110.6, 110.0, 69.2, 56.1, 55.9, 55.6, 31.9.
2-(4-n-Butylbenzenesulfonyl)-1-(3,4-dimethoxyphenyl)-5-phenylpent-4-en-1-one (4e). 4e was synthesized according to general synthetic procedure from 3e (376 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 83% (409 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C29H33O5S 493.2049, found 493.2052; 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 8.4 Hz, 2H), 7.58 (dd, J = 2.0, 8.4 Hz, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.29 (d, J = 8.4 Hz, 2H), 7.22–7.12 (m, 5H), 6.82 (d, J = 8.4 Hz, 1H), 6.40 (d, J = 15.6 Hz, 1H), 5.93 (dt, J = 7.2, 15.6 Hz, 1H), 5.17 (dd, J = 4.8, 9.6 Hz, 1H), 3.89 (s, 3H), 3.87 (s, 3H), 3.01–2.97 (m, 2H), 2.65 (t, J = 7.6 Hz, 2H), 1.61–1.54 (m, 2H), 1.37–1.24 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 189.8, 154.0, 150.0, 148.9, 136.4, 133.7 (2x), 133.6, 130.1, 129.6 (2x), 128.8, 128.3 (2x), 127.4, 126.0 (2x), 124.4, 123.3, 110.4, 109.9, 68.9, 56.0, 55.8, 35.5, 32.9, 31.5, 22.1, 13.7.
1-(3,4-Dimethoxyphenyl)-2-methanesulfonyl-5-phenylpent-4-en-1-one (4f). 4f was synthesized according to general synthetic procedure from 3f (258 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 84% (314 mg); colorless solid; mp = 154–155 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H23O5S 375.1266, found 375.1271; 1H NMR (400 MHz, CDCl3): δ 7.65 (dd, J = 2.0, 8.4 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.26–7.17 (m, 5H), 6.90 (d, J = 8.4 Hz, 1H), 6.51 (d, J = 16.0 Hz, 1H), 6.00 (dt, J = 7.2, 15.6 Hz, 1H), 4.95 (dd, J = 4.0, 10.4 Hz, 1H), 3.94 (s, 3H), 3.91 (s, 3H), 3.22–3.08 (m, 2H), 3.00 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 191.4, 154.7, 149.3, 136.3, 134.3, 129.7, 128.5 (2x), 127.8, 126.2 (2x), 124.8, 122.8, 110.6, 110.2, 68.2, 56.2, 55.9, 37.6, 32.4.
2-(n-Butane-1-sulfonyl)-1-(3,4-dimethoxyphenyl)-5-phenylpent-4-en-1-one (4g). 4g was synthesized according to general synthetic procedure from 3g (300 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 90% (375 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H29O5S 417.1736, found 417.1743; 1H NMR (400 MHz, CDCl3): δ 7.66 (dd, J = 2.0, 8.4 Hz, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.23–7.14 (m, 5H), 6.86 (d, J = 8.4 Hz, 1H), 6.48 (d, J = 15.6 Hz, 1H), 5.99 (dt, J = 7.2, 15.6 Hz, 1H), 5.03 (dd, J = 4.0, 10.8 Hz, 1H), 3.88 (s, 3H), 3.87 (s, 3H), 3.24–3.14 (m, 2H), 3.11–3.04 (m, 2H), 1.86–1.77 (m, 2H), 1.48–1.39 (m, 2H), 0.92 (t, J = 6.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 191.1, 154.4, 149.1, 136.2, 133.9, 129.7, 128.3 (2x), 127.5, 126.0 (2x), 124.7, 123.0, 110.5, 110.0, 68.1, 55.9, 55.7, 49.2, 31.8, 22.2, 21.6, 13.4.
1-Benzo[1,3]dioxol-5-yl-5-phenyl-2-(toluene-4-sulfonyl)pent-4-en-1-one (4h). 4h was synthesized according to general synthetic procedure from 3h (318 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 92% (399 mg); colorless solid; mp = 180–182 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H23O5S 435.1266, found 435.1270; 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.4 Hz, 2H), 7.59 (dd, J = 2.0, 8.4 Hz, 1H), 7.41 (d, J = 1.6 Hz, 1H), 7.34 (d, J = 8.8 Hz, 2H), 7.28–7.14 (m, 5H), 6.84 (d, J = 8.0 Hz, 1H), 6.40 (d, J = 16.0 Hz, 1H), 6.04 (d, J = 1.2 Hz, 1H), 6.03 (d, J = 1.6 Hz, 1H), 5.92 (dt, J = 7.2, 15.6 Hz, 1H), 5.07 (dd, J = 4.0, 10.4 Hz, 1H), 3.01–2.87 (m, 2H), 2.45 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 189.6, 152.7, 148.4, 145.4, 136.5, 134.0, 133.2, 132.0, 129.8 (2x), 129.5 (2x), 128.4 (2x), 127.5, 126.3, 126.1 (2x), 123.2, 108.5, 108.0, 102.1, 69.4, 31.8, 21.7.
5-Phenyl-2-(toluene-4-sulfonyl)-1-(3,4,5-trimethoxyphenyl)pent-4-en-1-one (4i). 4i was synthesized according to general synthetic procedure from 3i (364 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 90% (432 mg); colorless solid; mp = 98–100 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C27H29O6S 481.1685, found 481.1681; 1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 7.24–7.15 (m, 5H), 7.19 (s, 2H), 6.42 (d, J = 16.0 Hz, 1H), 5.92 (dt, J = 7.6, 16.0 Hz, 1H), 5.12 (dd, J = 4.0, 10.4 Hz, 1H), 3.91 (s, 3H), 3.87 (s, 6H), 3.07–2.89 (m, 2H), 2.44 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.6, 152.9, 145.5, 143.5, 136.4, 134.1 (2x), 133.2, 132.1, 129.8 (2x), 129.5 (2x), 128.4 (2x), 127.6, 126.1 (2x), 123.1, 106.7 (2x), 69.7, 60.9, 56.2 (2x), 31.9, 21.6.
2-Benzenesulfonyl-5-phenyl-1-(3,4,5-trimethoxyphenyl)pent-4-en-1-one (4j). 4j was synthesized according to general synthetic procedure from 3j (350 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 88% (410 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O6S 467.1528, found 467.1534; 1H NMR (400 MHz, CDCl3): δ 7.83–7.80 (m, 2H), 7.70–7.65 (m, 1H), 7.56–7.52 (m, 2H), 7.25–7.18 (m, 5H), 7.16 (s, 2H), 6.42 (d, J = 15.6 Hz, 1H), 5.92 (dt, J = 7.2, 15.6 Hz, 1H), 5.14 (dd, J = 4.0, 10.8 Hz, 1H), 3.91 (s, 3H), 3.87 (s, 6H), 3.08–2.90 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.5, 153.0 (2x), 143.5, 136.4, 136.2, 134.3, 134.2, 132.0, 129.8 (2x), 128.9 (2x), 128.5 (2x), 127.7, 126.1 (2x), 123.0, 106.7 (2x), 69.6, 60.9, 56.3 (2x), 31.9.
2-Methanesulfonyl-5-phenyl-1-(3,4,5-trimethoxyphenyl)pent-4-en-1-one (4k). 4k was synthesized according to general synthetic procedure from 3k (288 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 87% (352 mg); colorless solid; mp = 134–136 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H25O6S 405.1372, found 405.1378; 1H NMR (400 MHz, CDCl3): δ 7.25 (s, 2H), 7.22–7.13 (m, 5H), 6.47 (d, J = 16.0 Hz, 1H), 5.99 (dt, J = 7.2, 15.6 Hz, 1H), 5.04 (dd, J = 5.2, 9.2 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 6H), 3.13–3.09 (m, 2H), 2.99 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 192.0, 152.8 (2x), 143.6, 136.0, 134.1, 131.4, 128.3 (2x), 127.6, 125.9 (2x), 122.5, 106.5 (2x), 68.2, 60.6, 56.0 (2x), 37.5, 32.3.
1-(3-Methoxyphenyl)-5-phenyl-2-(toluene-4-sulfonyl)pent-4-en-1-one (4l). 4l was synthesized according to general synthetic procedure from 3l (304 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 84% (353 mg); colorless solid; mp = 119–120 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25O4S 421.1474, found 421.1478; 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 8.4 Hz, 2H), 7.53 (dt, J = 0.8, 7.6 Hz, 1H), 7.43 (t, J = 1.6 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 8.4 Hz, 2H), 7.24–7.16 (m, 5H), 7.11 (ddd, J = 0.8, 2.8, 8.4 Hz, 1H), 6.41 (d, J = 15.6 Hz, 1H), 5.94 (dt, J = 6.8, 16.0 Hz, 1H), 5.20 (dd, J = 4.4, 10.0 Hz, 1H), 3.80 (s, 3H), 3.07–2.93 (m, 2H), 2.42 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 191.8, 159.7, 145.4, 138.3, 136.4, 134.0, 133.4, 129.7 (2x), 129.6, 129.5 (2x), 128.3 (2x), 127.5, 126.1 (2x), 123.0, 121.7, 120.5, 112.9, 69.6, 55.3, 31.7, 21.5.
5-Phenyl-1-thiophen-2-yl-2-(toluene-4-sulfonyl)pent-4-en-1-one (4m). 4m was synthesized according to general synthetic procedure from 3m (280 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 87% (345 mg); colorless solid; mp = 110–112 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H21O3S2 397.0932, found 397.0936; 1H NMR (400 MHz, CDCl3): δ 7.78 (dd, J = 1.2, 4.0 Hz, 1H), 7.72–7.67 (m, 3H), 7.32 (d, J = 8.0 Hz, 2H), 7.25–7.16 (m, 5H), 7.12 (dd, J = 4.0, 4.8 Hz, 1H), 6.43 (d, J = 15.6 Hz, 1H), 5.95 (dt, J = 7.2, 15.6 Hz, 1H), 4.90 (dd, J = 4.0, 10.8 Hz, 1H), 3.04–2.88 (m, 2H), 2.44 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 183.8, 145.6, 144.3, 136.5, 136.1, 134.5, 134.2, 133.1, 129.8 (2x), 129.6 (2x), 128.6, 128.5 (2x), 127.6, 126.2 (2x), 123.1, 71.7, 31.3, 21.7.
1-Furan-2-yl-5-phenyl-2-(toluene-4-sulfonyl)pent-4-en-1-one (4n). 4n was synthesized according to general synthetic procedure from 3n (264 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 85% (323 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H21O4S 381.1161, found 381.1165; 1H NMR (400 MHz, CDCl3): δ 7.69 (d, J = 8.4 Hz, 2H), 7.58 (t, J = 0.8 Hz, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.29 (dd, J = 0.8, 3.6 Hz, 1H), 7.25–7.16 (m, 5H), 6.40 (dd, J = 1.6, 3.6 Hz, 1H), 6.42 (d, J = 15.6 Hz, 1H), 5.94 (dt, J = 7.2, 15.6 Hz, 1H), 4.97 (dd, J = 4.4, 10.8 Hz, 1H), 3.01–2.87 (m, 2H), 2.44 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 179.5, 152.7, 147.9, 145.5, 136.5, 134.1, 133.4, 129.7 (2x), 129.6 (2x), 128.4 (2x), 127.6, 126.2 (2x), 123.0, 119.9, 113.1, 70.2, 30.7, 21.7.
1-(3,4-Dimethoxyphenyl)-4-phenyl-2-(toluene-4-sulfonyl)pent-4-en-1-one (4o). 4o was synthesized according to general synthetic procedure from 3a (334 mg, 1.0 mmol) and 2-phenylallyl bromide (205 mg, 1.05 mmol); yield = 84% (378 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O5S 451.1579, found 451.1583; 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.4 Hz, 2H), 7.32–7.16 (m, 9H), 6.71 (d, J = 8.4 Hz, 1H), 5.16 (s, 1H), 5.12 (dd, J = 2.4, 11.6 Hz, 1H), 4.96 (s, 1H), 3.88 (s, 3H), 3.79 (s, 3H), 3.43 (d, J = 14.0 Hz, 1H), 3.16 (dd, J = 11.6, 14.0 Hz, 1H), 2.44 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.1, 153.9, 148.8, 145.2, 142.4, 138.8, 133.5, 130.3, 129.7 (2x), 129.4 (2x), 128.5 (2x), 127.9, 126.1 (2x), 123.9, 116.1, 110.3, 109.8, 67.6, 56.0, 55.7, 34.1, 21.6.
2-Benzenesulfonyl-1-(3,4-dimethoxyphenyl)4-phenylpent-4-en-1-one (4p). 4p was synthesized according to general synthetic procedure from 3b (320 mg, 1.0 mmol) and 2-phenylallyl bromide (205 mg, 1.05 mmol); yield = 85% (371 mg); colorless solid; mp = 127–129 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25O5S 437.1423, found 437.1428; 1H NMR (400 MHz, CDCl3): δ 7.82–7.80 (m, 2H), 7.67–7.63 (m, 1H), 7.54–7.50 (m, 2H), 7.29–7.26 (m, 3H), 7.23–7.20 (m, 3H), 7.15 (dd, J = 1.6, 8.4 Hz, 1H), 6.71 (d, J = 8.4 Hz, 1H), 5.17 (s, 1H), 5.13 (dd, J = 2.4, 11.2 Hz, 1H), 4.98 (s, 1H), 3.89 (s, 3H), 3.80 (s, 3H), 3.45 (d, J = 13.6 Hz, 1H), 3.18 (dd, J = 11.2, 13.6 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.0, 154.0, 148.9, 142.3, 138.8, 136.5, 134.2, 130.3, 129.7 (2x), 128.8 (2x), 128.6 (2x), 128.0, 126.2 (2x), 123.9, 116.3, 110.3, 109.8, 67.6, 55.0, 55.8, 34.1.
1-(3,4-Dimethoxyphenyl)-2-methanesulfonyl-4-phenylpent-4-en-1-one (4q). 4q was synthesized according to general synthetic procedure from 3f (258 mg, 1.0 mmol) and 2-phenylallyl bromide (205 mg, 1.05 mmol); yield = 84% (314 mg); colorless solid; mp = 114–115 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H23O5S 375.1266, found 375.1269; 1H NMR (400 MHz, CDCl3): δ 7.34–7.26 (m, 6H), 7.20 (dd, J = 2.0, 8.4 Hz, 1H), 6.74 (d, J = 8.4 Hz, 1H), 5.24 (s, 1H), 5.09 (s, 1H), 4.93 (dd, J = 2.4, 11.6 Hz, 1H), 3.91 (s, 3H), 3.81 (s, 3H), 3.56 (dd, J = 1.6, 13.6 Hz, 1H), 3.36 (dd, J = 11.6, 13.6 Hz, 1H), 3.01 (d, J = 0.8 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 191.5, 154.4, 149.0, 142.3, 138.4, 129.8, 128.7 (2x), 128.2, 126.3 (2x), 124.4, 116.5, 110.3, 110.0, 66.7, 56.1, 55.8, 37.6, 35.0.
1-(3,4-Dimethoxyphenyl)-4-phenyl-2-(toluene-3-sulfonyl)pent-4-en-1-one (4r). 4r was synthesized according to general synthetic procedure from 3o (334 mg, 1.0 mmol) and 2-phenylallyl bromide (205 mg, 1.05 mmol); yield = 80% (360 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O5S 451.1579, found 451.1584; 1H NMR (400 MHz, CDCl3): δ 7.60–7.58 (m, 2H), 7.42–7.35 (m, 2H), 7.27–7.24 (m, 3H), 7.22–7.18 (m, 3H), 7.13 (dd, J = 2.0, 8.4 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 5.16 (s, 1H), 5.10 (dd, J = 2.4, 11.2 Hz, 1H), 4.98 (s, 1H), 3.86 (s, 3H), 3.78 (s, 3H), 3.43 (d, J = 14.0 Hz, 1H), 3.22 (dd, J = 11.6, 14.0 Hz, 1H), 2.37 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 189.8, 153.8, 148.7, 142.4, 139.0, 138.7, 136.5, 134.8, 130.2, 129.7, 128.6, 128.5 (2x), 127.9, 126.6, 126.1 (2x), 123.8, 116.2, 110.1, 109.8, 67.5, 55.9, 55.7, 33.8, 21.1.
4-Phenyl-2-(toluene-4-sulfonyl)-1-(3,4,5-trimethoxyphenyl)pent-4-en-1-one (4s). 4s was synthesized according to general synthetic procedure from 3i (364 mg, 1.0 mmol) and 2-phenylallyl bromide (205 mg, 1.05 mmol); yield = 83% (399 mg); colorless solid; mp = 118–121 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C27H29O6S 481.1685, found 481.1680; 1H NMR (400 MHz, CDCl3): δ 7.64 (d, J = 8.4 Hz, 2H), 7.26–7.18 (m, 7H), 6.73 (s, 2H), 5.14 (s, 1H), 5.10 (dd, J = 2.4, 11.6 Hz, 1H), 4.94 (s, 1H), 3.81 (s, 3H), 3.62 (s, 6H), 3.46 (d, J = 13.2 Hz, 1H), 3.11 (dd, J = 11.6, 13.6 Hz, 1H), 2.36 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.7, 152.5 (2x), 145.2, 142.9, 141.8, 138.3, 133.3, 132.0, 129.4 (2x), 129.3 (2x), 128.4 (2x), 127.8, 125.9 (2x), 116.2, 105.7 (2x), 67.6, 60.5, 55.7 (2x), 34.0, 21.3.
1-(3,4-Dimethoxyphenyl)-2-(toluene-4-sulfonyl)hex-4-en-1-one (4t). 4t was synthesized according to general synthetic procedure from 3a (334 mg, 1.0 mmol) and crotyl bromide (141 mg, 1.05 mmol); two isomers (ratio: trans/cis = 8/1); yield = 80% (311 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H25O5S 389.1423, found 389.1428; for major product: 1H NMR (400 MHz, CDCl3): δ 7.67–7.56 (m, 3H), 7.46 (d, J = 2.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 2H), 6.88 (d, J = 8.8 Hz, 1H), 5.49–5.41 (m, 1H), 5.19–5.11 (m, 1H), 5.01 (dd, J = 4.0, 10.4 Hz, 1H), 3.94 (s, 3H), 3.91 (s, 3H), 2.82–2.64 (m, 2H), 2.41 (s, 3H), 1.48 (d, J = 6.4 Hz, 3H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 190.1, 154.1, 149.0, 145.2, 133.4, 130.4, 129.7 (2x), 129.6, 129.4 (2x), 124.5, 124.4, 110.6, 110.0, 69.3, 56.1, 55.9, 31.4, 21.6, 17.8.
1-(3,4-Dimethoxyphenyl)-2-(4-fluorobenzenesulfonyl)hex-4-en-1-one (4u). 4u was synthesized according to general synthetic procedure from 3c (338 mg, 1.0 mmol) and crotyl bromide (141 mg, 1.05 mmol); two isomers (ratio: trans/cis = 6/1); yield = 82% (322 mg); colorless solid; mp = 110–112 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H22FO5S 393.1172, found 393.1178; 1H NMR (400 MHz, CDCl3): δ 7.83–7.73 (m, 2H), 7.56 (dd, J = 2.0, 8.8 Hz, 1H), 7.43 (d, J = 2.0 Hz, 1H), 7.19–7.08 (m, 2H), 6.86 (d, J = 8.4 Hz, 1H), 5.48–5.39 (m, 1H), 5.17–5.06 (m, 1H), 5.03 (dd, J = 3.2, 10.4 Hz, 1H), 3.91 (s, 3H), 3.88 (s, 3H), 2.79–2.59 (m, 2H), 1.46 (dd, J = 1.2, 6.4 Hz, 3H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 190.0, 166.0 (d, J = 255.5 Hz), 154.2, 149.1, 132.6 (d, J = 9.9 Hz, 2x), 132.3 (d, J = 3.0 Hz), 130.1, 129.8, 124.4, 124.0, 116.0 (d, J = 22.0 Hz, 2x), 110.4, 110.0, 69.1, 56.0, 55.8, 31.4, 17.7.
1-(3,4-Dimethoxyphenyl)-2-(4-methoxybenzenesulfonyl)hex-4-en-1-one (4v). 4v was synthesized according to general synthetic procedure from 3d (350 mg, 1.0 mmol) and crotyl bromide (141 mg, 1.05 mmol); two isomers (ratio: trans/cis = 3/1); yield = 78% (315 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H25O6S 405.1372, found 405.1377; 1H NMR (400 MHz, CDCl3): δ 7.72–7.65 (m, 2H), 7.61–7.56 (m, 1H), 7.46–7.44 (m, 1H), 6.97–6.93 (m, 2H), 6.88–6.86 (m, 1H), 5.49–5.41 (m, 1H), 5.19–5.10 (m, 1H), 5.01 (dd, J = 3.2, 10.4 Hz, 1H), 3.934 (s, 9/4H), 3.928 (s, 3/4H), 3.91 (s, 9/4H), 3.90 (s, 3/4H), 3.85 (s, 3/4H), 3.84 (s, 9/4H), 2.80–2.62 (m, 2H), 1.54 (dd, J = 0.8, 6.8 Hz, 3/4H), 1.48 (d, J = 6.4 Hz, 9/4H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 190.3, 164.0, 154.0, 149.0, 131.9 (2x), 130.4, 129.6, 128.2, 124.41, 124.40, 113.9 (2x), 110.5, 110.0, 69.3, 56.1, 55.9, 55.6, 31.4, 17.8.
2-(4-n-Butylbenzenesulfonyl)-1-(3,4-dimethoxyphenyl)hex-4-en-1-one (4w). 4w was synthesized according to general synthetic procedure from 3e (376 mg, 1.0 mmol) and crotyl bromide (141 mg, 1.05 mmol); two isomers (ratio: trans/cis = 3/1); yield = 76% (327 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H31O5S 431.1892, found 431.1896; 1H NMR (400 MHz, CDCl3): δ 7.70–7.64 (m, 2H), 7.58–7.53 (m, 1H), 7.46–7.44 (m, 1H), 7.31–7.26 (m, 2H), 6.88–6.85 (m, 1H), 5.49–5.42 (m, 1H), 5.20–5.12 (m, 1H), 5.03–4.98 (m, 1H), 3.94 (s, 9/4H), 3.93 (s, 3/4H), 3.92 (s, 9/4H), 3.91 (s, 3/4H), 2.84–2.70 (m, 2H), 2.66 (t, J = 7.6 Hz, 2H), 1.62–1.56 (m, 2H), 1.49 (dt, J = 1.2, 6.4 Hz, 3H), 1.37–1.28 (m, 2H), 0.92 (d, J = 7.2 Hz, 3H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 190.2, 154.1, 150.0, 149.1, 133.7, 130.4, 129.8 (2x), 128.8 (2x), 128.3, 124.5, 124.4, 110.6, 109.9, 69.3, 56.1, 55.9, 35.6, 33.0, 31.3, 22.2, 17.8, 13.8.
1-(3,4-Dimethoxyphenyl)-5-methyl-2-(toluene-4-sulfonyl)hex-4-en-1-one (4x). 4x was synthesized according to general synthetic procedure from 3a (334 mg, 1.0 mmol) and prenyl bromide (155 mg, 1.05 mmol); yield = 74% (298 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H27O5S 403.1579, found 403.1584; 1H NMR (400 MHz, CDCl3): δ 7.63 (d, J = 8.0 Hz, 2H), 7.54 (dd, J = 2.0, 8.4 Hz, 1H), 7.42 (d, J = 2.0 Hz, 1H), 7.26 (d, J = 8.0 Hz, 2H), 6.84 (d, J = 8.4 Hz, 1H), 4.96 (dd, J = 4.8, 9.6 Hz, 1H), 4.85–4.81 (m, 1H), 3.90 (s, 3H), 3.87 (s, 3H), 2.74–2.69 (m, 2H), 2.37 (s, 3H), 1.51 (s, 3H), 1.49 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.4, 153.9, 148.9, 145.0, 136.0, 133.6, 130.3, 129.5 (2x), 129.3 (2x), 124.3, 117.5, 110.4, 109.9, 69.1, 56.0, 55.8, 27.1, 24.4, 21.5, 17.6.
1,5-Diphenyl-2-(toluene-4-sulfonyl)pent-4-en-1-one (4y)30. 4y was synthesized according to general synthetic procedure from 3p (274 mg, 1.0 mmol) and cinnamyl bromide (205 mg, 1.05 mmol); yield = 88% (343 mg); colorless solid; mp = 112–113 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H23O3S 391.1368, found 391.1374; 1H NMR (400 MHz, CDCl3): δ 7.96–7.94 (m, 2H), 7.68 (d, J = 8.4 Hz, 2H), 7.60–7.55 (m, 1H), 7.46–7.43 (m, 2H), 7.31 (d, J = 8.0 Hz, 2H), 7.24–7.15 (m, 5H), 6.41 (d, J = 16.0 Hz, 1H), 5.93 (dt, J = 7.2, 15.6 Hz, 1H), 5.21 (dd, J = 4.0, 10.4 Hz, 1H), 3.06–2.92 (m, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 192.0, 145.5, 137.0, 136.4, 134.0, 133.9, 133.2, 129.7 (2x), 129.5 (2x), 129.0 (2x), 128.7 (2x), 128.4 (2x), 127.5, 126.1 (2x), 123.1, 69.5, 31.7, 21.6.
A representative synthetic procedure of skeletons 5–7
In(OTf)3 (6 mg, 0.01 mmol) was added to a solution of 4 (1.0 mmol) in (CH2Cl)2 (20 mL) at 25 °C. The reaction mixture was stirred at reflux (84 °C) for 20 h. The reaction mixture was concentrated and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 10/1–2/1) afforded 5–7.
2,3-Dimethoxy-9-phenyl-6-(toluene-4-sulfonyl)-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5a). 5a was synthesized according to general synthetic procedure from 4a (450 mg, 1.0 mmol); two isomers (ratio: trans/cis = 2/1); yield = 91% (410 mg); colorless solid; mp = 176–178 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O5S 451.1579, found 451.1587; for major product, 1H NMR (400 MHz, CDCl3): δ 7.61 (d, J = 8.0 Hz, 2H), 7.27–7.20 (m, 5H), 7.17 (s, 1H), 7.00 (d, J = 7.2 Hz, 2H), 6.56 (s, 1H), 4.53 (dd, J = 2.4, 7.2 Hz, 1H), 4.39 (dd, J = 6.8, 10.4 Hz, 1H), 3.87 (s, 3H), 3.77 (s, 3H), 2.65–2.60 (m, 1H), 2.42 (s, 3H), 2.30–2.17 (m, 2H), 2.04–1.96 (m, 1H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 194.4, 152.0, 147.6, 144.5, 141.8, 137.0, 135.0, 131.0, 129.3 (2x), 128.8 (2x), 128.6 (2x), 127.3 (2x), 126.5, 113.7, 111.6, 73.2, 55.8 (2x), 48.7, 29.6, 22.2, 21.4. Single-crystal X-ray diagram: crystal of compound 5a was grown by slow diffusion of EtOAc into a solution of compound 5a in CH2Cl2 to yield colorless prisms. The compound crystallizes in the triclinic crystal system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.6479(4) Å, b = 11.7181(6) Å, c = 12.4402(7) Å, V = 1137.93(10) Å3, Z = 2, dcalcd = 1.315 g cm−3, F(000) = 476, 2θ range = 1.802–26.452°, R indices (all data) R1 = 0.0662, wR2 = 0.1132.
, a = 8.6479(4) Å, b = 11.7181(6) Å, c = 12.4402(7) Å, V = 1137.93(10) Å3, Z = 2, dcalcd = 1.315 g cm−3, F(000) = 476, 2θ range = 1.802–26.452°, R indices (all data) R1 = 0.0662, wR2 = 0.1132.
6-Benzenesulfonyl-2,3-dimethoxy-9-phenyl-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5b). 5b was synthesized according to general synthetic procedure from 4b (436 mg, 1.0 mmol); two isomers (ratio: trans/cis = 4/1); yield = 90% (393 mg); colorless solid; mp = 183–184 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25O5S 437.1423, found 437.1428; for major product: 1H NMR (400 MHz, CDCl3): δ 7.75–7.72 (m, 2H), 7.60–7.56 (m, 1H), 7.47–7.43 (m, 2H), 7.30–7.24 (m, 3H), 7.20 (s, 1H), 7.03–7.01 (m, 2H), 6.58 (s, 1H), 4.55 (dd, J = 2.4, 7.2 Hz, 1H), 4.44 (dd, J = 6.8, 10.4 Hz, 1H), 3.90 (s, 3H), 3.80 (s, 3H), 2.49–2.43 (m, 1H), 2.31–2.17 (m, 2H), 2.05–1.97 (m, 1H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 194.3, 152.1, 147.7, 141.7, 138.1, 137.1, 133.8, 133.6, 128.9 (2x), 128.8 (4x), 127.5 (2x), 126.7, 113.8, 111.7, 73.1, 56.0 (2x), 48.7, 29.7, 22.3. Single-crystal X-ray diagram: crystal of compound 5b was grown by slow diffusion of EtOAc into a solution of compound 5b in CH2Cl2 to yield colorless prisms. The compound crystallizes in the monoclinic crystal system, space group P21/c, a = 12.2830(19) Å, b = 19.038(4) Å, c = 9.2274(14) Å, V = 2059.2(6) Å3, Z = 4, dcalcd = 1.408 g cm−3, F(000) = 920, 2θ range = 1.737–26.458°, R indices (all data) R1 = 0.0745, wR2 = 0.1146.
6-(4-Fluorobenzenesulfonyl)-2,3-dimethoxy-9-phenyl-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5c). 5c was synthesized according to general synthetic procedure from 4c (454 mg, 1.0 mmol); two isomers (ratio: trans/cis = 5/1); yield = 86% (391 mg); colorless solid; mp = 205–206 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H24FO5S 455.1329, found 455.1332; for major product: 1H NMR (400 MHz, CDCl3): δ 7.81–7.76 (m, 2H), 7.30–7.21 (m, 4H), 7.16–7.09 (m, 2H), 7.05–7.03 (m, 2H), 6.59 (s, 1H), 4.58 (d, J = 5.2 Hz, 1H), 4.41 (dd, J = 7.2, 10.4 Hz, 1H), 3.91 (s, 3H), 3.82 (s, 3H), 2.55–2.48 (m, 1H), 2.29–2.13 (m, 2H), 2.08–1.99 (m, 1H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 194.1, 165.7 (d, J = 254.7 Hz), 152.3, 147.8, 141.5, 137.3, 134.1, 132.0 (d, J = 9.8 Hz, 2x), 130.9, 128.9 (2x), 127.5 (2x), 126.8, 116.1 (d, J = 22.7 Hz, 2x), 113.8, 111.7, 73.0, 56.0 (2x), 48.5, 29.6, 22.3.
2,3-Dimethoxy-6-(4-methoxybenzenesulfonyl)-9-phenyl-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5d). 5d was synthesized according to general synthetic procedure from 4d (466 mg, 1.0 mmol); two isomers (ratio: trans/cis = 1/1); yield = 89% (415 mg); colorless solid; mp = 158–160 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O6S 467.1528, found 467.1532; 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 9.2 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.35–7.11 (m, 6H), 7.01 (d, J = 8.8 Hz, 1H), 6.90 (d, J = 9.2 Hz, 1H), 6.56 (s, 1/2H), 6.11 (s, 1/2H), 4.54 (dd, J = 4.4, 6.8 Hz, 1/2H), 4.43 (dd, J = 6.8, 11.6 Hz, 1/2H), 4.38 (dd, J = 4.8, 12.0 Hz, 1/2H), 4.06 (dd, J = 4.0, 6.8 Hz, 1/2H), 3.89 (s, 3/2H), 3.86 (s, 3/2H), 3.84 (s, 3/2H), 3.83 (s, 3/2H), 3.78 (s, 3/2H), 3.51 (s, 3/2H), 2.68–2.02 (m, 1/2H), 2.46–2.39 (m, 1/2H), 2.32–1.97 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 195.7 (1/2x), 194.6 (1/2x), 163.8 (1/2x), 163.6 (1/2x), 152.0 (1/2x), 151.5 (1/2x), 147.6 (1/2x), 147.3 (1/2x), 143.2 (1/2x), 141.8 (1/2x), 138.0 (1/2x), 137.0 (1/2x), 131.9, 131.2 (2x), 131.1 (1/2x), 130.9 (1/2x), 129.9 (1/2x), 129.4 (1/2x), 128.71, 128.68, 128.5, 127.4, 127.0 (1/2x), 126.6 (1/2x), 113.92 (1/2x), 113.89 (1/2x), 113.7 (1/2x), 112.0 (1/2x), 111.6 (1/2x), 110.8 (1/2x), 73.4 (1/2x), 73.1 (1/2x), 55.9, 55.57, 55.55 (1/2x), 55.47 (1/2x), 48.8 (1/2x), 47.0 (1/2x) 32.2 (1/2x), 29.7 (1/2x), 23.1 (1/2x), 22.3 (1/2x).
6-(4-n-Butylbenzenesulfonyl)-2,3-dimethoxy-9-phenyl-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5e). 5e was synthesized according to general synthetic procedure from 4e (492 mg, 1.0 mmol); two isomers (ratio: trans/cis = 1/1); yield = 87% (428 mg); colorless solid; mp = 185–187 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C29H33O5S 493.2049, found 493.2056; 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 8.4 Hz, 1H), 7.37–7.72 (m, 7H), 7.20 (d, J = 8.8 Hz, 1/2H), 7.19–7.12 (m, 1/2H), 7.06 (s, 1/2H), 7.01 (d, J = 7.6 Hz, 1/2H), 6.57 (s, 1/2H), 6.11 (s. 1/2H), 4.54 (dd, J = 4.4, 6.8 Hz, 1/2H), 4.45 (dd, J = 6.8, 11.6 Hz, 1/2H), 4.40 (dd, J = 4.8, 12.0 Hz, 1/2H), 4.09 (dd, J = 4.0, 6.8 Hz, 1/2H), 3.89 (s, 3/2H), 3.82 (s, 3/2H), 3.79 (s, 3/2H), 3.52 (s, 3/2H), 2.69 (t, J = 7.6 Hz, 1H), 2.65 (t, J = 7.6 Hz, 1H), 2.45–2.41 (m, 1/2H), 2.30–2.11 (m, 2H), 2.02–1.95 (m, 1/2H), 1.65–1.55 (m, 2H), 1.39–1.30 (m, 2H), 0.92 (t, J = 7.6 Hz, 3/2H), 0.91 (t, J = 7.2 Hz, 3/2H); 13C{1H} NMR (100 MHz, CDCl3): δ 195.6 (1/2x), 194.4 (1/2x), 152.0 (1/2x), 151.6 (1/2x), 149.6 (1/2x), 149.4 (1/2x), 147.6 (1/2x), 147.3 (1/2x), 143.1 (1/2x), 141.8 (1/2x), 137.9 (1/2x), 137.0 (1/2x), 135.8 (1/2x), 135.2 (1/2x), 131.1 (1/2x), 130.8 (1/2x), 129.6, 128.9, 128.8, 128.72 (2x), 128.67, 128.5, 127.4, 127.1 (1/2x), 126.6 (1/2x), 113.7 (1/2x), 112.0 (1/2x), 111.6 (1/2x), 110.8 (1/2x), 73.1 (1/2x), 73.0 (1/2x), 55.9, 55.5, 48.7 (1/2x), 47.0 (1/2x), 35.51 (1/2x), 35.45 (1/2x), 33.0, 32.1 (1/2x) 29.7 (1/2x), 22.8 (1/2x), 22.3 (1/2x), 22.2 (1/2x), 22.1 (1/2x), 13.8. Single-crystal X-ray diagram: crystal of compound 5e was grown by slow diffusion of EtOAc into a solution of compound 5e in CH2Cl2 to yield colorless prisms. The compound crystallizes in the monoclinic crystal system, space group P21/c, a = 12.2830(19) Å, b = 19.038(4) Å, c = 9.2274(14) Å, V = 2059.2(6) Å3, Z = 4, dcalcd = 1.408 g cm−3, F(000) = 920, 2θ range = 1.737–26.458°, R indices (all data) R1 = 0.0745, wR2 = 0.1146.
6-Methanesulfonyl-2,3-dimethoxy-9-phenyl-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5f). 5f was synthesized according to general synthetic procedure from 4f (374 mg, 1.0 mmol); two isomers (ratio: trans/cis = 3/1); yield = 90% (337 mg); colorless solid; mp = 187–188 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H23O5S 375.1266, found 375.1269; for major product, 1H NMR (400 MHz, CDCl3): δ 7.41–7.28 (m, 3H), 7.26–7.19 (m, 2H), 7.25 (s, 1H), 6.17 (s, 1H), 4.19–4.14 (m, 2H), 3.90 (s, 3H), 3.57 (s, 3H), 3.31 (s, 3H), 2.81–2.73 (m, 1H), 2.37–2.32 (m, 2H), 2.14–2.04 (m, 1H); for major product, 13C{1H} NMR (100 MHz, CDCl3): δ 197.1, 152.3, 147.6, 142.3, 137.7, 130.5, 128.9 (2x), 128.6 (2x), 127.3, 111.8, 111.2, 71.5, 56.1, 55.7, 46.7, 41.8, 31.4, 21.3.
6-(n-Butane-1-sulfonyl)-2,3-dimethoxy-9-phenyl-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5g). 5g was synthesized according to general synthetic procedure from 4g (416 mg, 1.0 mmol); two isomers (ratio: trans/cis = 1/1); yield = 94% (391 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H29O5S 417.1736, found 417.1740; 1H NMR (400 MHz, CDCl3): δ 7.36–7.18 (m, 5H), 7.06 (d, J = 7.6 Hz, 1H), 6.56 (s, 1/2H), 6.12 (s, 1/2H), 4.63–4.60 (m, 1/2H), 4.17–4.11 (m, 3/2H), 3.88 (s, 3/2H), 3.84 (s, 3/2H), 3.77 (s, 3/2H), 3.58–3.51 (m, 1/2H), 3.53 (s, 3/2H), 3.44–3.37 (m, 1/2H), 3.26–3.19 (m, 1/2H), 3.09–3.02 (m, 1/2H), 2.75–2.68 (m, 1/2H), 2.50–2.42 (m, 1/2H), 2.36–2.20 (m, 2H), 2.11–2.01 (m, 1/2H), 1.91–1.81 (m, 2H), 1.68–1.60 (m, 1/2H), 1.59–1.45 (m, 1H), 1.41–1.30 (m, 1H), 0.96 (t, J = 7.2 Hz, 3/2H), 0.88 (t, J = 7.2 Hz, 3/2H); 13C{1H} NMR (100 MHz, CDCl3): δ 197.7 (1/2x), 196.0 (1/2x), 152.3 (1/2x), 152.1 (1/2x), 147.7 (1/2x), 147.4 (1/2x), 141.9 (1/2x), 141.8 (1/2x), 137.1 (1/2x), 136.8 (1/2x), 130.7 (1/2x), 130.4 (1/2x), 128.7, 128.6, 128.5, 127.5, 127.1 (1/2x), 126.6 (1/2x), 113.4 (1/2x), 111.6 (1/2x), 111.2 (1/2x), 110.9 (1/2x), 69.9 (1/2x), 69.5 (1/2x), 55.8, 55.4, 53.5 (1/2x), 52.3 (1/2x), 48.2 (1/2x), 46.1 (1/2x), 30.7 (1/2x), 29.4 (1/2x), 23.3 (1/2x), 23.2 (1/2x), 21.6 (1/2x), 21.5 (1/2x), 20.8 (1/2x), 20.2 (1/2x), 13.4 (1/2x), 13.3 (1/2x).
9-Phenyl-6-(toluene-4-sulfonyl)-6,7,8,9-tetrahydro-1,3-dioxacyclohepta[f]inden-5-one (5h). 5h was synthesized according to general synthetic procedure from 4h (435 mg, 1.0 mmol); two isomers (ratio: trans/cis = 1/1); yield = 93% (404 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H23O5S 435.1266, found 435.1269; 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.38–7.11 (m, 5H), 7.14 (d, J = 8.4 Hz, 1H), 7.04 (s, 1/2H), 7.02 (d, J = 7.6 Hz, 1H), 6.92 (s, 1/2H), 6.52 (s, 1/2H), 6.11 (s, 1/2H), 5.99 (s, 1H), 5.91 (d, J = 1.2 Hz, 1/2H), 5.89 (d, J = 1.6 Hz, 1/2H), 4.52 (t, J = 6.0 Hz, 1/2H), 4.40–4.31 (m, 1H), 4.02 (t, J = 8.4 Hz, 1/2H), 2.72–2.65 (m, 1/2H), 2.46 (s, 3/2H), 2.43 (s, 3/2H), 2.33–2.20 (m, 5/2H), 2.19–2.04 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 196.4 (1/2x), 195.0 (1/2x), 151.0 (1/2x), 150.7 (1/2x), 146.8 (1/2x), 146.4 (1/2x), 144.9 (1/2x), 144.8 (1/2x), 142.4 (1/2x), 142.2 (1/2x), 139.1 (1/2x), 138.7 (1/2x), 135.7 (1/2x), 135.2 (1/2x), 132.7 (1/2x), 132.5 (1/2x), 129.52, 129.50, 129.46, 128.93, 128.86, 128.7, 128.5, 127.6, 127.2 (1/2x), 126.7 (1/2x), 111.0 (1/2x), 109.0 (1/2x), 108.9 (1/2x), 108.0 (1/2x), 101.9 (1/2x), 101.7 (1/2x), 73.9 (1/2x), 72.7 (1/2x), 49.4 (1/2x), 46.8 (1/2x), 31.3 (1/2x), 29.8 (1/2x), 22.5 (1/2x), 22.0 (1/2x), 21.64 (1/2x), 21.60 (1/2x).
1,2,3-Trimethoxy-9-phenyl-6-(toluene-4-sulfonyl)-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5i). 5i was synthesized according to general synthetic procedure from 4i (480 mg, 1.0 mmol); two isomers (ratio: trans/cis = 3/1); yield = 90% (432 mg); colorless solid; mp = 182–183 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C27H29O6S 481.1685, found 481.1692; for major product, 1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 8.0 Hz, 2H), 7.25–7.14 (m, 5H), 7.03 (s, 1H), 7.00 (d, J = 7.6 Hz, 2H), 4.86 (dd, J = 2.4, 7.6 Hz, 1H), 4.43 (dd, J = 7.2, 10.8 Hz, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.51 (s, 3H), 2.41 (s, 3H), 2.46–2.36 (m, 1H), 2.18–2.07 (m, 2H), 1.96–1.90 (m, 1H); for major product, 13C{1H} NMR (100 MHz, CDCl3): δ 195.1, 152.3, 151.8, 145.6, 144.6, 141.8, 134.9, 134.2, 129.9, 129.4 (2x), 129.0 (2x), 128.5 (2x), 127.1 (2x), 126.4, 107.7, 73.1, 60.7, 60.6, 56.0, 42.0, 29.8, 22.6, 21.5.
6-Benzenesulfonyl-1,2,3-trimethoxy-9-phenyl-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5j). 5j was synthesized according to general synthetic procedure from 4j (466 mg, 1.0 mmol); two isomers (ratio: trans/cis = 3/1); yield = 95% (443 mg); colorless solid; mp = 138–139 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O6S 467.1528, found 467.1523; for major product, 1H NMR (400 MHz, CDCl3): δ 7.73–7.71 (m, 2H), 7.59–7.55 (m, 1H), 7.46–7.42 (m, 2H), 7.23–7.13 (m, 3H), 7.04 (s, 1H), 7.00 (d, J = 7.6 Hz, 2H), 4.87 (dd, J = 2.4, 7.6 Hz, 1H), 4.46 (dd, J = 7.2, 10.8 Hz, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.52 (s, 3H), 2.45–2.38 (m, 1H), 2.17–2.09 (m, 2H), 1.93–1.85 (m, 1H); for major product, 13C{1H} NMR (100 MHz, CDCl3): δ 194.7, 152.4, 151.8, 145.7, 140.5, 137.9, 134.1, 133.6, 129.9, 128.9 (2x), 128.8 (2x), 128.5 (2x), 127.0 (2x), 126.4, 107.7, 72.8, 60.7, 60.6, 56.0, 41.8, 29.7, 22.5.
6-Methanesulfonyl-1,2,3-trimethoxy-9-phenyl-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5k). 5k was synthesized according to general synthetic procedure from 4k (404 mg, 1.0 mmol); two isomers (ratio: trans/cis = 3/1); yield = 92% (372 mg); colorless solid; mp = 163–164 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H25O6S 405.1372, found 405.1375; for major product, 1H NMR (400 MHz, CDCl3): δ 7.29–7.19 (m, 3H), 7.10–7.08 (m, 2H), 7.09 (s, 1H), 4.96 (dd, J = 2.4, 7.6 Hz, 1H), 4.24 (dd, J = 6.4, 11.6 Hz, 1H), 3.91 (s, 3H), 3.90 (s, 3H), 3.55 (s, 3H), 3.03 (s, 3H), 2.58–2.52 (m, 1H), 2.28–2.18 (m, 2H), 2.12–2.05 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 196.3, 152.5, 151.9, 146.0, 141.3, 134.0, 130.0, 128.7 (2x), 127.1 (2x), 126.7, 107.7, 71.8, 60.8, 60.7, 56.0, 41.5, 39.8, 29.8, 22.1.
3-Methoxy-9-phenyl-6-(toluene-4-sulfonyl)-6,7,8,9-tetrahydrobenzocyclohepten-5-one (5l). 5l was synthesized according to general synthetic procedure from 4l (420 mg, 1.0 mmol); two isomers (ratio: trans/cis = 1/1); yield = 84% (353 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25O4S 421.1474, found 421.1478; 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.38–6.91 (m, 9H), 6.80 (dd, J = 2.8, 8.8 Hz, 1/2H), 6.60 (d, J = 8.8 Hz, 1/2H), 4.54 (dd, J = 4.4, 6.8 Hz, 1/2H), 4.41 (dd, J = 6.8, 11.6 Hz, 1/2H), 4.38 (dd, J = 4.8, 12.0 Hz, 1/2H), 4.04 (t, J = 6.8 Hz, 1/2H), 3.82 (s, 3/2H), 3.75 (s, 3/2H), 2.67–2.62 (m, 1/2H), 2.46 (s, 3/2H), 2.43 (s, 3/2H), 2.35–2.17 (m, 5/2H), 2.15–2.06 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 197.9 (1/2x), 196.6 (1/2x), 158.2 (1/2x), 158.1 (1/2x), 144.9 (1/2x), 144.8 (1/2x), 142.8 (1/2x), 142.5 (1/2x), 139.61 (1/2x), 139.57 (1/2x), 135.4 (1/2x), 135.1 (1/2x), 134.5 (1/2x), 134.2 (1/2x), 132.9 (1/2x), 130.2 (1/2x), 129.54, 129.49 (2x), 128.9, 128.7, 128.6, 128.5, 127.6, 126.9 (1/2x), 126.5 (1/2x), 119.2 (1/2x) 118.2 (1/2x), 112.7 (1/2x), 111.8 (1/2x), 73.8 (1/2x), 72.9 (1/2x), 55.39 (1/2x), 55.37 (1/2x), 48.3 (1/2x), 46.5 (1/2x), 31.8 (1/2x), 29.8 (1/2x), 22.9 (1/2x), 22.5 (1/2x), 21.61 (1/2x), 21.56 (1/2x).
6,7-Dimethoxy-4-methyl-4-phenyl-2-(toluene-4-sulfonyl)-3,4-dihydro-2H-naphthalen-1-one (6a). 6a was synthesized according to general synthetic procedure from 4o (450 mg, 1.0 mmol); yield = 84% (378 mg); colorless solid; mp = 129–131 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O5S 451.1579, found 451.1583; 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 8.0 Hz, 2H), 7.49 (s, 1H), 7.31 (d, J = 8.0 Hz, 2H), 7.27–7.18 (m, 3H), 6.94–6.91 (m, 2H), 6.81 (s, 1H), 3.92 (s, 3H), 3.90 (s, 3H), 3.80 (dd, J = 4.4, 13.6 Hz, 1H), 3.07 (dd, J = 4.4, 13.2 Hz, 1H), 2.70 (dd, J = 13.2, 13.6 Hz, 1H), 2.41 (s, 3H), 1.89 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 187.7, 154.5, 148.4, 145.2, 144.6, 143.5, 136.5, 129.4 (2x), 129.3 (2x), 128.7 (2x), 127.0, 126.6 (2x), 125.7, 109.1, 108.8, 66.9, 56.1, 56.0, 42.4, 38.2, 30.1, 21.6. Single-crystal X-ray diagram: crystal of compound 6a was grown by slow diffusion of EtOAc into a solution of compound 6a in CH2Cl2 to yield colorless prisms. The compound crystallizes in the monoclinic crystal system, space group P21/c, a = 17.4857(9) Å, b = 9.6877(5) Å, c = 17.1467(9) Å, V = 2547.4(2) Å3, Z = 4, dcalcd = 1.175 g cm−3, F(000) = 952, 2θ range = 2.376–26.419°, R indices (all data) R1 = 0.0524, wR2 = 0.1009.
2-Benzenesulfonyl-6,7-dimethoxy-4-methyl-4-phenyl-3,4-dihydro-2H-naphthalen-1-one (6b). 6b was synthesized according to general synthetic procedure from 4p (436 mg, 1.0 mmol); yield = 83% (362 mg); colorless solid; mp = 136–138 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25O5S 437.1423, found 437.1430; 1H NMR (400 MHz, CDCl3): δ 7.95–7.92 (m, 2H), 7.64–7.59 (m, 1H), 7.55–7.50 (m, 2H), 7.49 (s, 1H), 7.28–7.19 (m, 3H), 6.94–6.92 (m, 2H), 6.82 (s, 1H), 3.92 (s, 3H), 3.91 (s, 3H), 3.83 (dd, J = 4.4, 14.0 Hz, 1H), 3.08 (dd, J = 4.4, 13.2 Hz, 1H), 2.72 (dd, J = 13.2, 14.0 Hz, 1H), 1.89 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 187.6, 154.6, 148.5, 145.2, 143.5, 139.5, 133.6, 129.3 (2x), 128.7 (4x), 127.0, 126.6 (2x), 125.7, 109.1, 108.9, 66.9, 56.2, 56.0, 42.4, 38.2, 30.2.
2-Methanesulfonyl-6,7-dimethoxy-4-methyl-4-phenyl-3,4-dihydro-2H-naphthalen-1-one (6c). 6c was synthesized according to general synthetic procedure from 4q (374 mg, 1.0 mmol); yield = 84% (314 mg); colorless solid; mp = 155–156 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H23O5S 375.1266, found 375.1275; 1H NMR (400 MHz, CDCl3): δ 7.59 (s, 1H), 7.28–7.18 (m, 3H), 6.95–6.92 (m, 2H), 6.86 (s, 1H), 3.954 (s, 3H), 3.948 (s, 3H), 3.57 (dd, J = 4.4, 13.6 Hz, 1H), 3.23 (s, 3H), 2.99 (dd, J = 4.4, 13.2 Hz, 1H), 2.68 (dd, J = 13.2, 13.6 Hz, 1H), 1.90 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 188.4, 154.9, 148.6, 145.3, 143.9, 128.7 (2x), 127.0, 126.5 (2x), 125.5, 109.2, 108.9, 65.3, 56.2, 56.0, 43.1, 41.9, 35.8, 30.0.
6,7-Dimethoxy-4-methyl-4-phenyl-2-(toluene-3-sulfonyl)-3,4-dihydro-2H-naphthalen-1-one (6d). 6d was synthesized according to general synthetic procedure from 4r (450 mg, 1.0 mmol); yield = 84% (378 mg); colorless solid; mp = 175–176 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27O5S 451.1579, found 451.1586; 1H NMR (400 MHz, CDCl3): δ 7.74–7.71 (m, 2H), 7.50 (s, 1H), 7.41–7.40 (m, 2H), 7.27–7.19 (m, 3H), 6.94–6.91 (m, 2H), 6.82 (s, 1H), 3.92 (s, 3H), 3.91 (s, 3H), 3.83 (dd, J = 4.0, 13.6 Hz, 1H), 3.05 (dd, J = 4.0, 12.8 Hz, 1H), 2.71 (dd, J = 13.6, 13.6 Hz, 1H), 2.41 (s, 3H), 1.89 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 187.6, 154.5, 148.5, 145.2, 143.4, 139.4, 138.9, 134.4, 129.3, 128.7 (2x), 128.6, 127.0, 126.6 (2x), 126.4, 125.8, 109.1, 108.9, 66.7, 56.1, 56.0, 42.4, 38.2, 30.2, 21.3.
5,6,7-Trimethoxy-4-methyl-4-phenyl-2-(toluene-4-sulfonyl)-3,4-dihydro-2H-naphthalen-1-one (6e). 6e was synthesized according to general synthetic procedure from 4s (480 mg, 1.0 mmol); yield = 88% (423 mg); colorless solid; mp = 245–247 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C27H29O6S 481.1685, found 481.1689; 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.32–7.26 (m, 3H), 7.29 (s, 1H), 7.20–7.17 (m, 2H), 4.36 (dd, J = 6.0, 12.8 Hz, 1H), 3.84 (s, 3H), 3.78 (s, 3H), 2.86 (s, 3H), 2.68–2.60 (m, 2H), 2.44 (s, 3H), 1.90 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 188.2, 152.9, 151.0, 150.3, 148.6, 144.8, 137.8, 136.1, 129.6 (2x), 129.4 (4x), 128.3 (2x), 126.1, 125.7, 104.7, 66.6, 60.5, 59.1, 55.9, 42.0, 41.0, 22.2, 21.7.
1-(3,4-Dimethoxyphenyl)-4-hydroxy-2-(toluene-4-sulfonyl)hexan-1-one (7a). 7a was synthesized according to general synthetic procedure from 4t (388 mg, 1.0 mmol); two isomers (ratio: trans/cis = 2/1); yield = 60% (244 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H27O6S 407.1528, found 407.1532; 1H NMR (400 MHz, CDCl3): δ 7.69–7.61 (m, 3H), 7.49 (d, J = 2.0 Hz, 2/3H), 7.45 (dd, J = 2.0 Hz, 1/3H), 7.28 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 8.4 Hz, 2/3H), 6.87 (d, J = 8.8 Hz, 1/3H), 5.47 (dd, J = 2.8, 11.2 Hz, 2/3H), 5.25 (dd, J = 4.4, 8.4 Hz, 1/3H), 3.944 (s, 2H), 2.40–2.04 (m, 1H), 3.92 (s, 2H), 3.91 (s, 1H), 3.78–3.72 (m, 1/3H), 3.26 (br s, 2/3H), 2.42 (s, 3H), 2.37–2.20 (m, 2H), 1.52–1.36 (m, 2H), 1.43 (br s, 1H), 0.89 (t, J = 7.6 Hz, 1H), 0.86 (t, J = 7.2 Hz, 2H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 191.0, 154.2, 149.1, 145.1, 133.8, 130.4, 129.6 (2x), 129.5 (2x), 124.7, 110.6, 110.1, 69.9, 66.5, 56.1, 56.0, 35.1, 31.0, 21.6, 9.7.
1-(3,4-Dimethoxyphenyl)-2-(4-fluorobenzenesulfonyl)-4-hydroxyhexan-1-one (7b). 7b was synthesized according to general synthetic procedure from 4u (392 mg, 1.0 mmol); two isomers (ratio: trans/cis = 2/1); yield = 50% (205 mg); colorless solid; mp = 87–89 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H24FO6S 411.1278, found 411.1283; 1H NMR (400 MHz, CDCl3): δ 7.80–7.75 (m, 2H), 7.65 (dd, J = 2.0, 8.4 Hz, 2/3H), 7.60 (dd, J = 2.0, 8.4 Hz, 1/3H), 7.48 (d, J = 2.4 Hz, 2/3H), 7.44 (d, J = 2.4 Hz, 1/3H), 7.19–7.14 (m, 2H), 6.87 (d, J = 8.4 Hz, 2/3H), 6.76 (d, J = 8.4 Hz, 1/3H), 5.50 (dd, J = 2.4, 7.6 Hz, 2/3H), 5.27 (dd, J = 4.4, 8.4 Hz, 1/3H), 3.94 (s, 2H), 3.93 (s, 1H), 3.91 (s, 2H), 3.90 (s, 1H), 3.80–3.72 (m, 1/3H), 3.26–3.22 (m, 2/3H), 2.37–2.20 (m, 1H), 2.16–2.06 (m, 1H), 1.56 (br s, 1H), 1.51–1.35 (m, 2H), 0.88 (t, J = 7.6 Hz, 1H), 0.86 (t, J = 7.2 Hz, 2H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 190.9, 166.0 (d, J = 255.4 Hz), 154.4, 154.1, 149.2, 132.5 (d, J = 9.9 Hz, 2x), 130.2, 124.7, 116.1 (d, J = 22.7 Hz, 2x), 110.5, 110.1, 69.8, 66.5, 56.2, 56.0, 35.2, 30.9, 9.6.
1-(3,4-Dimethoxyphenyl)-4-hydroxy-2-(4-methoxybenzenesulfonyl)hexan-1-one (7c). 7c was synthesized according to general synthetic procedure from 4v (404 mg, 1.0 mmol); two isomers (ratio: trans/cis = 2/1); yield = 50% (211 mg); colorless solid; mp = 95–97 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H27O7S 423.1478, found 423.1488; 1H NMR (400 MHz, CDCl3): δ 7.68–7.64 (m, 8/3H), 7.61 (dd, J = 2.0, 8.4 Hz, 1/3H), 7.47 (d, J = 2.0 Hz, 2/3H), 7.44 (d, J = 2.0 Hz, 1/3H), 6.94–6.91 (m, 2H), 6.86 (d, J = 8.4 Hz, 2/3H), 6.85 (d, J = 8.4 Hz, 1/3H), 5.46 (dd, J = 3.2, 11.6 Hz, 2/3H), 5.25 (dd, J = 3.6, 8.4 Hz, 1/3H), 3.922 (s, 2H), 3.918 (s, 1H), 3.90 (s, 2H), 3.89 (s, 1H), 3.84 (s, 1H), 3.83 (s, 2H), 3.76–3.72 (br s, 1/3H), 3.25–3.22 (m, 2/3H), 2.35–2.22 (m, 1H), 2.16–2.02 (m, 1H), 1.66 (br s, 1H), 1.50–1.32 (m, 2H), 0.88 (t, J = 7.6 Hz, 1H), 0.85 (t, J = 7.2 Hz, 2H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 191.2, 164.0, 154.1, 149.0, 131.7 (2x), 130.4, 128.2, 124.6, 114.0 (2x), 110.5, 110.1, 69.9, 66.5, 56.1, 55.9, 55.6, 35.1, 30.9, 9.6.
2-(4-t-Butylbenzenesulfonyl)-1-(3,4-dimethoxyphenyl)-4-hydroxyhexan-1-one (7d). 7d was synthesized according to general synthetic procedure from 4w (430 mg, 1.0 mmol); two isomers (ratio: trans/cis = 2/1); yield = 58% (260 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H33O6S 449.1998, found 449.2005; 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.4 Hz, 2H), 7.57 (dd, J = 2.0, 8.4 Hz, 1H), 7.45 (dd, J = 2.0, 8.4 Hz, 1H), 7.26 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 8.4 Hz, 2/3H), 6.83 (d, J = 8.4 Hz, 1/3H), 5.47 (dd, J = 2.8, 7.6 Hz, 2/3H), 5.25 (dd, J = 4.4, 8.4 Hz, 1/3H), 3.93 (s, 2H), 7.92 (s, 1H), 3.91 (s, 2H), 3.89 (s, 1H), 3.77–3.75 (s, 1/3H), 3.29–3.24 (s, 2/3H), 2.64 (t, J = 7.6 Hz, 2H), 2.40–2.28 (m, 2/3H), 2.20–2.07 (m, 4/3H), 1.59–1.25 (m, 7H), 0.93–0.84 (m, 6H); for major product: 13C{1H} NMR (100 MHz, CDCl3): δ 191.0, 153.9, 149.9, 149.0, 134.1, 130.4, 129.6 (2x), 128.8 (2x), 124.5, 110.5, 110.0, 70.0, 66.4, 56.1, 55.9, 35.6, 34.9, 33.0, 31.0, 22.2, 13.8, 9.6.
A representative synthetic procedure of skeleton 10
NaBH4 (68 mg, 2.0 mmol) was added to a solution of 5a (225 mg, 0.5 mmol) in MeOH (15 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 3 h. The reaction mixture was concentrated and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Without further purification, the freshly prepared arylmagnesium bromide (Ar′MgBr, 1.0 mmol) in THF (10 mL) was added to a solution of CuI (190 mg, 1.0 mmol) in THF (10 mL) at 25 °C. Then, a solution of the resulting vinyl sulfone (130 mg, 0.3 mmol) in THF (5 mL) was added to the reaction mixture at 25 °C. The reaction mixture was stirred at 25 °C for 20 h. The reaction mixture was concentrated and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 10/1–2/1) afforded 10.
5-(4-Fluorophenyl)-2,3-dimethoxy-9-phenyl-6-(toluene-4-sulfonyl)-6,7,8,9-tetrahydro-5H-benzocycloheptene (10a). Yield = 83% (132 mg); colorless solid; mp = 214–215 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C32H32FO4S 531.2005, found 531.2011; 1H NMR (400 MHz, CDCl3): δ 7.50 (d, J = 8.0 Hz, 2H), 7.41 (br s, 2H), 7.40 (br s, 2H), 7.33–7.28 (m, 1H), 7.06 (d, J = 8.0 Hz, 2H), 7.05–7.01 (m, 2H), 6.87–6.83 (m, 2H), 6.25 (s, 1H), 5.99 (s, 1H), 5.25 (d, J = 9.2 Hz, 1H), 4.88 (dd, J = 6.4, 11.6 Hz, 1H), 3.90–3.86 (m, 1H), 3.54 (s, 3H), 3.48 (s, 3H), 2.63–2.53 (m, 2H), 2.34 (s, 3H), 2.04–1.96 (m, 1H), 1.56–1.47 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 161.5 (d, J = 244.9 Hz), 147.8, 146.8, 144.2, 142.5, 136.8, 136.5 (d, J = 3.0 Hz), 134.7, 130.7 (d, J = 8.4 Hz, 2x), 130.3, 129.3 (2x), 129.1 (2x), 128.4 (2x), 128.4 (2x), 126.7, 115.2 (d, J = 21.2 Hz, 2x), 111.9, 111.2, 65.9, 55.7, 55.7, 45.8, 44.0, 26.8, 24.4, 21.4.
2,3-Dimethoxy-9-phenyl-6-(toluene-4-sulfonyl)-5-p-tolyl-6,7,8,9-tetrahydro-5H-benzocycloheptene (10b). Yield = 76% (120 mg); colorless solid; mp = 194–195 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C33H35O4S 527.2256, found 527.2263; 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 7.30–7.20 (m, 5H), 7.14 (d, J = 8.0 Hz, 2H), 7.05 (d, J = 7.2 Hz, 2H), 6.50 (s, 1H), 6.13 (s, 1H), 4.86 (d, J = 3.6 Hz, 1H), 3.88–3.83 (m, 1H), 3.77 (s, 3H), 3.75–3.71 (m, 1H), 3.56 (s, 3H), 2.44 (s, 3H), 2.34 (s, 3H), 2.26–2.21 (m, 1H), 2.05–1.97 (m, 1H), 1.83–1.74 (m, 1H), 1.41–1.31 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 148.1, 147.2, 144.7, 141.9, 139.6, 136.3, 134.6, 134.3, 129.7 (2x), 129.3 (2x), 129.13 (2x), 129.06, 128.3 (4x), 127.2 (2x), 126.7, 114.5, 111.3, 68.4, 55.8, 55.6, 47.0, 44.1, 28.9, 25.8, 21.6, 20.9.
2,3-Dimethoxy-5-naphthalen-2-yl-9-phenyl-6-(toluene-4-sulfonyl)-6,7,8,9-tetrahydro-5H-benzocycloheptene (10c). Yield = 74% (125 mg); colorless solid; mp = 197–198 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C36H35O4S 563.2256, found 563.2264; 1H NMR (400 MHz, CDCl3): δ 7.78–7.76 (m, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.60–7.58 (m, 1H), 7.48–7.39 (m, 8H), 7.35 (s, 1H), 7.34–7.29 (m, 1H), 7.22 (d, J = 1.6, 8.4 Hz, 1H), 6.68 (d, J = 8.0 Hz, 2H), 6.29 (s, 1H), 6.11 (s, 1H), 5.37 (d, J = 9.2 Hz, 1H), 4.98 (dd, J = 6.8, 11.6 Hz, 1H), 4.13–4.06 (m, 1H), 3.55 (s, 3H), 3.37 (s, 3H), 2.77–2.73 (m, 1H), 2.70–2.60 (m, 1H), 2.11–2.03 (m, 1H), 1.84 (s, 3H), 1.67–1.57 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 147.8, 146.8, 144.0, 142.6, 137.7, 136.7, 134.8, 133.1, 132.1, 130.5, 129.2 (2x), 128.9 (2x), 128.3 (3x), 128.1 (2x), 128.0, 127.6, 127.4, 127.1, 126.7, 126.1, 125.8, 112.2, 111.2, 65.5, 55.8, 55.7, 46.9, 44.1, 26.9, 24.2, 20.9. Single-crystal X-ray diagram: crystal of compound 10c was grown by slow diffusion of EtOAc into a solution of compound 10c in CH2Cl2 to yield colorless prisms. The compound crystallizes in the monoclinic crystal system, space group P21/c, a = 15.699(4) Å, b = 7.434(2) Å, c = 24.908(8) Å, V = 2874.6(14) Å3, Z = 4, dcalcd = 1.300 g cm−3, F(000) = 1192, 2θ range = 1.312–25.146°, R indices (all data) R1 = 0.1166, wR2 = 0.1989.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank the Ministry of Science and Technology of the Republic of China for its financial support (MOST 106-2628-M-037-001-MY3).
Notes and references
-
(a) H.-L. Qin, J. Leng, C.-P. Zhang, I. Jantan, M. W. Amjad, M. Sher, M. Naeem-ul-Hassan, M. Z. Hussain and S. N. A. Bukhari, J. Med. Chem., 2016, 59, 3549–3561 CrossRef PubMed;
(b) S. W. Yee, L. Jarno, M. S. Gomaa, C. Elford, L.-L. Ooi, M. P. Coogan, R. McClelland, R. I. Nicholson, B. A. J. Evans, A. Brancale and C. Simons, J. Med. Chem., 2005, 48, 7123–7131 CrossRef PubMed.
-
(a) H. Niu, T. E. Strecker, J. L. Gerberich, J. E. Campbell III, D. Saha, D. Mondal, E. Hamel, D. J. Chaplin, R. P. Mason, M. L. Trawick and K. G. Pinney, J. Med. Chem., 2019, 62, 5594–5615 CrossRef PubMed;
(b) P. Y. Maximove, D. J. Fernandes, R. E. McDaniel, C. B. Myers, R. F. Curpan and V. C. Jordan, J. Med. Chem., 2014, 57, 4569–4583 CrossRef PubMed;
(c) Y. Yadagiri, U. D. Holagunda, R. Bantu, L. Nagarapu, C. G. Kumar, S. Pombala and B. Sridhar, Eur. J. Med. Chem., 2014, 79, 260–265 CrossRef PubMed;
(d) R. McCague, R. Kuroda, G. Leclercq and S. Stoessel, J. Med. Chem., 1986, 29, 2053–2059 CrossRef CAS PubMed.
-
(a) H.-L. Li, X.-M. Li, X. Li, C.-Y. Wang, H. Liu, M. U. Kassack, L. H. Meng and B.-G. Wang, J. Nat. Prod., 2017, 80, 162–168 CrossRef CAS PubMed;
(b) K. P. Devkota, D. Covell, T. Ransom, J. B. McMahon and J. A. Beutler, J. Nat. Prod., 2013, 76, 710–714 CrossRef CAS PubMed;
(c) Y. Tezuka, K. Morikawa, F. Li, L. Auw, S. Awale, T. Nobukawa and S. Kadota, J. Nat. Prod., 2011, 74, 102–105 CrossRef CAS PubMed.
-
(a) G. Majetich and G. Zou, Org. Lett., 2008, 10, 81–83 CrossRef CAS PubMed;
(b) Z.-W. Jiao, Y.-Q. Tu, Q. Zhang, W.-X. Liu, S.-Y. Zhang, S.-H. Wang, F.-M. Zhang and S. Jiang, Nat. Commun., 2015, 6, 7332 CrossRef CAS PubMed.
-
(a) C. Zhao, F. D. Toste and R. G. Bergman, J. Am. Chem. Soc., 2011, 133, 10787–10789 CrossRef CAS PubMed;
(b) S. K. Thummanapelli, S. Hosseyni, Y. Su, N. G. Akhmedov and X. Shi, Chem. Commun., 2016, 52, 7687–7690 RSC;
(c) E. V. Beletskiy, C. Sudheer and C. J. Douglas, J. Org. Chem., 2012, 77, 5884–5893 CrossRef CAS PubMed;
(d) Y. Qin, J. Lv, S. Luo and J.-P. Cheng, Org. Lett., 2014, 16, 5032–5035 CrossRef CAS PubMed.
- For a review, see:
(a) T. A. Farghaly, S. M. Gomha, K. M. Dawood and M. R. Shaaban, RSC Adv., 2016, 6, 17955–17979 RSC and cited references therein. For a recent example, see:
(b) D. Cao, K. Zhang, R. An, H. Xu, S. Hao, X. Yang, Z. Hou and C. Guo, Org. Lett., 2019, 21, 8948–8951 CrossRef CAS PubMed.
- M.-Y. Chang and Y.-C. Cheng, Org. Lett., 2016, 18, 608–611 CrossRef CAS PubMed.
- M.-Y. Chang, Y.-T. Hsiao and K.-H. Lai, J. Org. Chem., 2018, 83, 14110–14119 CrossRef CAS PubMed.
- E. Hasegawa, K. Nemoto, R. Nagumo, E. Tayama and H. Iwamoto, J. Org. Chem., 2016, 81, 2692–2703 CrossRef CAS PubMed.
- E. V. Beletskiy, C. Sudheer and C. J. Douglas, J. Org. Chem., 2012, 77, 5884–5893 CrossRef CAS PubMed.
- E. Fillion, D. Fishlock, A. Wilsily and J. M. Goll, J. Org. Chem., 2005, 70, 1316–1327 CrossRef CAS PubMed.
-
(a) C. Jia, W. Lu, J. Oyamada, T. Kitamura, K. Matsuda, M. Irie and Y. Fujiwara, J. Am. Chem. Soc., 2000, 122, 7252–7263 CrossRef CAS;
(b) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992–1995 CrossRef CAS PubMed.
- For iridium complex, see:
(a) M. Nagamoto, H. Yorimitsu and T. Nishimura, Org. Lett., 2018, 20, 828–831 CrossRef CAS PubMed;
(b) M. Nagamoto, J.-i. Fukuda, M. Hatano, H. Yorimitsu and T. Nishimura, Org. Lett., 2017, 19, 5952–5955 CrossRef CAS PubMed.
- For cobalt complex, see:
(a) W. Xu and N. Yoshikai, Org. Lett., 2018, 20, 1392–1395 CrossRef CAS PubMed;
(b) F. Schroeter, S. Lerch, M. Kaliner and T. Strassner, Org. Lett., 2018, 20, 6215–6219 CrossRef CAS PubMed;
(c) H. Shigehisa, T. Ano, H. Honma, K. Ebisawa and K. Hiroya, Org. Lett., 2016, 18, 3622–3625 CrossRef PubMed.
- For platinium complex, see:
(a) D. Kang, J. Kim, S. Oh and P. H. Lee, Org. Lett., 2012, 14, 5636–5639 CrossRef PubMed;
(b) X. Han and R. A. Widenhoefer, Org. Lett., 2006, 8, 3801–3804 CrossRef PubMed;
(c) S. J. Pastine, S. W. Youn and D. Sames, Org. Lett., 2003, 5, 1055–1058 CrossRef PubMed.
- For ruthenium complex, see:
(a) A. Mandal, H. Sahoo, S. Dana and M. Baidya, Org. Lett., 2017, 19, 4138–4141 CrossRef PubMed;
(b) A. Biafora, B. A. Khan, J. Bahri, J. M. Hewer and L. J. Goossen, Org. Lett., 2017, 19, 1232–1235 CrossRef PubMed;
(c) M.-O. Simon, J.-P. Genet and S. Darses, Org. Lett., 2010, 12, 3038–3041 CrossRef PubMed.
- For bismuth complex, see:
(a) G. Lemière, B. Cacciuttolo, E. Belhassen and E. Duñach, Org. Lett., 2012, 14, 2750–2753 CrossRef PubMed;
(b) M. Rueping, B. J. Nachtsheim and T. Scheidt, Org. Lett., 2006, 8, 3717–3719 CrossRef PubMed.
- For gold complex, see:
(a) R. Babouri, L. Traore, Y. A. Bekro, V. I. Matveeva, Y. M. Sadykova, J. K. Voronina, A. R. Burilov, T. Ayad, J. N. Volle, D. Virieux and J. L. Pirat, Org. Lett., 2019, 21, 45–49 CrossRef PubMed;
(b) M. Jha, S. Dhiman, T. S. Cameron, D. Kumar and A. Kumar, Org. Lett., 2017, 19, 2038–2041 CrossRef CAS PubMed;
(c) J. C. Timmerman, W. W. Schmitt and R. A. Widenhoefer, Org. Lett., 2016, 18, 4966–4969 CrossRef CAS PubMed.
- For rhodium complex, see:
(a) D.-Y. Li, Z.-L. Huang and P.-N. Liu, Org. Lett., 2018, 20, 2028–2032 CrossRef CAS PubMed;
(b) K. Shibata, S. Natsui and N. Chatani, Org. Lett., 2017, 19, 2234–2237 CrossRef CAS PubMed;
(c) F. Wang, Z. Qi, J. Sun, X. Zhang and X. Li, Org. Lett., 2013, 15, 6290–6293 CrossRef CAS PubMed.
- For zinc complex, see: S. Y. Lee, A. Villani-Gale and C. C. Eichman, Org. Lett., 2016, 18, 5034–5037 CrossRef CAS PubMed.
- For indium complex, see:
(a) V. K. Rao, G. M. Shelke, R. Tiwari, K. Parang and A. Kumar, Org. Lett., 2013, 15, 2190–2193 CrossRef CAS PubMed;
(b) Y. Kwon, H. Cho and S. Kim, Org. Lett., 2013, 15, 920–923 CrossRef CAS PubMed.
- For acids mediated routes, see:
(a) X. Cai, A. Tohti, C. Ramirez, H. Harb, J. C. Fettinger, H. P. Hratchian and B. J. Stokes, Org. Lett., 2019, 21, 1574–1577 CrossRef CAS PubMed;
(b) W. Zhu, Q. Sun, Y. Wang, D. Yuan and Y. Yao, Org. Lett., 2018, 20, 3101–3104 CrossRef CAS PubMed;
(c) Y. Jung and I. Kim, Org. Lett., 2015, 17, 4600–4603 CrossRef CAS PubMed;
(d) X. Cai, A. Keshavarz, J. D. Omaque and B. J. Stokes, Org. Lett., 2017, 19, 2626–2629 CrossRef CAS PubMed.
- Recent examples on synthetic applications of β-ketosulfones by the authors, see:
(a) M.-Y. Chang, Y.-C. Cheng and Y.-J. Lu, Org. Lett., 2014, 16, 6252–6255 CrossRef CAS PubMed;
(b) M.-Y. Chang, Y.-C. Cheng and Y.-J. Lu, Org. Lett., 2015, 17, 1264–1267 CrossRef CAS PubMed;
(c) M.-Y. Chang and Y.-C. Cheng, Org. Lett., 2016, 18, 1682–1685 CrossRef CAS PubMed;
(d) M.-Y. Chang, H.-Y. Chen and Y.-L. Tsai, J. Org. Chem., 2019, 84, 326–337 CrossRef CAS PubMed.
- M.-Y. Chang, Y.-J. Lu and Y.-C. Cheng, Tetrahedron, 2015, 71, 6840–6845 CrossRef CAS.
-
(a) M.-Y. Chang, Y.-H. Chen and Y.-C. Cheng, Tetrahedron, 2016, 72, 518–524 CrossRef CAS;
(b) C.-K. Chan, H.-S. Wang, R.-T. Hsu and M.-Y. Chang, Synthesis, 2017, 49, 2045–2056 CrossRef CAS;
(c) C.-K. Chan, H.-S. Wang, Y.-L. Tsai and M.-Y. Chang, RSC Adv., 2017, 7, 29321–29329 RSC.
- CCDC 1915762 (5a), 1915763 (5b), 1915765 (6a) and 1915767 (10c) contain the ESI crystallographic data for this paper.†.
- K. Namba, H. Yamamoto, I. Sasaki, K. Mori, H. Imagawa and M. Nishizawa, Org. Lett., 2008, 10, 1767–1770 CrossRef CAS PubMed.
- H2O should come from the (CH2Cl)2 solvent. The purity of (CH2Cl)2 is 97% and it contains at least 2–3% of H2O.
-
(a) Z. Cao, R. Zhang, X. Meng, H. Li, J. Li, H. Zhu, G. Chen, X. Sun and J. You, RSC Adv., 2016, 6, 74582–74585 RSC;
(b) J.-J. Yun, M.-L. Zhi, W.-X. Shi, X.-Q. Chu, Z.-L. Shen and T.-P. Loh, Adv. Synth. Catal., 2018, 360, 2632–2637 CrossRef CAS.
- M.-Y. Chang, Y.-J. Lu and Y.-C. Cheng, Tetrahedron, 2015, 71, 1192–1201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scanned photocopies of NMR spectral data for all compounds and X-ray analysis data of compounds 5a, 5b, 6a and 10c. CCDC 1915762, 1915763, 1915765 and 1915767. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01962c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Kai-Xiang Lai and
Yu-Lun Chang
*,
Kai-Xiang Lai and
Yu-Lun Chang