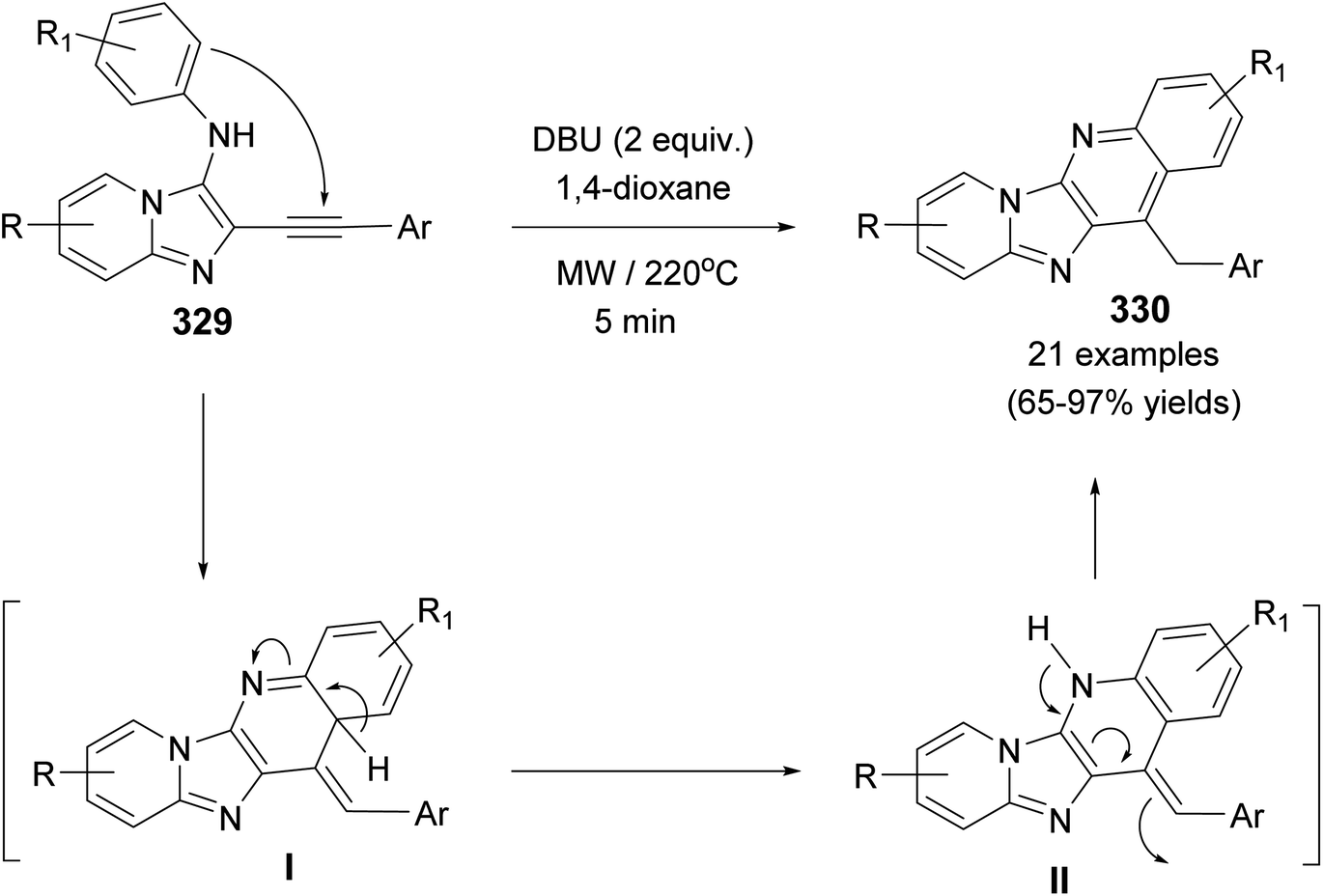
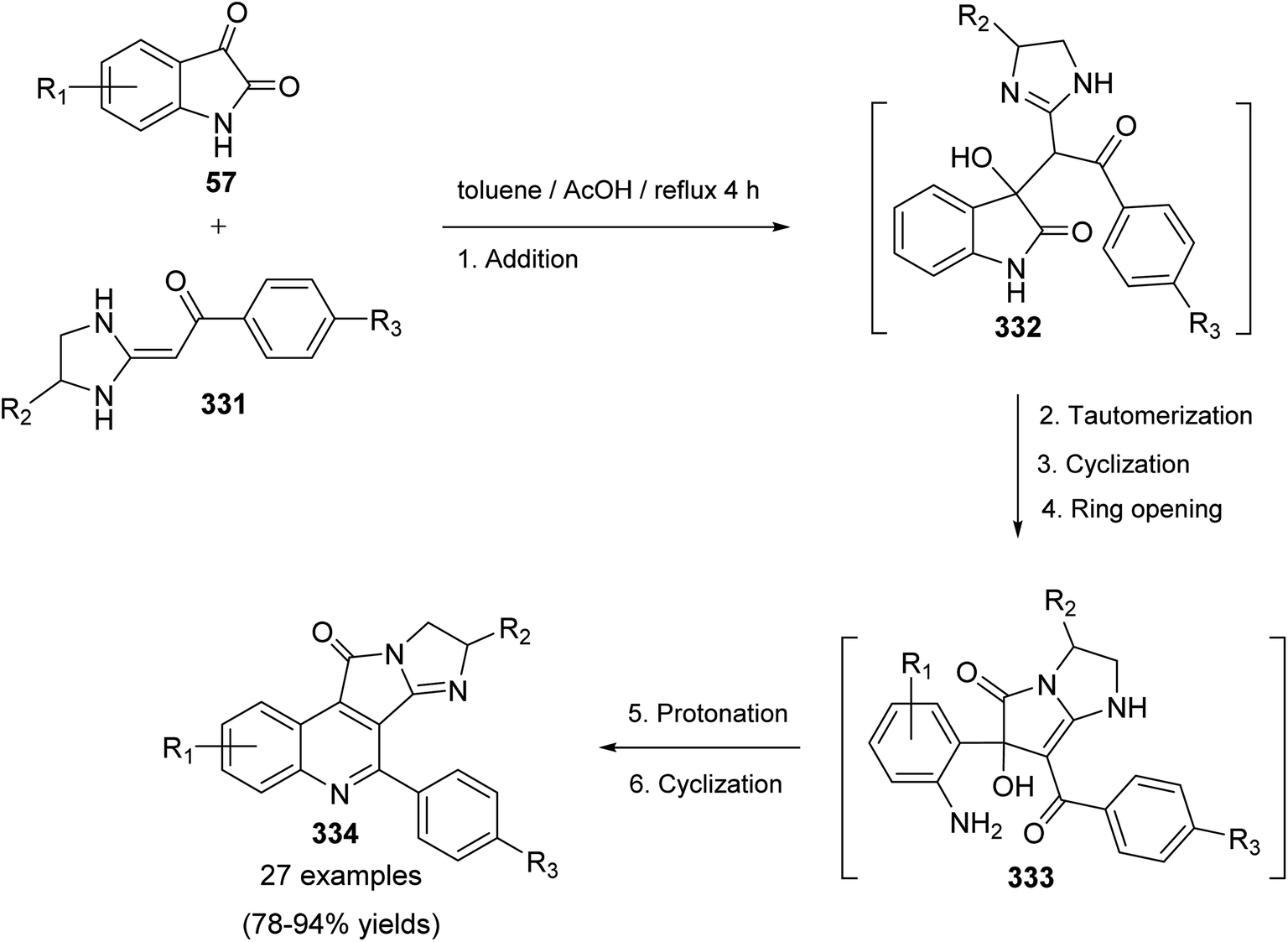
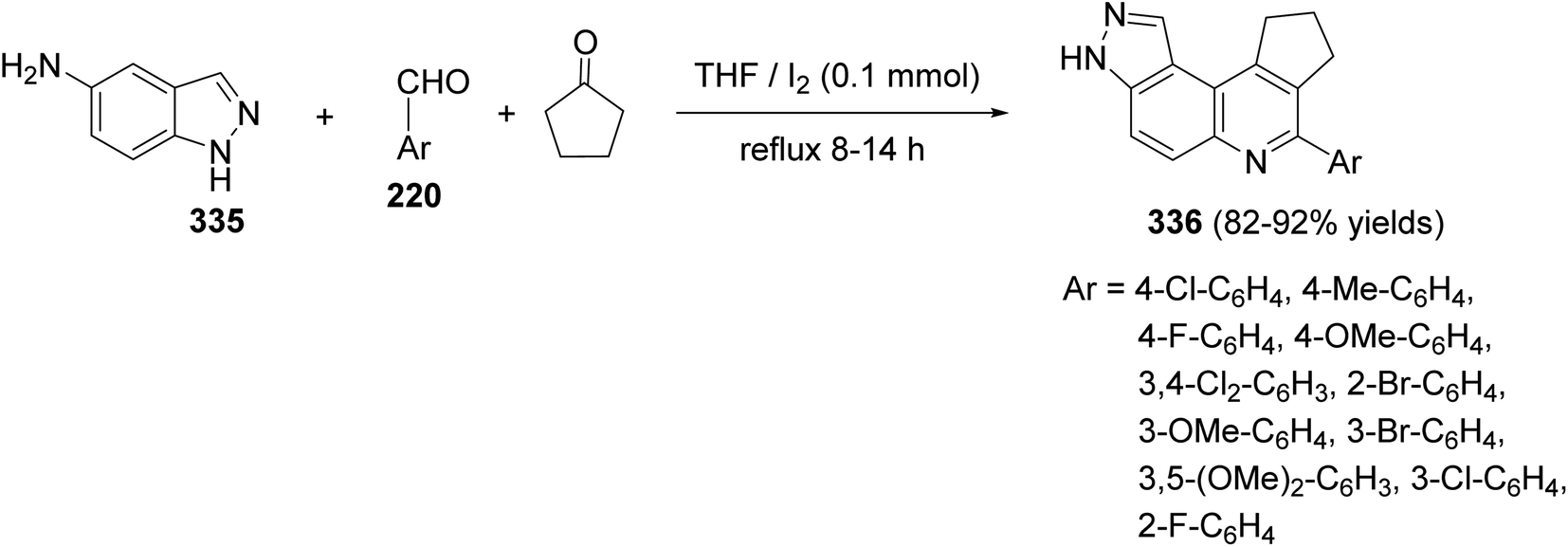
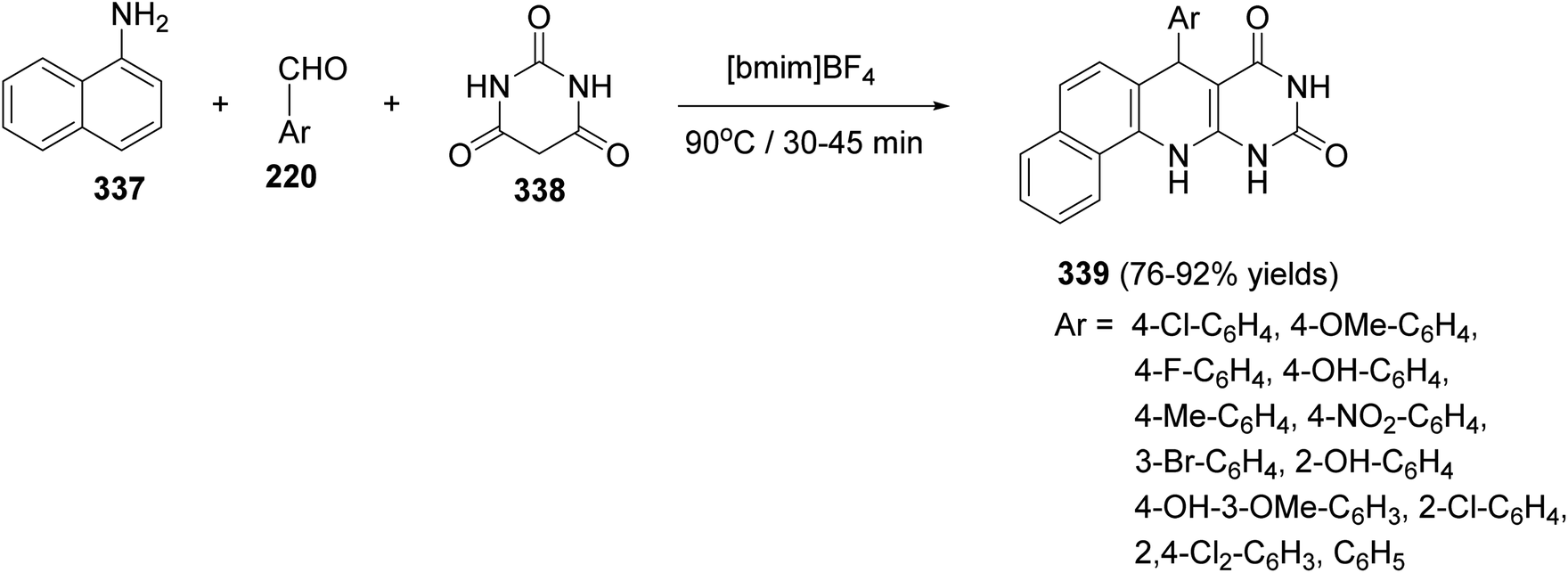
DOI:
10.1039/D0RA02786C
(Review Article)
RSC Adv., 2020,
10, 19867-19935
Advancements in the synthesis of fused tetracyclic quinoline derivatives
Received
26th March 2020
, Accepted 7th May 2020
First published on 27th May 2020
Abstract
Fused tetracyclic systems containing a quinoline nucleus represent an important class of heterocyclic bioactive natural products and pharmaceuticals because of their significant and wide-spectrum biological properties. Several of these compounds have been obtained with diverse pharmacological and biological activities, such as antiplasmodial, antifungal, antibacterial, potent antiparasitic, antiproliferative, anti-tumor and anti-inflammatory activities. This information will be beneficial for medicinal chemists in the field of drug discovery to design and synthesize new fused tetracyclic quinolines as potent therapeutical agents. This review article provides a comprehensive report regarding the methods developed for the synthesis of fused tetracyclic quinolines reported so far (till October 2019). The article includes synthesis by one-pot domino reaction, microwave synthesis using a catalyst, using ionic liquids, photocatalytic synthesis (UV radiation), Pfitzinger reaction, I2-catalyzed cyclization reaction, Wittig reaction, cascade reaction, imino Diels–Alder reaction, Friedel–Crafts reaction, CDC reaction, solvent-free reactions and using small chiral organic molecules as catalysts. To the best of our knowledge, this is the first review focused on the synthesis of fused tetracyclic quinolines along with mechanistic aspects.
 Ramadan A. Mekheimer | Ramadan Ahmed Mekheimer was born in El-Minia, Egypt, on March 9, 1960 and graduated in chemistry with distinction degree at the university of Minia. He received his B.Sc. (1982) degree from Minia University. In 1987–1989, the University of Minia awarded him a channel system program to complete his PhD thesis under the supervision of Prof. Dr Thomas Kappe in Institute of organic chemistry, Karl-Franzens University, Graz, Austria, and he received his PhD (1990) degrees from Chemistry Department, Faculty of Science, Minia University. After his PhD degree he take time in establishing his organic synthesis lab. In, 1994 the University of Minia awarded him the University Prize designated for the outstanding organic researcher scientist. In 1995, he became assistant Professor. During a postdoctoral stay in 1997–1998 at the University of Connecticut, Storrs, USA, he joined the group of Professor M. Smith. In 2000, the Academy of Science and Technology, Cairo, Egypt awarded him “The State's Encouragement National Prize in Organic Chemistry”. Since 2001, he has been Full Professor of Organic Chemistry at the University of Minia. At the beginning of his career, he was interested in the development of new methodologies for the synthesis of new nitrones, heterocyclic azides and studying their chemical behavior, synthesis of amino-quinolines as anti-malaria and the synthesis of novel fused-heterocyclic ring systems, especially those containing pyridine or quinoline nucleus, of biological and pharmacological interest. His current research interests include the design of efficient environmental benign synthetic approaches for the synthesis of potentially bioactive heterocyclic compounds utilizing microwave and ultrasound irradiations and solar energy as energy sources besides the use of eco-friendly catalysts and solvents (particularly water) in organic synthesis. To date, he has synthesized with his research group more than 500 new organic substances, mainly heterocyclic structure. |
| Mariam Abdullah AL-Sheikh was born in 1972 in Makkah-Kingdom of Saudi Arabia. She received her B.Sc. degree in pure chemistry and MSc degree in Organic Chemistry from the University of King AbdulAziz-Jeddah-Saudi Arabia, involving a dissertation entitled “Novel synthesis of heterocyclic compounds containing nitrogen and/ or sulphur” in 1999. Also, she obtained her PhD in 2003 from the University of King AbdulAziz on utility of α-enones in the synthesis of some new heterocyclic compounds under the supervision of Prof. Mohamed Hilmy Elnagdi and Prof. Ebtisam Hafez. In, 2009 and 2013 the University of King AbdulAziz awarded her the University Prize designated for research and academic science. This prize has been given to the best organic researcher scientist in the University. She is currently working as Associate Professor of Organic Chemistry at University of King AbdulAziz. Her current research interest is focused in the development of new synthetic methodology in organic chemistry, which includes the chemistry of nitrogen- and sulfur-containing compounds for the synthesis of biologically active heterocycles. |
| Hanadi Yousef Medrasi was born in 1976 in Makkah-Kingdom of Saudi Arabia. She obtained her MSc degree in Organic Chemistry from the University of King AbdulAziz-Jeddah-Saudi Arabia, involving a dissertation entitled “Novel approaches to functionally substituted condensed azoles and azines” in 2003. She received her PhD degree from the University of King AbdulAziz-Jeddah-Saudi Arabia, completing her doctoral thesis on alkylation reactions of arenes in 2010. In, 2013 the University of King AbdulAziz awarded her the University Prize designated for the outstanding organic researcher scientist. She is currently working as Assistant Professor of Organic Chemistry at the University of Jeddah, Saudi Arabia. Her research interests include the study of synthesis and reactivity of biologically important heterocycles and organic chemistry education. |
 Mariam A. Al-Sheikh | Kamal Usef Sadek was born in El-Minia (1947) and has received B.Sc. degree (honour) in Applied Chemistry from Assiut University (1969) followed by MSc and PhD from Cairo University (1980) under the supervision of Prof. M. H. Elnagdi. He was appointed as a demonstrator of Chemistry in Assiut University then he shifted to the Department of Chemistry, El-Minia University (1975). Since then he was appointed as lecturer (1980), associate Prof. (1985) and Full Professor (1990). In 1987 he was awarded the Alexander von Hmboldt fellowship with Prof. W. Weigrebe in Pegensburg University and have several studies leaves with Prof. M. Regitz of Kuierslautern University and Prof. H. H. Otto in Freiburg University. Currently, he is working in developing green technologies for the synthesis of biologically active heterocycles. |
1. Introduction
Quinoline, a well known heterocyclic compound, itself has few applications in chemical domains, but many of its derivatives are useful in diverse applications including pharmaceuticals, agrochemicals, materials and dyestuffs. Today they are available as drugs. The prominent ones are the fluoroquinolone antibiotics (Ciprofloxacin and its analogs), antimalarials (Quinine, Chloroquine, Quinidine, Mefloquine, Primaquine, Amodiaquinine), antibacterials (Sparfloxacin, Gatifloxacin), a cholesterol lowering agent (Pitavastatin), an antiviral agent (Saquinavir), an anthelmintic agent (Oxamniquine), an antifungal–antiprotozoal agent (Clioquinol), a local anesthetic (Dibucaine), an antiretroviral agent (Saquinavir), an antiasthmatic (Montelukast), antipsychotics (Aripiprazole, Brexpiprazole), an anti-TB agent (Bedaquiline), anticancer agents (Camptothecin, Irinotecan, Topotecan), antiglaucoma (Cartiolol) and cardiotonic (Vesnarinone) agents, protein kinase inhibitors (Lenvatinib, Bosutinib and Cabozantinib), and a farnesyl transferase inhibitor for leukemia (Tipifarnib). Moreover, several quinolines have been reported to display various useful biological and pharmacological activities as antiinflammatory agents,1,2 antipsychotics,3 antiprotozoals,4–9 antituberculosis agents,10–12 anti-Alzheimers agents,13 anti-HIV agents,14–16 antiasthmatics,17 potent melanin-concentrating hormone 1 receptor (MCH1R) antagonists,18–22 antioxidants,23 antivirals,24 antifungals,25,26 Src kinase inhibitors,27 antihypertensive agents,28 anti-microbials,29–31 antibiotics,32 tyrokinase PDGF-RTK inhibiting agents,33 agents for treatment of lupus34,35 and neurodegenerative diseases36 and efflux pump inhibitors.37
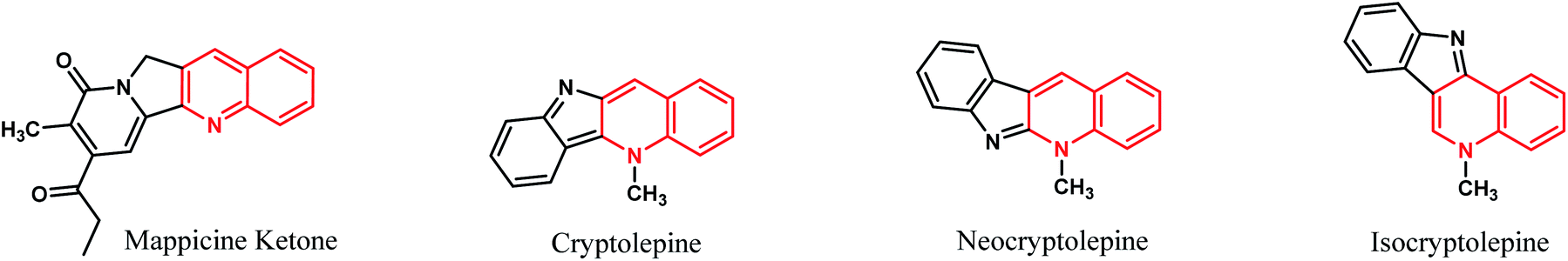
Fused tetracyclic systems containing a quinoline moiety are an important class of organic molecules since many of them exhibit excellent biological activities. They have been well established to be useful as potent topoisomerase I and II inhibitor, antifungal, antiplasmodial, antibacterial, potent antiparasitic, antiproliferative, anti-inflammatory and anti-tumor agents.38–43 In addition, mappicine ketone, as fused quinoline natural product, is an antiviral lead compound with selective activities against herpes viruses HSV-1 and HSV-2 and human cytomegalovirus (HCMV).44 A fused quinoline alkaloids cryptolepine, neocryptolepine, and isocryptolepine are antimalarial natural products having cytotoxic activities.45,46 The chemical structures of these natural products are shown in Fig. 1.
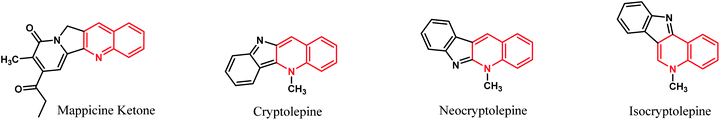 |
| | Fig. 1 Structures of natural products containing quinoline ring. | |
Due to their tremendous pharmacological and biological importance, chemists have developed a large number of protocols for the synthesis of fused tetracyclic systems comprising quinoline nucleus, to the best of our knowledge, this has never been reviewed. The present review article provides, for the first time, a comprehensive compilation of synthetic methods on the synthesis of these tetracyclic ring systems, as a significant family in the field of organic chemistry.
2. Synthesis of tetracyclic quinolines
2.1. Tetracyclic quinolines with one heteroatom
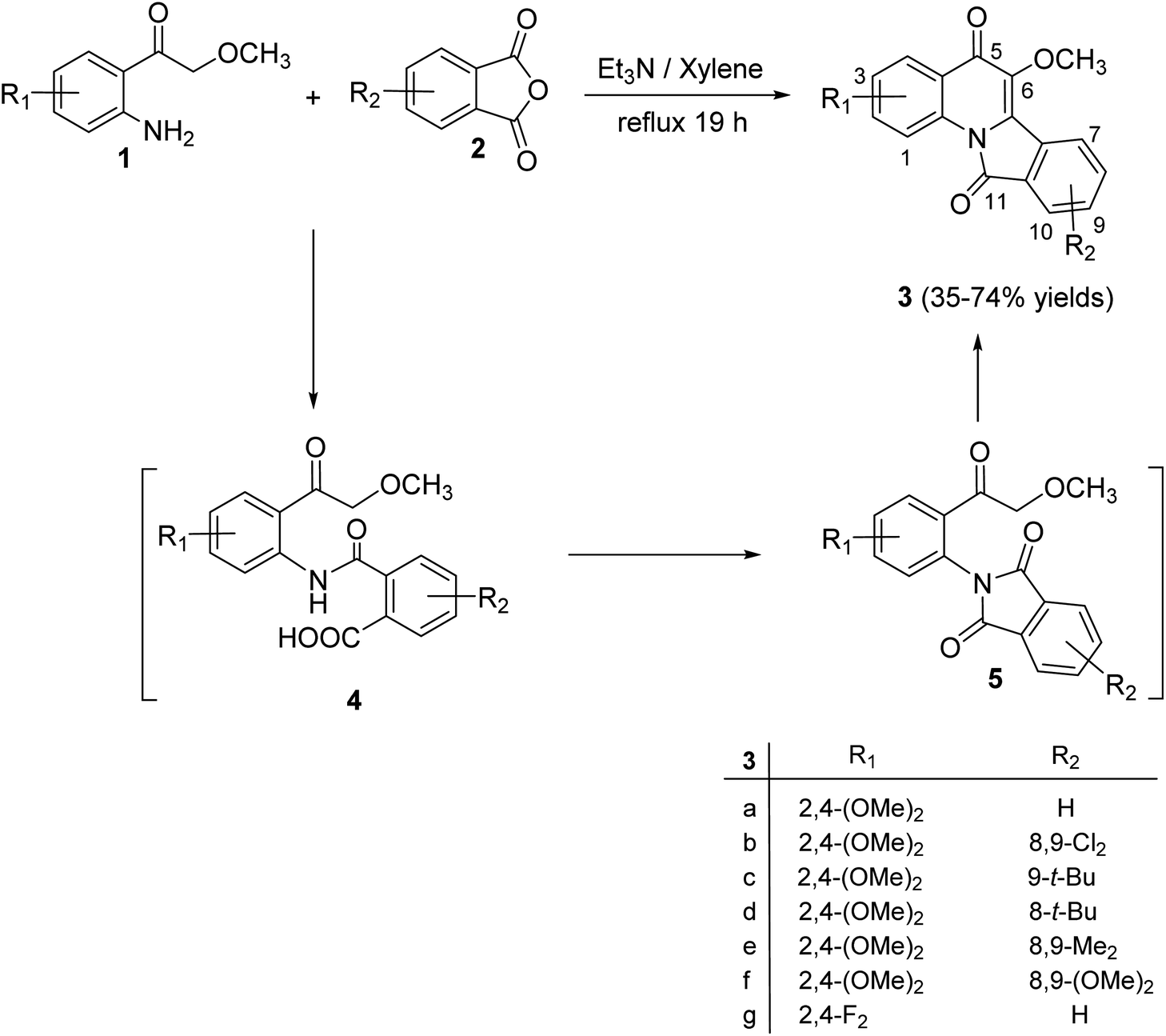
2.1.1. Isoindolo[2,1-a]quinolines. A novel series of tetracyclic isoindolo[2,1-a]quinoline-5,11-dione derivatives 3 have been synthesized and evaluated as potent inhibitors of human topoisomerase-II and DNA-gyrase by Sui et al.47 On heating 2-aminoacetophenones 1 with phthalic anhydrides 2 at reflux in xylene in the presence of TEA, as a base catalyst, the tetracyclic products 3 were obtained in 35–74% yields (Scheme 1). Only in one case the imide intermediate 5 was isolated. The isolation of 5 confirmed that imide formation occurs prior to intramolecular cyclization of the amide in 4 to form the quinoline nucleous (Scheme 1).
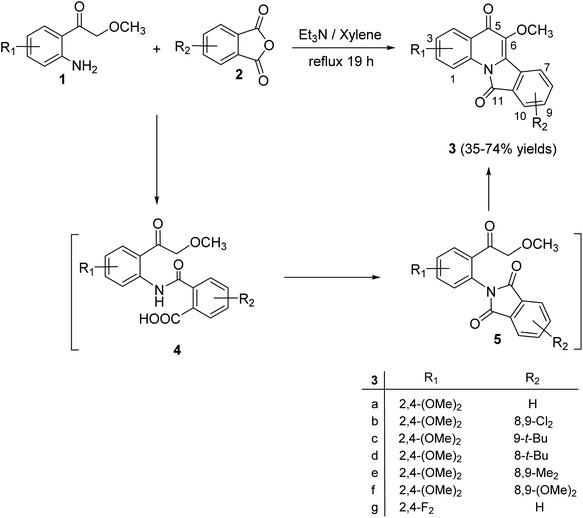 |
| | Scheme 1 Synthesis of isoindolo[2,1-a]quinoline-5,11-diones 3. | |
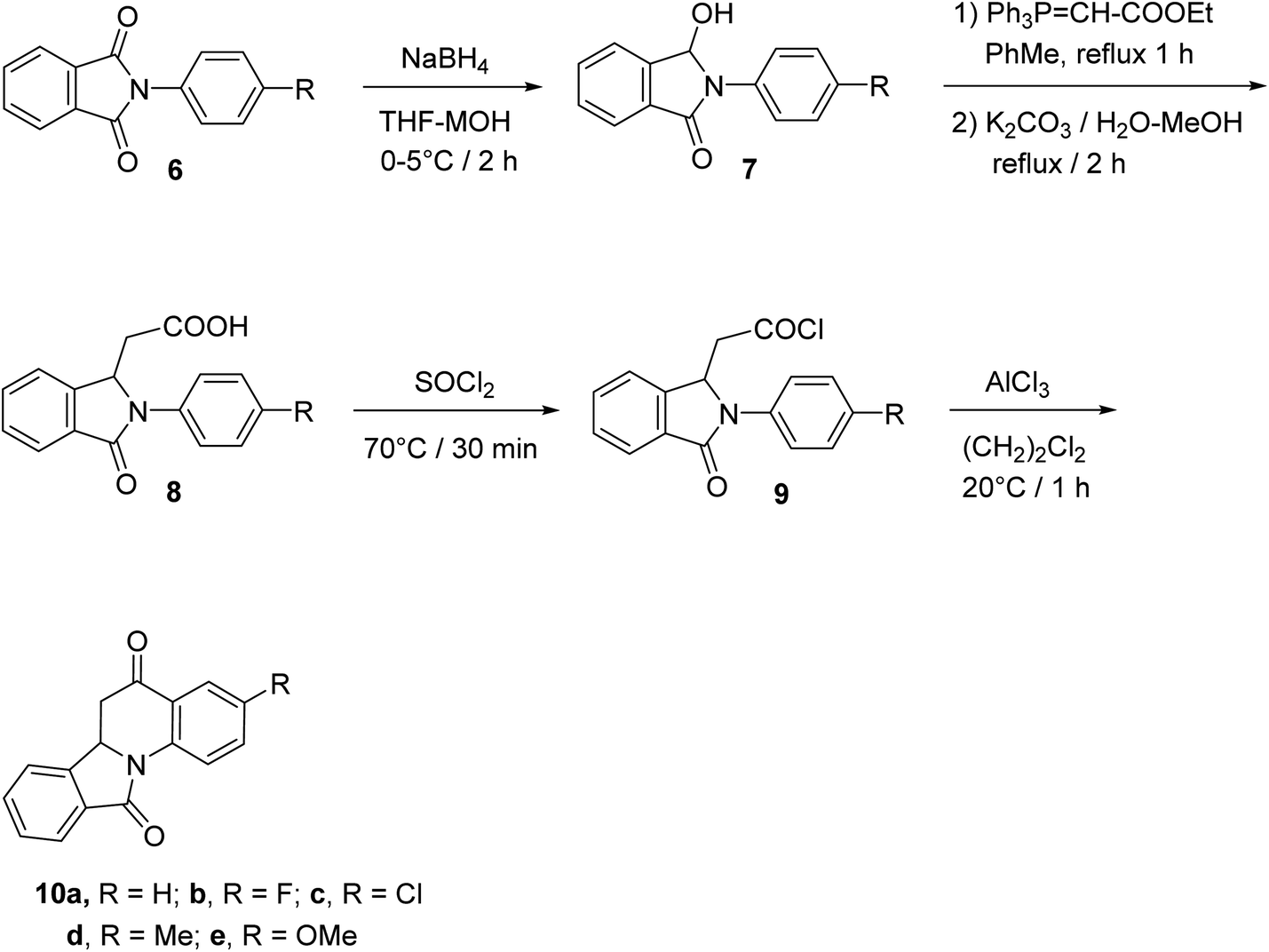
The synthetic method to tetracyclic 3-substituted-6,6a-dihydroisoindolo[2,1-a]quinoline-5,11-diones 10 is outlined in Scheme 2.48 Reduction of 2-arylisoindolo-1,3(2H)-diones 6 with sodium borohydride (NaBH4) in THF/MeOH at 0–5 °C produced 2-aryl-3-hydroxy-isoindolin-1-ones 7, which converted into 2-aryl-3-oxo-isoindole-1-acetic acids 8 by a Wittig reaction and subsequent hydrolysis. Treatment 8 with thionyl chloride at 70 °C gave the corresponding 2-aryl-3-oxo-isoindole-1-acetyl chlorides 9, which underwent an intramolecular Friedel–Crafts reaction in 1,2-dichloroethane with AlCl3 as a catalyst to give the desired isoindolo[2,1-a]quinolines 10. These synthesized products exhibited a protective effect against N2-induced hypoxia.
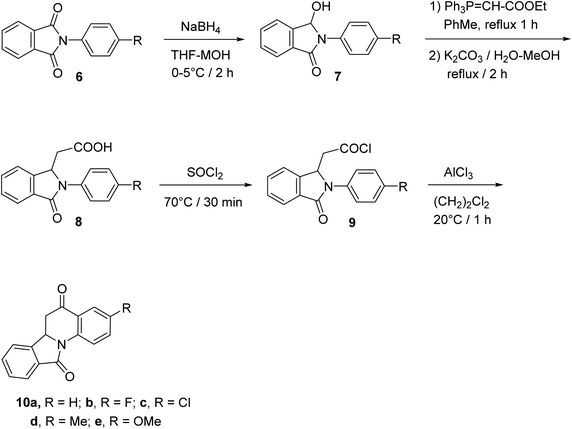 |
| | Scheme 2 The synthetic route to tetracyclic isoindolo[2,1-a]quinolines 10. | |
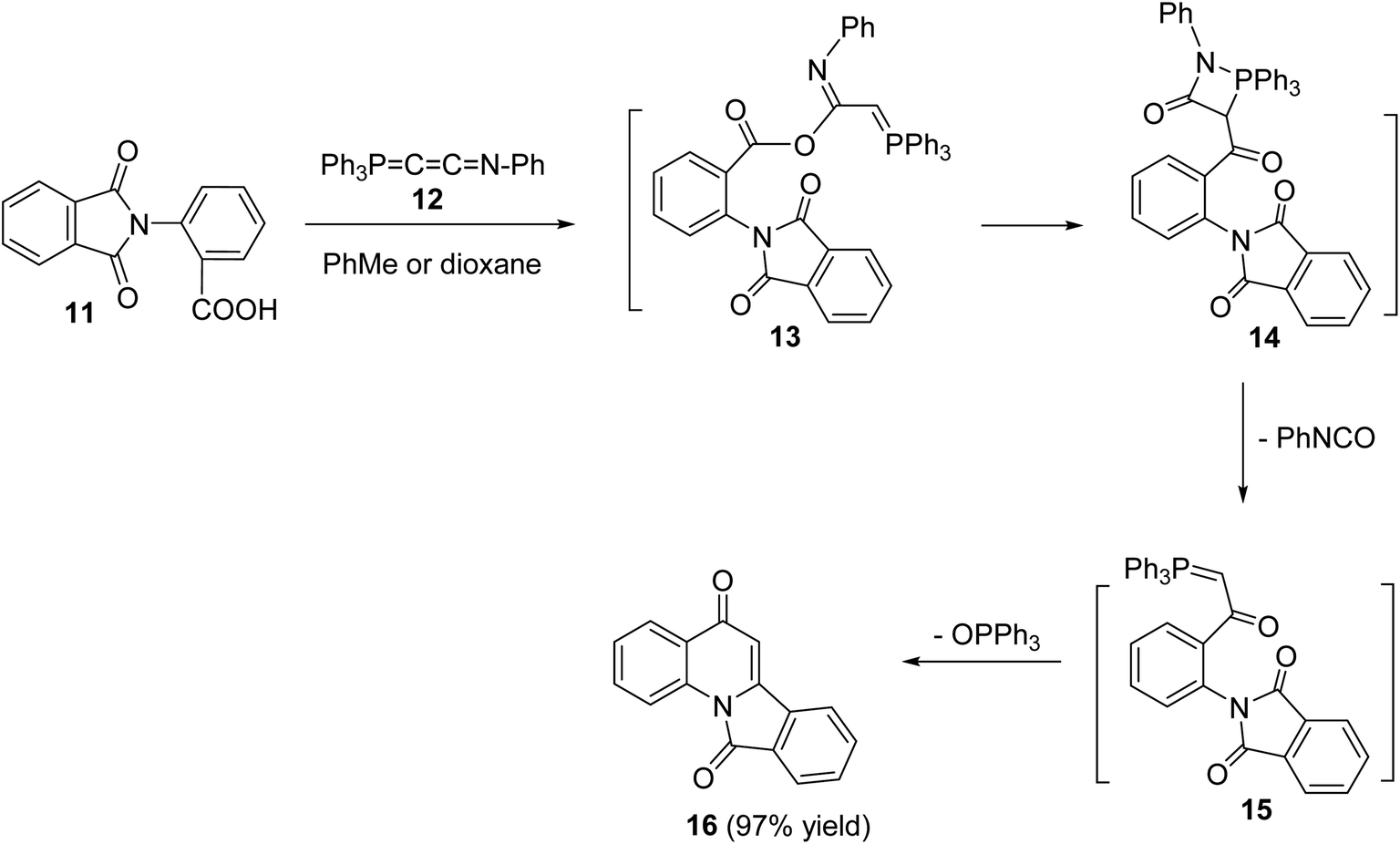
In 1994, Kumar and coworkers49 have reported an efficient synthesis of isoindolo[2,1-a]quinoline-5,11-dione (16) via the reaction of 2-(1,3-dioxoisoindolin-2-yl)benzoic acid (11) with N-phenyl(triphenylphosphoranylidine)ethenimine (12) (Scheme 3). When a mixture of 11 and 12 was refluxed in toluene or dioxane, the desired isoindolo[2,1-a]quinoline-5,11-dione (16) was obtained in 97% yields. The time of reaction was not reported. The plausible mechanism of this reaction includes the initial protonation of N-phenyl(triphenylphosphoranylidine)ethenimine (12) by 11 to give the O-acyl-imidate 13. Then, a migration of the ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group from oxygen to C-7 led to the formation of the intermediate 14, which eliminates a molecule of phenyl isocyanate to afford the acylphosphorane 15. Subsequent cyclization of 15 via the intramolecular Wittig reaction on the imide CO gave the tetracyclic product 16 (Scheme 3).
O group from oxygen to C-7 led to the formation of the intermediate 14, which eliminates a molecule of phenyl isocyanate to afford the acylphosphorane 15. Subsequent cyclization of 15 via the intramolecular Wittig reaction on the imide CO gave the tetracyclic product 16 (Scheme 3).
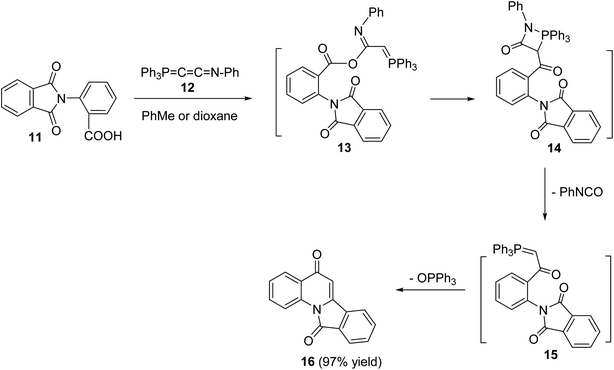 |
| | Scheme 3 An efficient synthesis of isoindolo[2,1-a]quinolines 16 from 11. | |
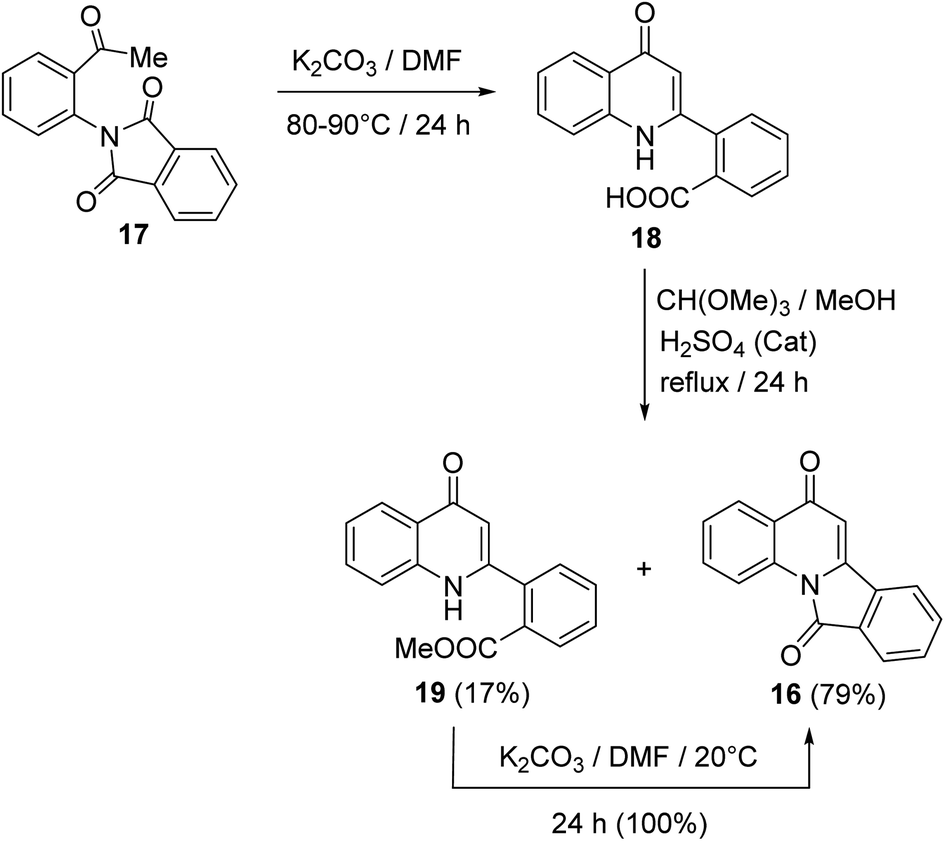
In 1997, Kim and Keum50 developed another method for the synthesis of isoindolo[2,1-a]quinolines 16 utilizing 2-(2-acetylphenyl)isoindoline-1,3-dione (17) as starting material. When 17 was heated in DMF in the presence of a catalytic amount of K2CO3 at 80–90 °C, it underwent intramolecular cyclization to produce the 2-(4-oxo-1,4-dihydro-quinolin-2-yl)benzoic acid (18), in 74% yield. Refluxing 18 with trimethyl orthoformate in MeOH in the presence of H2SO4 as an acidic catalyst gave a mixture of methyl 2-(4-oxo-1,4-dihydroquinolin-2-yl)benzoate (19) (17% yield) and tetracyclic isoindolo[2,1-a]quinoline-5,11-dione (16) (79% yield). On the other hand, when 19 was treated with K2CO3 in DMF at room temperature, it was converted quantitatively into the desired product 16 (Scheme 4).
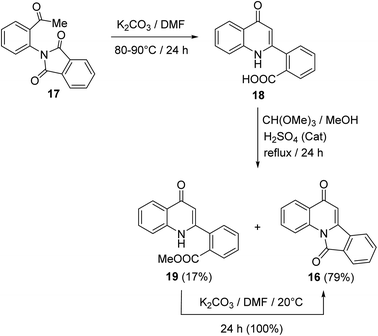 |
| | Scheme 4 Synthesis of isoindolo[2,1-a]quinolines 16 from 17. | |
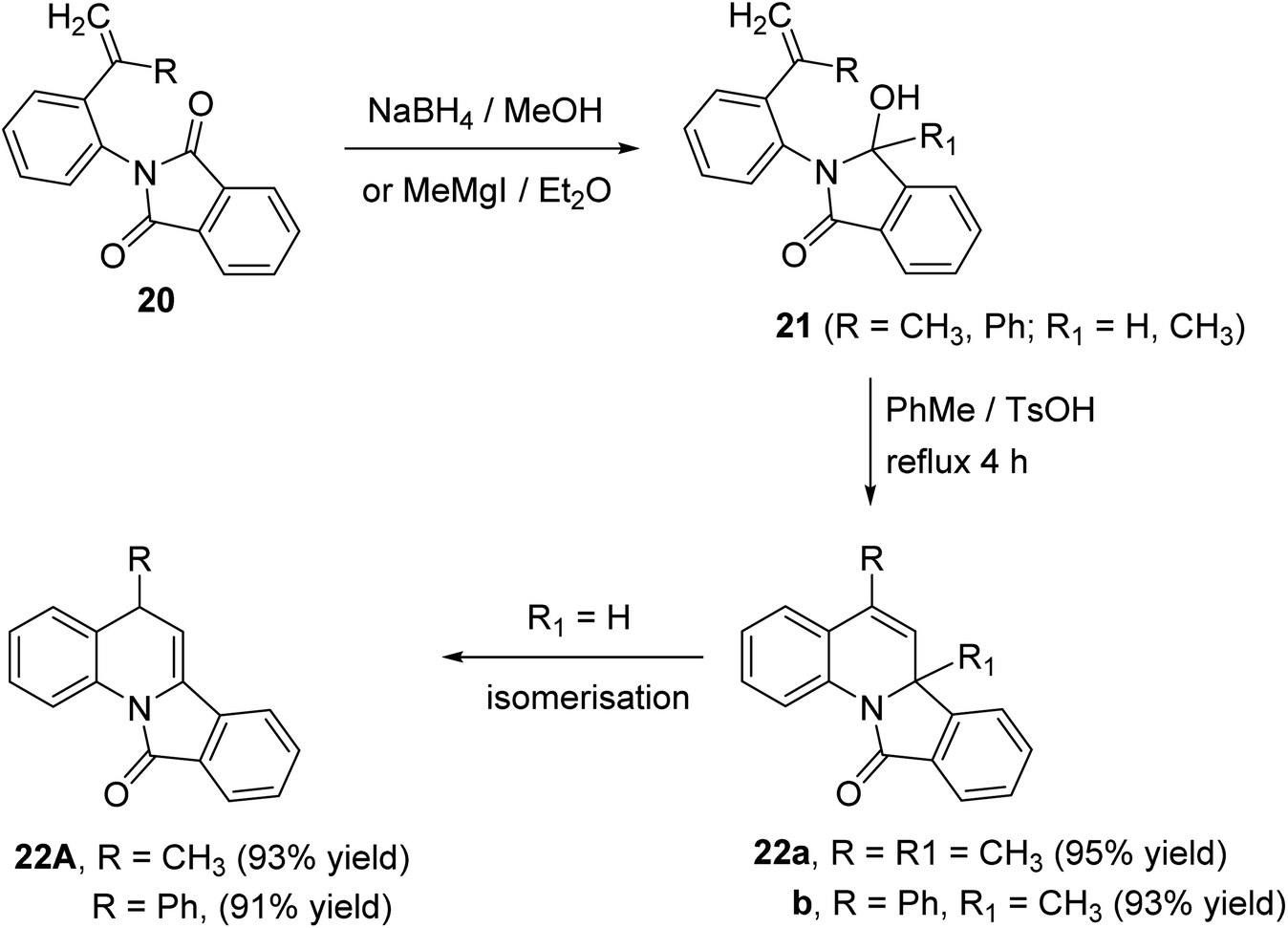
A novel route for the synthesis of 6a-methyl-5-substituted-isoindolo[2,1-a]quinoline-11(6aH)-ones 22 has been developed utilizing N-aryl-phthalimides 20 as the key precursors.51 The reaction of 20 with sodium borohydride in dry MeOH at 10 °C or the Grignard reagent (MeMgI) in dry ether afforded the corresponding alcohols 21. When compounds 21 (R1 = CH3) were refluxed in toluene in the presence of p-toluene-sulfonic acid (TsOH), as catalyst, they underwent intramolecular ring closure to the isoindolo[2,1-a]quinolines 22 (Scheme 5). In case of 21 (R1 = H), the isoindoloquinolines 22 were obtained which were rapidly isomerized, under these reaction conditions, to give the stable enamides 22A, namely, 5-substituted-isoindolo[2,1-a]quinolin-11(5H)-ones.
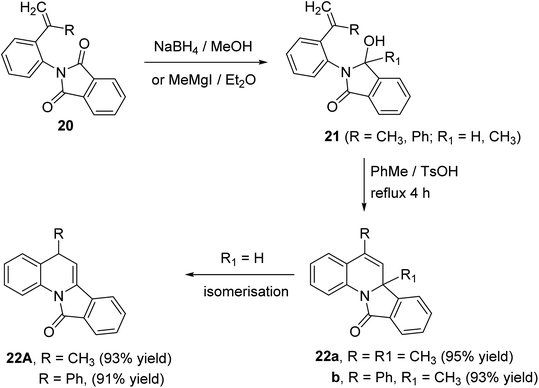 |
| | Scheme 5 Synthesis of isoindolo[2,1-a]quinolines 22 from phthalimides 20. | |
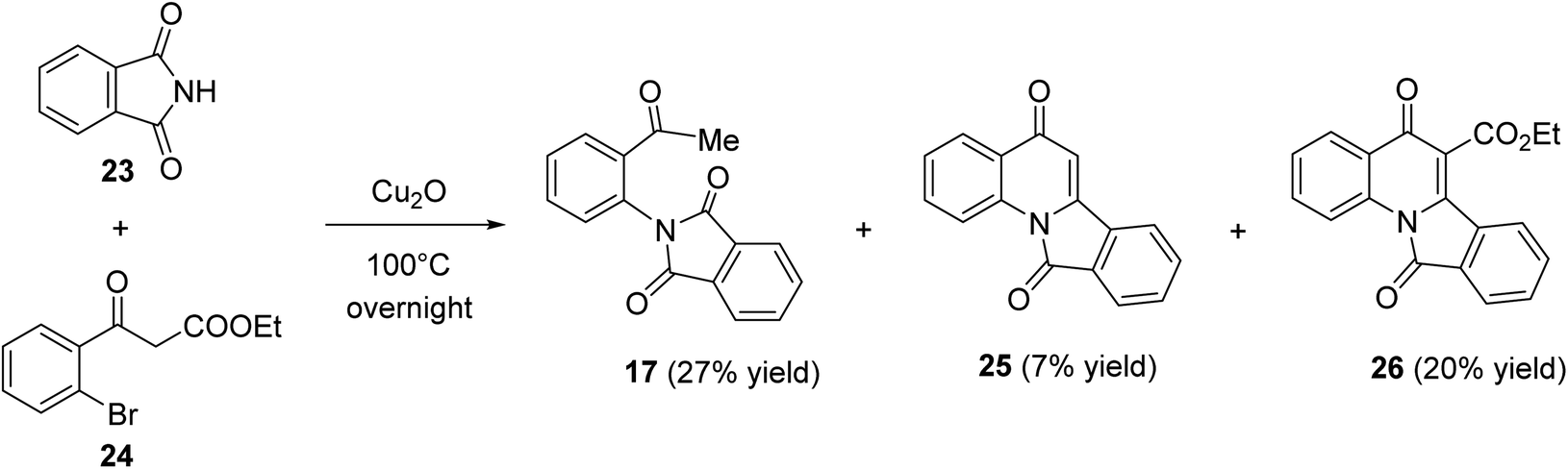
Heating a neat mixture of phthalimide (23) with ethyl bromobenzoylacetate (24) in the presence of Cu(I) oxide at 100 °C produced the 2-acetylphenylphthalimide (17) as the major product (27% yield), via the coupling reaction followed by the deethoxycarbonylation process, along with isoindolo[2,1-a]quinoline-5,11-dione (25) (7% yield) which formed via the coupling reaction followed by the deethoxycarbonylation and intramolecular cyclo-dehydration process, and ethyl 5,11-dioxo-5,11-dihydroisoindolo[2,1-a]quinoline-6-carboxylate (26) (20% yield) (Scheme 6). The product 26 was assumed to be formed through the coupling reaction followed by a thermal cyclodehydration reaction.50
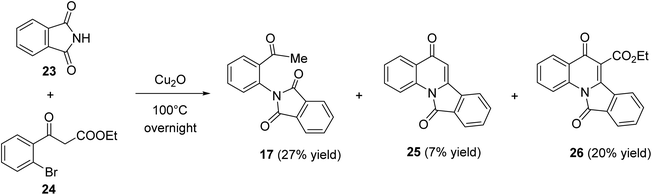 |
| | Scheme 6 Synthesis of 5,11-dihydroisoindolo[2,1-a]quinoline-5,11-diones 25 and 26. | |
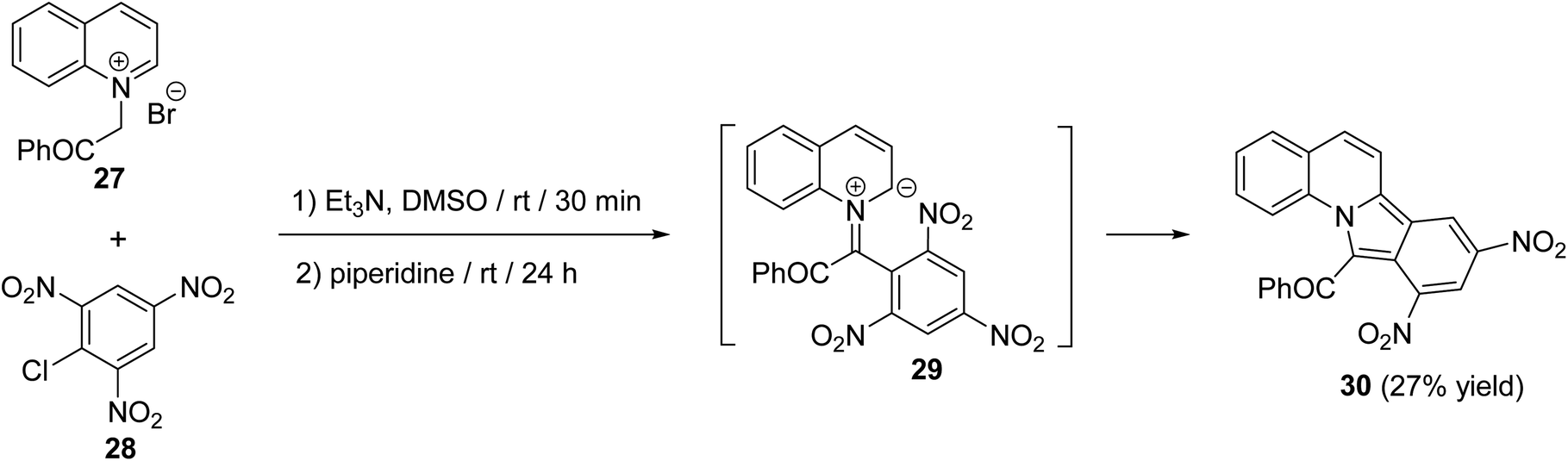
A simple route for the synthesis of 11-benzoyl-8,10-dinitroisoindolo[2,1-a]quinoline (30) has been reported by Reuschling and Kröhnke.52 Reacting N-phenacylquinolinium bromide (27) with picryl chloride (28) at room temperature, under basic conditions, afforded the respective tetracyclic product 30 in low yield (27%), via intermediacy 29 (Scheme 7).
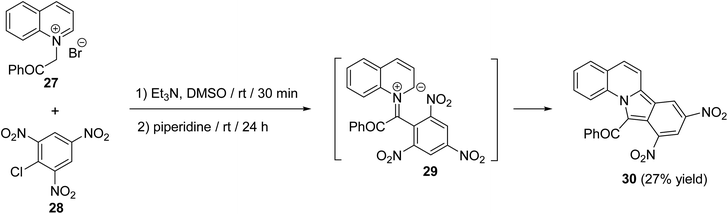 |
| | Scheme 7 Synthesis of 11-benzoyl-8,10-dinitroisoindolo[2,1-a]quinoline (30). | |
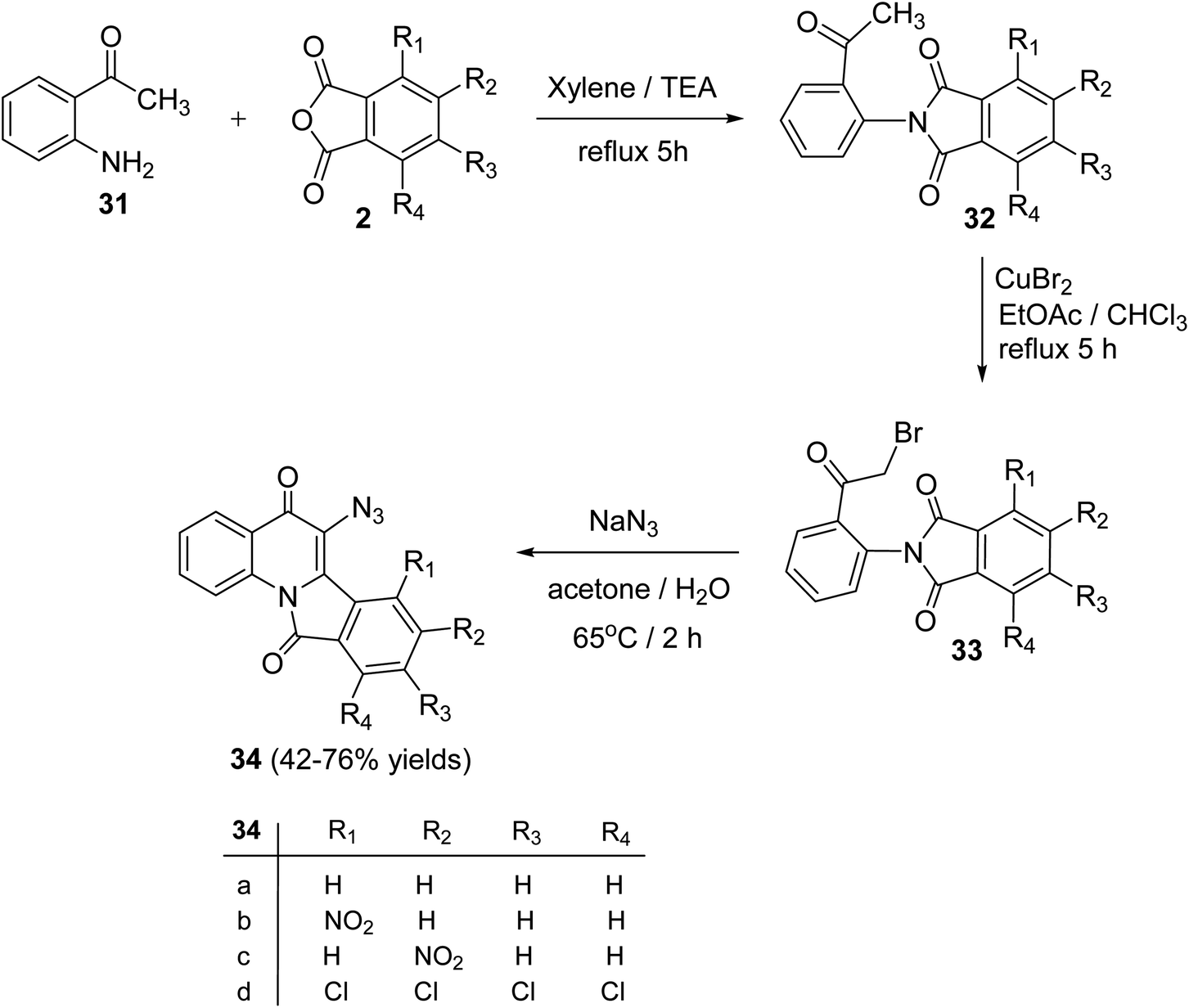
Alkhathlan and his group53 have described the synthesis of 6-azido-isoindolo[2,1-a]quinoline,11-diones 34. On reacting 2-aminoacetophenone (31) with phthalic anhydride derivatives 2 in xylene in the presence of a catalytic amount of Et3N under reflux for 5 h, 2-(2-acetylphenyl)-isoindolo-1,3-diones 32 were obtained. Bromination of 32 with CuBr2 in a EtOAc/CHCl3 mixture under reflux for 5 h gave the corresponding bromo derivatives 33. When the bromo compounds 33 were heated with sodium azide in a mixture of acetone/H2O at 65 °C for 2 h, they underwent intramolecular cyclocondensation to furnish directly the new tetracyclic ring system 34 in 42–76% yields (Scheme 8). Attempts to isolate the azido analogs of 34 by conducting the reaction at different reaction conditions were failed.
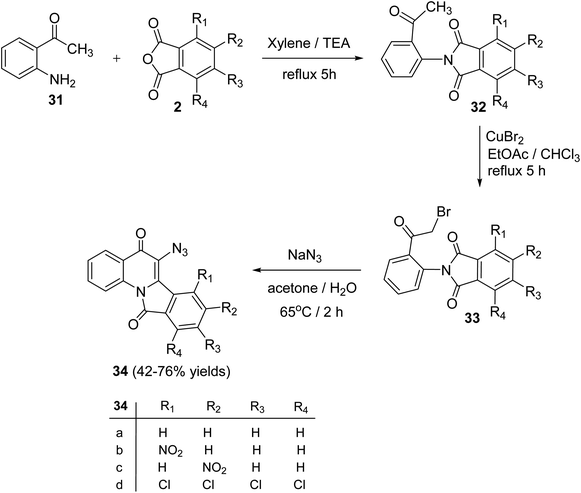 |
| | Scheme 8 Synthesis of 6-azido-isoindolo[2,1-a]quinoline-5,11-diones 34. | |
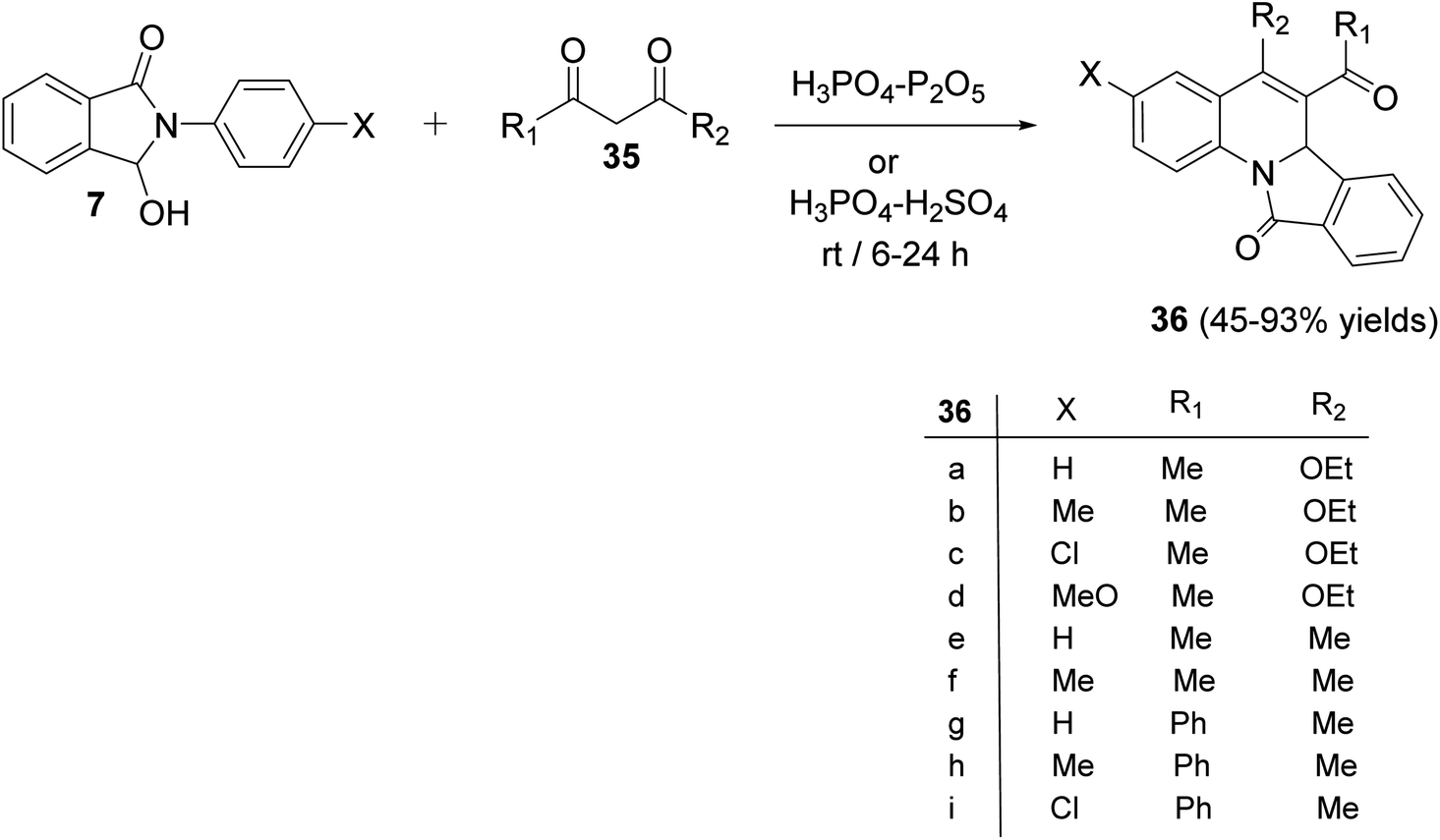
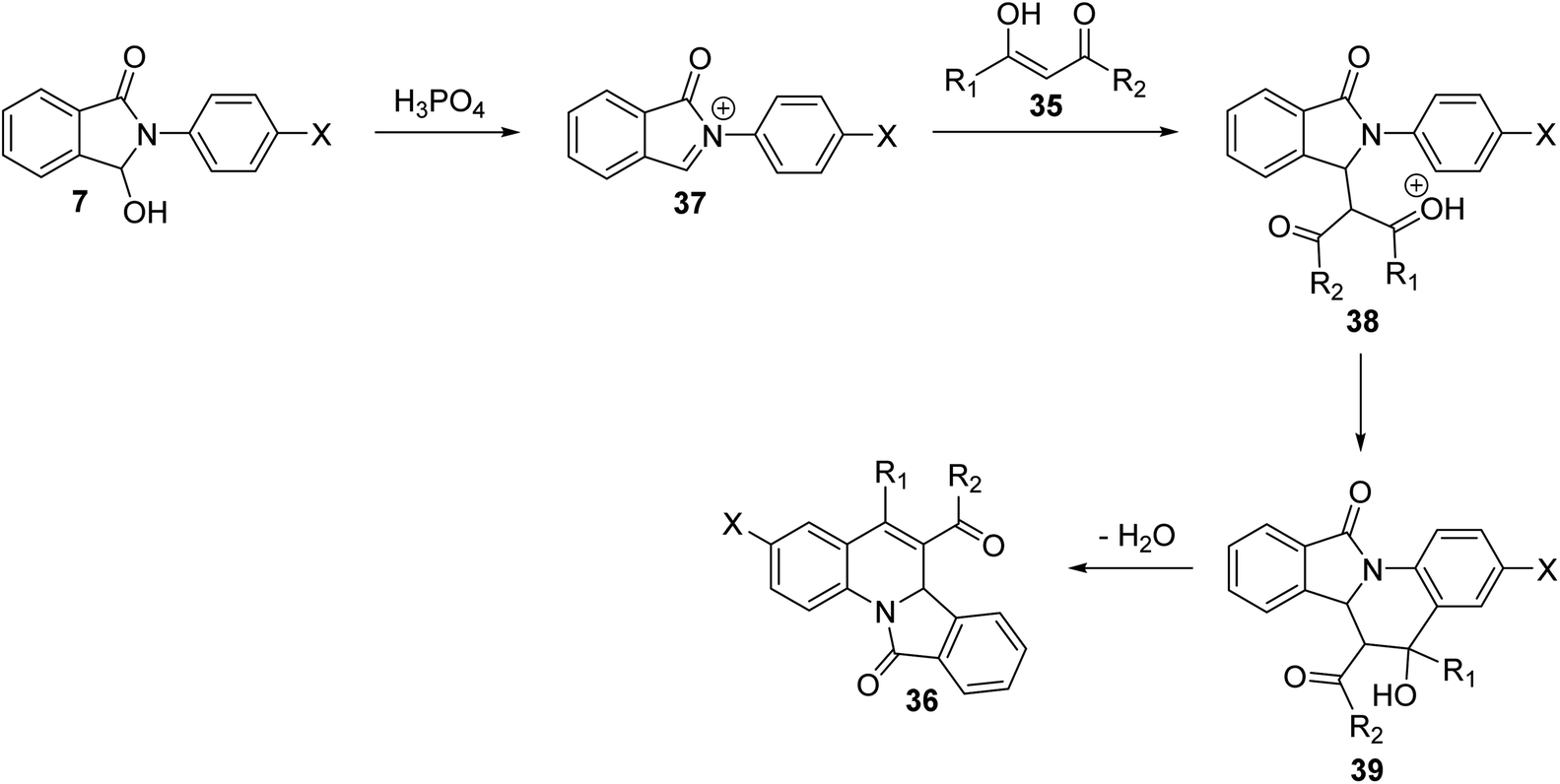
In 2009, Zhang et al.54 have described as the first one-pot synthesis of isoindolo[2,1-a]quinolin-11-ones 36 by reacting 2-aryl-3-hydroxy-isoindol-1-ones 7 with 1,3-dicarbonyls 35 via coupling of N-acyliminium ion intermediates 37 with 35 followed by intramolecular Friedel–Crafts reaction catalyzed by H3PO4–P2O5 or H3PO4–H2SO4. Reactions were carried out by stirring 7 with 35 under the catalysis of H3PO4–P2O5 or H3PO4–H2SO4 at room temperature for 6–24 h to give the desired isoindolo[2,1-a]quinoline-11-ones 36 in 45–93% yields (Scheme 9). The structures of all the newly synthesized compounds were fully characterized by 1H NMR, 13C NMR, and MS. Furthermore, X-ray crystallographic analysis was performed to determine the absolute configuration of the products 36. A plausible mechanism for the formation of 36 is given in Scheme 10. The reaction was initiated by the formation of an N-acyliminium intermediate 37 which was reacted with 35 to give the intermediate 38. Intramolecular Friedel–Crafts reaction between the protected aniline and the ketone gave the desired tetracyclic product 36.
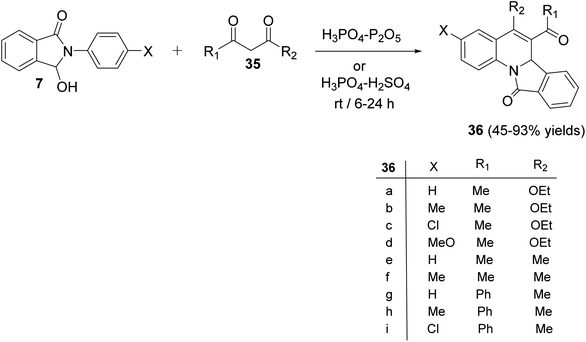 |
| | Scheme 9 One-pot synthesis of isoindolo[2,1-a]quinolin-11-ones 36 by reacting 7 with 35. | |
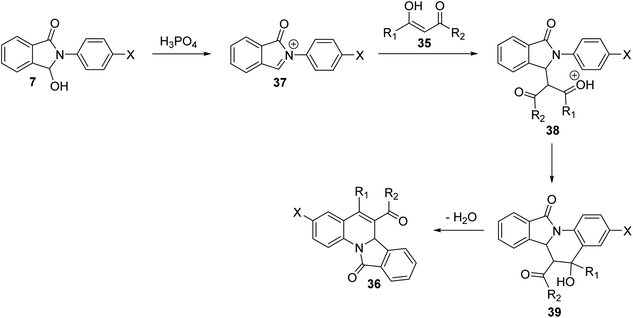 |
| | Scheme 10 A plausible mechanism for the formation of isoindolo[2,1-a]quinolin-11-ones 36. | |
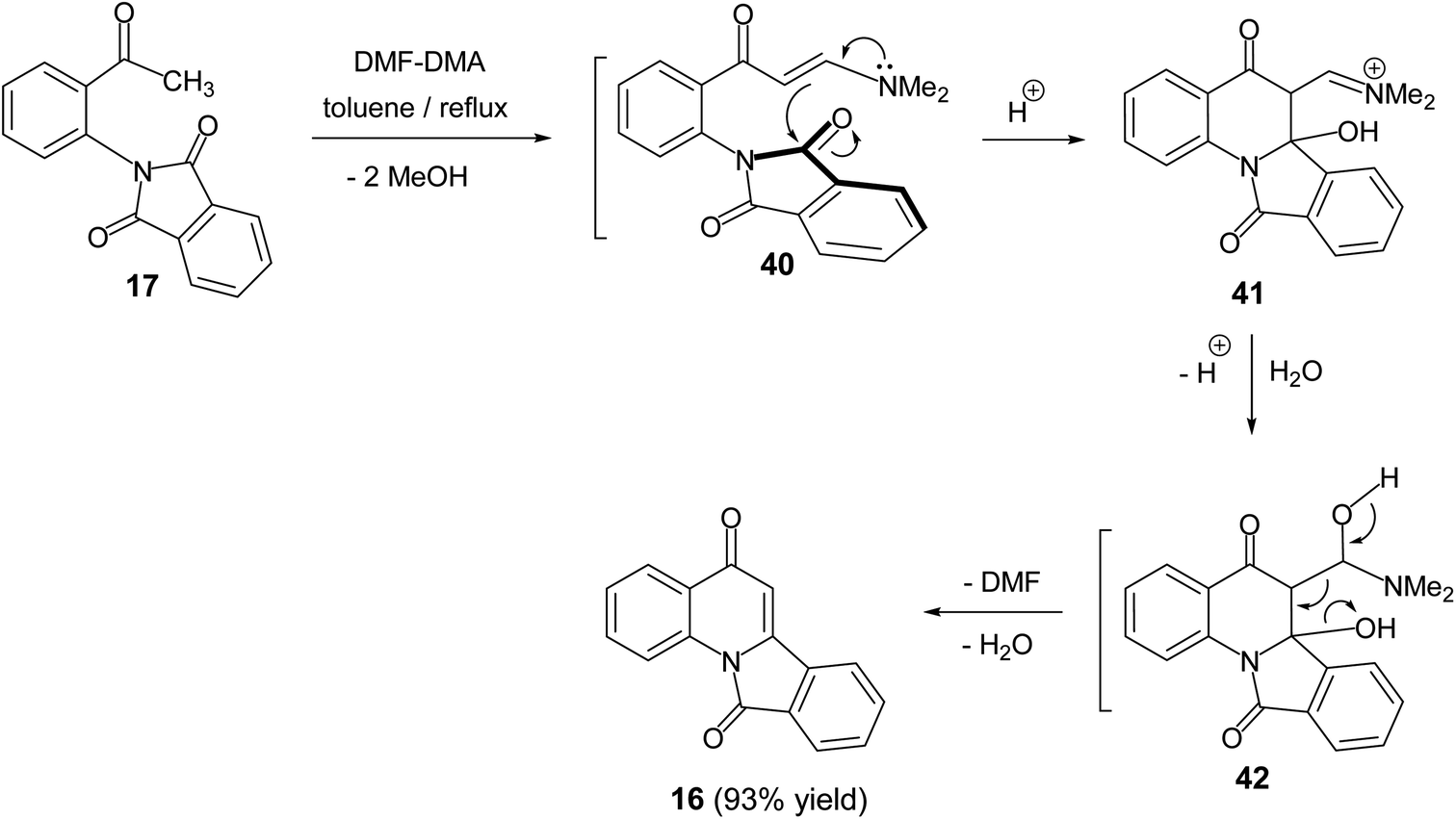
In 2011, Svete and his co-workers55 reported that the treatment of 2-(2-acetylphenyl)-1H-isoindole-1,3(2H)-dione (17) with N,N-dimethylformamide dimethyl acetal (DMF-DMA) in refluxing toluene resulted in the formation of the isoindolo[2,1-a]quinoline-5,11-dione (16) in 93% yield (Scheme 11). The mechanism of this reaction began with the formation of enaminone 40, which next underwent intramolecular cyclization to give the intermediate 41. The formed adduct 41 was subsequently underwent β-elimination of DMF and H2O to furnish the desired tetracyclic 16 (Scheme 11).
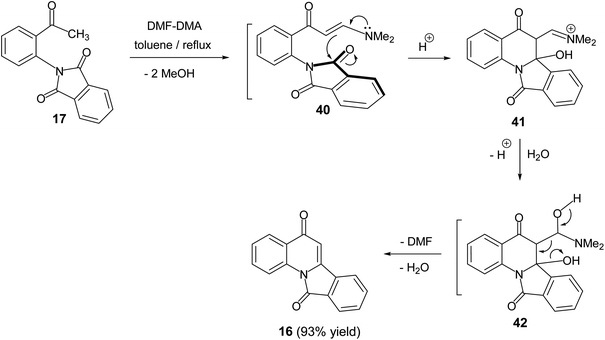 |
| | Scheme 11 Synthesis of isoindolo[2,1-a]quinoline-5,11-dione (16). | |
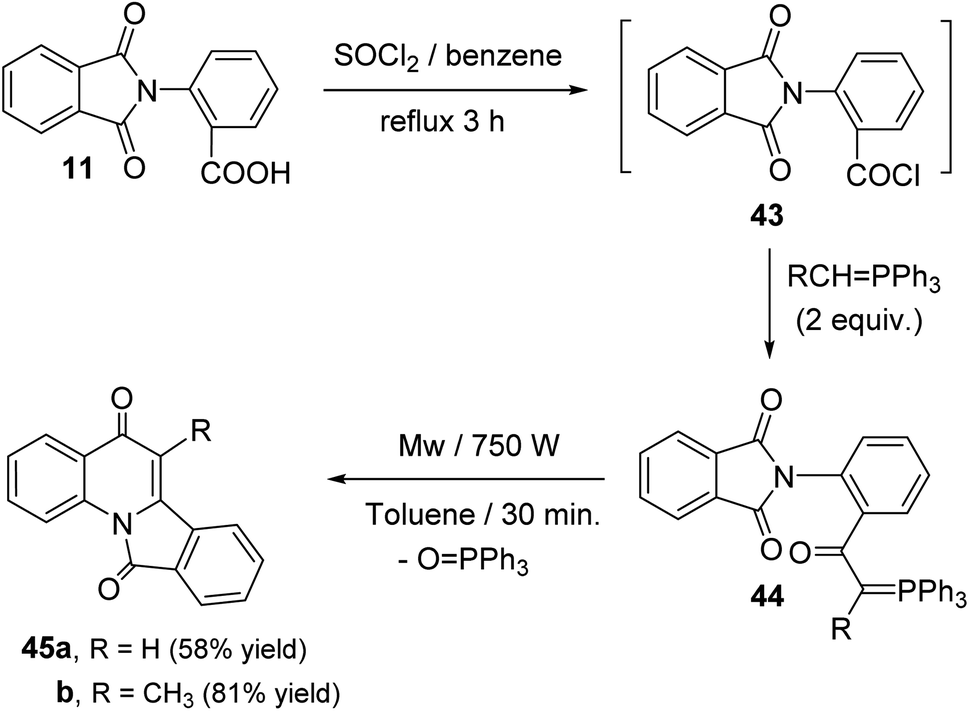
Sakhautdinov et al.56 have described the synthesis of isoindolo[2,1-a]quinoline-5,11-diones 45 via intramolecular cyclization of N-[2-(triphenyl-λ5-phosphanylidene)acetyl]-phthalimides 44 under microwave-heating. Refluxing a mixture of o-phthalimidobenzoic acid (11) and SOCl2 in dry benzene for 3 h afforded the corresponding acid chlorides intermediate 43, and the latter was reacted in situ with alkylidenephosphorane (2 equiv.) to give keto-stabilized phosphorus ylides 44. The compounds 44 when heated in dry toluene in the presence of a catalytic amount of benzoic acid (5 mol%) at reflux temperature under argon atmosphere for 56 h, they underwent intramolecular cyclization to afford the tetracyclic products 45 in low yields (15–27% yields). Interestingly, it was observed that use of microwave irradiation gave much faster reaction and improved the yields, thereby allowing rapid access to this class of compounds 45. Thus, the ylides 44 when heated in dry toluene for 30 min under microwaves, they underwent intramolecular cyclization followed by elimination of a triphenylphosphine oxide (OPPh3) molecule to afford tetracyclic isoindolo[2,1-a]quinoline-5,11-diones 45, in good yields (Scheme 12). However, one drawback of this methodology is electron-acceptor substituents on the ylide carbon atom hamper the intramolecular cyclization process.
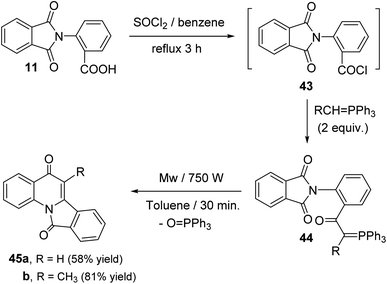 |
| | Scheme 12 Microwave-assisted synthesis of isoindolo[2,1-a]quinoline-5,11-diones 45. | |
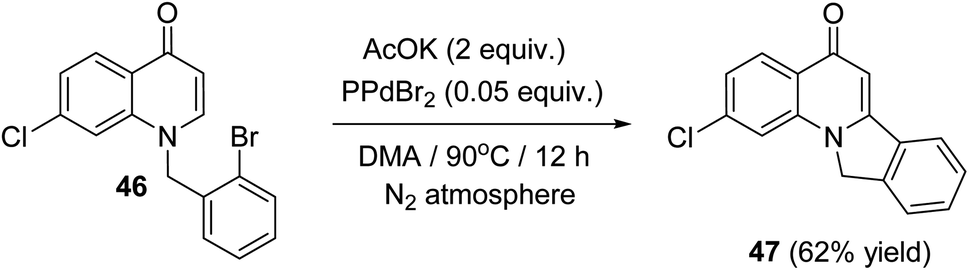
In 2014, Fu et al.57 have described a new and efficient synthesis of 2-chloro-isoindolo[2,1-a]quinoline-5(11H)-one (47) through the intramolecular Heck coupling cyclization of 1-(2-bromobenzyl)-7-chloroquinolin-4(1H)-one (46). The reaction was carried out by heating 46 (1 equiv.) with AcOK (2 equiv.) as the base and PdBr2 (0.05 equiv.) as the catalyst in DMA at 90 °C under N2 atmosphere for 12 h to provide the tetracyclic product 47 in 62% yield (Scheme 13). Noticeably, a definite mechanism for the synthesis of compound 47 has not been reported.
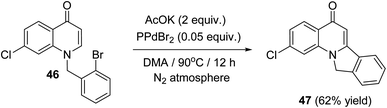 |
| | Scheme 13 Synthesis of 2-chloro-isoindolo[2,1-a]quinoline-5(11H)-one (47) via intramolecular Heck coupling cyclization of 46. | |
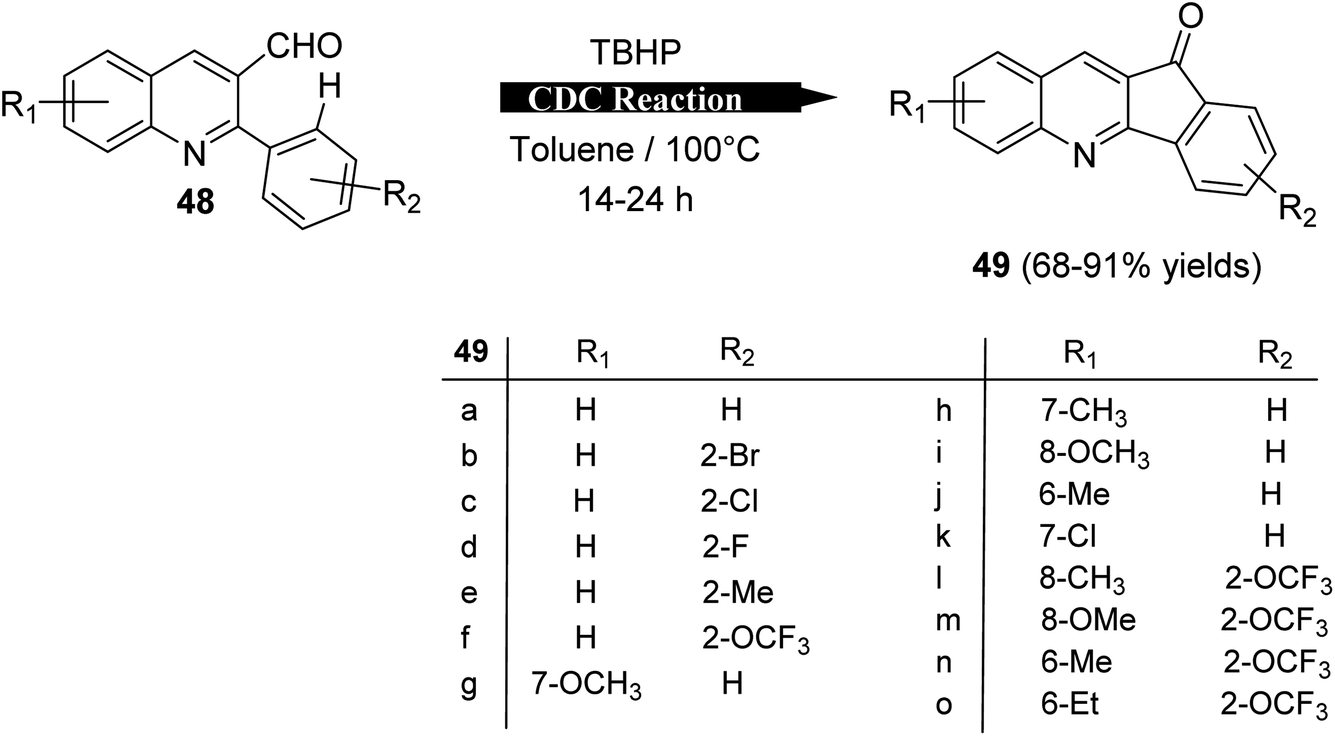
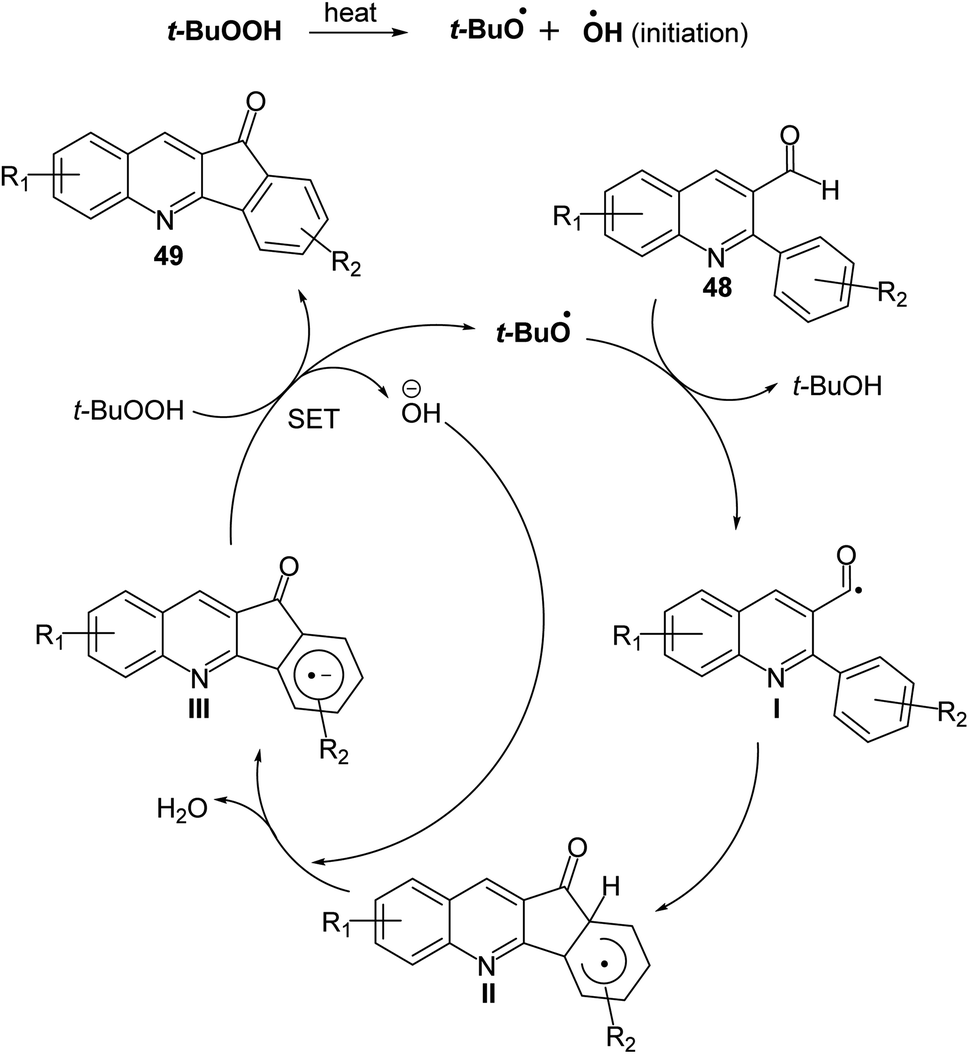
2.1.2. Indino[1,2-b]quinolines. Recently, Mishra et al.58 have developed an eco-friendly, metal-free and TBHP-promoted economical method for the synthesis of 11H-indeno[1,2-b]quinolin-11-ones 49, as antibacterial agent. The reactions were carried out by heating a solution of 2-aryl-quinoline-3-carbaldehyde derivatives 48 (1 equiv.) and tert-butyl hydroperoxide (TBHP) (3 equiv.) in toluene at 100 °C for 14–24 h and furnished the desired tetracyclic indenoquinolinones 49 in 68–91% yields (Scheme 14). Depending on product formation, they suggested the free radical mechanism of this reaction in Scheme 15.
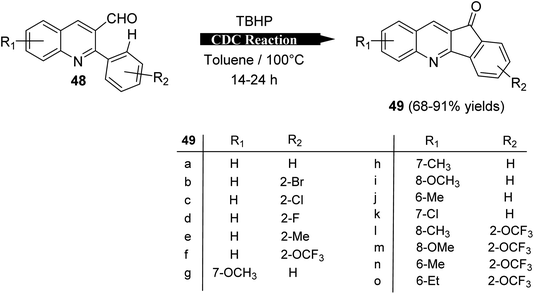 |
| | Scheme 14 Synthesis of indeno[1,2-b]quinolin-11-ones 49 via intramolecular CDC reaction. | |
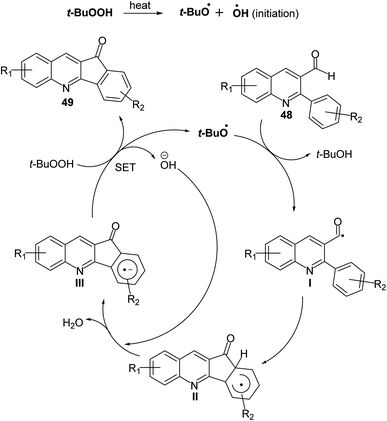 |
| | Scheme 15 Proposed mechanism for the formation of indeno[1,2-b]quinolin-11-ones 49 via intramolecular CDC reaction. | |
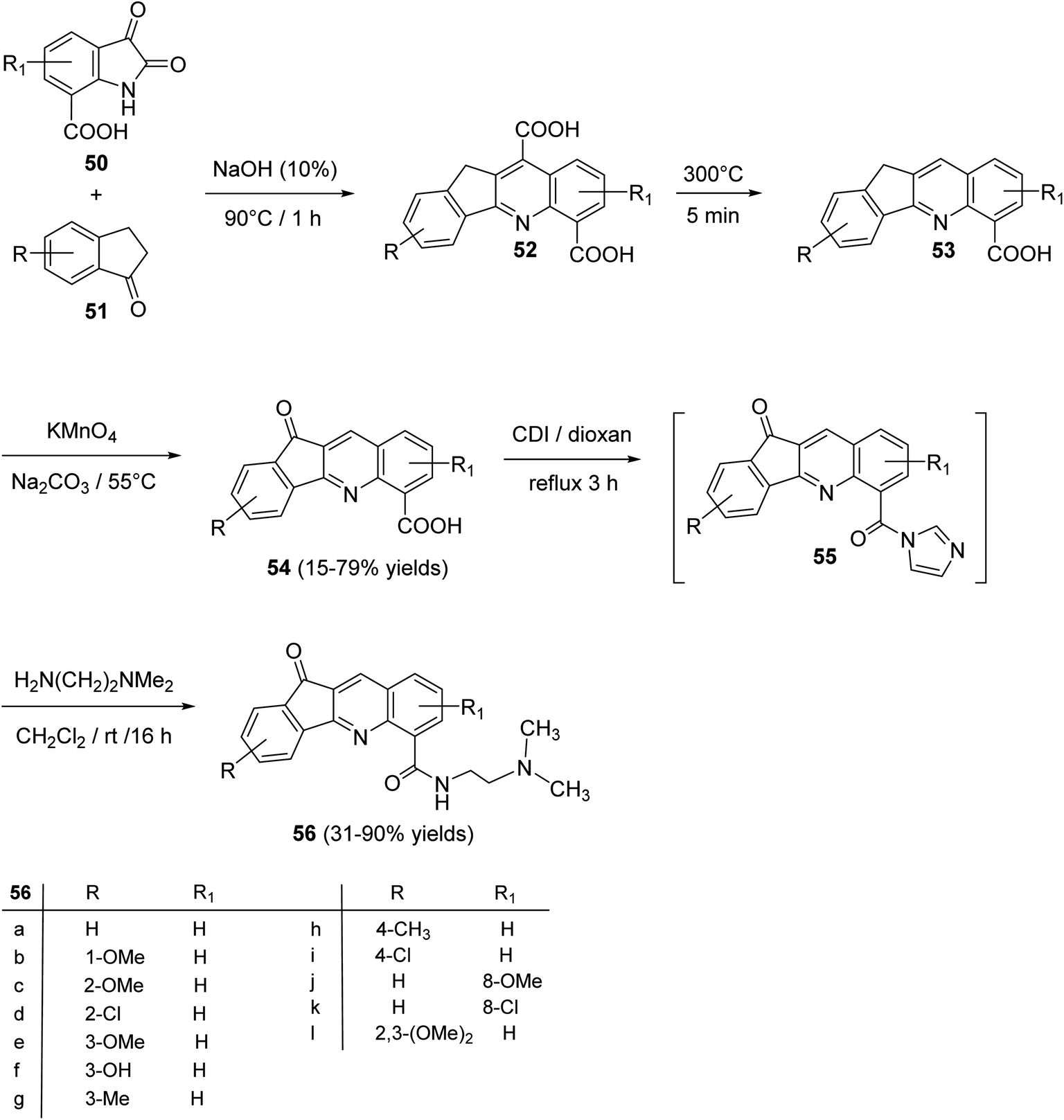
An interesting utility of Pfitzinger reaction for construction of tetracyclic quinolines has been reported by Deady et al.59 This protocol implies Pfitzinger reaction of isatin-7-carboxylic acids 50 and 1-indanones 51 to prepare analogues of the topoisomerase inhibitor 11-oxo-11H-indeno[1,2-b]quinoline-6-carboamides 56. The one-pot reaction between 50 and 51 in NaOH (10%) at 90 °C under nitrogen atmosphere afforded tetracyclic 11H-indeno-[1,2-b]quinoline-6,10-dicarboxylic acids 52, in 64–78% yields. Selective decarboxylation of 52, followed by oxidation of CH2 group in 53 with KMnO4 produced 11-oxo-11H-indeno-[1,2-b]quinoline-6-carboxylic acids 54, in 15–79% yields. Refluxing 54 with 1,1′-carbonyl-diimidazole (CDI) in dioxan gave the corresponding imidazolide intermediate 55, which was coupled, in situ, with N,N-dimethylethylenediamine in CH2Cl2 at room temperature to give the required tetracyclic 11-oxo-11H-indeno[1,2-b]quinoline-6-carboxamides 56, in 31–90% yields (Scheme 16).
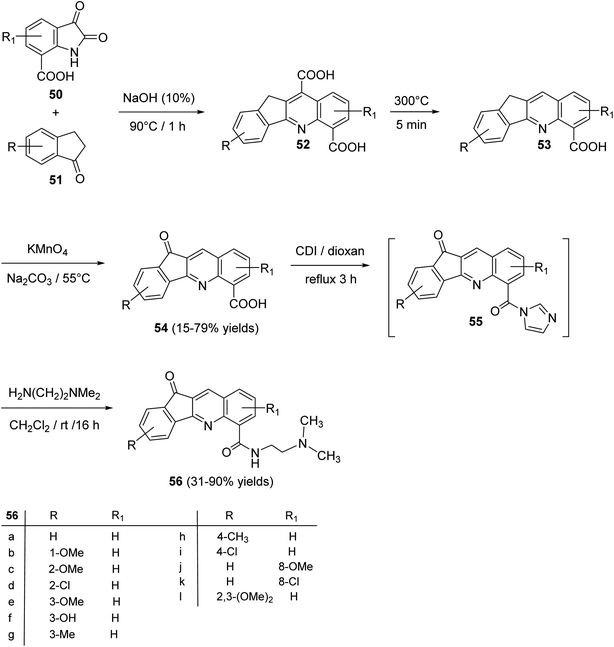 |
| | Scheme 16 Synthesis of 11-oxo-11H-indeno[1,2-b]quinoline-6-carboxamides 56. | |
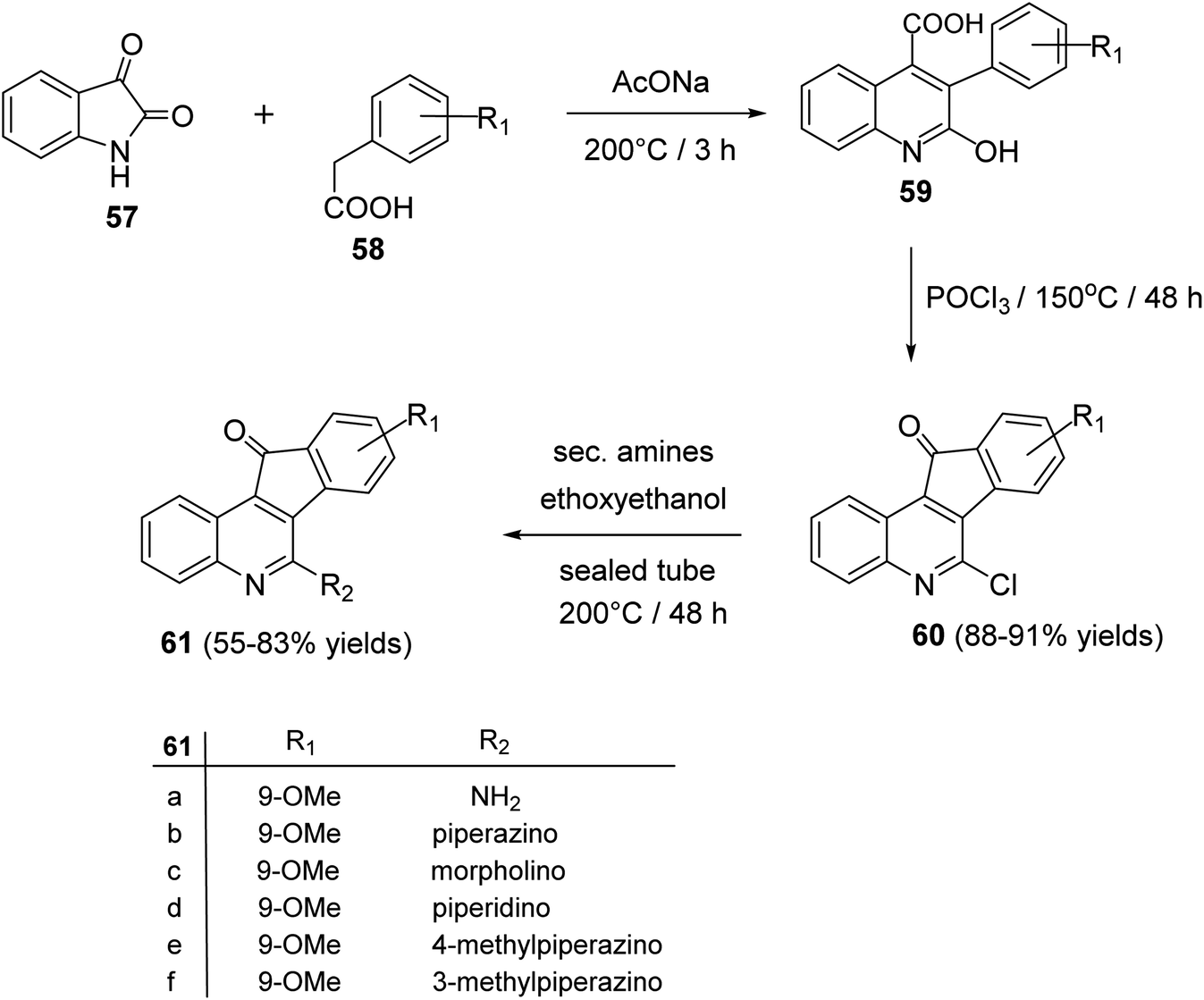
2.1.3. Indino[1,2-c]quinolines. Tseng et al.60 reported synthesis and antiproliferative evaluation of some 6-amino-11H-indeno[1,2-c]quinolin-11-ones 61 from reaction of isatin (57) with substituted phenylacetic acids 58. Heating a mixture of 57 and 58 in the presence of a catalytic amount of CH3CO2Na at 200 °C produced 3-aryl-2-hydroxy-quinoline-4-carboxylic acid derivatives 59, which were chlorinated with POCl3 at 150 °C to give 6-chloro-indeno[1,2-c]quinoline-11-ones 60, in 88–91% yields. On heating 60 with cyclic secondary amines in ethoxyethanol at 200 °C, the corresponding 6-amino-indeno[1,2-c]quinoline-11-ones 61 were obtained in 55–83% yields (Scheme 17).
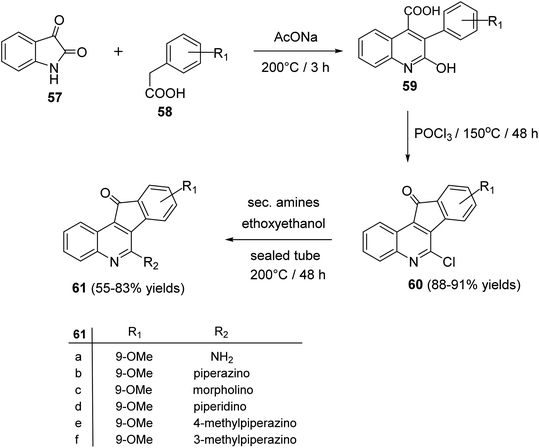 |
| | Scheme 17 Synthesis of indeno[1,2-c]quinolines 61. | |
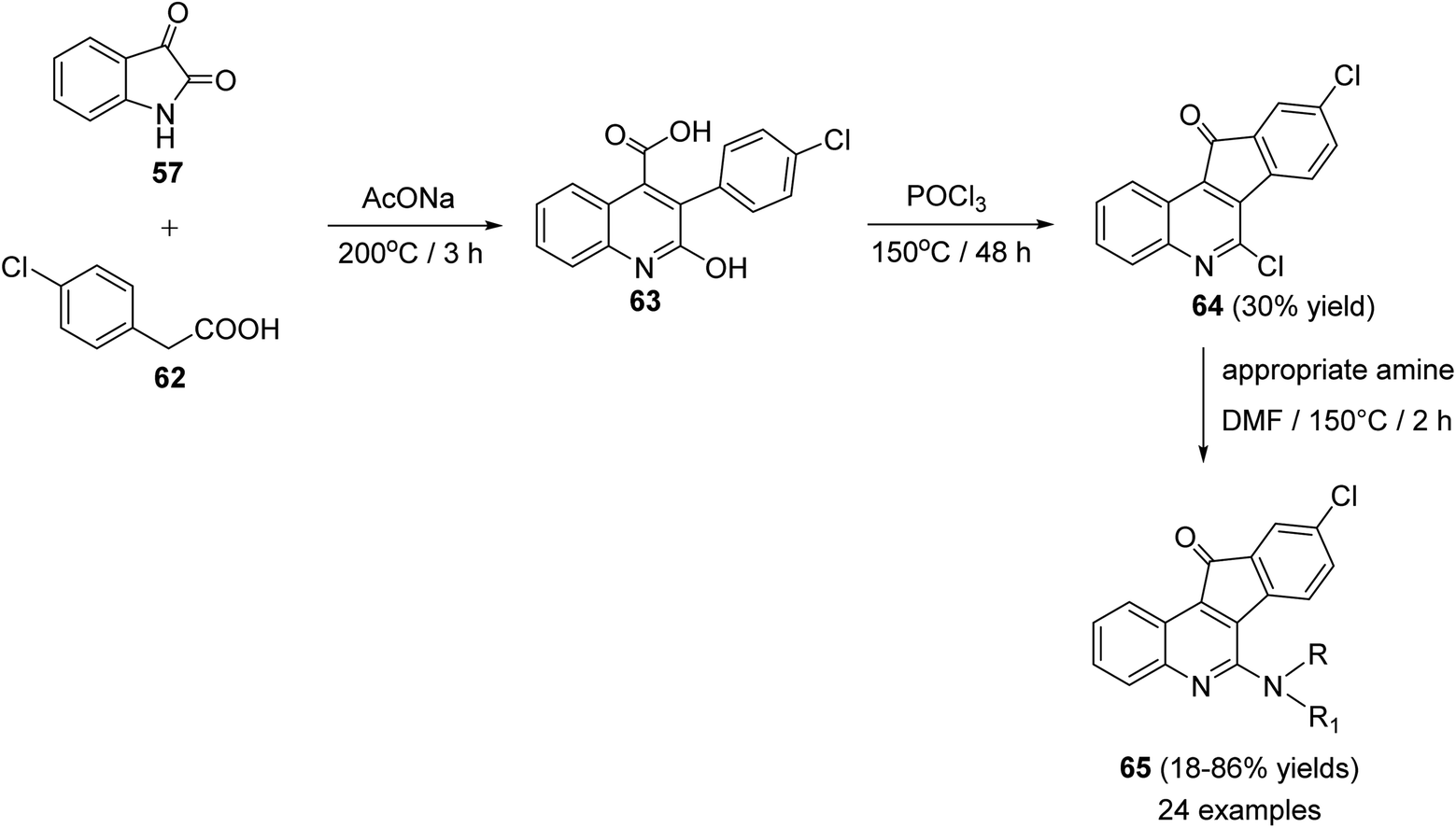
Chen et al.61 reported synthesis of a series of novel tetracyclic 6-amino-9-chloro-11H-indeno[1,2-c]quinoline-11-ones 65, as potential anticancer agents, through the Pfitzinger synthetic reaction of isatin (57) and 4-chlorophenyl-acetic acid (62). At first, a mixture of 57 and 62 was stirred at 200 °C with sodium acetate to produce 3-(4-chlorophenyl)-2-hydroxy-quinolin-4-carboxylic acid (63) as a key intermediate. Treatment of 63 with POCl3 at 150 °C afforded 6,9-dichloro-11H-indeno[1,2-c]quinoline-11-one (64), in 30% yield. Reaction of 64 with an appropriate primary and secondary amines in DMF at 150 °C gave the corresponding side chain tetracyclic compounds 65 in 18–86% yields (Scheme 18).
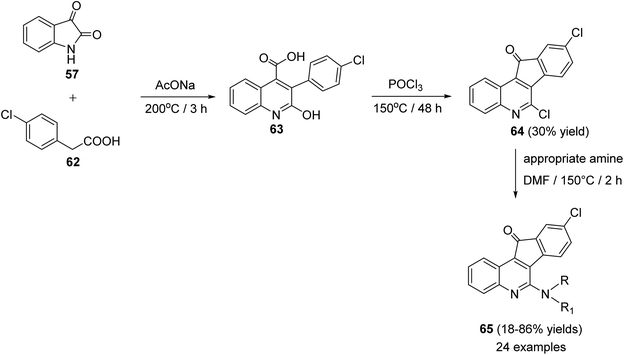 |
| | Scheme 18 Synthesis of 6-amino-9-chloro-11H-indeno[1,2-c]quinoline-11-ones 65. | |
2.2. Tetracyclic quinolines with two heteroatoms
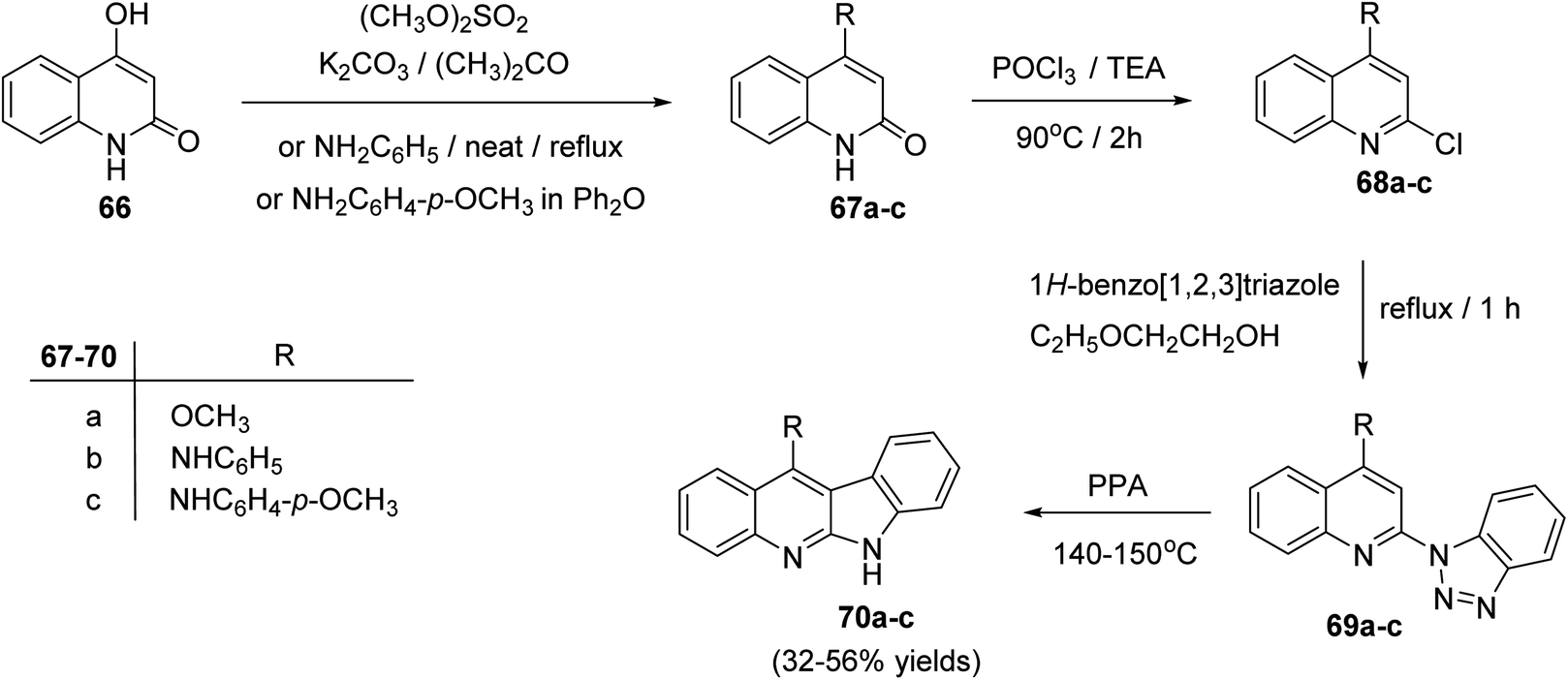
2.2.1. Indolo[2,3-b]quinolines. In 2004, Chen's group62 reported an efficient synthesis of some 11-substituted-6H-indolo[2,3-b]quinolines 70a–c, starting from 4-hydroxy-quinolin-2(1H)-one (66). In this cascade reaction, the hydroxyquinoline 66 was methylated with Me2SO4 in acetone in the presence of K2CO3 to produce 4-methoxy-quinoline-2(1H)-one (67a). Refluxing 66 with aniline or p-anisidine in Ph2O yielded 4-anilino-quinolin-2(1H)-one (67b) or its 4-methoxy derivative 67c, respectively. Heating 67a–c with POCl3 at 90 °C afforded the 2-chloro-quinolines 68a–c. Treatment of 68a–c with 1H-benzo[1,2,3]triazole in ethoxyethanol at reflux temperature gave the corresponding triazoles 69a–c, which underwent decomposition reaction in polyphosphoric acid (PPA) to afford the respective tetracyclic indolo[2,3-b]quinolines 70a–c, in 32–56% yields (Scheme 19). The products exhibited selective cytotoxicities for K-562, HL-60, RPMI-8226 and MOLT-4.
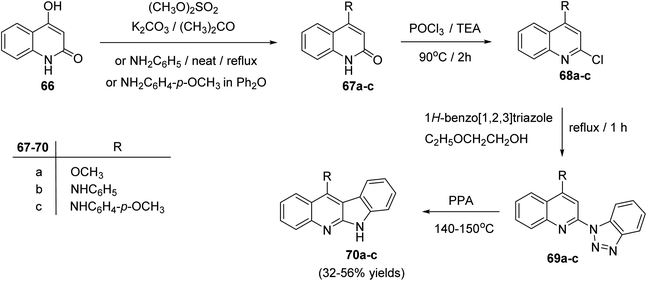 |
| | Scheme 19 Synthesis of indolo[2,3-b]quinoline derivatives 70a–c. | |
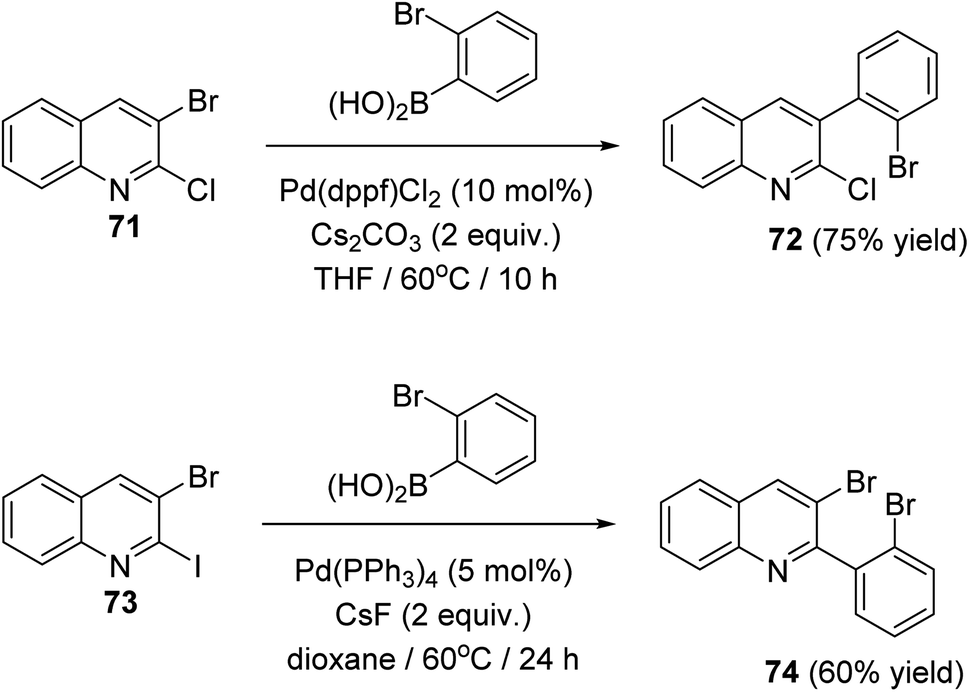
In 2018, Salman et al.63 have developed a simple and regioselective synthesis of two series of 10H-indolo[3,2-b]quinolines 75 and 6H-indolo[2,3-b]quinolines 76 via palladium catalyzed Suzuki reaction of 2,3-dihaloquinolines with 2-bromophenylboronic acid, followed by double C–N coupling. The Suzuki reaction of 2-chloro-3-bromoquinoline (71) and 3-bromo-2-iodoquinoline (73) with 2-bromophenylboronic acid gave 3-(2-bromophenyl)-2-chloroquinoline (72) and 3-bromo-2-(2-bromophenyl)quinoline (74) in 75 and 60% yield, respectively (Scheme 20).
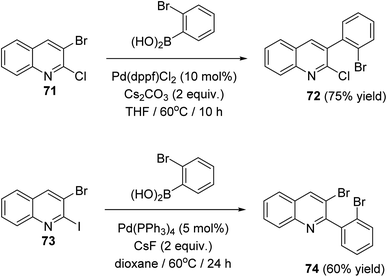 |
| | Scheme 20 Chemoselective Suzuki reaction of 2,3-dihaloquinsolines 71 and 73. | |
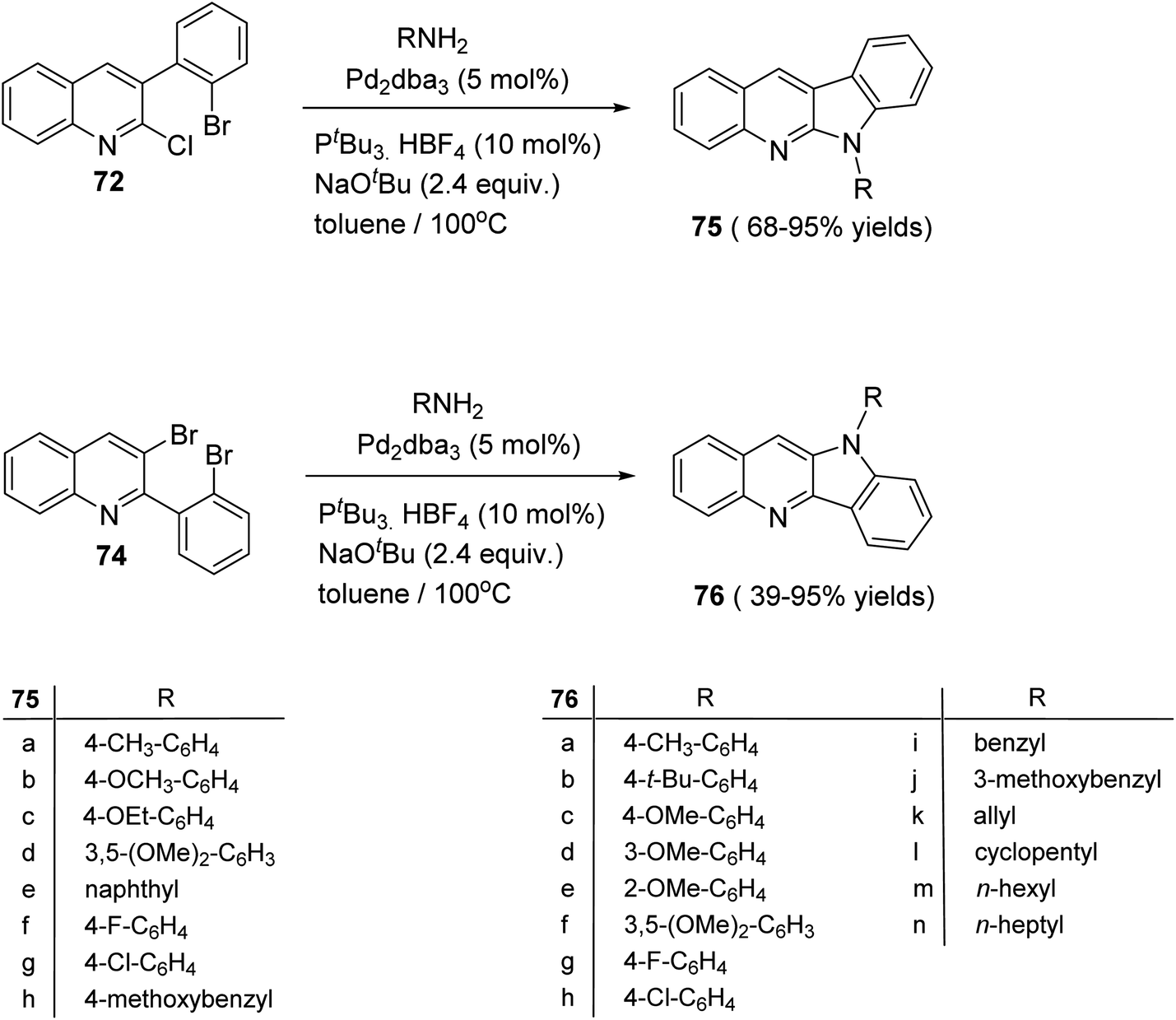
The double carbon–nitrogen coupling reaction was performed by heating 72 and 74 with various amines in the presence of the catalyst tris(dibenzylideneacetone)-dipalladium [Pd2(dba)3] with the base NaOtBu in toluene at 100 °C to give the desired 10H-indolo[3,2-b]quinolines 75 and 6H-indolo[2,3-b]quinoline 76, in moderate to excellent yields (Scheme 21).63
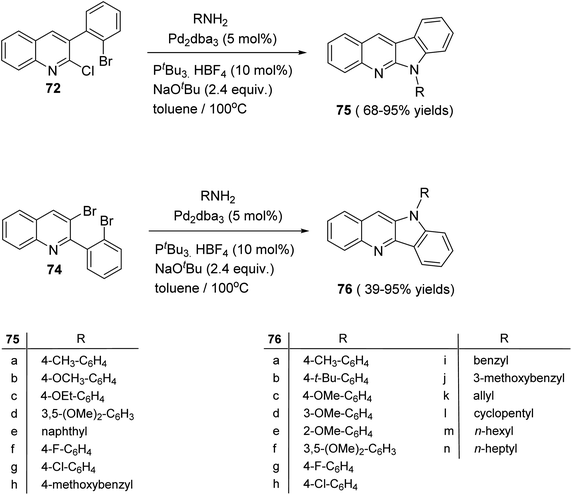 |
| | Scheme 21 Synthesis of 10H-indolo[3,2-b]quinolines 75 and 6H-indolo[2,3-b]quinolines 76. | |
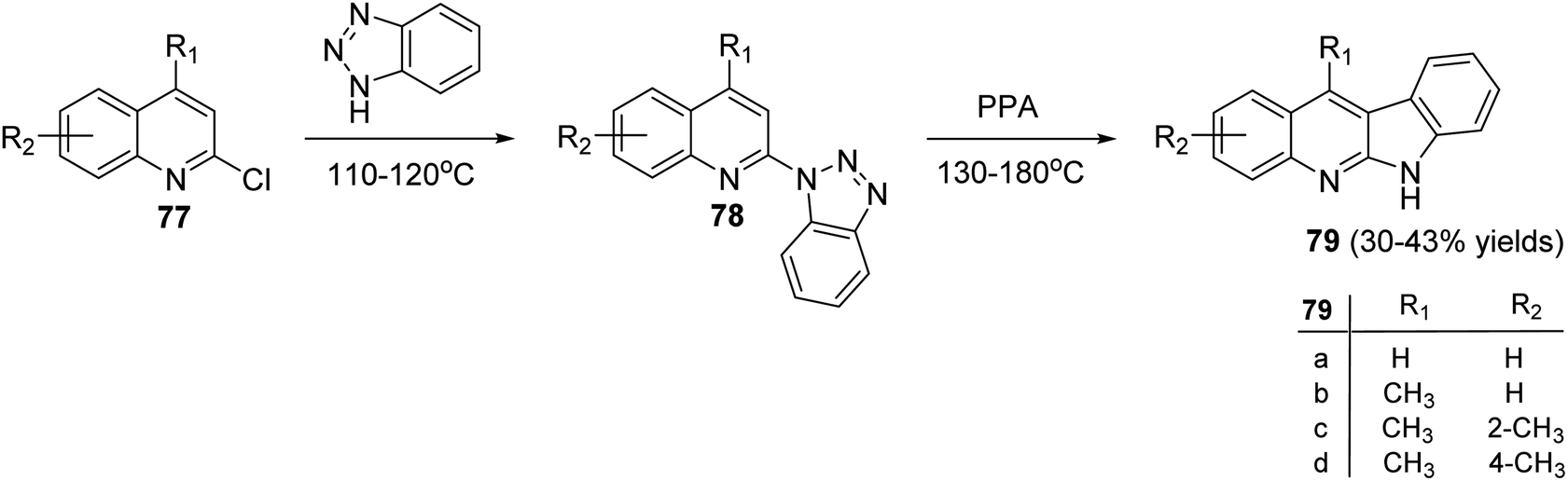
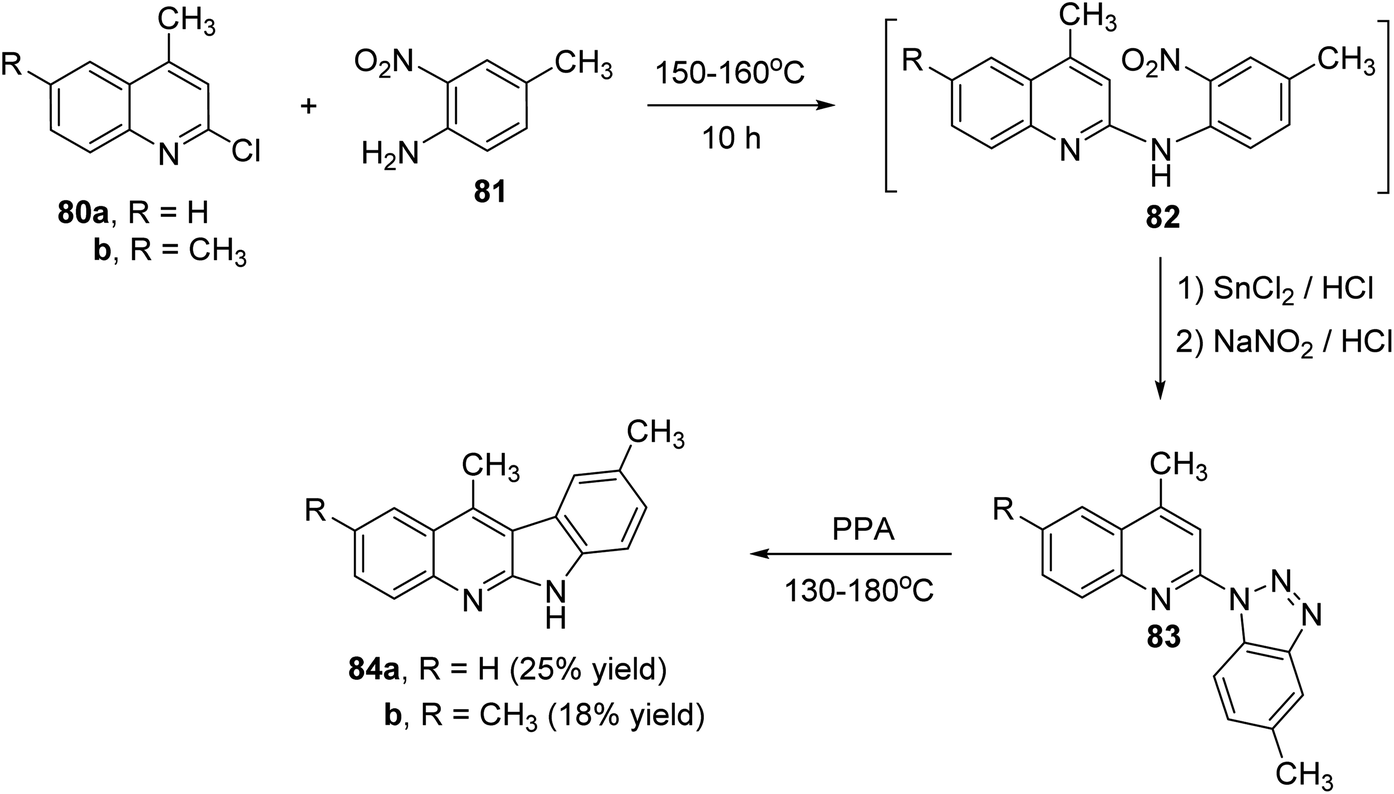
Peczyńska-Czoch and his group64a,b have described the synthesis of 6H-indolo[2,3-b]-quinolines 79 and 84, as novel and more efficient DNA Topoisomerase I1 inhibitors, via the methods showed in Schemes 22 and 23. Heating the chloroquinolines 77 with 1H-benzo[1,2,3]triazole at 110–120 °C gave the corresponding triazoles 78 (Scheme 22). However, compounds 83 were prepared by heating chloroquinoline derivatives 80 with 4-methyl-3-nitroaniline (81) at 150–160 °C, followed by the reduction of the NO2 group in intermediate 82 and diazotization of the amino obtained (Scheme 23). Subsequent decomposition of triazoles 78 and 83 in polyphosphoric acid (PPA) at 130–180 °C afforded the respective indolo[2,3-b]quinolines 79 and 84, respectively, in low yields.
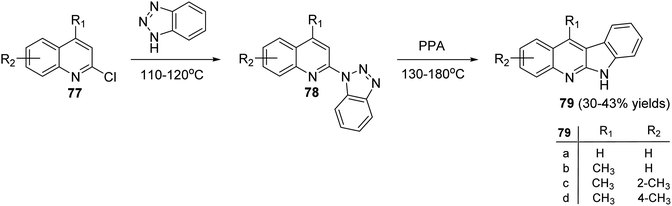 |
| | Scheme 22 Synthesis of 6H-indolo[2,3-b]quinolines 79. | |
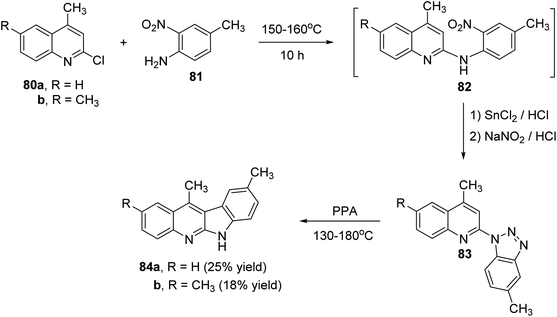 |
| | Scheme 23 Synthesis of 6H-indolo[2,3-b]quinolines 84. | |
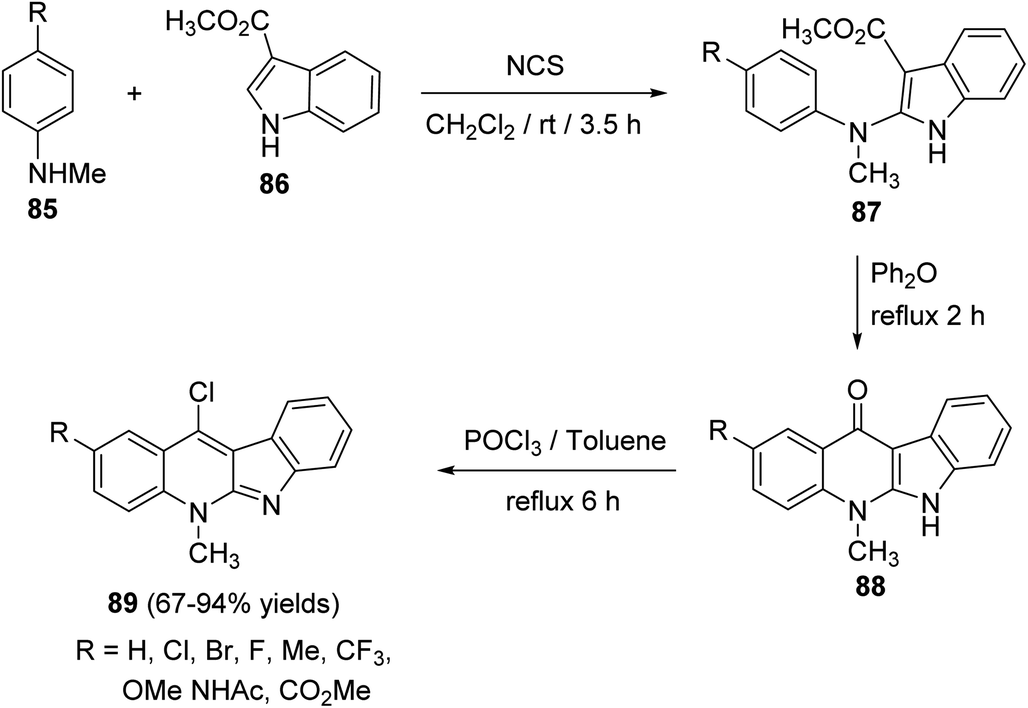
Wang et al. and Lu et al.43,65a–d developed an efficient protocol for the synthesis of a series of 11-chloro-5-methyl-5H-indolo[2,3-b]quinolines (neocryptolepines) 89 with different substituents on the quinoline ring, as useful scaffold for the synthesis of anticancer and antimalaria agents. In this cascade reaction, various N-methyl-anilines 85 were treated with N-chlorosuccinimide (NCS) in CH2Cl2 at room temperature, followed by the addition of methyl 1H-indole-3-carboxylate (86) producing the corresponding methyl 2-((4-substituted phenyl)(methyl)amino)indole-3-carboxylates 87. Upon refluxing 87 in diphenyl ether at 250 °C, it underwent intramolecular cyclization to give the tetracyclic ketones 88 in 68–98% yields. Chlorination of 88 was carried out in POCl3 under reflux conditions to yield the desired 11-chloroneocryptolepines 89 in 67–94% yields, with high purity (Scheme 24).
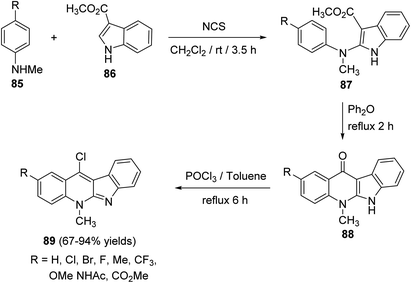 |
| | Scheme 24 Synthesis of 11-chloroneocryptolepines 89. | |
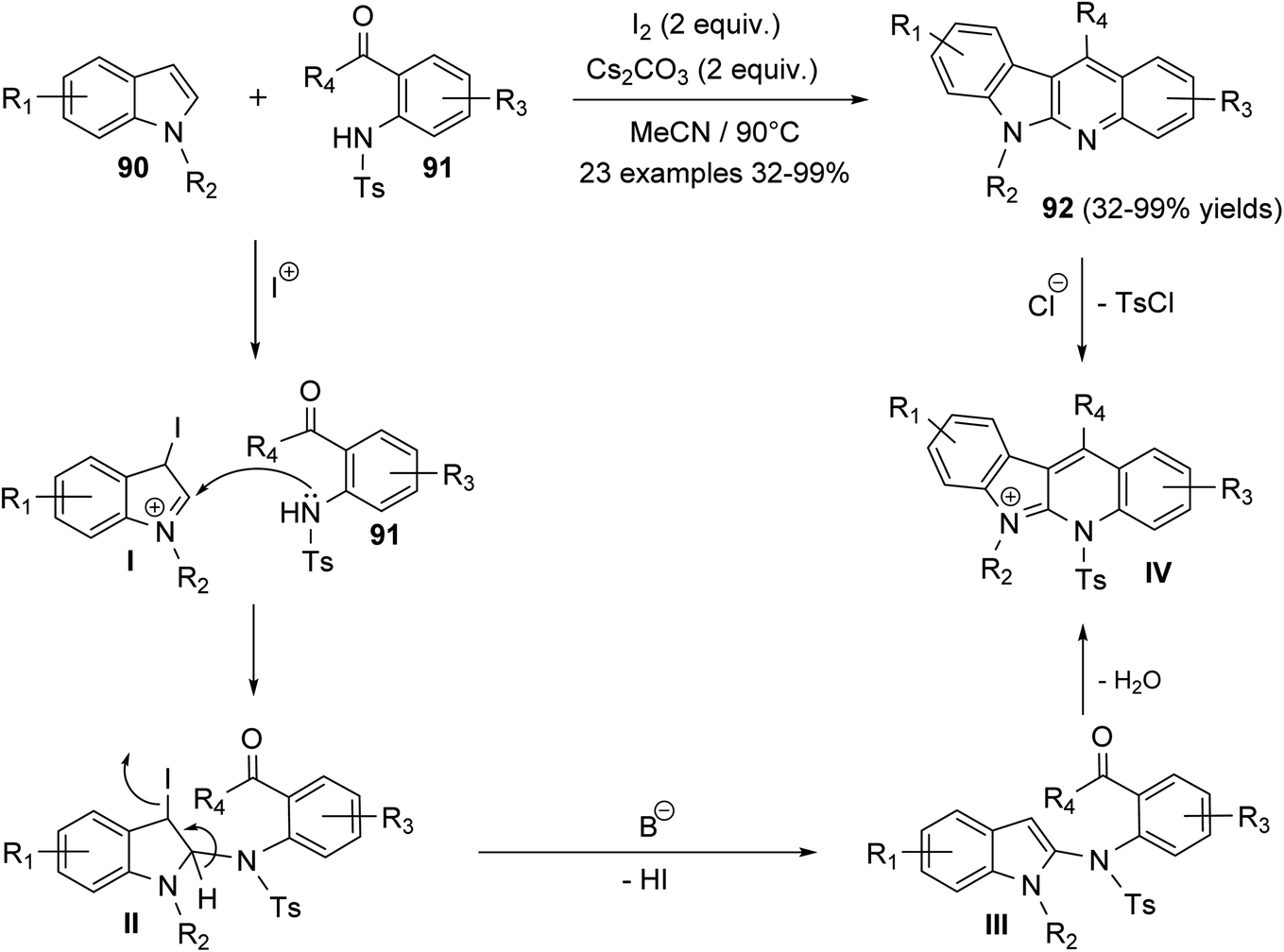
Ali et al.66 reported synthesis of highly substituted indolo[2,3-b]quinolines 92 via metal-free, one-pot annulation reaction of substituted indoles 90 with 1-(2-tosylaminophenyl)-ketones 91. The reaction occurs via an activation of C-2 and C-3 of indoles 90 by molecular iodine and Cs2CO3, as a base catalyst, followed by in situ reaction with 1-(2-tosylamino-phenyl)ketones 91 in acetonitrile at 90 °C to afford polyfunctionalized indolo[2,3-b]quinolines 92, in moderate to excellent yields (Scheme 25). In this reaction, electrophilic addition of iodonium ion to the 3-position of indoles 90 afforded cation intermediate I. 2-Amination of I with 91 gives II, which undergoes elimination of one molecule of HI in the presence of base to furnish III. Subsequent alkylation, followed by detosylation of III in the presence of HCl (12 M) affords 92 (Scheme 25).
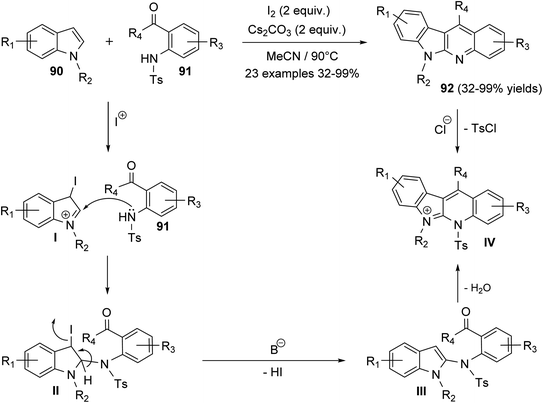 |
| | Scheme 25 Molecular iodine catalyzed synthesis of polyfunctionalized indolo[2,3-b]-quinolines 92. | |
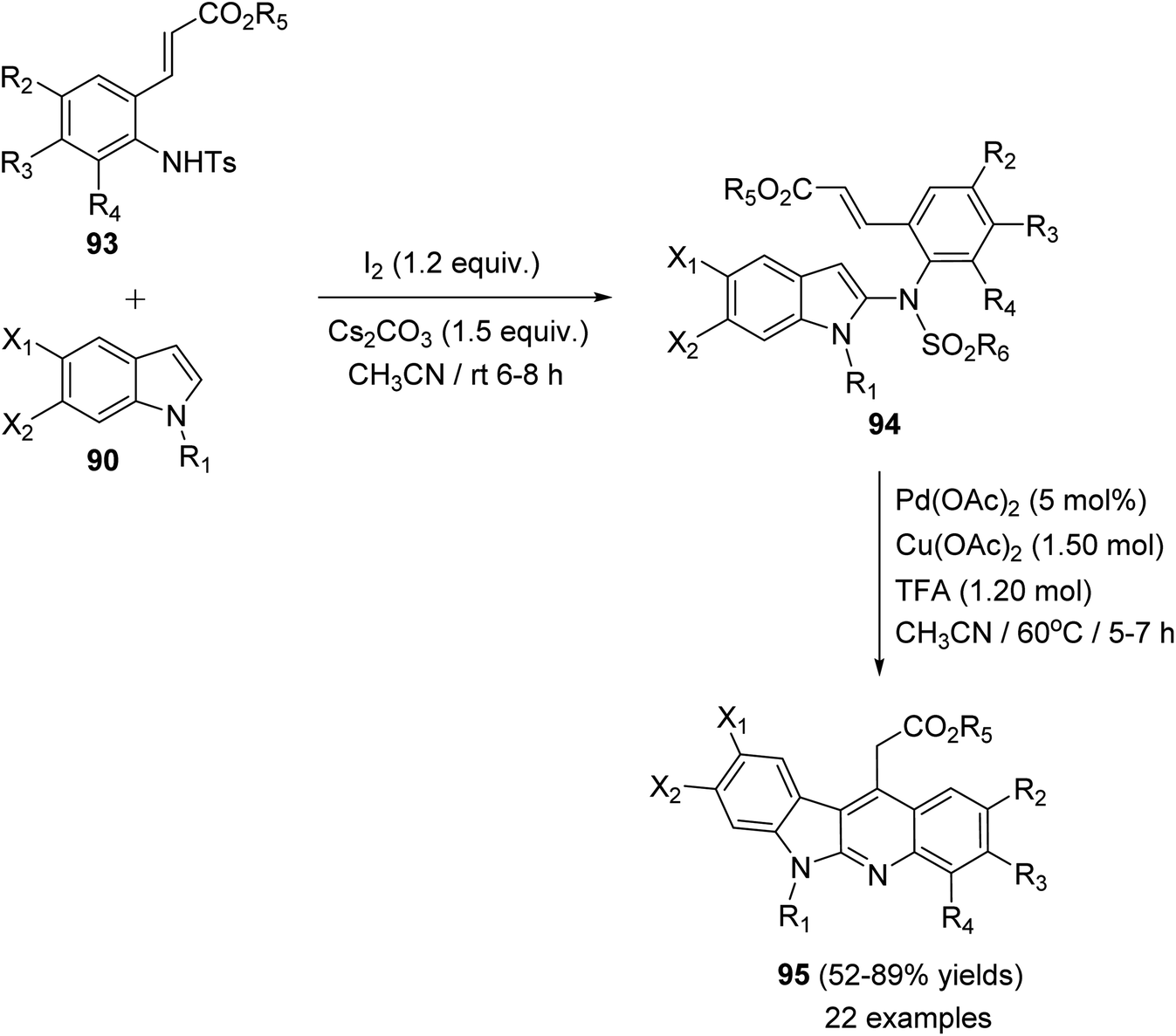
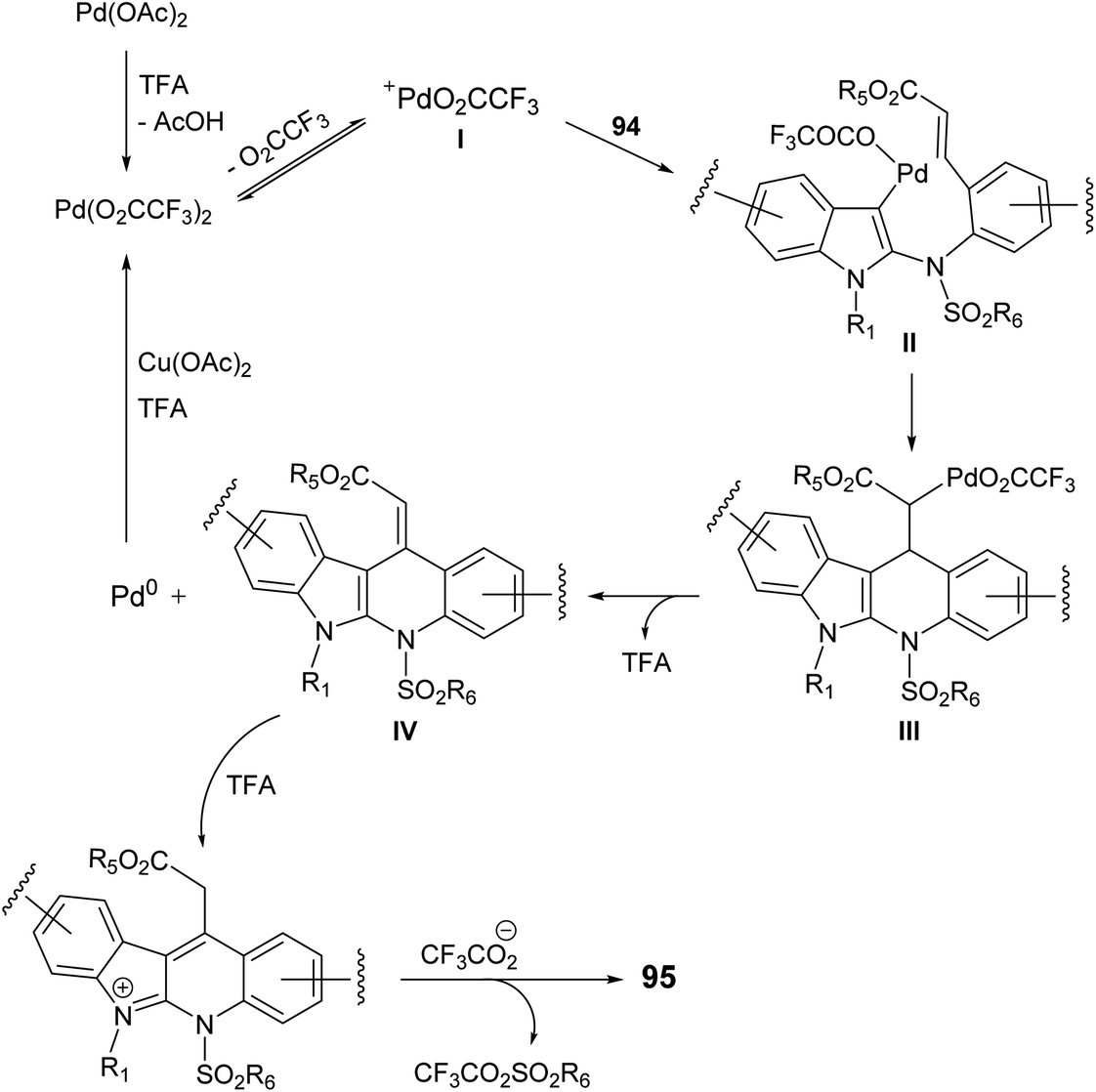
In 2015, Pal et al.67 have developed a new one-pot strategy for the synthesis of novel 11-carboxymethyl substituted 6H-indolo[2,3-b]quinolines 95, as potential inducers of apoptosis, involving Pd(II)-catalyzed intramolecular oxidative C3–H alkenylation of the indole ring of (E)-alkyl-3-(2-(1H-indol-2-ylamino)phenyl)acrylates 94 followed by desulfonylation. In the present study, the starting material 94 was prepared via I2-mediated addition of sulfonamide derivative 93 to indoles 90. Heating a mixture of 93 (1 mmol), Pd(OAc)2 (5 mol%), Cu(OAc)2 (1.50 mol) and trifluoroacetic acid (TFA) (1.20 mol) in CH3CN at 60 °C under an aerobic atmosphere for 5–7 h afforded the target tetracyclic products 95 in 52–89% yields (Scheme 26). A mechanism for the Pd(II)-catalyzed formation of 95 is outlined in Scheme 27.
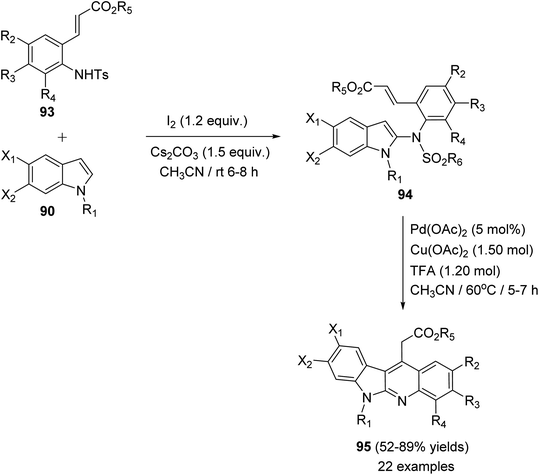 |
| | Scheme 26 Pd-mediated synthesis of 11-carboxymethyl substituted 6H-indolo[2,3-b]-quinolines 95. | |
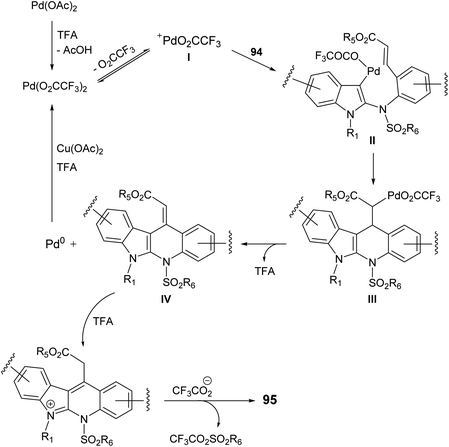 |
| | Scheme 27 Proposed reaction mechanism for the formation of 11-carboxymethyl substituted 6H-indolo[2,3-b]quinolines 95. | |
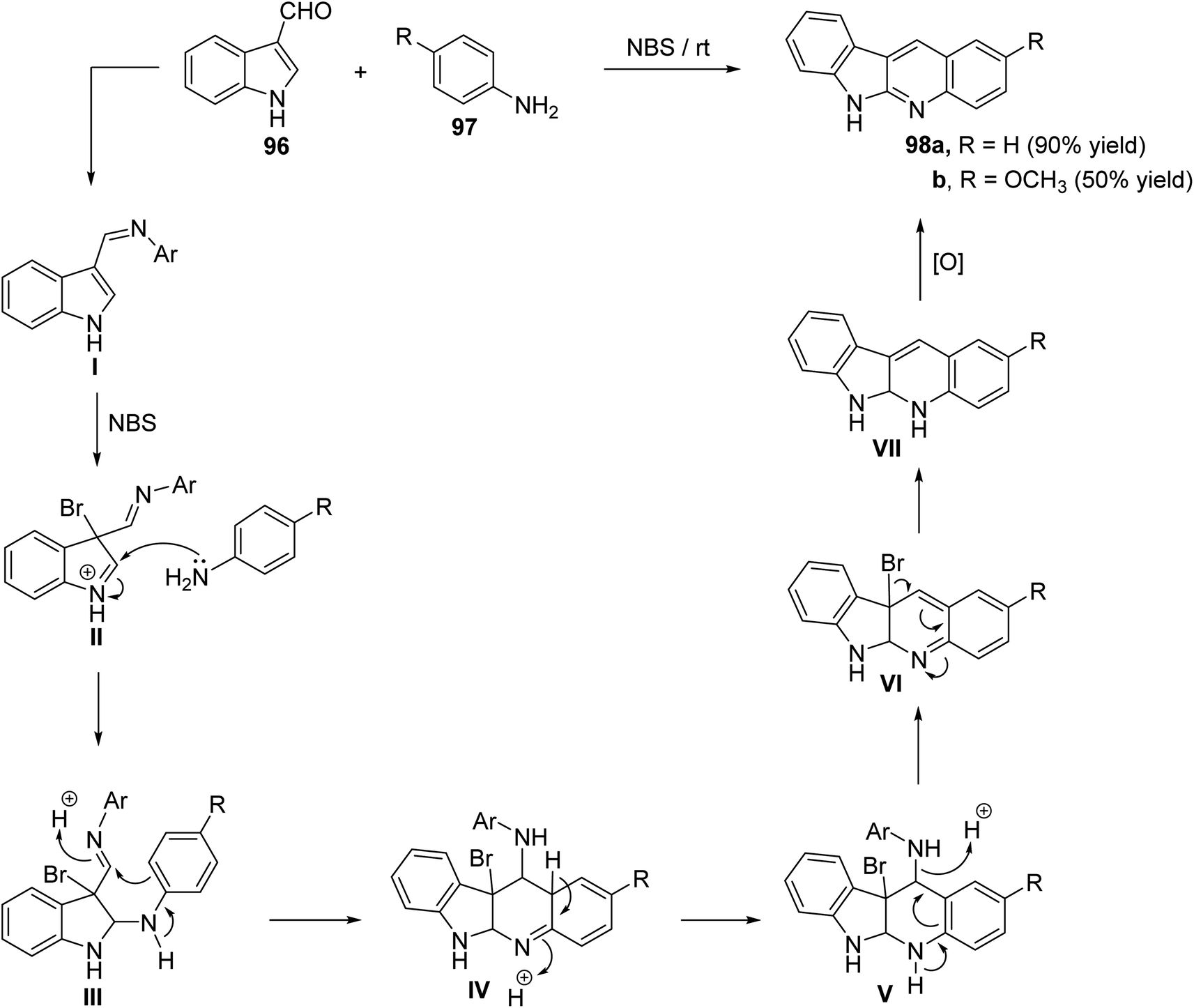
Ghorbani-Vaghei and Malaekehpoor68 described a simple and one-pot synthesis of novel 6H-indolo[2,3-b]quinolines 98 from the reaction of indole-3-carbaldehyde (96) with various aryl amines 97, under mild conditions, utilizing N-bromo-succinimide (NBS) as an efficient catalyst. NBS initially catalyzed the formation of Schiff base I and then a 3-bromo-indolinium cation intermediate II. The intermediate II on the subsequent nucleophilic attack by an another mole of aniline, followed by intramolecular cyclization and oxidation produces 98. Reaction is outlined in Scheme 28.
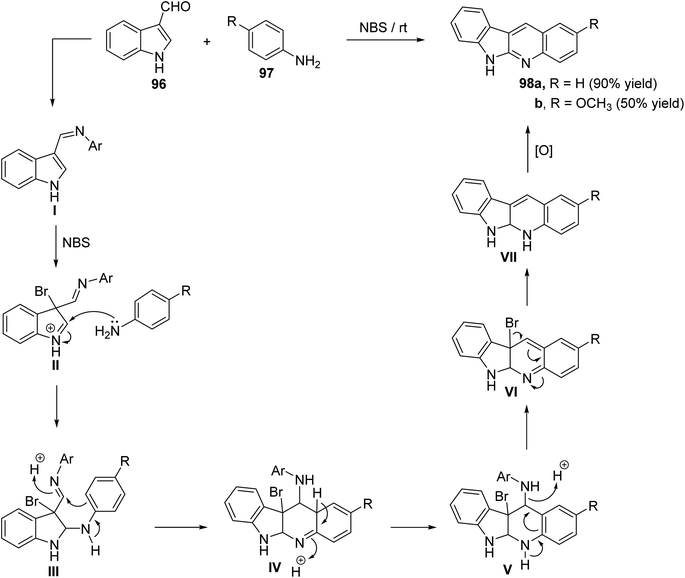 |
| | Scheme 28 NBS catalyzed synthesis of novel indolo[2,3-b]quinolines 98. | |
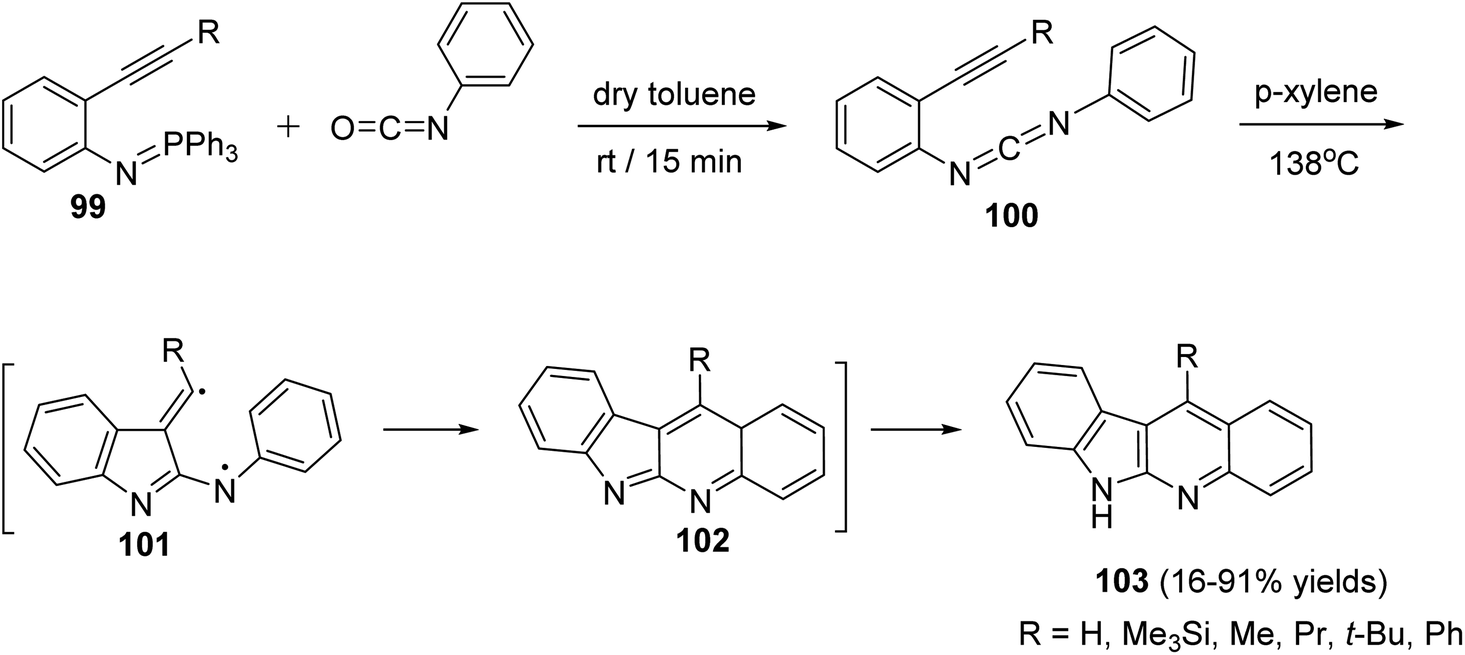
A novel and efficient method for the synthesis of 11-substituted-6H-indolo[2,3-b]quinolines 103 via thermolysis of the carbodiimide 100, which represents a new way of generating biradicals from unsaturated molecules having two nitrogen atoms in the conjugated system, is reported by Wang et al.69 Thus, thermolysis of the carbodiimide 100, having a different substituents at the acetylenic terminus, in refluxing p-xylene at 138 °C produced 103 in 16–91% yields. Apparently, a two-step biradical pathway through 101 or a one-step intramolecular Diels–Alder reaction produced the intermediate 102, which on subsequent tautomerization under the reaction conditions affords the final product 103 (Scheme 29). An especially attractive feature of this synthetic method is the possibility of placing a wide variety of substituents at different positions of the 6H-indolo[2,3-b]quinoline structure by selecting suitable fragments.
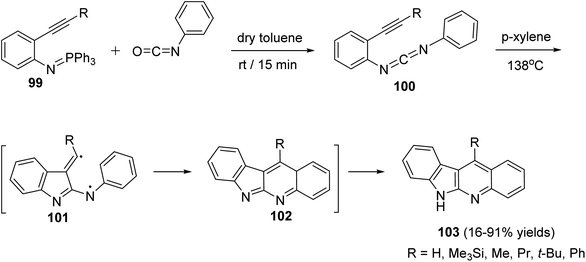 |
| | Scheme 29 Synthesis of 6H-indolo[2,3-b]quinolines 103 via thermolysis of N-[2-(1-alkynyl)-phenyl]-N-phenylcarbodiimides 100. | |
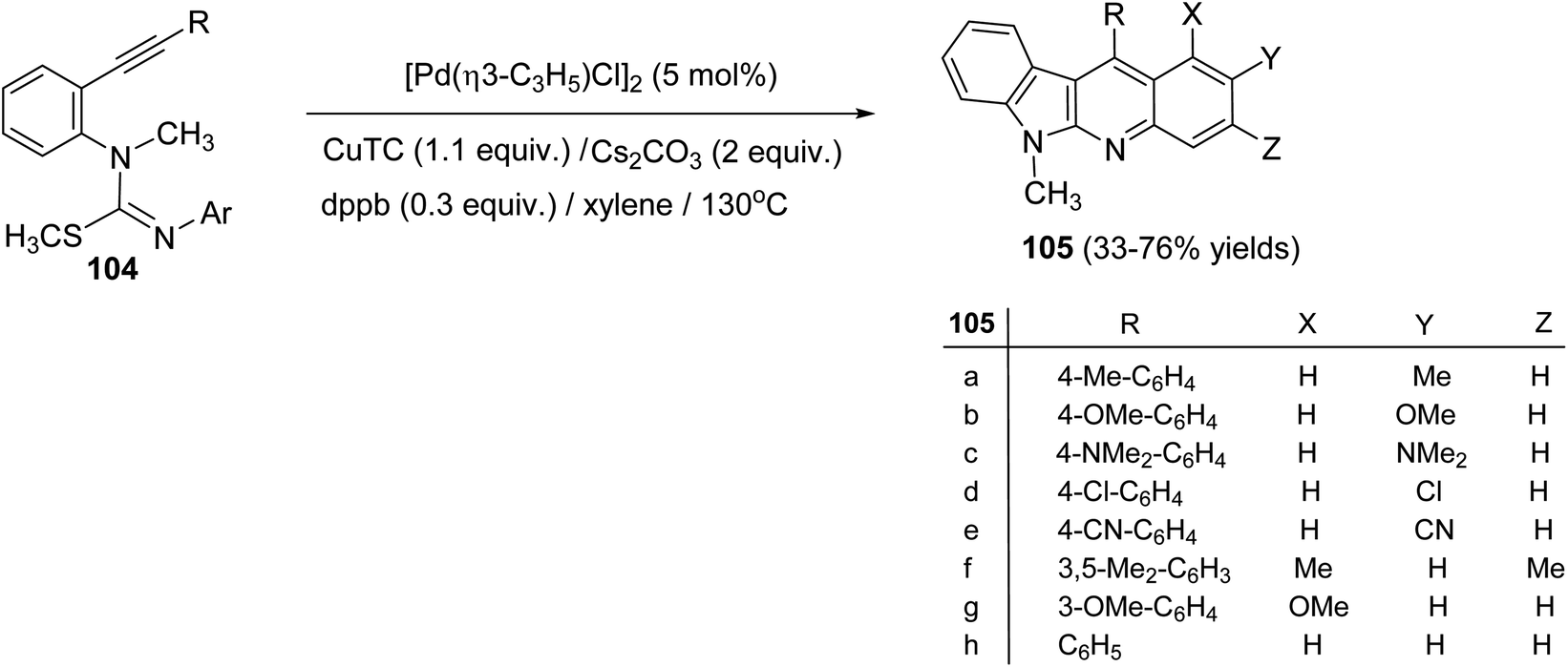
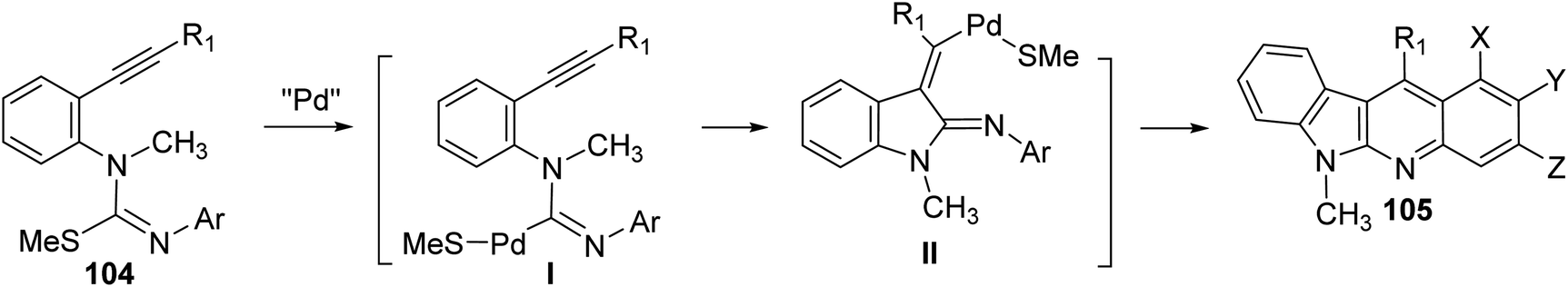
On heating 1-(2-alkynyl)phenyl-3-aryl-2-methylisothioureas 104 with [Pd(η3-C3H5)–Cl]2 (5 mol%) in the presence of Cs2CO3 (2 equiv.), CuTC (copper thiophene-carboxylate) (1.1 equiv.) and 1,4-bis(diphenylphosphino)butane (dppb) (0.3 equiv.) in xylene at 130 °C, a wide range of the desired tetracyclic adduct 6-methyl-indolo[2,3-b]quinolines 105 were obtained in 33–76% yields (Scheme 30).70 Importantly, a variety of electron-rich aryl groups on the aryl ring (Ar) promoted this process. However, substrates bearing an electron-withdrawing group decreased the chemical yields. In fact, the reaction of isothiourea bearing a 3,5-bis(trifluoromethyl)phenyl group afforded no cyclized adduct. Respecting to the substituent (R) on the alkynyl group, both phenyl and alkyl groups were tolerated under these reaction conditions. In the suggested reaction mechanism, the oxidative addition of MeS–C bond to Pd(0), followed by ligand exchange with CuTC gave the alkenylpalladium complex II. Subsequent nucleophilic attack of the aryl group to the Pd(II) complex followed by reductive elimination of the resulting Pd-complex provided the tetracyclic product 105 (Scheme 31).
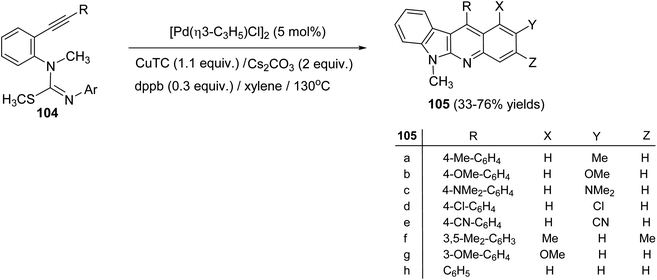 |
| | Scheme 30 Synthesis of 6-methyl-indolo[2,3-b]quinolines 105 by Pd-catalyzed annulation of unsaturated isothioureas. | |
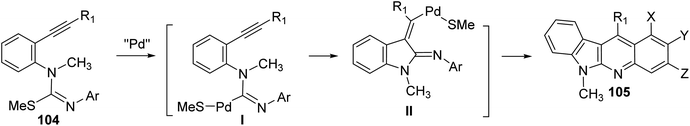 |
| | Scheme 31 A suggested mechanism for the Pd-catalyzed formation of 6-methyl-indolo[2,3-b]quinolines 105. | |
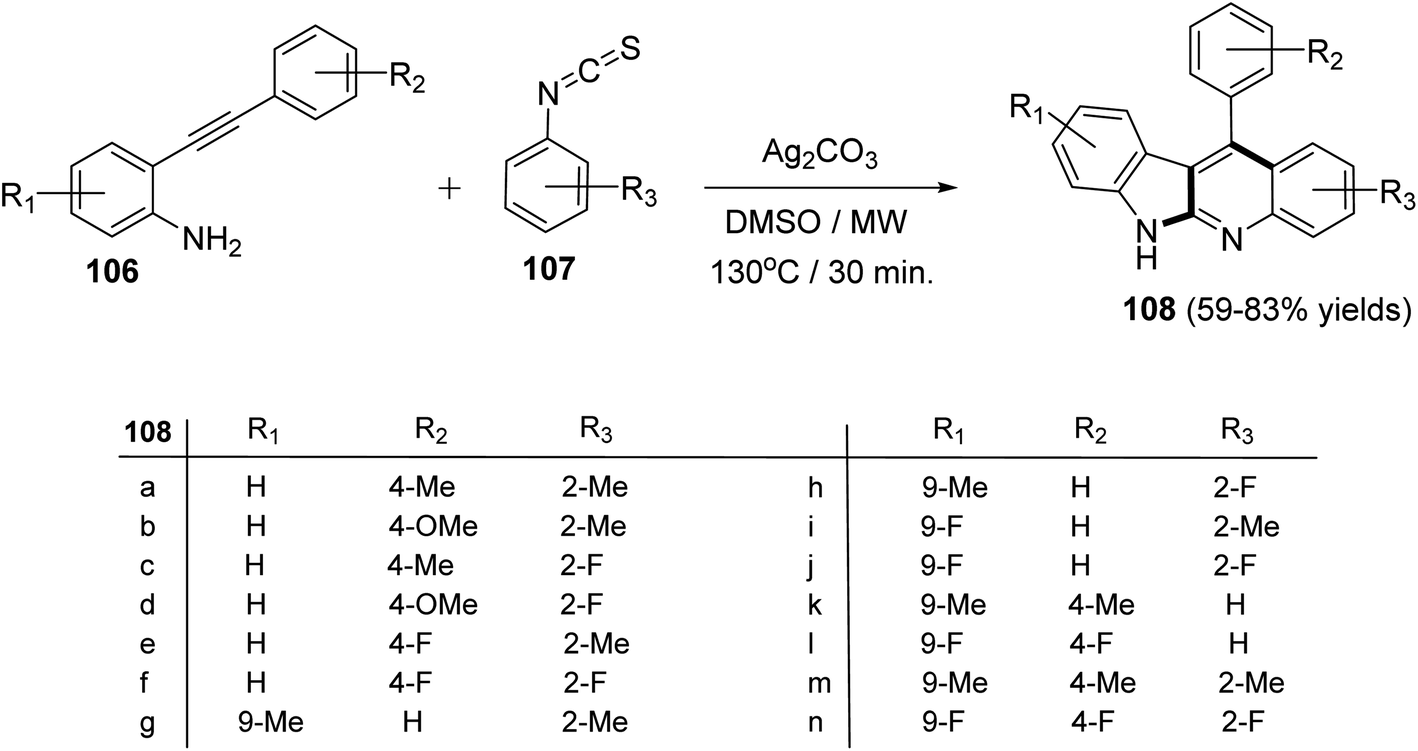
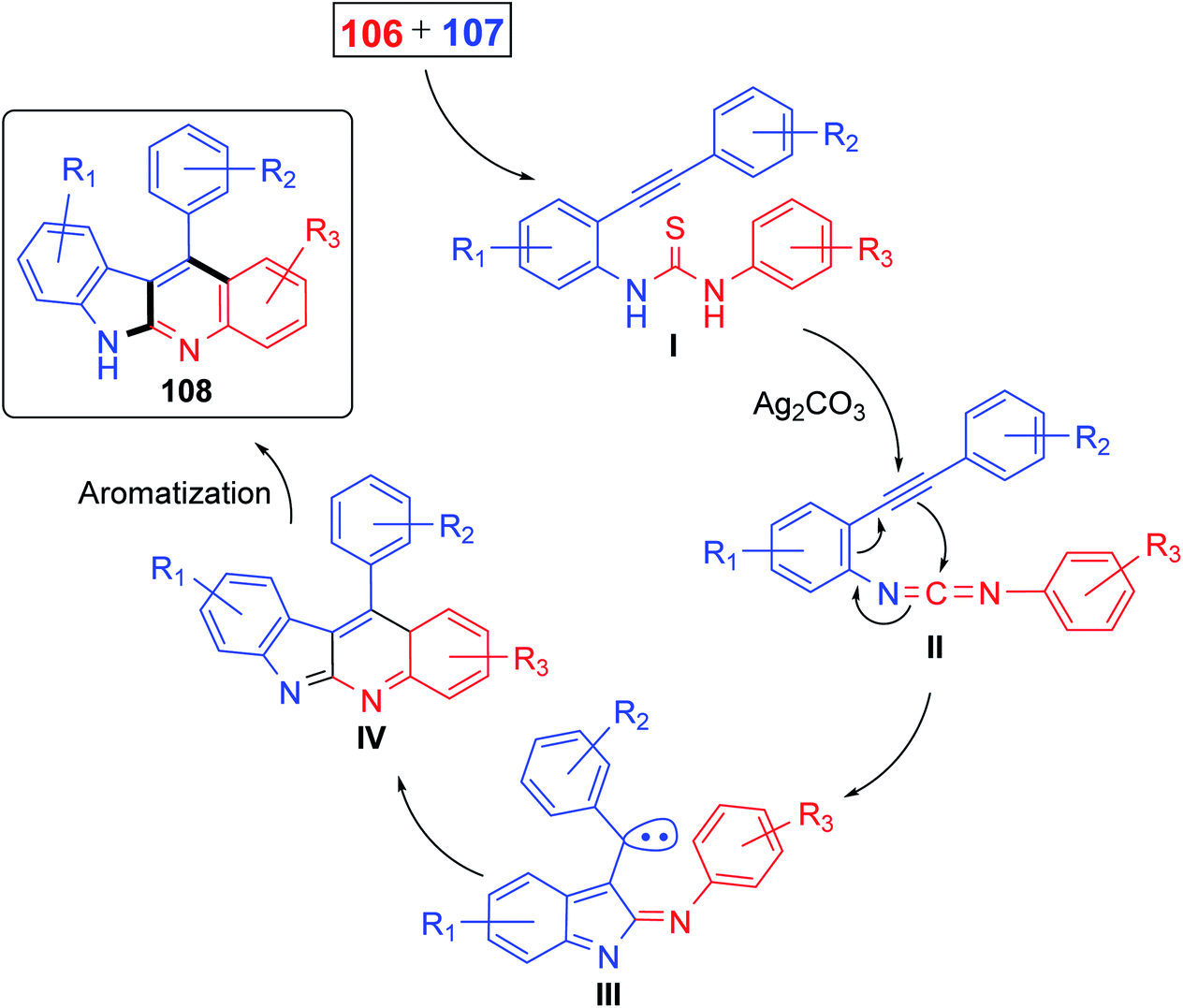
Recently, Patel et al.71 have described an elegant and green approach for the synthesis of 11-aryl-6H-indolo[2,3-b]quinolines 108 from 2-(phenylethynyl)anilines 106 and aryl isothiocynates 107. The in situ generated o-alkynylthioureas, obtained by reacting 106 and 107, underwent efficient cascade cyclization in the presence of Ag2CO3, as the catalyst, in DMSO to give 11-aryl-6H-indolo[2,3-b]quinolines 108, in 59–83% yields, under microwave conditions (30 min reaction time at 130 °C, 150 W, closed vial) (Scheme 32). The authors examined the electronic effect of substituents R3 on the aryl isothiocynates 107, R2 present on the other phenyl ring of 2-(phenylethynyl)anilines 106 and R1 substituents present on the amine bearing ring of 2-(phenylethynyl)anilines 106 on the product yields and they found that when substituents R2 and R3 are electron-donating groups the products were obtained in better yields compared to electron-withdrawing substituents. On the other hand, R1 substituents showed a trend on the product yields opposite to that of substituents R2 and R3. A plausible reaction mechanism has been proposed as outlined in Scheme 33. Reaction of 106 and 107 gives thiourea intermediate I, which is desulfurized in the presence of Ag2CO3 to a carbodiimide intermediate II. Intramolecular cyclization of II formed a carbene-type intermediate III. The latter III undergoes further cyclization via carbene C–H insertion to afford a non-aromatic tetracyclic intermediate IV, which is aromatized to furnish the desired tetracyclic product 108.
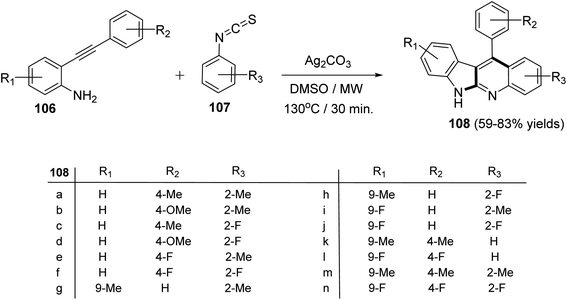 |
| | Scheme 32 A green method for the synthesis of 11-aryl-6H-indolo[2,3-b]quinolines 108 under microwave heating. | |
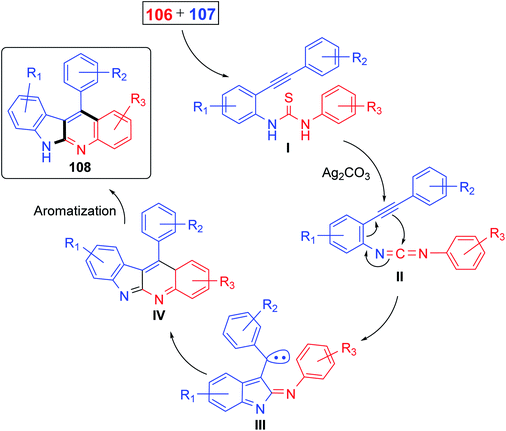 |
| | Scheme 33 Proposed mechanistic pathway. | |
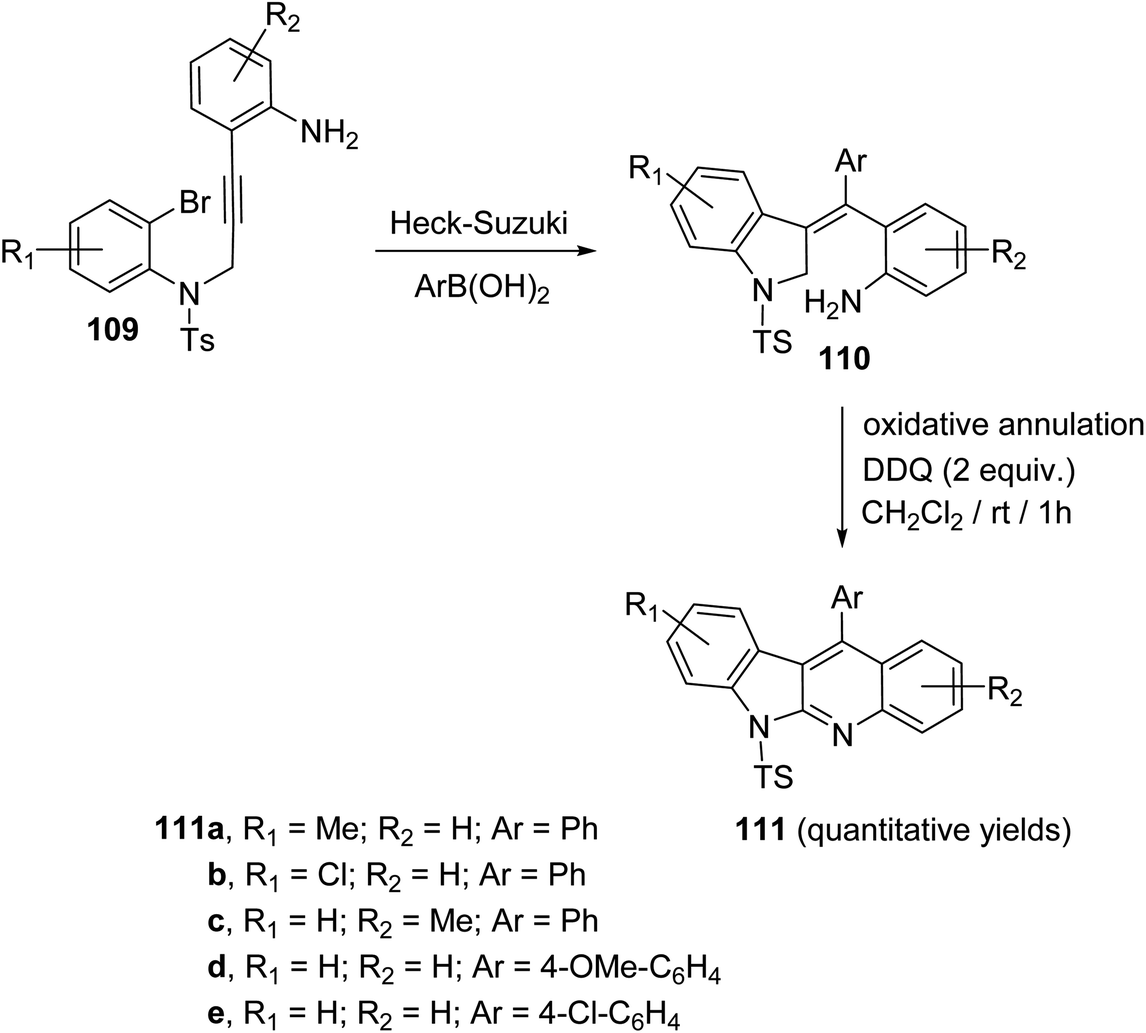
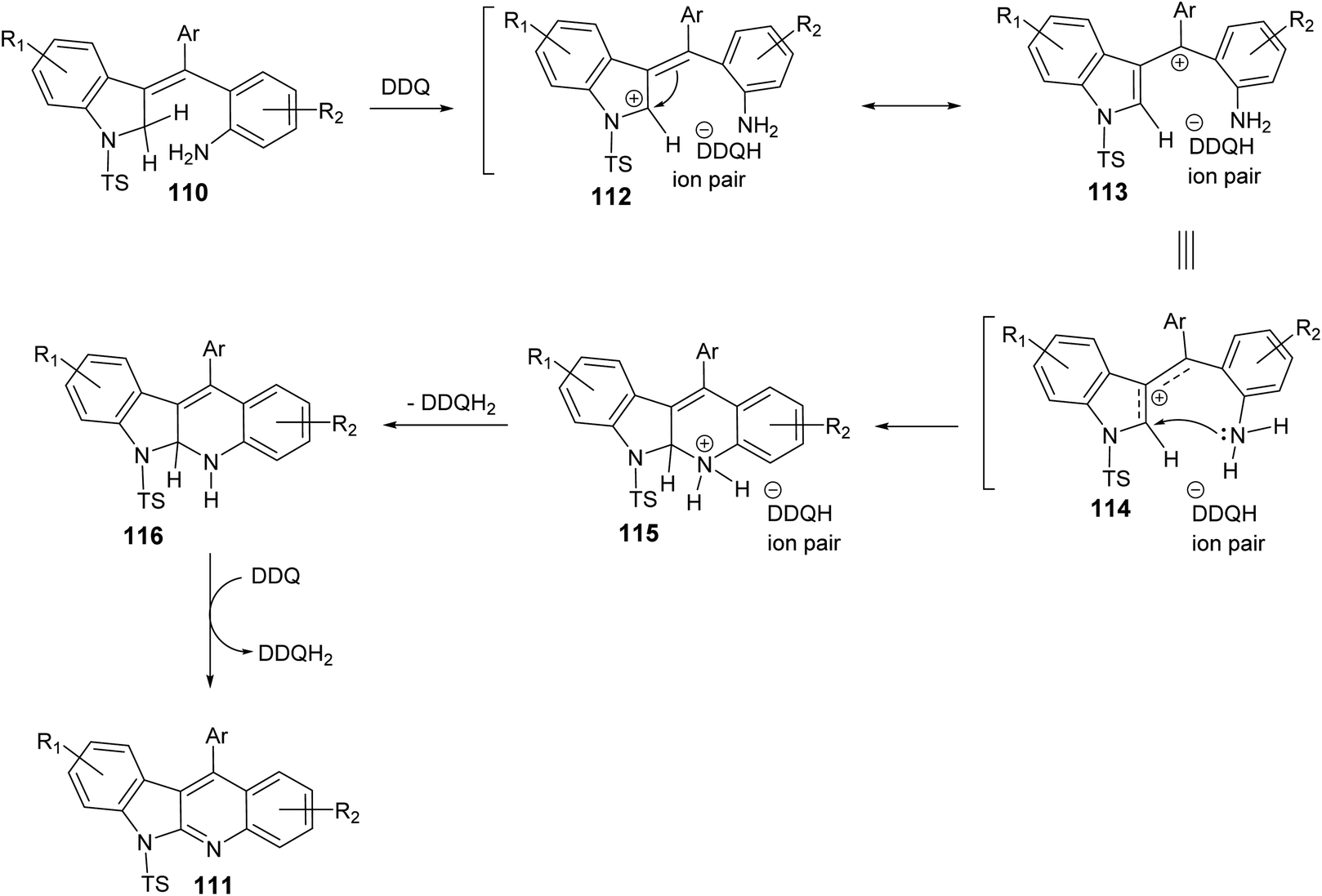
In 2019, Kundal et al.72 reported a novel, general and efficient method for the synthesis of diversely substituted indolo[2,3-b]quinolines 111 through a two-step domino reaction. First, the starting 3-indolines 110 were prepared by Pd-catalyzed domino Heck–Suzuki coupling of 2-bromo-N-propargylanilides 109 with aryl boronic acids (Scheme 34). Next, when the 3-indolines 110 were subjected to the oxidative cross-dehydrogenative coupling (CDC) between allylic C sp3–H of the indoline ring and the free NH2 group in the presence of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (2 equiv.) in CH2Cl2 at room temperature, the fused tetracyclic indolo[2,3-b]quinolines 111 were produced in quantitative yields within one hour (Scheme 34). Gratifyingly, it was found that there was no significant electronic effect of the substituent on the aryl ring on the olefinic motif during the formation of C–N bond. DDQ has been proven as an efficient metal-free oxidant for CDC reactions of C–C bond formation reactions, but the formation of the C–N bond via DDQ-mediated cross-dehydrogenative coupling (CDC) has been relatively rare. Therefore, this strategy for the preparation of indolo[2,3-b]quinolines 111 should be very attractive in the modern context of organic chemistry research. A plausible mechanism for the DDQ-mediated oxidative C–H amination is outlined in Scheme 35.
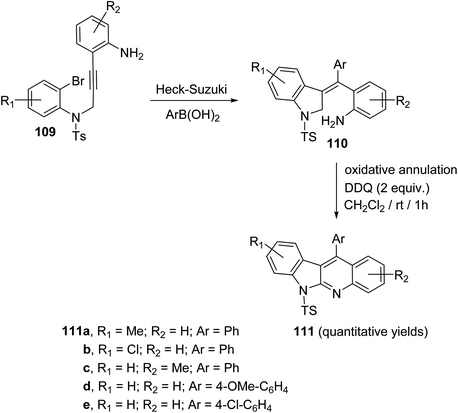 |
| | Scheme 34 Synthesis of indolo[2,3-b]quinolines 111 via Heck–Suzuki coupling and DDQ-mediated C–H amination. | |
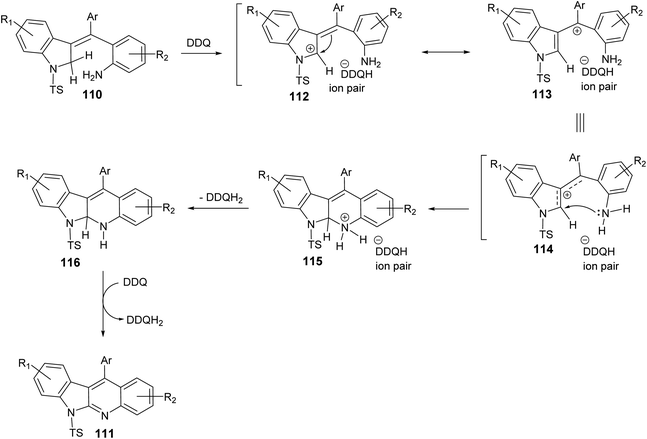 |
| | Scheme 35 A suggested mechanism for the DDQ-mediated oxidative C–H cycloamination. | |
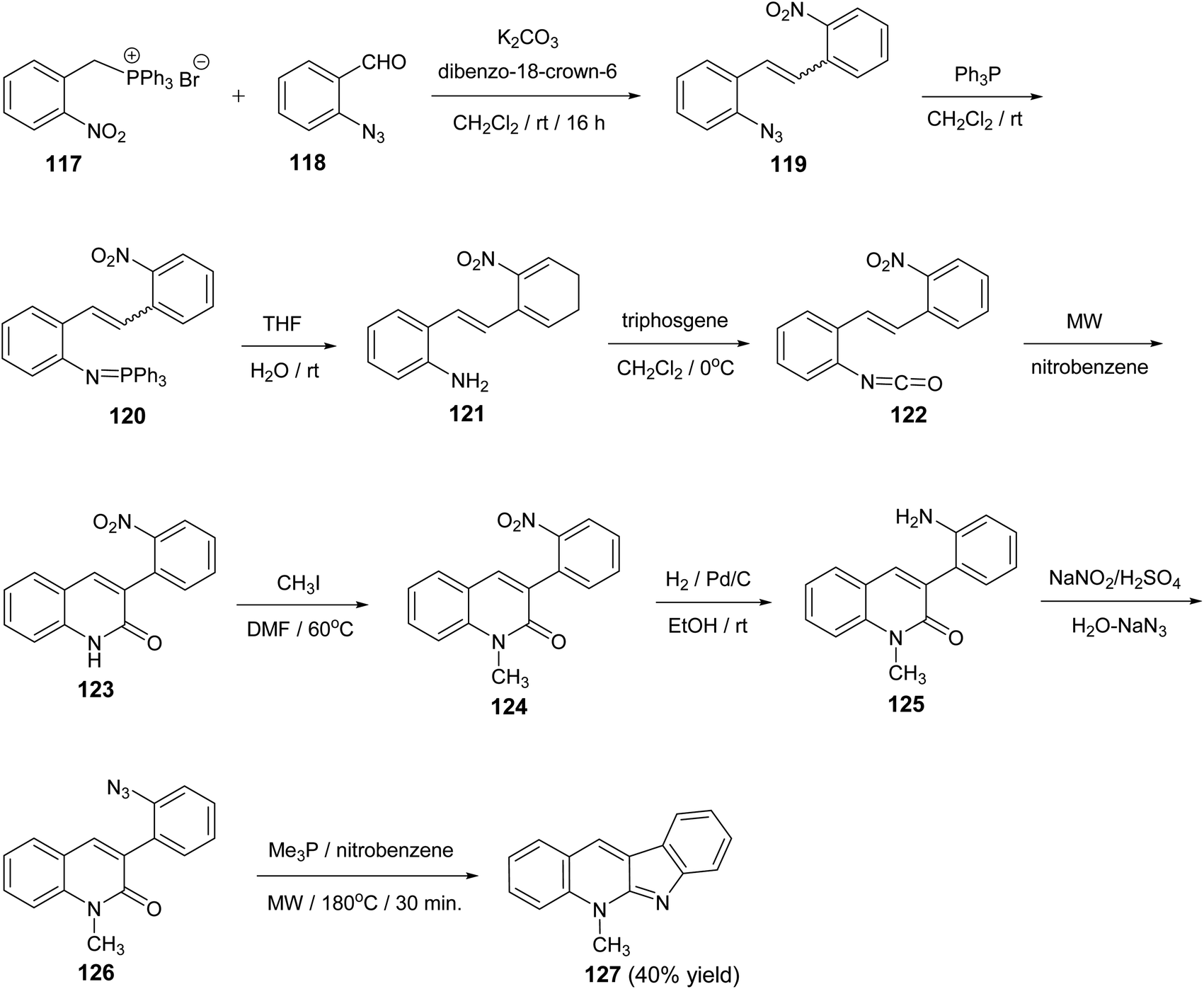
A novel and divergent approach for the synthesis of 5-methyl-5H-indolo[2,3-b]quinoline alkaloid cryptotackieine 127 was reported.73 The reaction was performed in eight steps, starting from (2-nitrobenzyl)triphenylphosphonium bromide (117). First, condensation of 117 with o-azidobenzaldehyde (118) in the presence of anhydrous K2CO3 and catalytic amounts of dibenzo-18-crown-6 gave the 1-azido-2-(2-nitrostyryl)benzene (119). Staudinger reaction between the azide 119 and triphenylphosphine in dry CH2Cl2 at room temperature for 5 h followed by hydrolysis of the resulting iminophosphorane 120 provided the 2-(2-nitrostyryl)aniline (121). One-pot conversion of 121 into the 3-(2-nitrophenyl)quinoline-2(1H)-one (123) was performed by sequential treatment with bis(trichloromethyl)carbonate (triphosgene) and further microwave-promoted cyclization of the resulting 1-isocyanate-2-(2-nitrostyryl)-benzene (122). Conversion of the quinoline 123 into 1-methylquinoline-2-one 126 was performed by the three-step sequence: (i) methylation with CH3I in DMF at 60 °C for 2 h to give 1-methyl-3-(2-nitrophenyl)quinoline-2(1H)-one (124); (ii) reduction of the nitro group in 124 with H2 in the presence of Pd/c (10%) in EtOH at room temperature for 5 h to give 3-(2-amiophenyl)-1-methyl-quinoline-2(1H)-one (125) and (iii) diazotization followed by the reaction with NaN3 furnished 3-(2-azidophenyl)-1-methyl-quinoline-2(1H)-one (126). Finally, when a solution of the iminophosphorane derived from 126 and Me3P in nitrobenzene was heated under microwave irradiation at 180 °C for 30 min, the required 5-methyl-5H-indolo-[2,3-b]quinoline (127) was obtained in 40% yield (Scheme 36). The conversion of 126 into 127 represents the first example of an intramolecular aza-Wittig reaction involving a 2-pyridone carbonyls group.
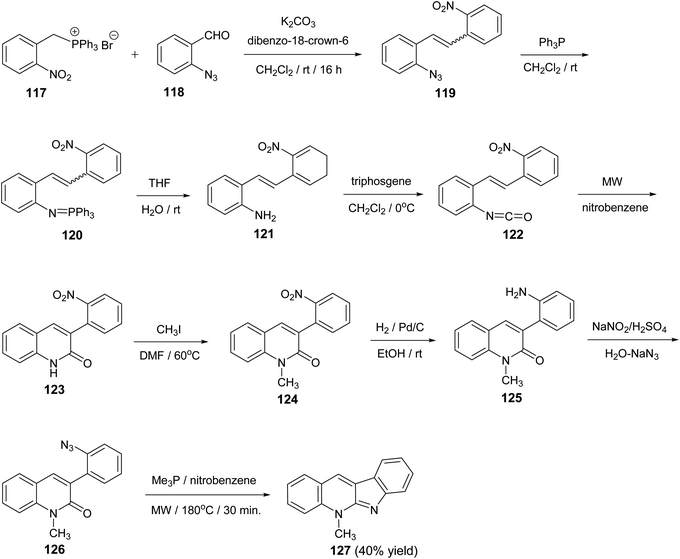 |
| | Scheme 36 A novel approach for the synthesis of 5-methyl-5H-indolo[2,3-b]quinoline (127). | |
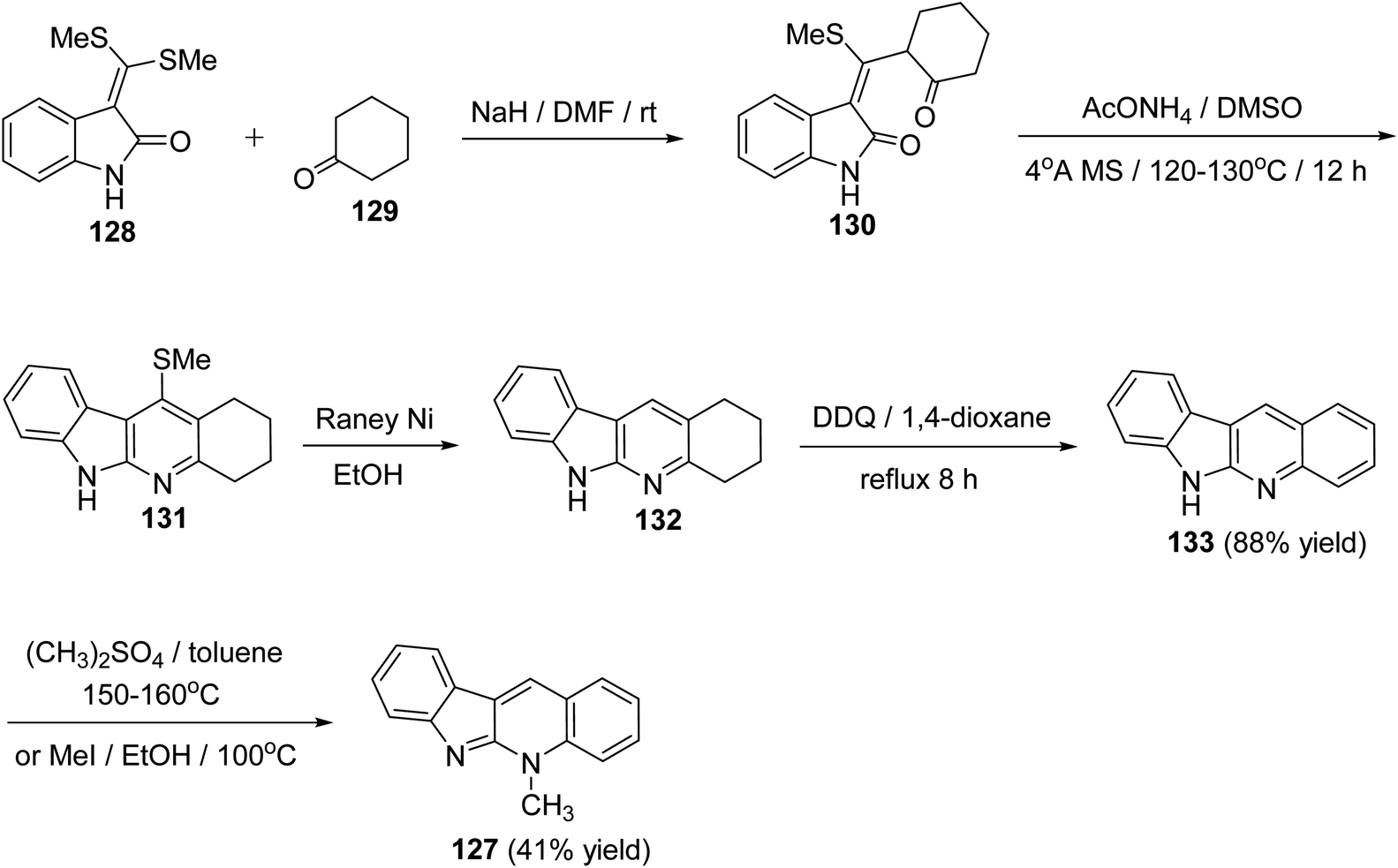
A new, efficient and four-step formal synthesis of alkaloid cryptotackiene 127, a linear 5-N-methyl-5H-indolo[2,3-b]quinoline alkaloid, isolated from the West African shrub Cryptolepis sanguinolenta, is reported.74 It should be noted that the cryptotackiene has been reported to display strong antiplasmodial activity. First, the starting 3-bis[(methylsulfanyl)-methylene]-2-oxindole (128) was subjected to conjugate addition with cyclohexanone (129) in DMF in the presence of NaH to afford the required 3-[(methylthio)(2-oxocyclohexyl)-methylene]indolin-2-one (130). Heterocyclization of the latter adduct 130 was performed by heating with AcONH4 in dry DMSO in the presence of 4 Å molecular sieves at 120–130 °C for 12 h to give the 11-(methylthio)-2,3,4,6-tetrahydro-1H-indolo[2,3-b]quinoline (131). Dethiomethylation of 131 with Raney Ni in EtOH at reflux temperature for 6 h gave 2,3,4,6-tetrahydro-1H-indolo[2,3-b]quinoline (132). Subsequent dehydrogenation of 132 with DDQ in refluxing 1,4-dioxane furnished the requested 6H-indolo[2,3-b]quinoline (133) in 88% yield (Scheme 37). Finally, indoloquinoline 133 was converted into 5-methyl-5H-indolo[2,3-b]quinoline (cryptotackiene) (127) by heating with dimethylsulfate in toluene in sealed tube at 150–160 °C. Compound 127 was found to be identical in all respects with the one prepared by heating 133 with methyl iodide in EtOH in closed glass tube at 100 °C (Scheme 37).
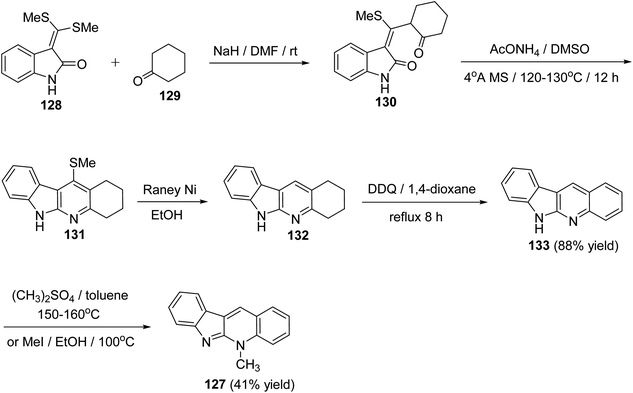 |
| | Scheme 37 A four-step formal synthesis of 5-methsyl-5H-indolo[2,3-b]quinoline (127). | |
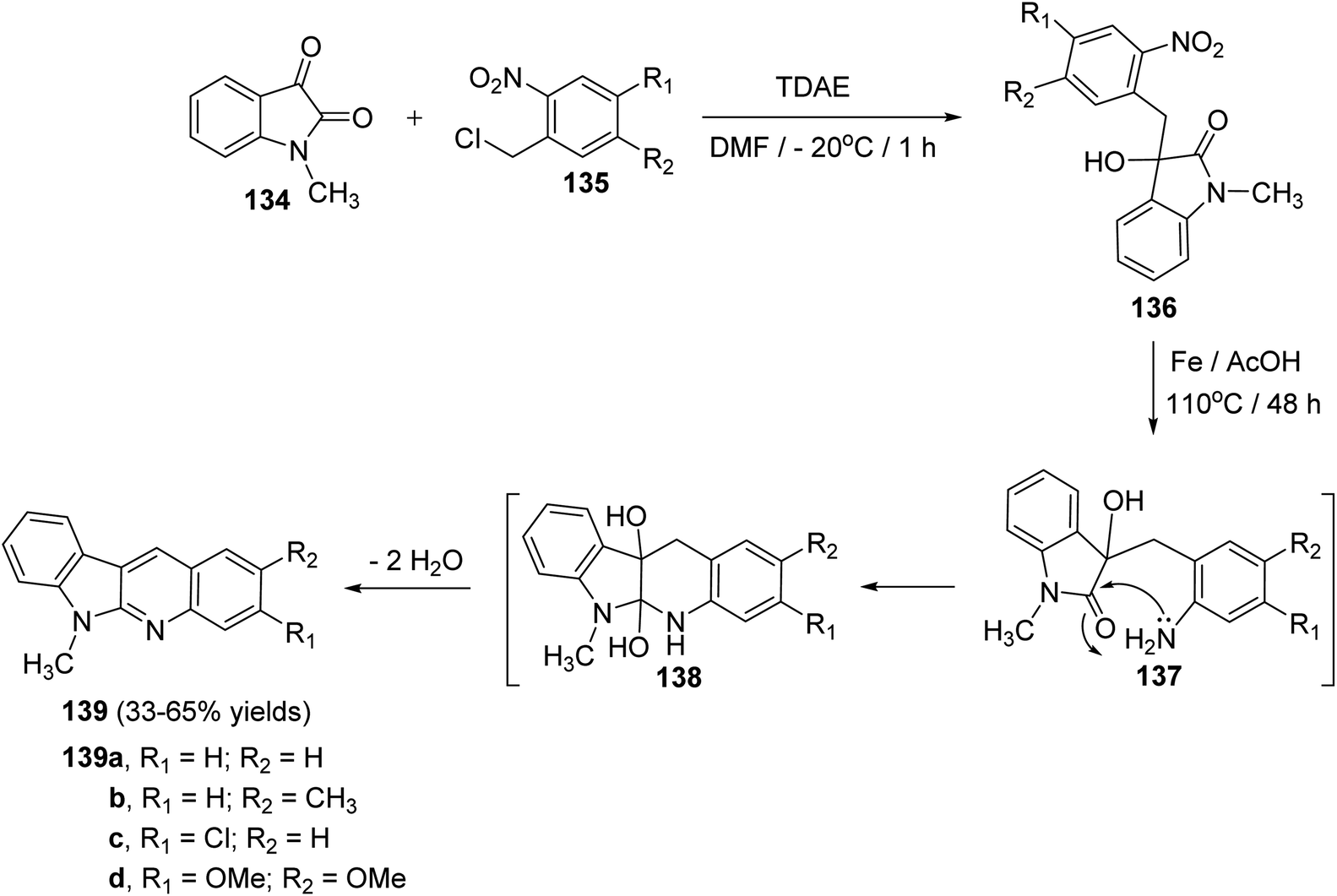
Vanelle et al.75 have reported an original, rapid and easy two-step synthesis of new substituted indolo[2,3-b]quinolines based on TDAE strategy from reaction between 1-methylisatin (134) and substituted o-nitrobenzyl chlorides 135 followed by a one-pot reduction-intramolecular cyclization-double-dehydration reaction. First, reaction of 134 (3 equiv.) with 135 in DMF in the presence of TDAE at −20 °C for 1 h gave the corresponding α-hydroxy lactame 136 (Scheme 38). Reduction of the nitro aromatic group in 136 with iron in AcOH acid at 110 °C for 48 h gave the desired 6-methyl-6H-indolo[2,3-b]quinolines 139 in 33–65% yields (Scheme 38). The latter step involves the nucleophilic attack of NH2 group on lactam CO group in intermediate 137, followed by the acid-promoted double dehydration of the resulting 5a,6,10b,11-tetrahydro-6-methyl-5H-indolo[2,3-b]quinoline-5a,10b-diols intermediate 138 (Scheme 38).
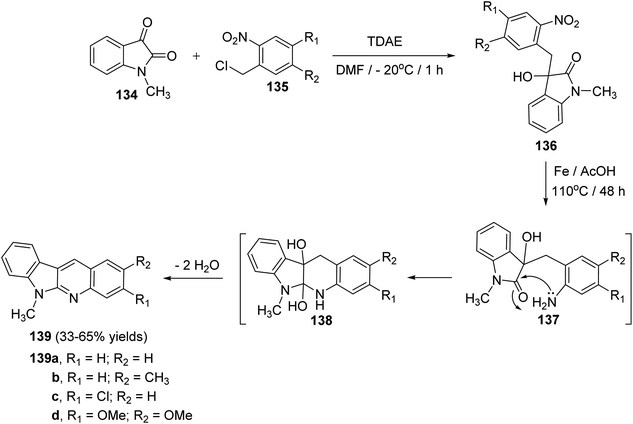 |
| | Scheme 38 Synthesis of 6-methyl-6H-indolo[2,3-b]quinolines 139 via one-pot reduction–cyclization–dehydration reactions of α-hydroxy lactams 136. | |
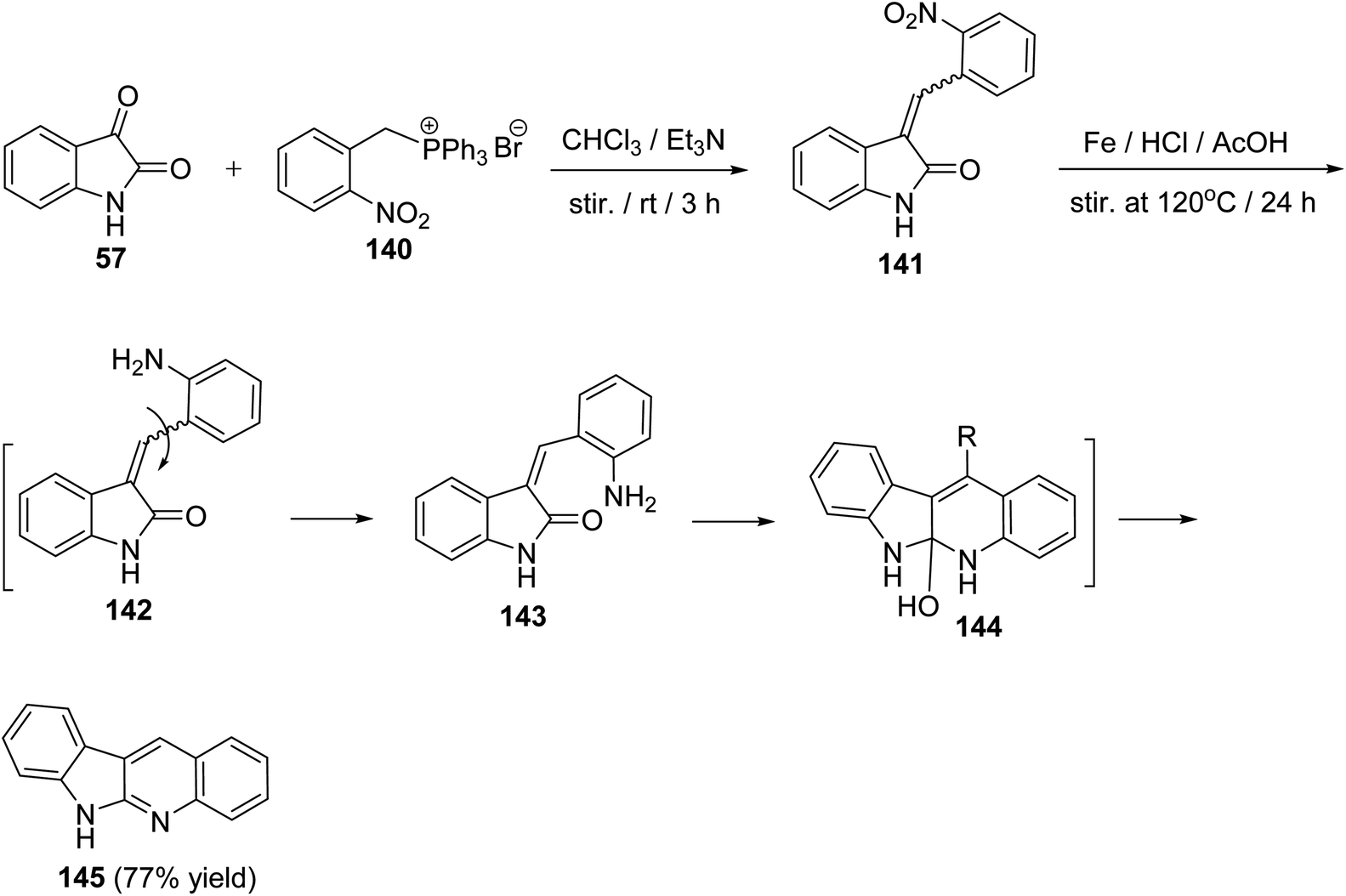
A convenient, short and high yielding approach for the synthesis of 6H-indolo[2,3-b]quinoline (145) is described utilizing Wittig reaction and one-pot reduction–cyclization–dehydration method as the key steps.76 By condensation of isatin (57) with (2-nitrobenzyl)-triphenylphosphonium bromide (140) in CHCl3 in the presence of Et3N at room temperature for 3 h, the corresponding Wittig product, namely 3-(2-nitrobenzylidene)indolin-2-one (141), was formed. Reduction of 141 with Fe/AcOH in the presence of a catalytic amount of HCl at 120 °C for 24 h gave 6H-indolo[2,3-b]quinoline (145) in 77% yield (Scheme 39). In the last step, reduction of NO2 group followed by isomerization of C–C double bond, cyclization and then dehydration took place in one-pot to afford the aromatized tetracyclic product 145 through intermediates 142–144.
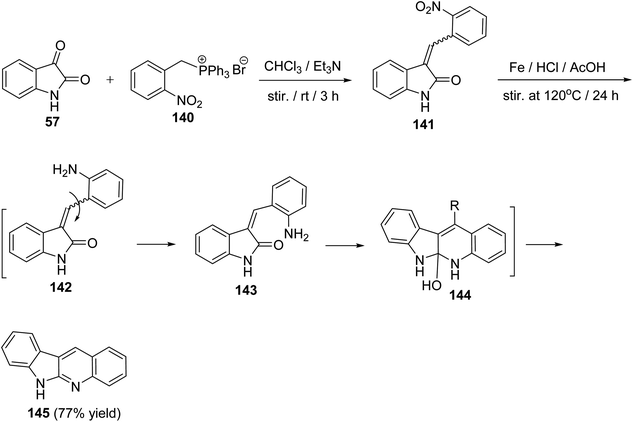 |
| | Scheme 39 A short and convenient method for the synthesis of 6H-indolo[2,3-b]quinoline (145). | |
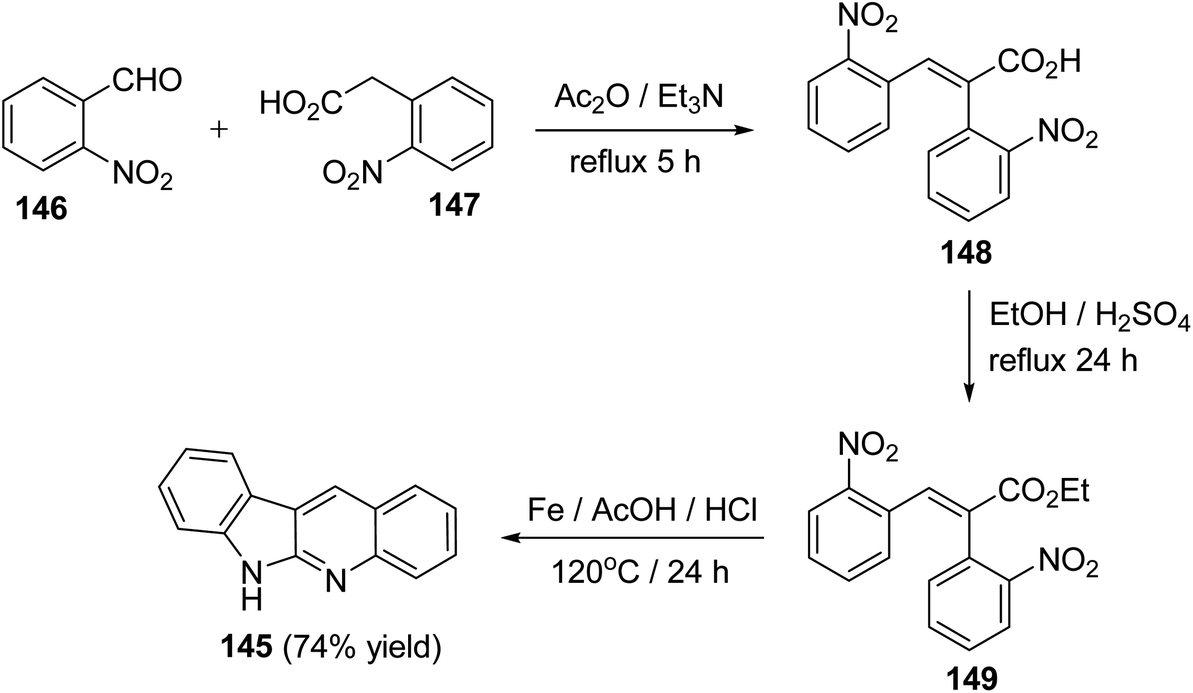
A facile synthesis of 6H-indolo[2,3-b]quinolines 145 utilizing a Perkin reaction followed by a one-pot double reduction, double cyclization and isomerization has been reported by Tilve et al.77 Condensation of o-nitrobenzaldehyde (146) with o-nitrophenyl-acetic acid (147) in the presence of Ac2O and Et3N under reflux for 5 h afforded the corresponding 2,3-bis(2-nitrophenyl)acrylic acid (148), which on esterification gave the ester 149. Reduction of 149 with Fe/AcOH in presence of HCl at 120 °C for 24 h furnished the required 6H-indolo[2,3-b]quinoline (145) in 74% yield (Scheme 40). In this step, four reactions had happened in a tandem manner, that is, reduction of both the NO2 groups, cyclization, isomerization of the intermediate E-amide to the Z-amide followed by a second cyclization.
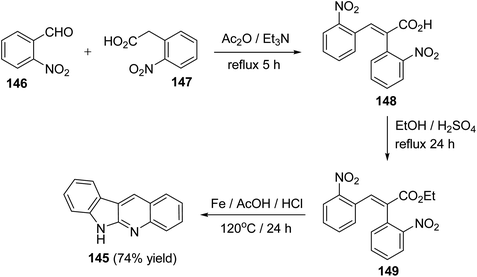 |
| | Scheme 40 A new synthesis of 6H-indolo[2,3-b]quinolines 145. | |
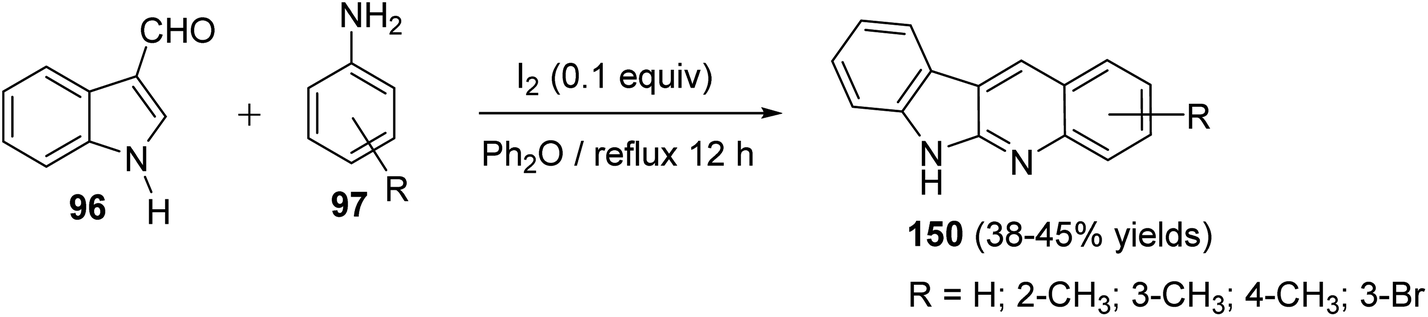
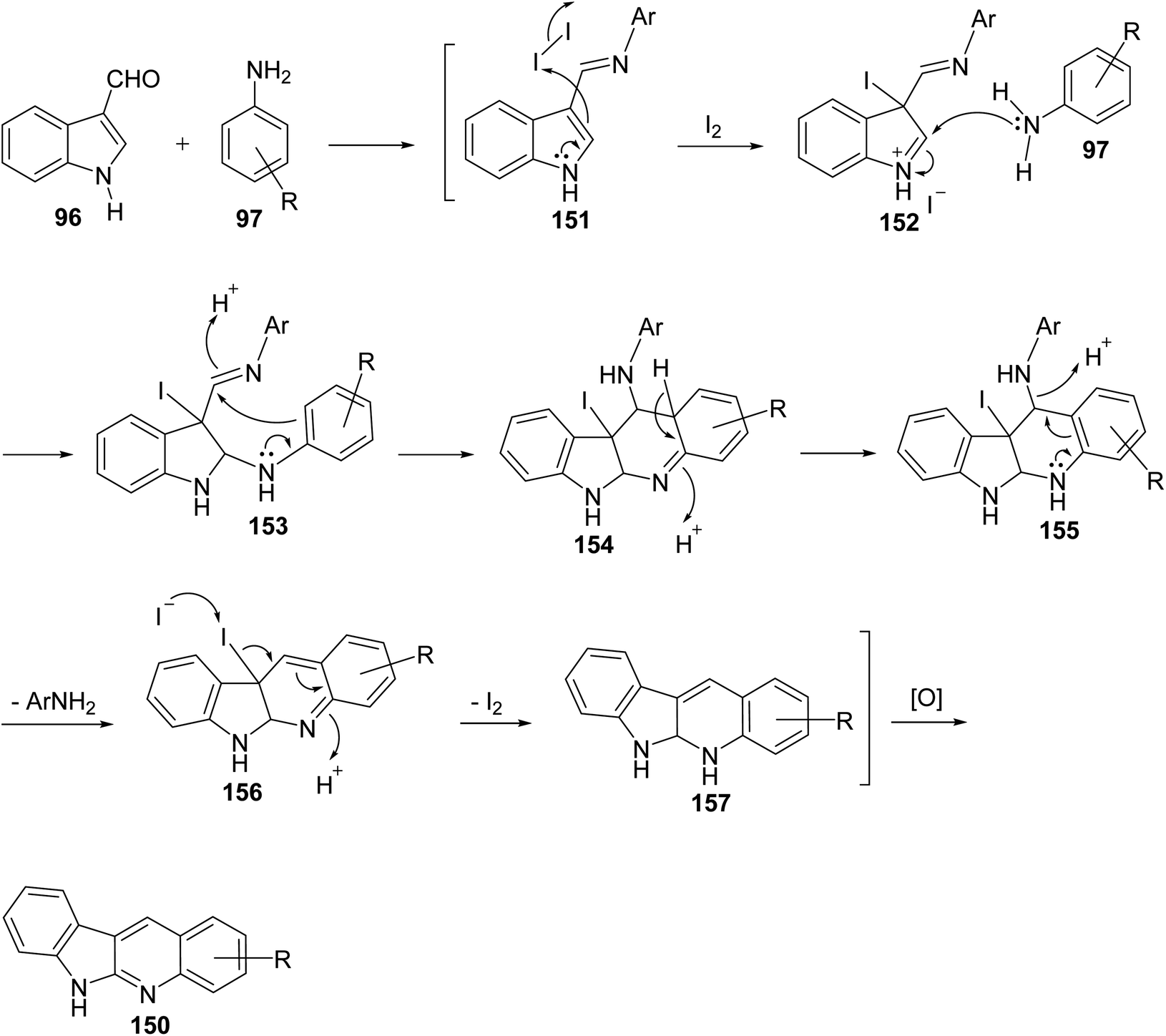
An easy and one-pot procedure for the synthesis of a series of novel 6H-indolo[2,3-b]quinolines 150 with different substituents on quinoline ring was reported by Parvatkar et al.78 This synthetic method involves refluxing of indole-3-carboxyaldehyde (96) (1 equiv.) with aryl amines 97 (2 equiv.) in diphenyl ether in the presence of a catalytic amount of I2 (0.1 equiv.) for 12 h to afford 6H-indolo[2,3-b]quinolines 150 in 38–45% yields (Scheme 41). A suggested mechanism for the formation of 150 is given in Scheme 42.
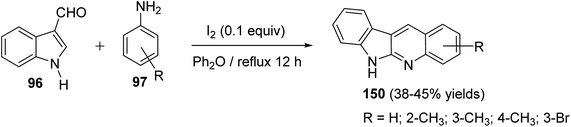 |
| | Scheme 41 A novel and one-pot synthesis of different 6H-indolo[2,3-b]quinolines 150 using I2 as a catalyst. | |
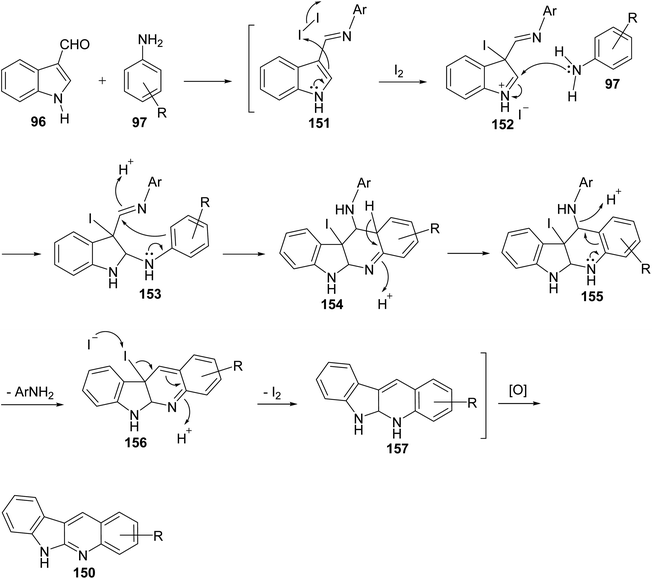 |
| | Scheme 42 Postulated mechanism for the formation of 150. | |
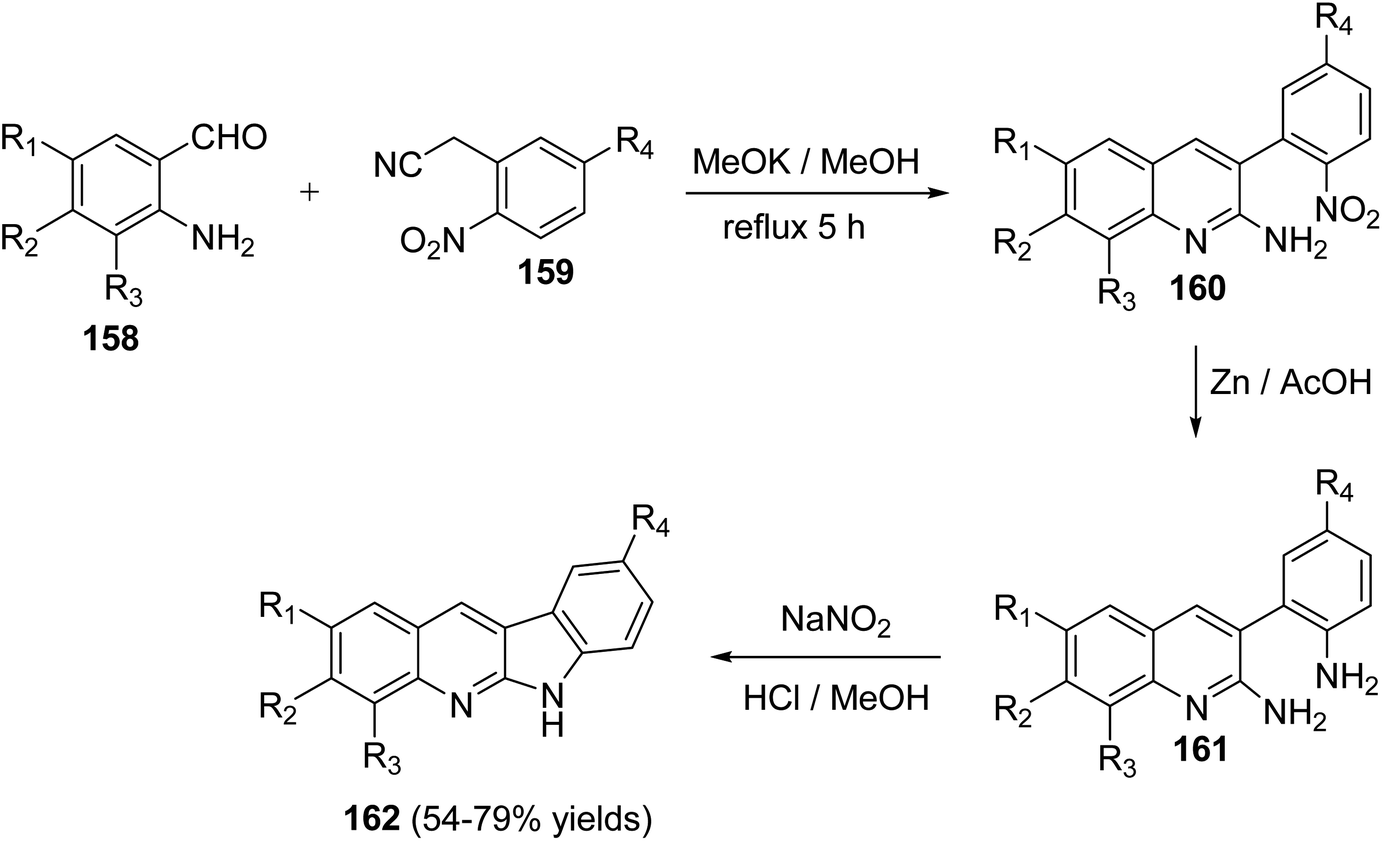
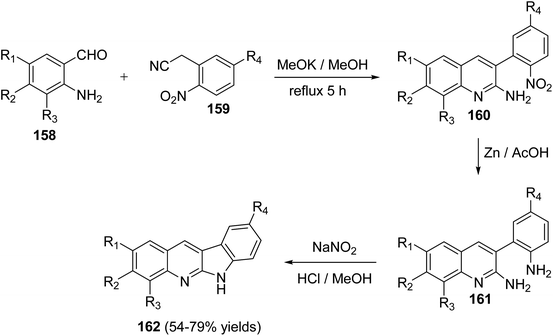
In 2010, Haddadin and his co-workers79 have described a highly efficient, easy and three-step protocol for the synthesis of biologically active 6H-indolo[2,3-b]quinolines 162. An optimized route to 162 is outlined in Scheme 43. Condensation of o-aminobenzaldehydes 158 with o-nitroaryl acetonitriles 159 in refluxing methanolic KOH afforded a series of novel 2-amino-3-(2-nitroaryl)quinolines 160. Reduction of 160 with Zn/AcOH afforded the corresponding 3-(2-aminoaryl)quinolin-2-amines 161. Diazotization of 161 followed by intramolecular cyclization under acidic conditions gave the desired tetracyclic 6H-indolo- [2,3-b]quinolines 162 in 54–79% yields (Scheme 43).
 |
| | Scheme 43 Route toward the synthesis of 162. | |
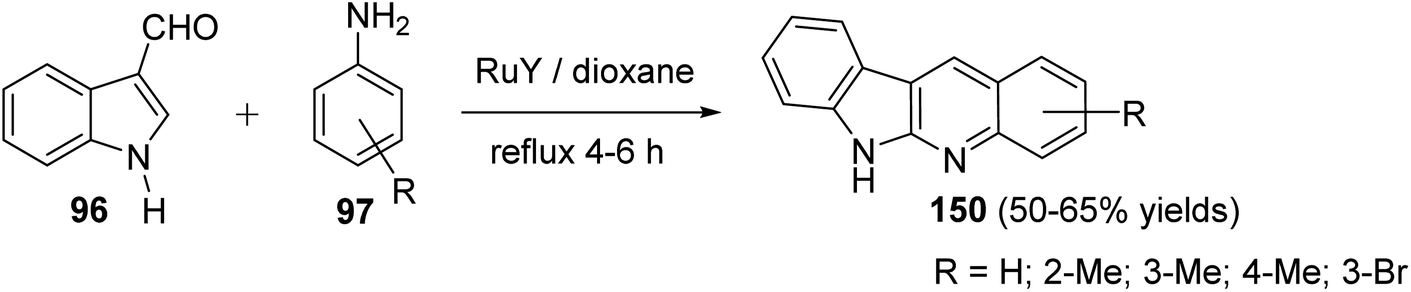
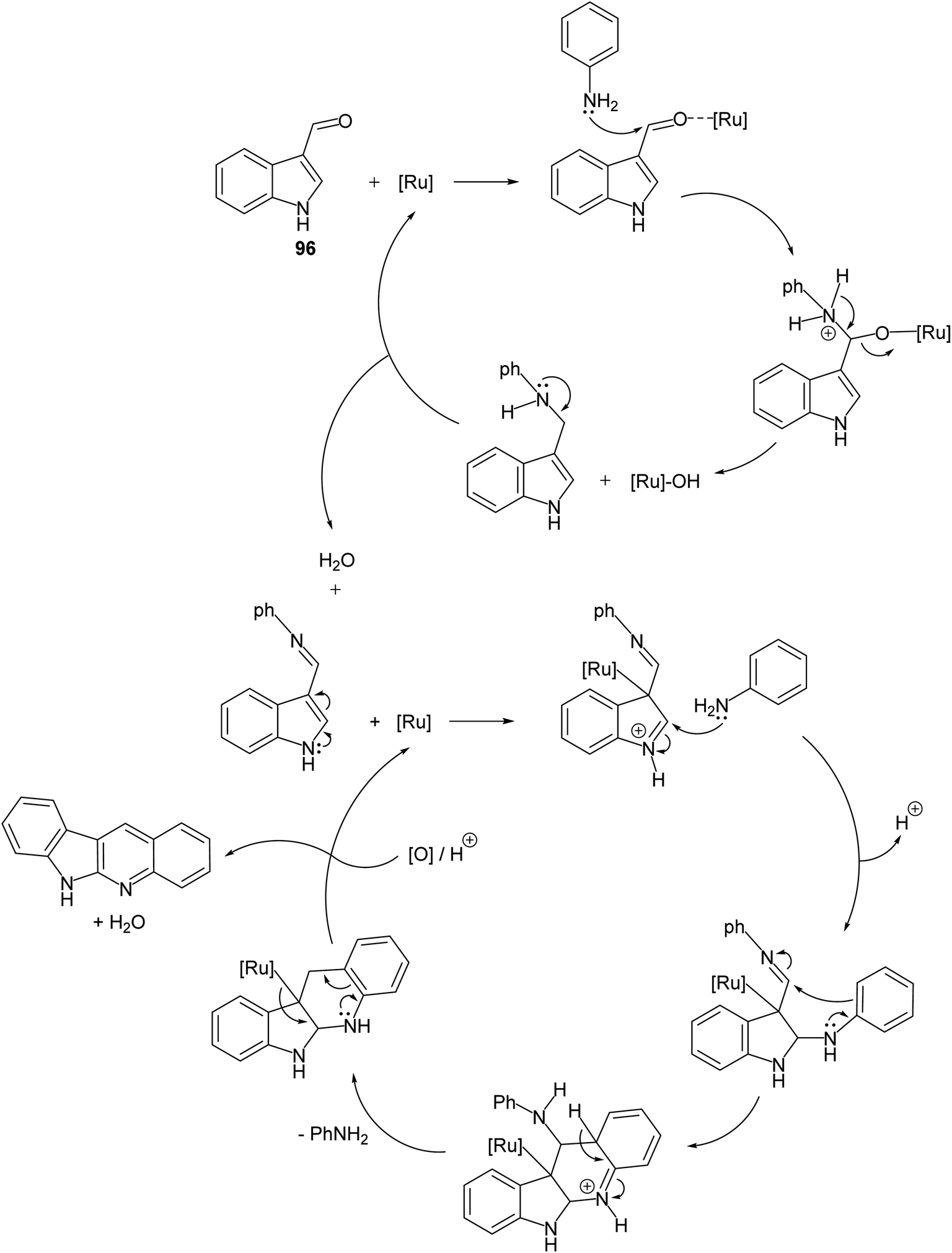
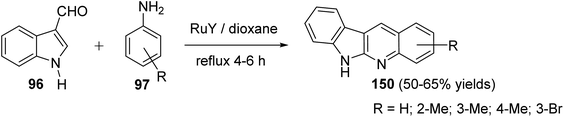
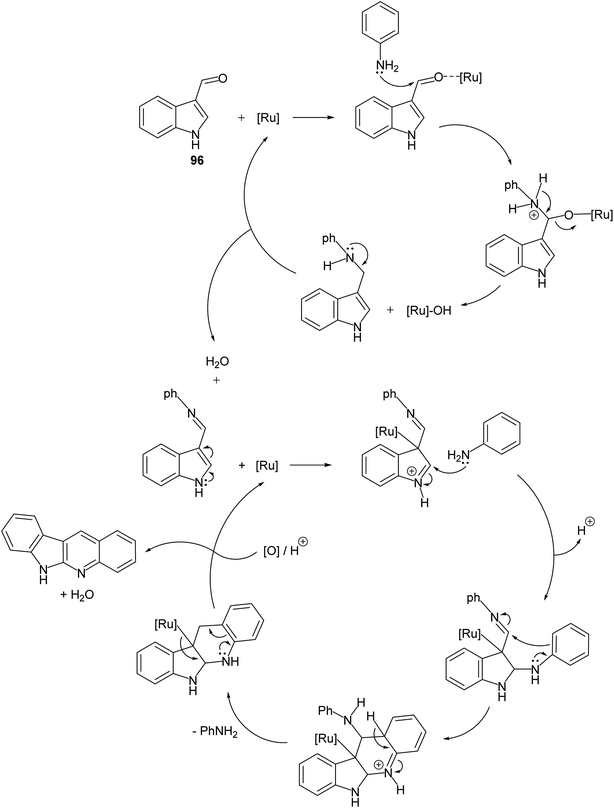
A convenient method for preparation of 6H-indolo[2,3-b]quinolines 150 utilizing ruthenium-exchanged FAU-Y zeolite (RuY) as a recyclable heterogeneous catalyst is reported.80 Reaction of indole-3-carbaldehyde (96) (1 equiv.) with aryl amines 97 (1 equiv.) in refluxing dioxane in the presence of Ru3+ exchanged FAU-Y zeolite (RuY, 0.1 g, activated at 550 °C for 3 h) for 4–6 h gave 6H-indolo[2,3-b]quinolines 150 in 50–65% isolated yields (Scheme 44). A mechanism for the RuY catalyzed formation of the 6H-indolo[2,3-b]-quinolines 150 is outlined in Scheme 45.
 |
| | Scheme 44 RuY catalyzed synthesis of 6H-indolo[2,3-b]quinolines 150. | |
 |
| | Scheme 45 Proposed mechanistic pathway for the RuY catalyzed formation of 6H-indolo- [2,3-b]quinoline 150. | |
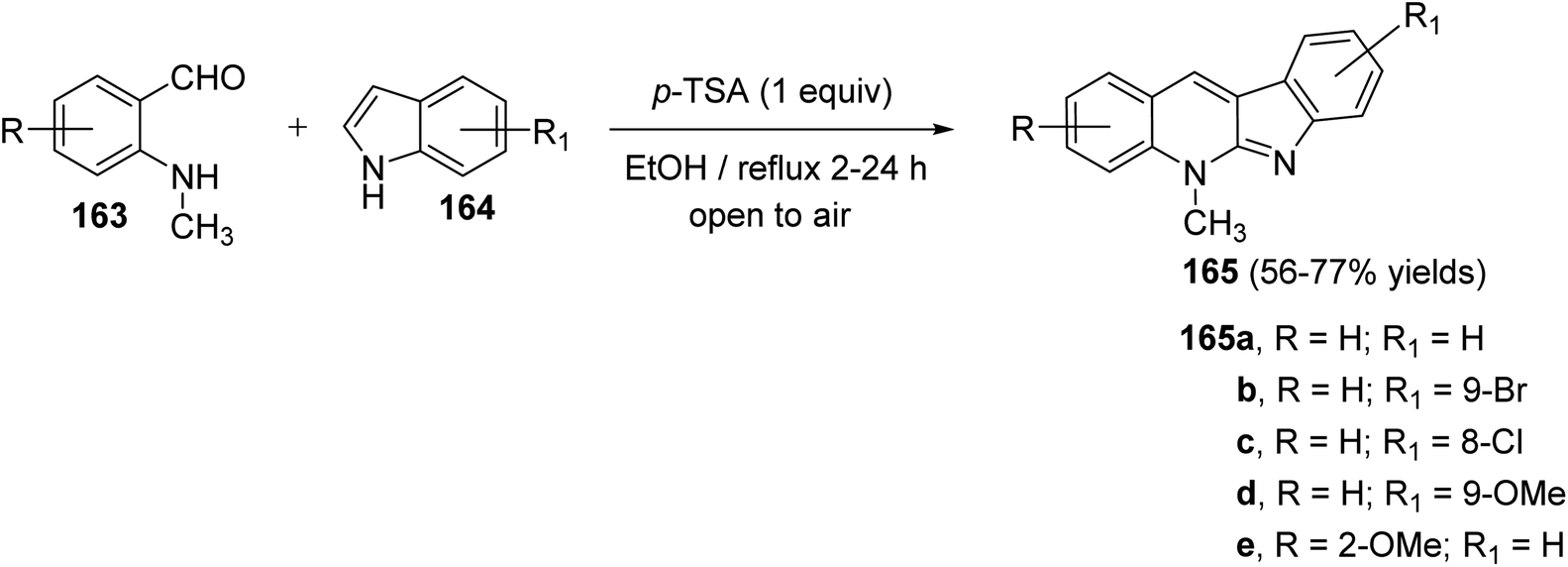
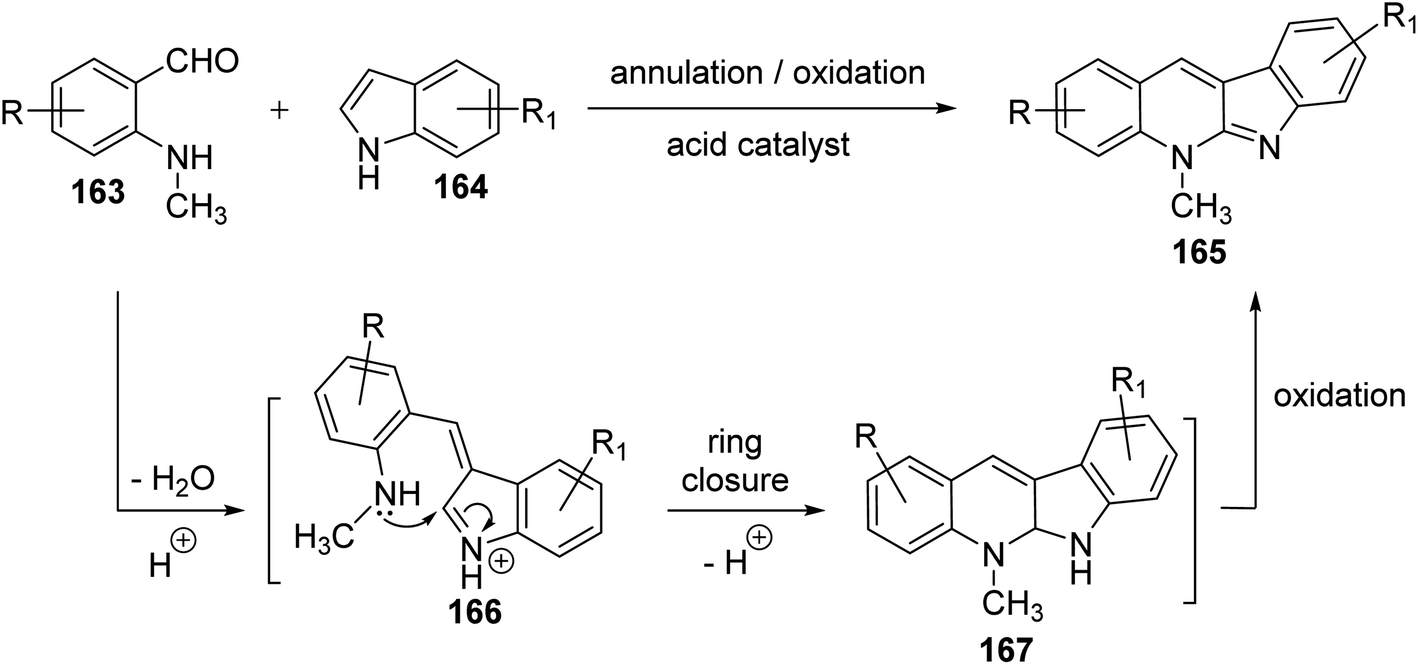
Seidel et al.81 have described a new acid-catalyzed indole annulation with secondary aminobenzaldehydes 163 for the rapid and one-step synthesis of neocryptolepine and various analogues, a bioactive materials as promising lead for new antimalarial agents. Condensation of 163 with indoles 164 in the presence of one equivalent of para-toluene sulfonic acid (p-TSA) in EtOH under reflux gave neocryptolepine analogues (5-methyl-5H-indolo[2,3-b]-quinolines) 165 in 56–77% yields (Scheme 46). Condensation of 163 with 164 is expected to form azafulvenium ion 166 which undergoes direct ring-closure to furnish 167. Subsequent oxidation of 167 completes the synthesis of the desired products 165 (Scheme 47). Interestingly, the oxidized products 165 were obtained directly and the putative intermediate 167 was not isolated, presumably because it undergoes facile oxidation by air.
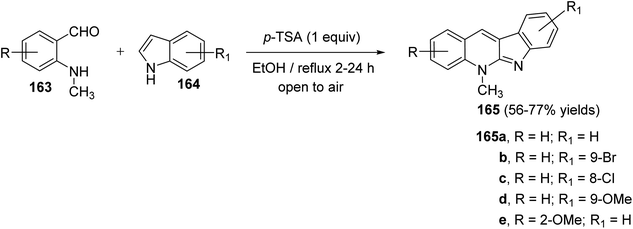 |
| | Scheme 46 One-step synthesis of 5-methyls-5H-indolo[2,3-b]quinolines 165. | |
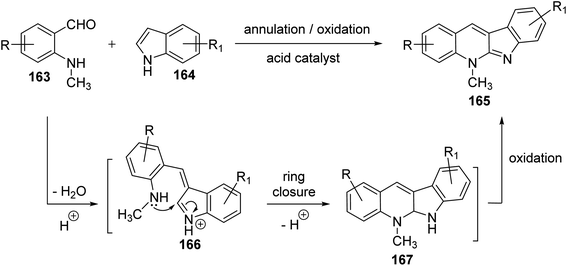 |
| | Scheme 47 Proposed synthesis of 5-methyl-5H-indolo[2,3-b]quinolines 165. | |
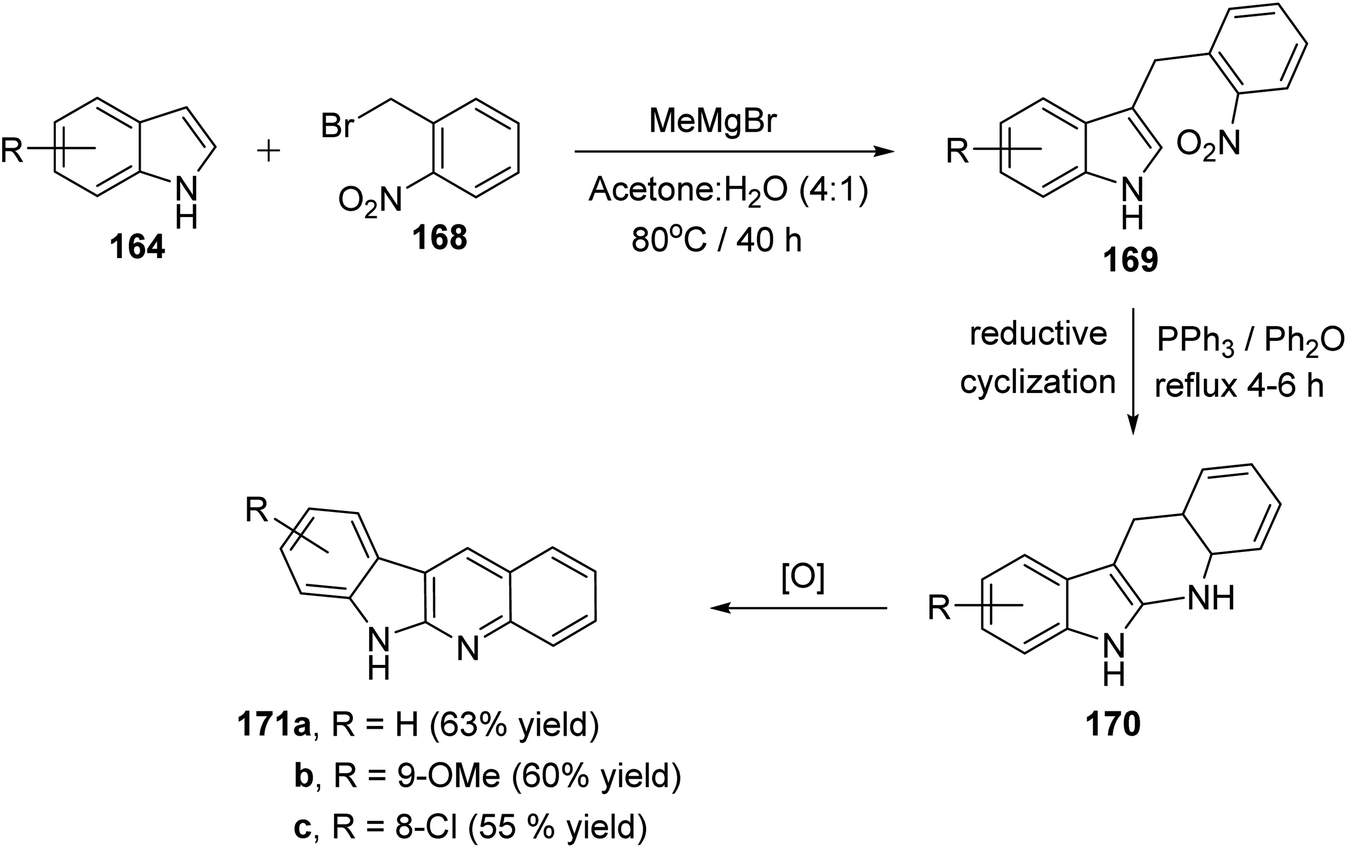
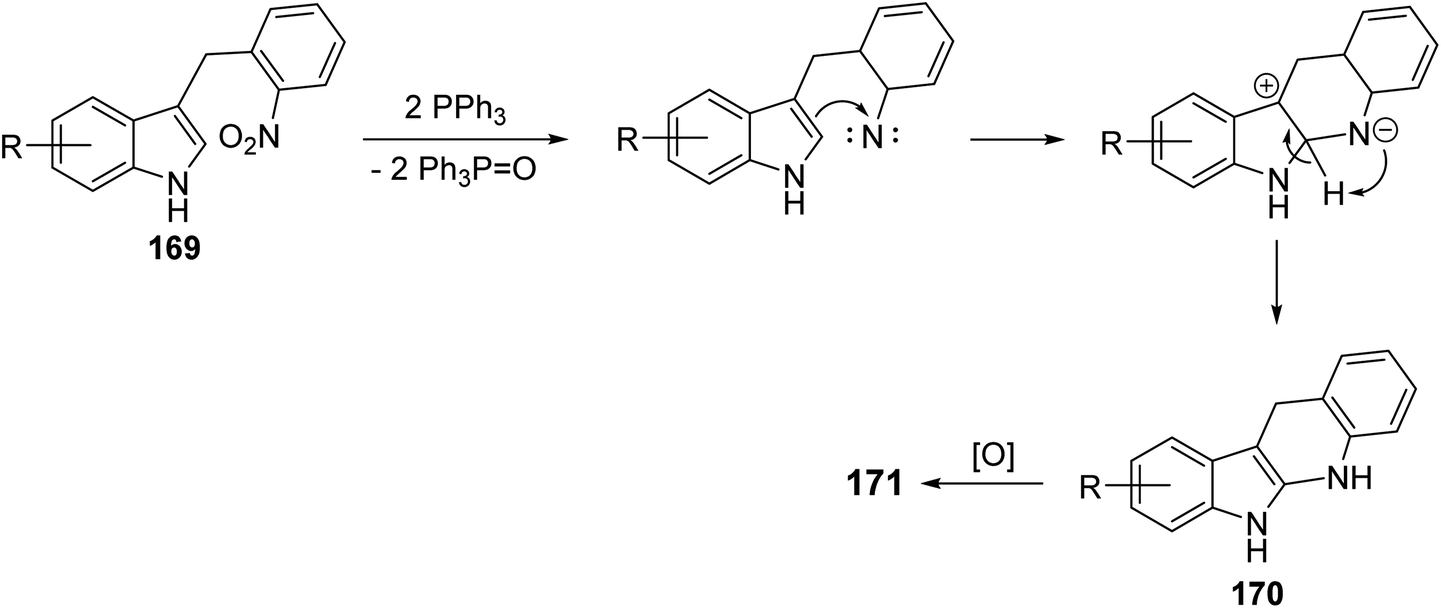
A simple and concise two-step approach for the synthesis of 6H-indolo[2,3-b]-quinolines 171 is reported using an alkylation of 1H-indoles 164 at its more reactive 3-position and domino reduction–intramolecular cyclization–aromatization approach.82 Alkylation of indoles 164 was performed using 2-nitrobenzyl bromide (168) in the presence of methylmagnesium bromide (MeMgBr), as the base, in a mixture of acetone–H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 80 °C for 40 h to yield 3-(2-nitrobenzyl)-1H-indoles 169. Reductive cyclization of 169 with triphenylphosphine in refluxing diphenyl ether for 4–6 h provided the tetracyclic compound 170, which underwent oxidation followed by aromatization under reaction conditions to give the desired products 171 in 55–63% yields (Scheme 48). A suggested mechanism for the formation of 171 is depicted in Scheme 49.
:1) at 80 °C for 40 h to yield 3-(2-nitrobenzyl)-1H-indoles 169. Reductive cyclization of 169 with triphenylphosphine in refluxing diphenyl ether for 4–6 h provided the tetracyclic compound 170, which underwent oxidation followed by aromatization under reaction conditions to give the desired products 171 in 55–63% yields (Scheme 48). A suggested mechanism for the formation of 171 is depicted in Scheme 49.
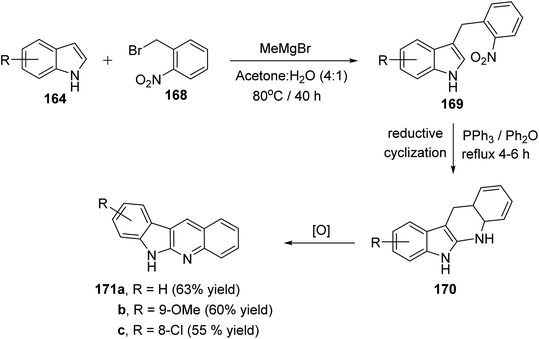 |
| | Scheme 48 A concise synthesis of 6H-indolo[2,3-b]quinolines 171. | |
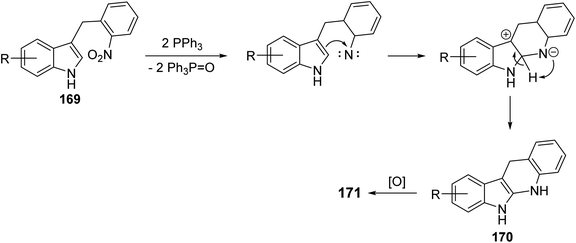 |
| | Scheme 49 Proposed mechanism for the formation of 6H-indolo[2,3-b]quinolines 171. | |
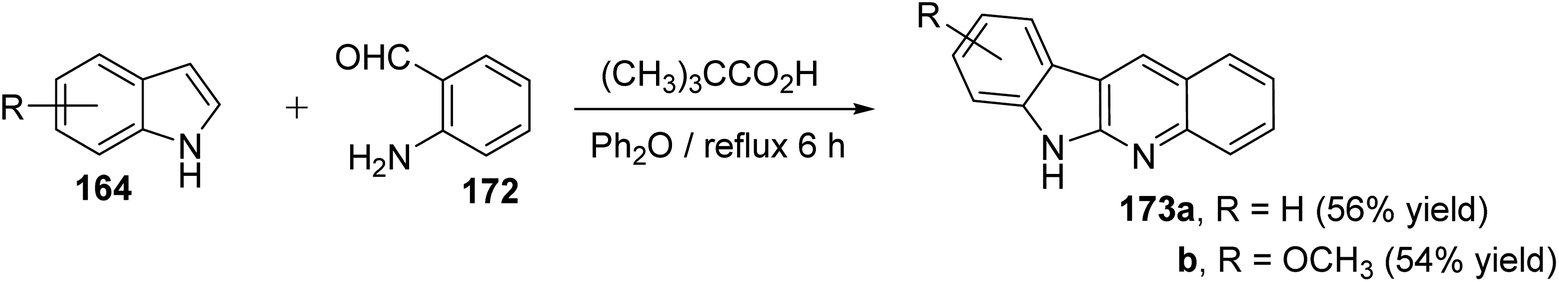
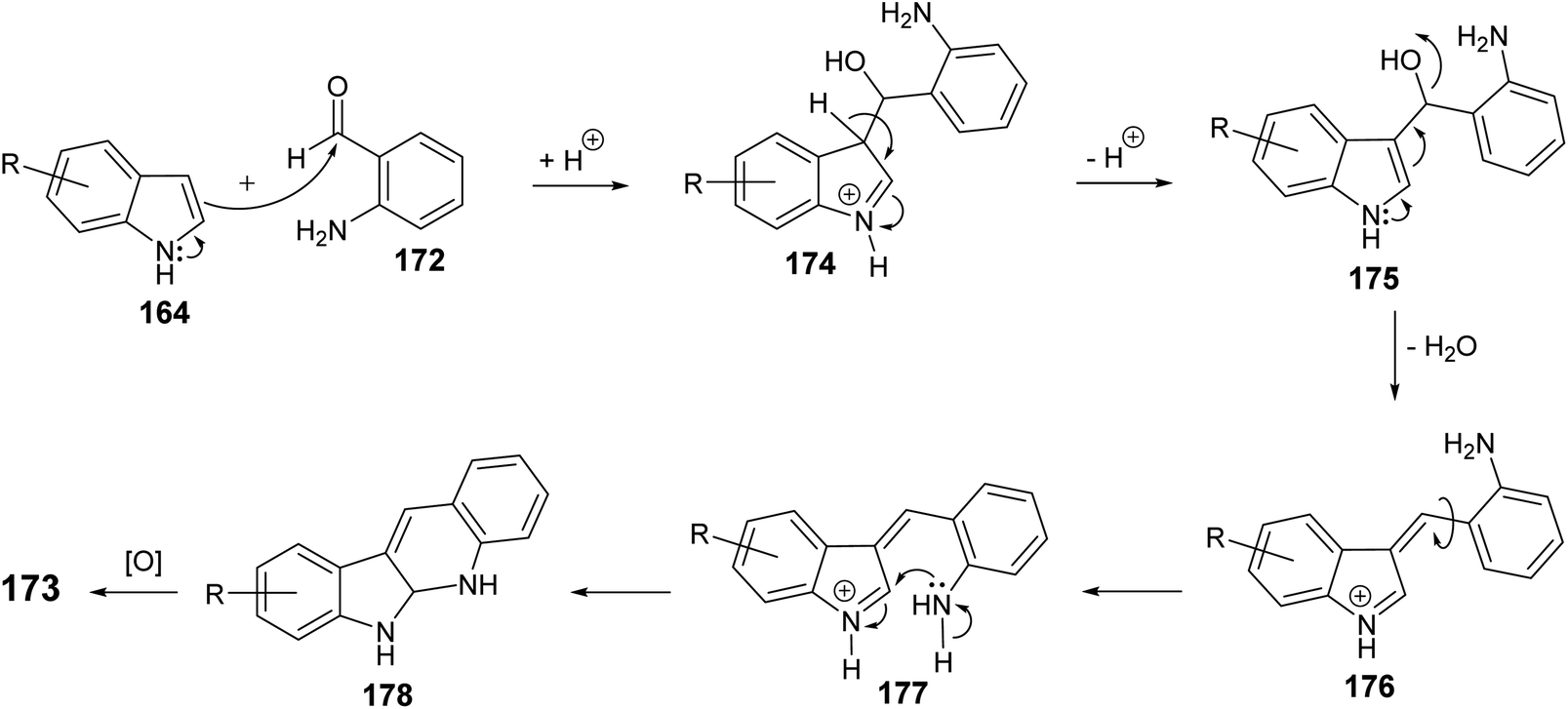
An efficient and straightforward synthesis of 6H-indolo[2,3-b]quinolines 173 in one-pot by alkylation–dehydration–cyclization–aromatization method utilizing pivalic acid with indoles 164 and o-aminobenzaldehyde (172) is described by Kadam and Tilve.83 Thus, refluxing 164 with 172 in the presence of pivalic acid in Ph2O for 6 h gave the tetracyclic 6H-indolo[2,3-b]quinolines 173 (Scheme 50). In the proposed reaction mechanism, the alkylation of 164 with 172, followed by dehydration and cyclization gives dihydroindolo-quinoline intermediate 178, which oxidizes under reaction conditions to furnish the final product indoloquinolines 173 (Scheme 51).
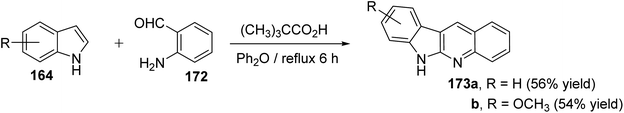 |
| | Scheme 50 A pivalic-acid assisted one-pot alkylation–dehydration–cyclization–aromatization method for the synthesis of 6H-indolo[2,3-b]quinolines 173. | |
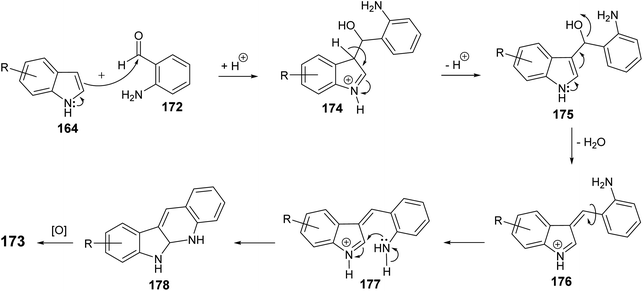 |
| | Scheme 51 A plausible mechanism for the synthesis of 6H-indolo[2,3-b]quinolines 173. | |
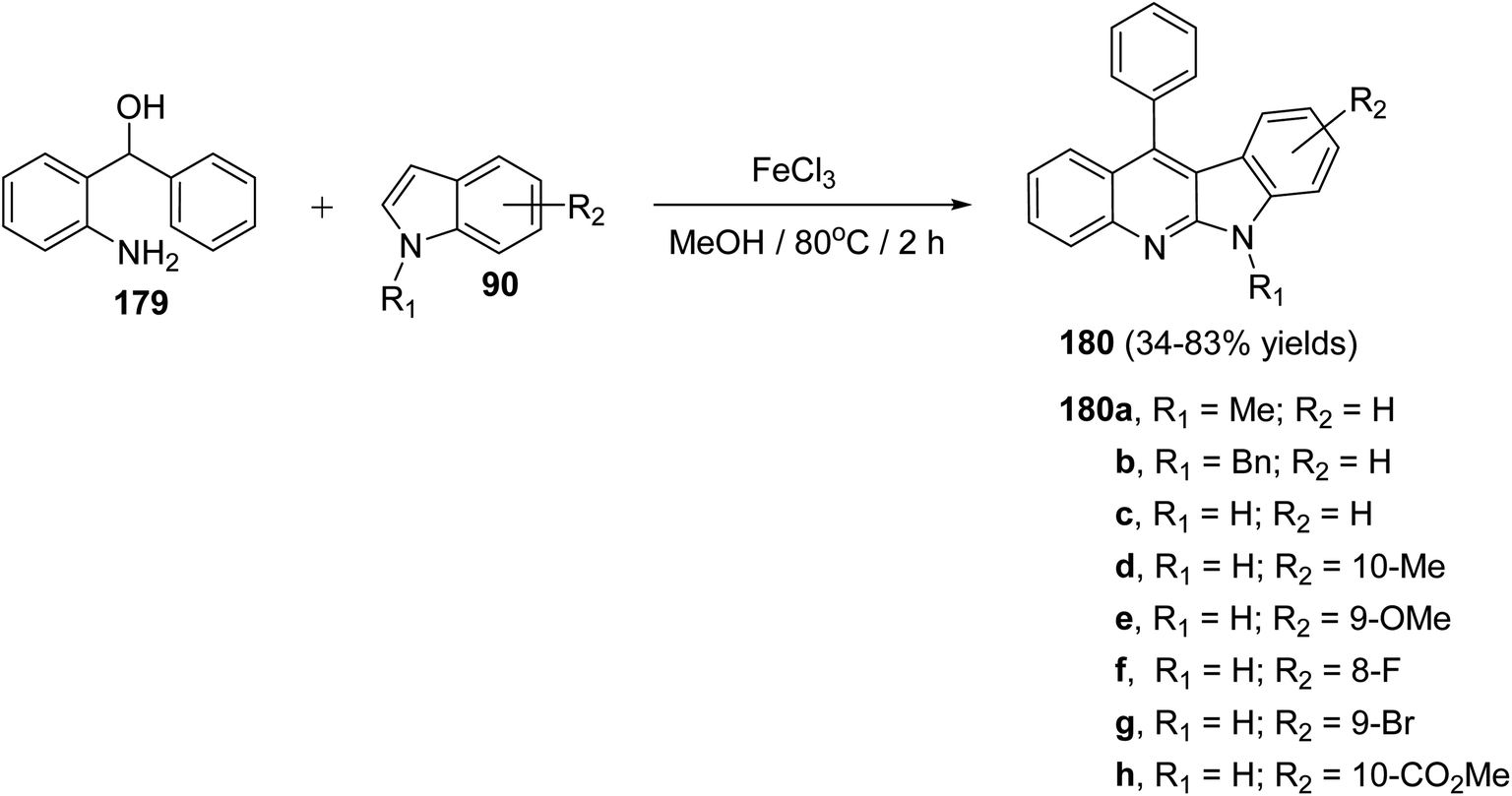
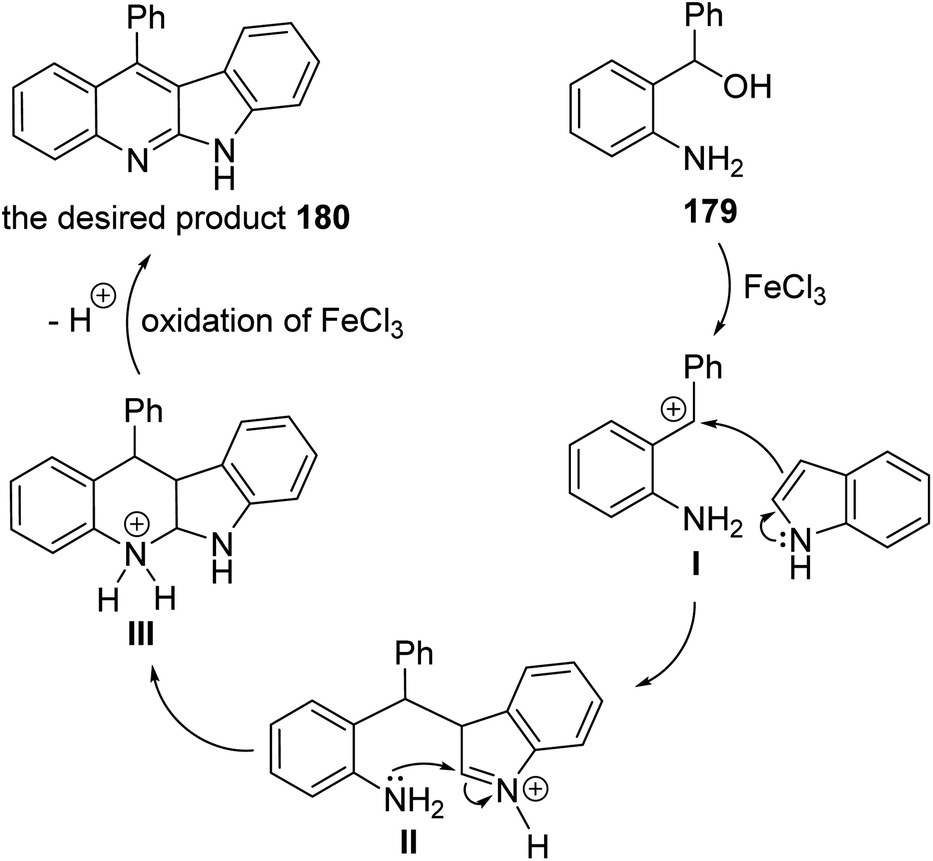
A facile and iron-mediated synthesis of 6H-indolo[2,3-b]quinolines 180 by reaction of 2-amino-α-phenylbenzene methanol 179 and various indoles 90 was developed by Wang et al.84 This synthetic approach proceeded by heating a mixture of 179 (1.5 equiv.), 90 (1 equiv.) and FeCl3 (2.5 equiv.) in MeOH at 80 °C for 2 h (Scheme 52). In the present study, the target products have been prepared through a new approach in 34–83% yields. When the metal salt was replaced with FeCl2 or absent, the desired products 180 were not obtained. This indicated that FeCl3 played the role of an oxidant in the process of reaction rather than the function of catalysis. The authors also studied the influence of electronic effects on these reactions. The indoles bearing electron-withdrawing groups (EWG) on the benzene ring (R2) seemed to be less efficient than electron-donating groups (EDG) and resulted in low yields. A mechanism for the iron-promoted synthesis of 6H-indolo[2,3-b]quinolines 180 is proposed as shown in Scheme 53. Firstly, FeCl3 may activate 179 to give intermediate I, which undergoes the Friedel–Crafts reaction to furnish intermediate II. The latter intermediate II undergoes intramolecular cyclization to afford intermediate III, which subsequently undergoes deprotonation and oxidation by FeCl3 to afford the desired product 180.
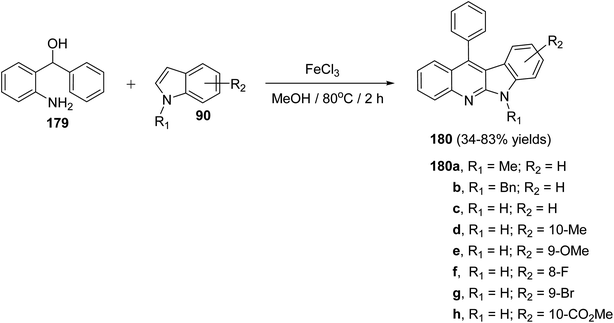 |
| | Scheme 52 Iron-promoted synthesis of 6H-indolo[2,3-b]quinolines 180 from the reaction of 179 with different indoles 90. | |
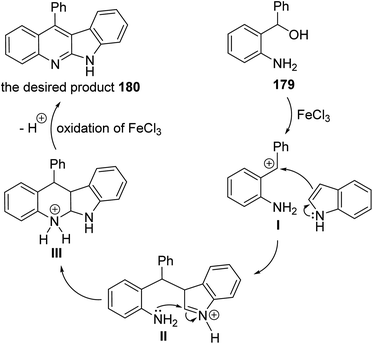 |
| | Scheme 53 Postulated mechanism for the construction of indolo[2,3-b]quinolines 180 from 179 and 90. | |
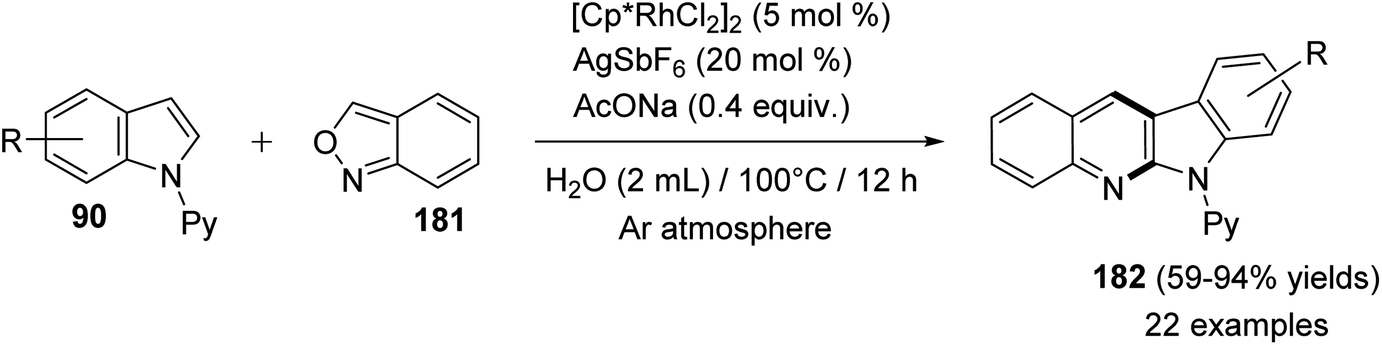
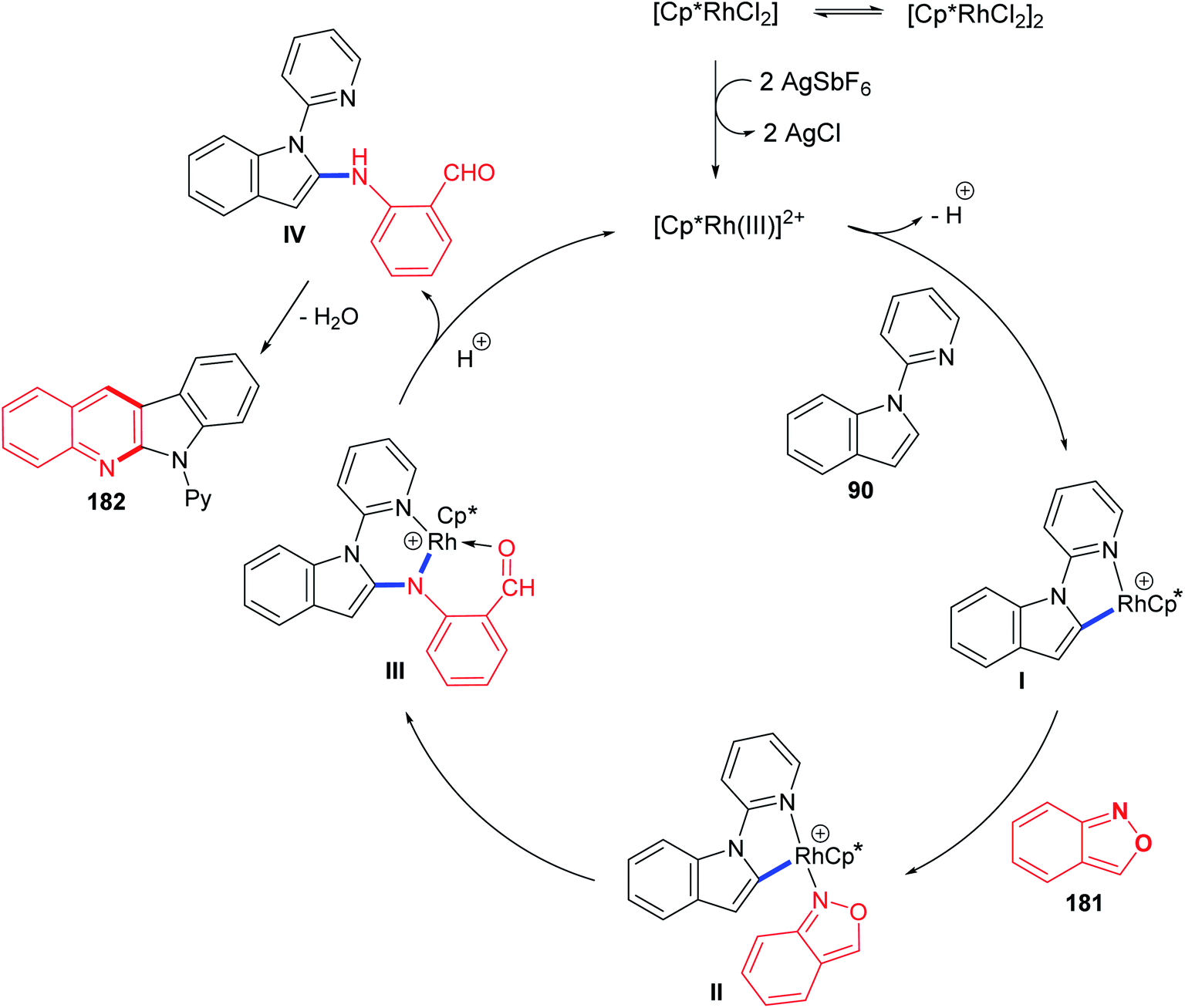
In 2016, Shi and Wang85 have developed an environmentally friendly and efficient Rh(III)-catalyzed synthetic approach for 6H-indolo[2,3-b]quinolines 182 from various indoles 90 and benzo[c]isoxazole (181) using H2O as an efficient solvent. This annulation procedure undergoes tandem C–H activation, cyclization, and condensation steps. The reaction was carried out by reacting 1-(pyridin-2-yl)-1H-indoles 90 (1.0 equiv.) with 181 (1.5 equiv.) in the presence of a catalytic amount of [Cp*RhCl2]2 (5 mol%), AgSbF6 (20 mol%) and AcONa (0.4 equiv.) in H2O at 100 °C under Ar atmosphere for 12 h. The methodology furnished the desired tetracyclic 182 in 59–94% yields (Scheme 54). A control experiment showed that both [Cp*RhCl2]2 and AgSbF6 were essential catalyst system for this transformation as their omission led to no formation of tetracyclic product 182. In this work, the authors investigated the effect of additives in the present reaction and they have noted that the use of AcONa gave the best result as the yields of products 182 were increased. The mechanistic pathway for the reaction of 90 with 181, using Rh(III) as a catalyst, is outlined in Scheme 55. First, indoles 90 reacted with Cp*Rh(III) via directed C–H cleavage to give intermediate I. The coordination of 181 to I yields intermediate II. Subsequently, the migration insertion of the coordinated 181 into the Rh–C bond gives intermediate III. Protonation of III provides the further intermediate IV and releases the Rh(III) species for the next catalytic cycle. Finally, the latter intermediate IV undergoes intramolecular cyclization via elimination of one molecule of H2O to afford the final product 182.
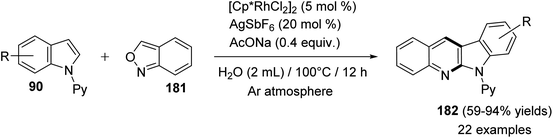 |
| | Scheme 54 Rh(III)-catalyzed C–H amination/annulation of substituted indole derivatives 90 with 181. | |
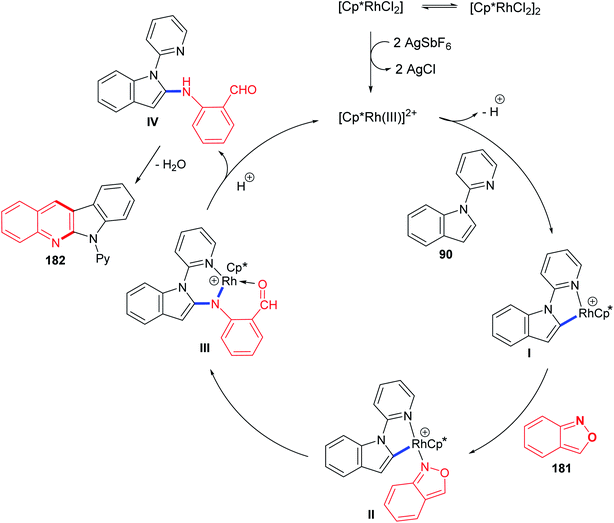 |
| | Scheme 55 Plausible catalytic cycle. | |
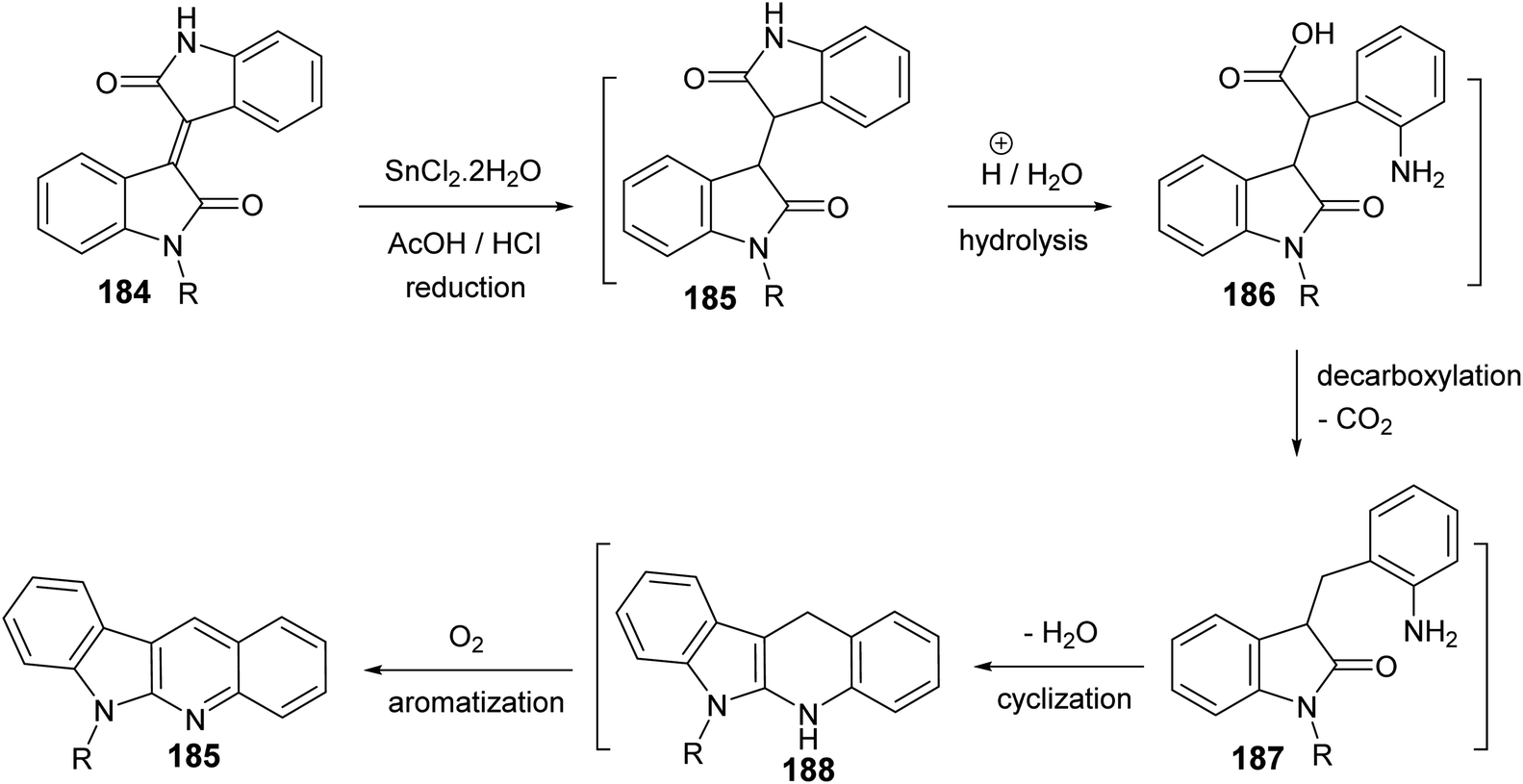
Yin et al.86 developed a facile and efficient method for the synthesis of 6-substituted 6H-indolo[2,3-b]quinolines 185 from isoindigo derivatives 184, prepared from isatins 57 and indolin-2-ones 183, in the presence of SnCl2, in combination with AcOH/HCl under heating at 120 °C for 12–24 h in moderate yields (60–74% yields) (Scheme 56). Pyrrole and pyridine rings are synchronously constructed in one-pot for these tetracyclic products. A possible reaction mechanism for the formation of 185 is presented as shown in Scheme 57. First, reduction of the carbon–carbon double bond in 184 by SnCl2 in an acid medium affords the saturated 1,4-diketone intermediate 185. Hydrolysis of the amide bond of 185, followed by a decarboxylation yields the further intermediate 187. Subsequent intramolecular cyclization/aromatization furnishes the target tetracyclic molecule 185 via intermediate 188.
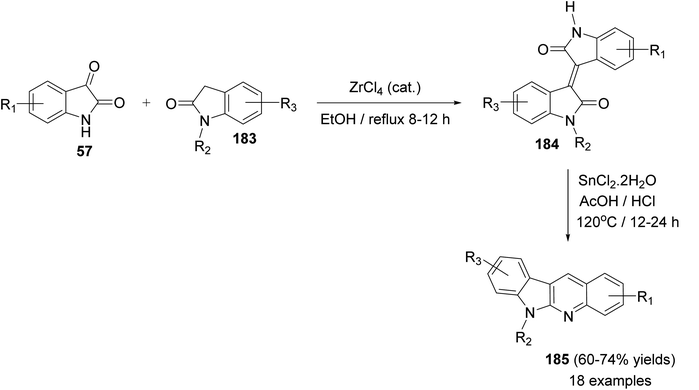 |
| | Scheme 56 Synthesis of 6-substituted-6H-indolo[2,3-b]quinolines 185 from isoindigo derivatives 184 in the presence of SnCl2 in acidic media. | |
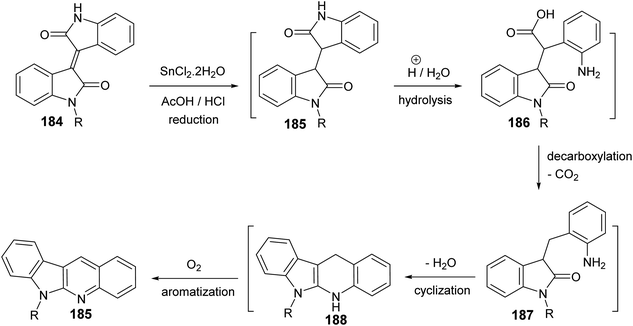 |
| | Scheme 57 A possible reaction mechanism for the formation of 6-substituted-6H-indolo-[2,3-b]quinolines 185. | |
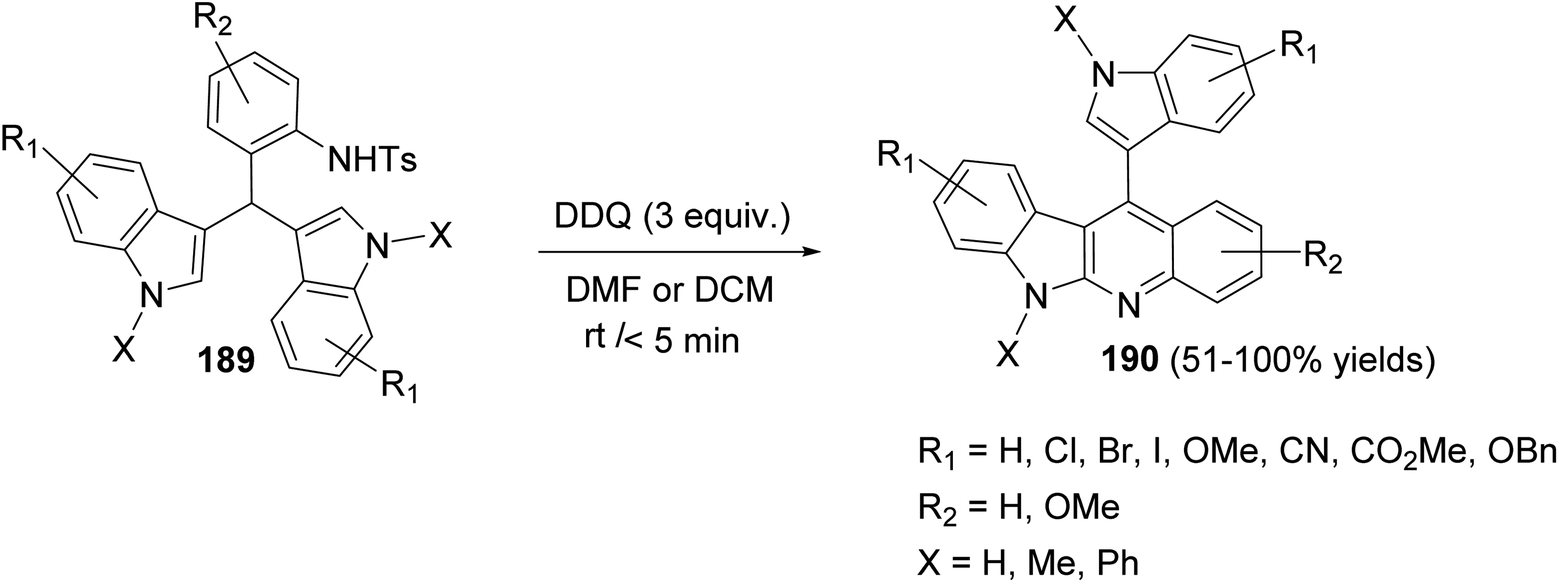
In 2017, Challa et al.87 reported an efficient metal-free method for the synthesis of indolo[2,3-b]quinolines 190 under DDQ-mediated oxidative conditions from the easily accessible 3,3′-diindolylmethanes (DIMs). Treatment of 3,3′-diindolylphenylmethanes (DIPMs) 189 with DDQ (3 equiv.) at room temperature for <5 min using DCM or DMF as the solvent afforded the desired indolo[2,3-b]quinolines 190 in 51–100% yields (Scheme 58).
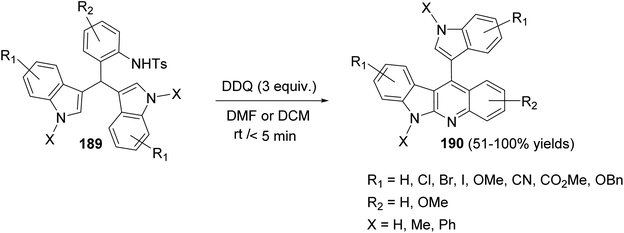 |
| | Scheme 58 Scope of DDQ-mediated intramolecular C2–N bond formation for the synthesis of indolo[2,3-b]quinolines 190. | |
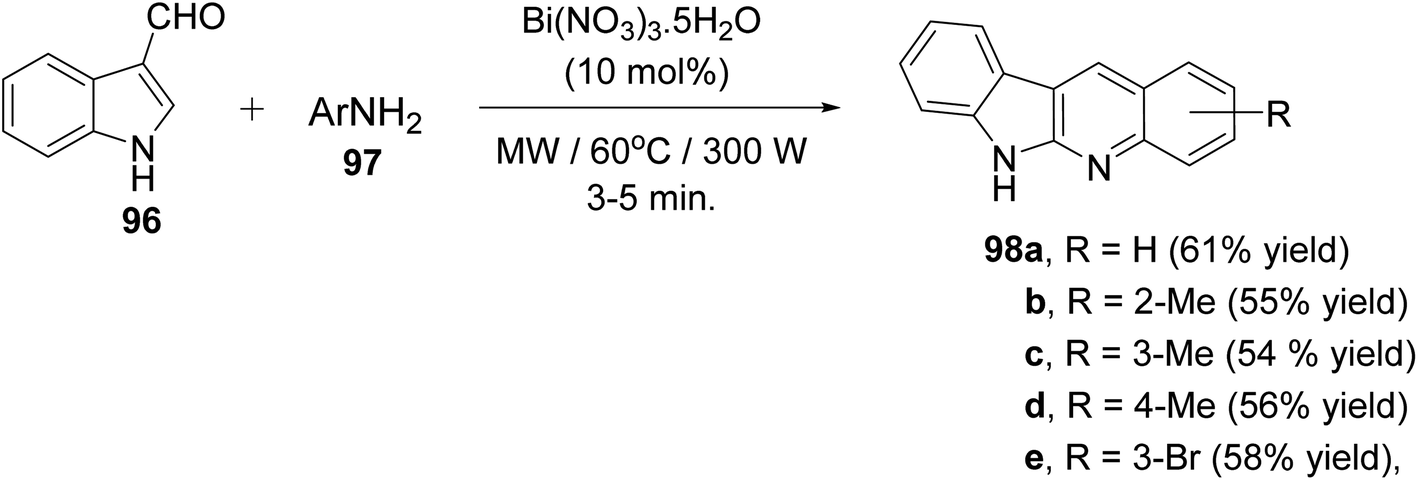
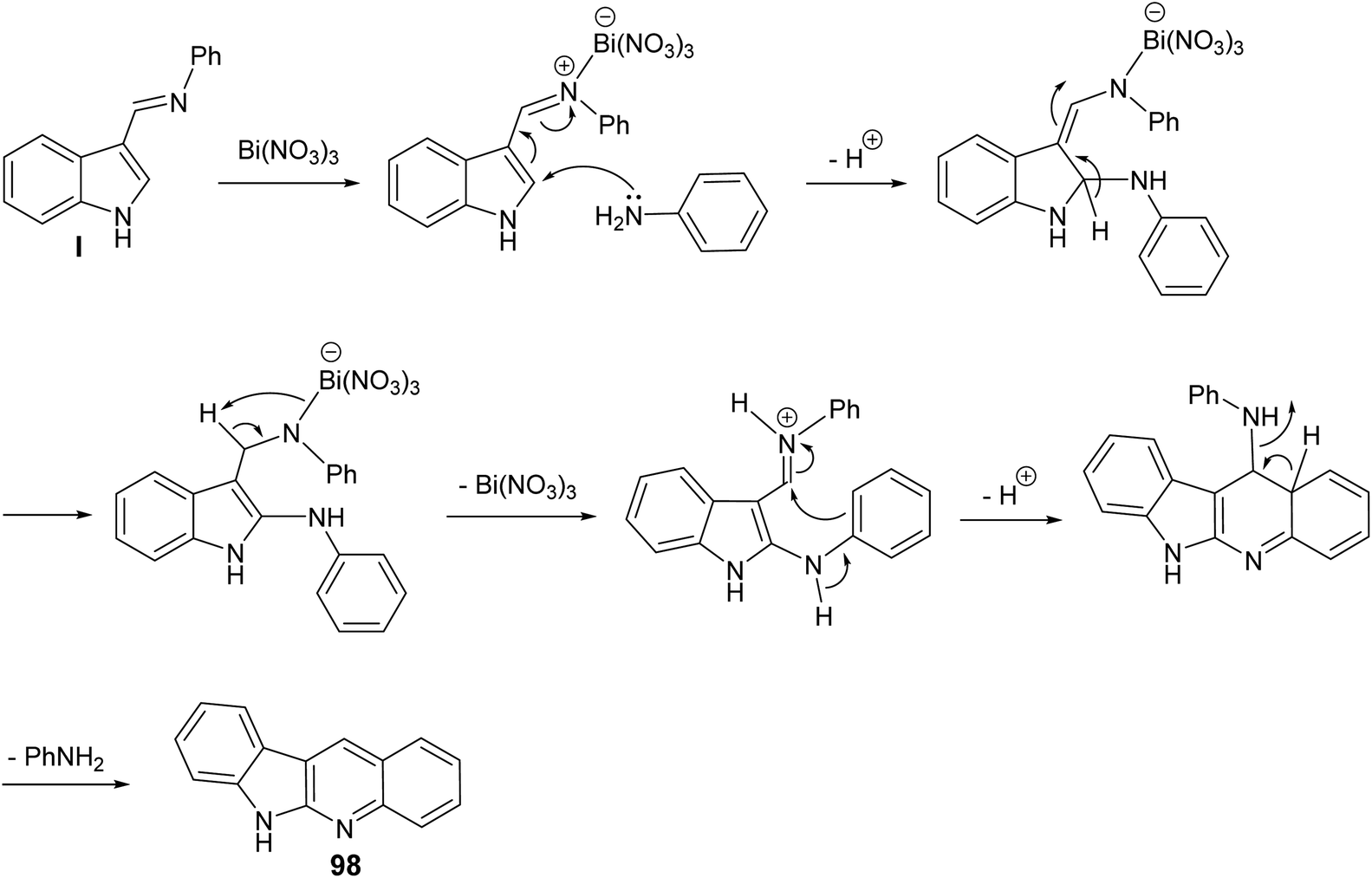
A clean and expeditious microwave-mediated one-pot methodology for the synthesis of a series of 6H-indolo[2,3-b]quinolines 98 using eco- and user-friendly bismuth-nitrate [Bi(NO3)3·5H2O] as a catalyst under mild reaction conditions is reported.88 A mixture of indole-3-carboxyaldehyde 96 (1 equiv.), aryl amines 97 (2 equiv.) and Bi(NO3)3·5H2O (10 mol%, 0.1 equiv.) in tightly sealed vessel under solvent-free condition were irradiated in microwave at 60 °C (300 W) for 3–5 min to provide different linear 6H-indolo[2,3-b]quinolines 98 in good yields (52–61% yields) (Scheme 59). Aryl amines containing electron-withdrawing or electron-donating groups and heteroaryl moiety are compatible with these reaction conditions yielding the desired tetracyclic products in good yields within short period of time. In contrast to all available approaches, this is the fastest method reported so far for the synthesis of linear indolo[2,3-b]quinolines. A proposed mechanism for the Bi(III)-catalyzed for the formation of the indolo[2,3-b]quinolines 98 is given in Scheme 60.
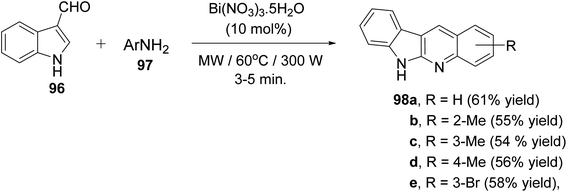 |
| | Scheme 59 Microwave-mediated Bi(NO3)3-catalyzed synthesis of indolo[2,3-b]quinolines 98. | |
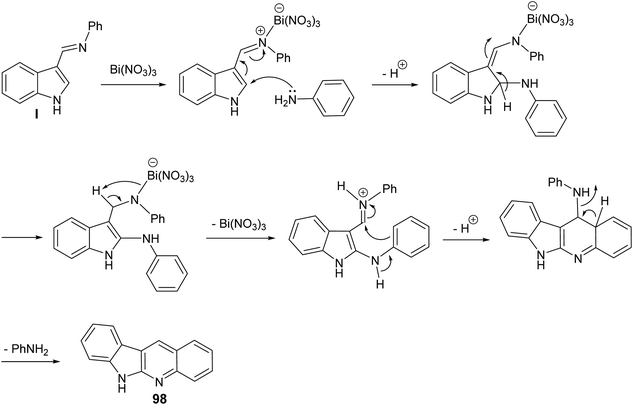 |
| | Scheme 60 Plausible reaction mechanism for Bi(III)-catalyzed synthesis of indolo[2,3-b]-quinolines 98 through activation of the in situ formed imine. | |
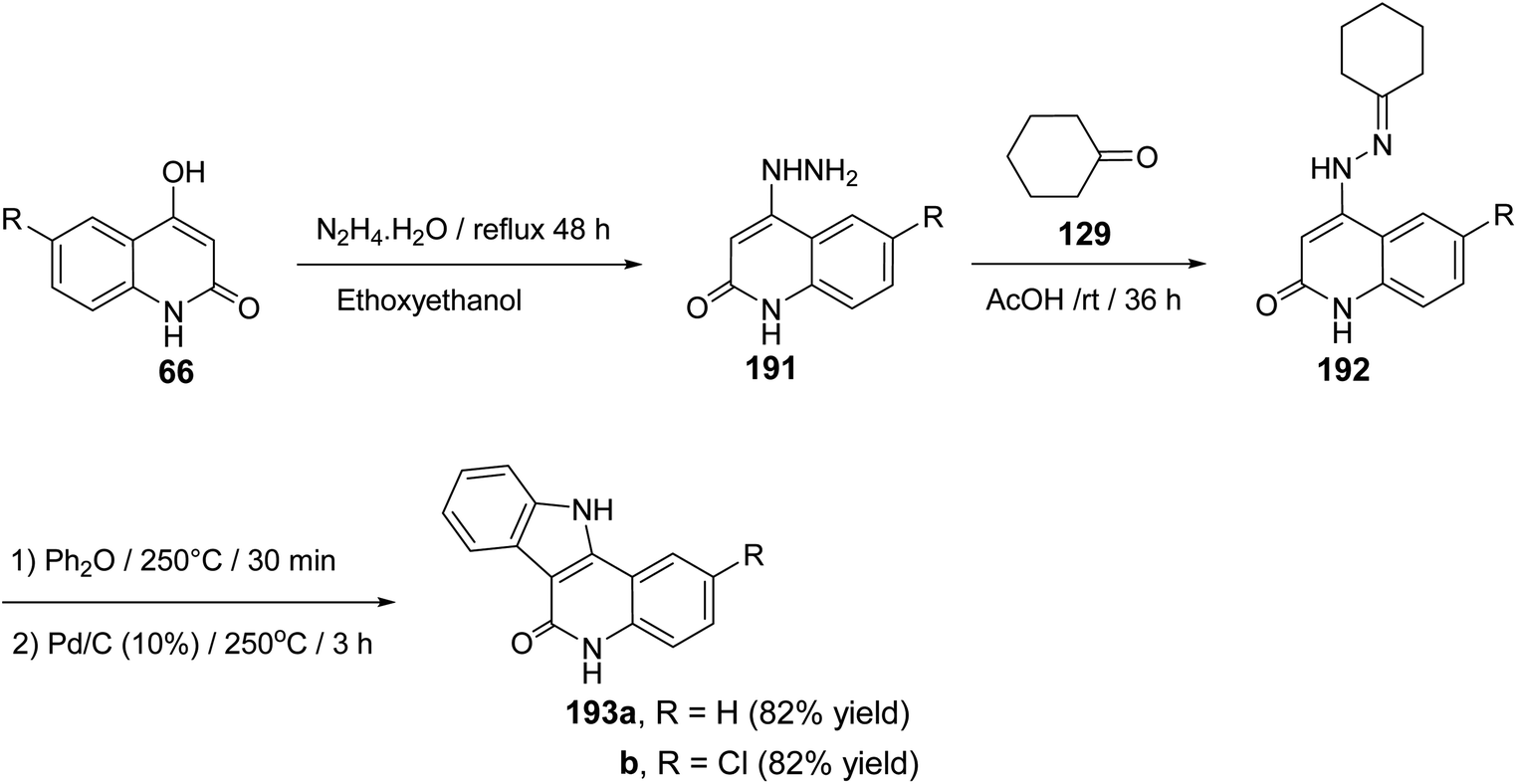
2.2.2. Indolo[3,2-c]quinolines. The synthesis and cytotoxic activity evaluation of 5,11-dihydro-6H-indolo[3,2-c]quinolin-6-ones 193 were reported by Chen and his co-workers89 in 2002. The starting 4-hydrazinoquinolin-2(1H)-ones 191 were synthesized by refluxing 4-hydroxy-quinolin-2(1H)-ones 66 with hydrazine hydrate in ethoxyethanol as a solvent. Stirring 191 with cyclohexanone (129) in glacial AcOH at room temperature afforded the corresponding hydrazones 192. The thermal Fischer indolization of 192 followed by the dehydrogenation gave the fused heterocyclic compounds 193, in good yields (Scheme 61).
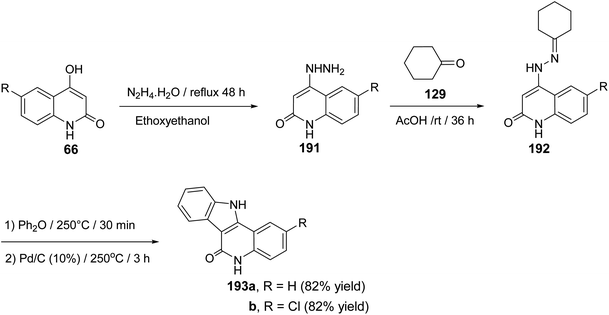 |
| | Scheme 61 Synthesis of 5,11-dihydro-6H-indolo[3,2-c]quinolin-6-ones 193. | |
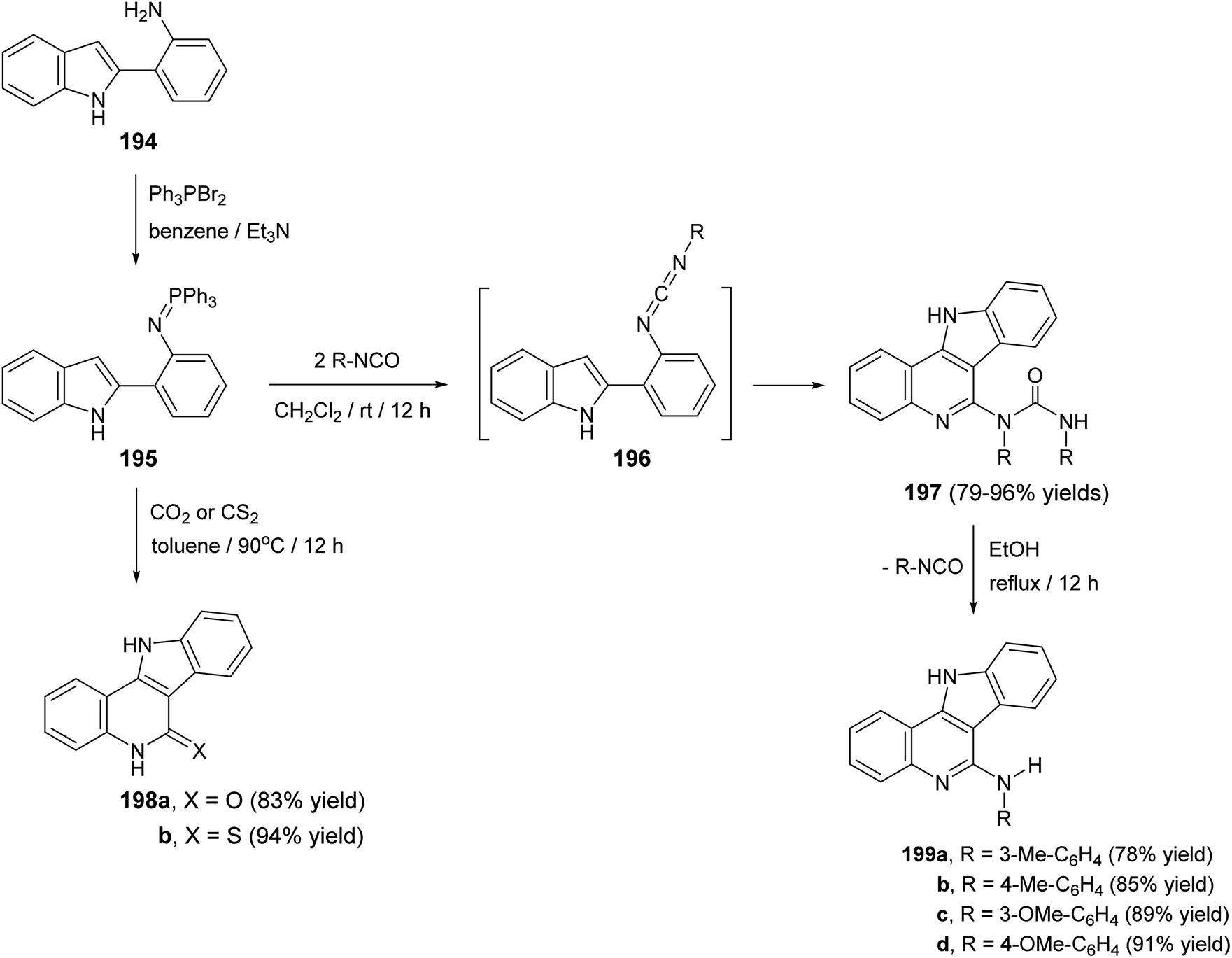
Molina et al.90 have developed a new, simple and general methodology for the synthesis of indolo[3,2-c]quinolines 197–199, bearing an amino, oxygen, or a sulfur atom at the 5-position. This approach based on the aza-Wittig type reaction of iminophosphoranes with hetero-cumulenes to afford the 2-azahexatriene moiety containing a cumulated double bond at one end and the CC bond of the pyrrole ring at the other, which subsequently underwent ring closure to give the fused tetracyclic products 197–199. Thus, iminophosphorane 195, which was obtained from 2-(o-aminophenyl)indole (194) and triphenylphosphine dibromide, reacted with aromatic isocyanates, (1:2) molar ratio, in dry CH2Cl2 at room temperature for 12 h to afford the new tetracyclic 5-[N-aryl-N(arylcarbamoyl)]-amino-11H-indolo[3,2-c]quinolines 197 in 79–96% yields (Scheme 62). The compounds 197 when heated in EtOH at reflux temperature for 12 h, they underwent elimination of the isocyanate to give 5-arylamino-11H-indolo[3,2-c]quinolines 199 in 78–91% yields (Scheme 62). The authors believed that the mechanism of the conversion of 195 into 197 involves initial aza-Wittig reaction to afford a carbodiimide 196, as highly reactive intermediate, which underwent electrocyclic ring closure followed by 1,3-H shift with concomitant addition of the formed exocyclic NH group to the second molecule of the isocyanate. On the other hand, treatment of iminophosphorane 195 with an excess of CO2 at 90 °C in a sealed vessel or CS2 in toluene at 90 °C led to the formation of indolo-[3,2-c]quinolin-6-one 198a and indolo[3,2-c]quinolin-6-thione 198b in 83 and 94% yield, respectively (Scheme 62).
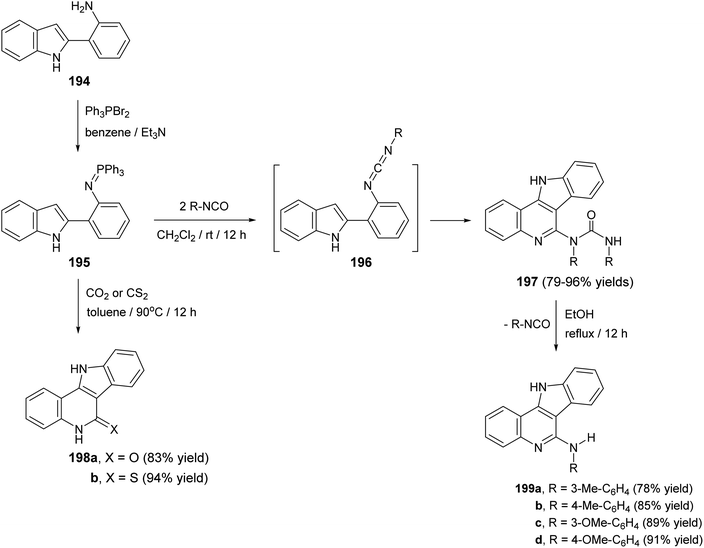 |
| | Scheme 62 A new methodology for the synthesis of indolo[3,2-c]quinolines 197–199. | |
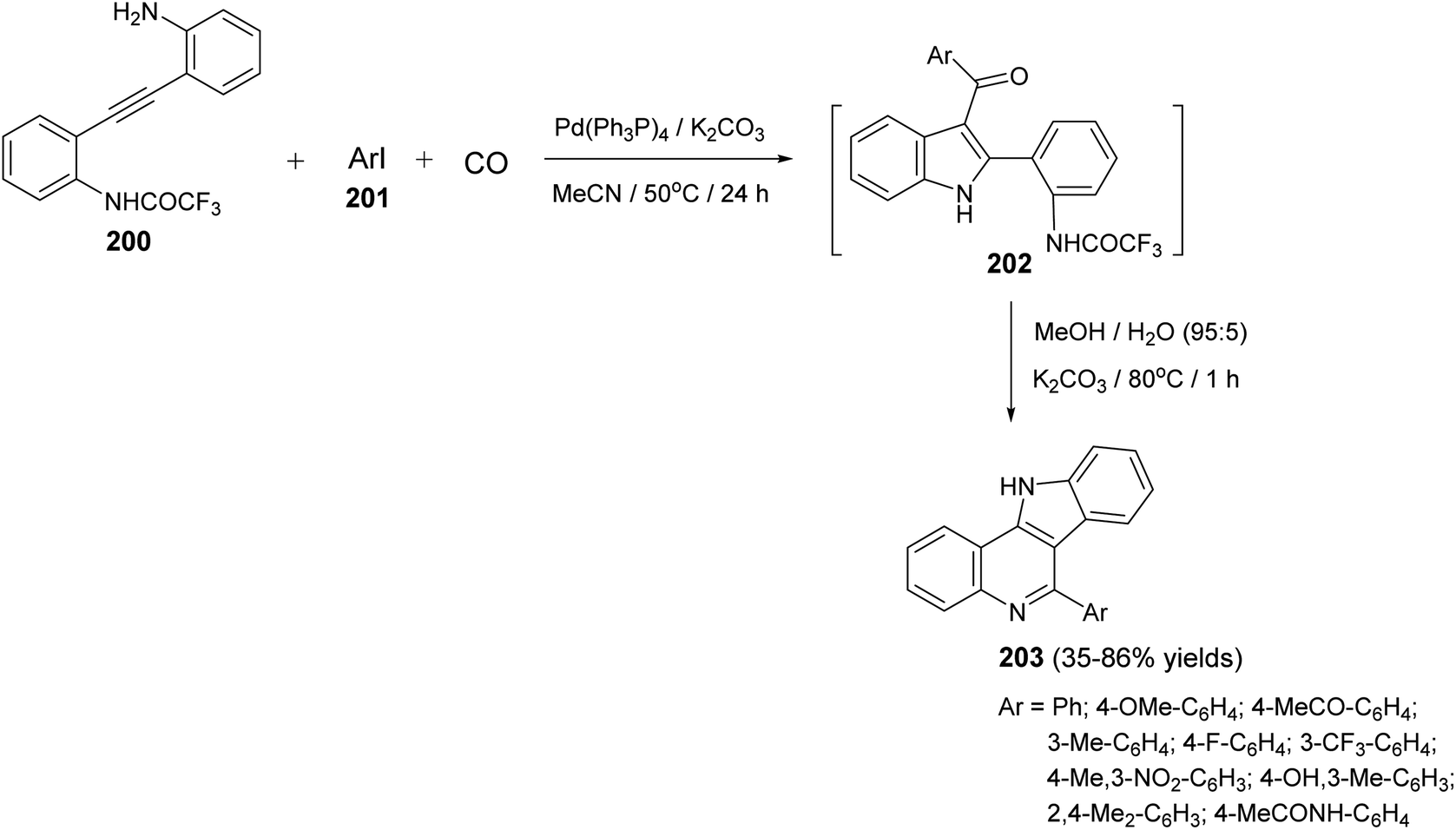
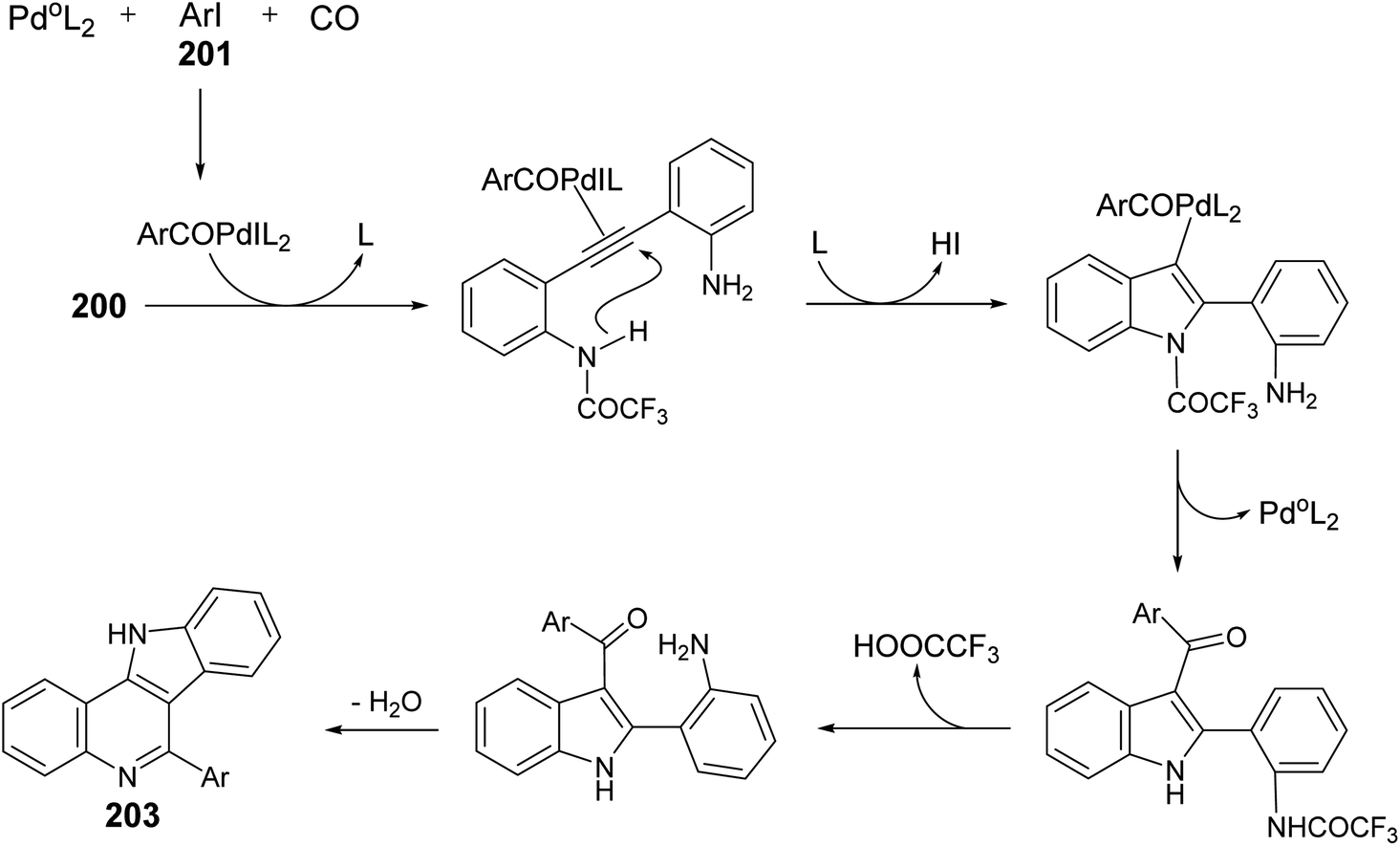
A one-pot and highly efficient procedure for the preparation of 6-aryl-11H-indolo[3,2-c]quinolines 203 via Pd-catalyzed carbonylative cyclization of o-(o-aminophenyl)-ethynyltrifluoroacetanilide 200 with aryl iodides 201 followed by the cyclization of the resultant 3-aroylindoles 202 was described.91 By reaction of 200 with 201 in the presence of a catalytic amount of Pd(Ph3P)4 and K2CO3 in MeCN at 50 °C for 24 h, under a balloon of carbon monoxide, the corresponding N-(2-(3-aroyl-1H-indol-2-yl)phenyl)-2,2,2-trifluoro-acetamides 202 were obtained, in situ, which underwent basic hydrolysis followed by intramolecular cyclization to give the desired indolo[3,2-c]quinolines 203 in 35–86% yields (Scheme 63). A suggested mechanism for the formation of 203 is illustrated in Scheme 64.
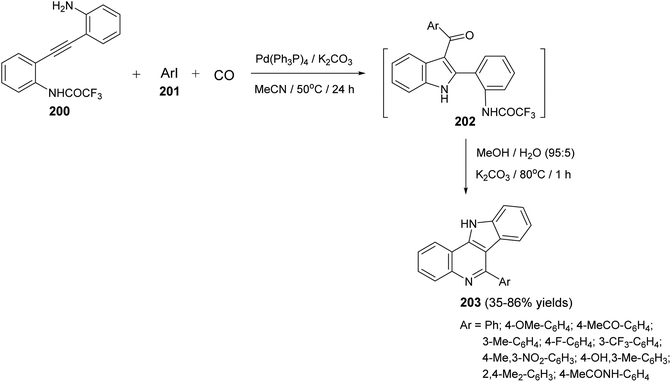 |
| | Scheme 63 A one-pot procedure for the synthesis of 6-aryl-11H-indolo[3,2-c]quinolines 203. | |
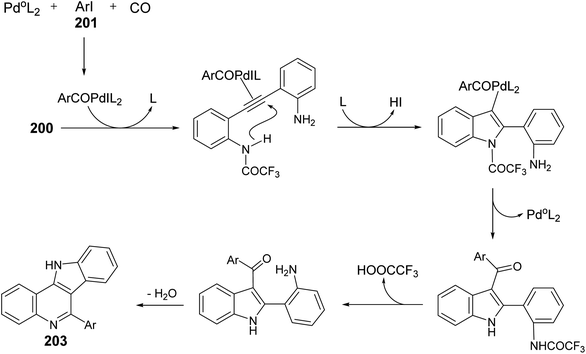 |
| | Scheme 64 A plausible reaction mechanism for the formation of 203. | |
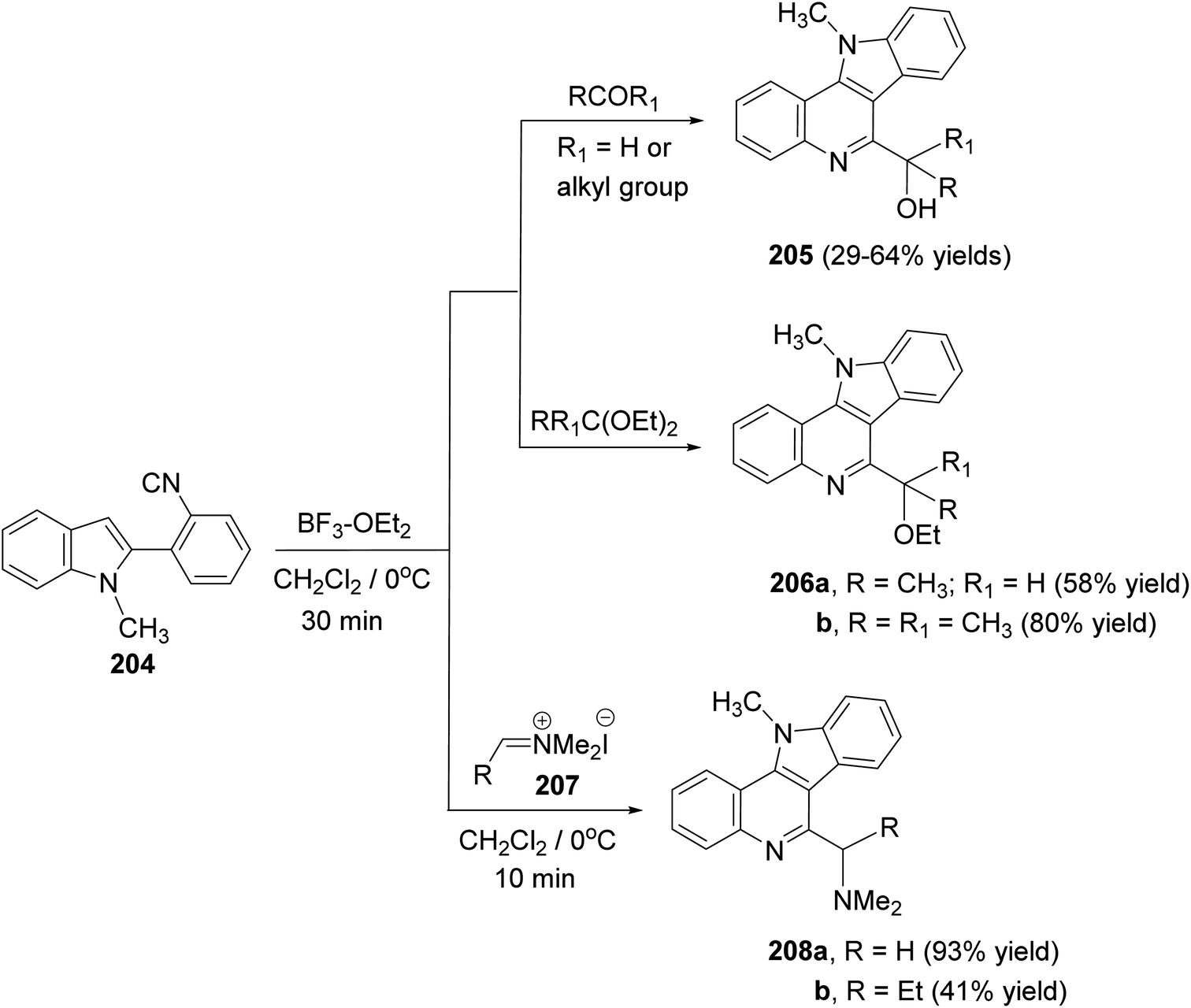
A convenient synthetic method for the synthesis of 11H-indolo[3,2-c]quinolines carrying a different substituents at the 6-position, by electrophile-mediated cyclization reactions of 2-(2-isocyanophenyl)-1-methyl-1H-indole (204), was reported by Kobayashi and his group92 in 2005. Treatment of 204 with various aldehydes (or ketones) and acetals in the presence of a catalytic amount of boron trifluoride–diethyl ether (BF3–OEt2) in CH2Cl2 at 0 °C for 30 min afforded 6-(1-hydroxy)-11-methyl-11H-indolo[3,2-c]quinolines 205 and 6-(1-ethoxyalkyl)-11-methyl-11H-indolo[3,2-c]quinolines 206, respectively, in moderate to good yields (Scheme 65). However, the reaction of 204 with iminium salts 207 was carried out in the absence of catalyst in CH2Cl2 at 0 °C for 10 min to give 6-dimethylaminomethyl-11-methyl-11H-indolo[3,2-c]quinolines 208 in moderate to excellent yields.
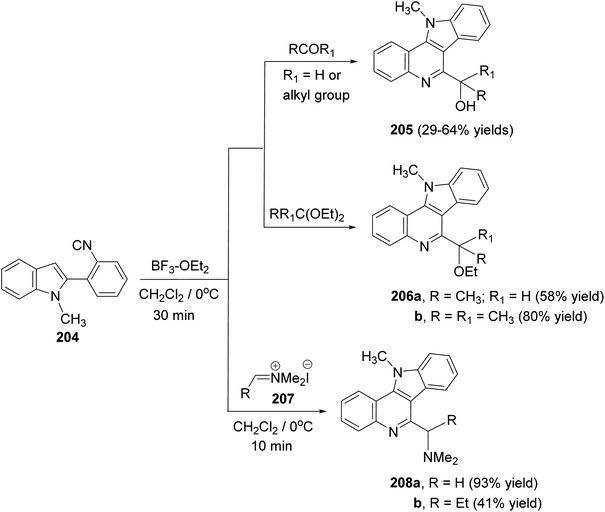 |
| | Scheme 65 A convenient synthesis of 6-substituted-11-methyl-11H-indolo[3,2-c]quinolines 205, 206 and 208. | |
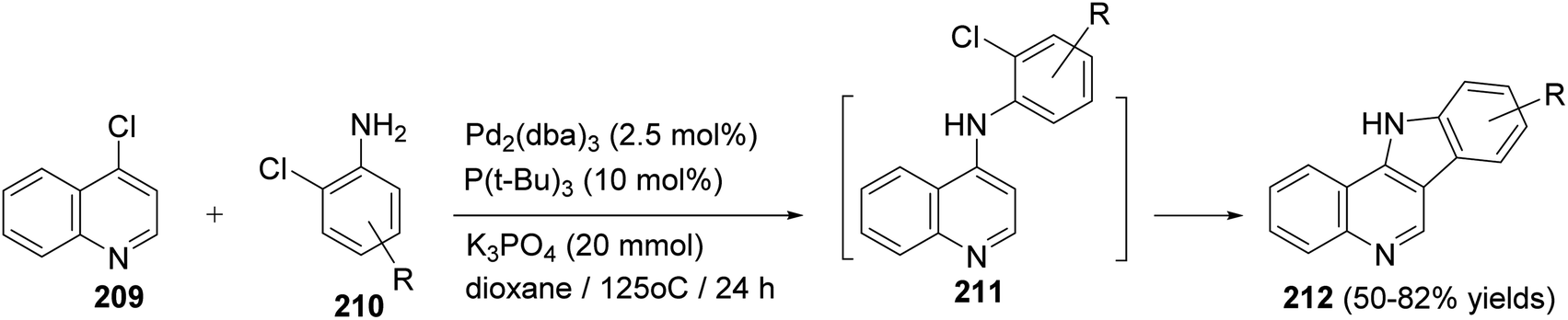
Maes and his group93 have developed the first synthesis of D-ring substituted 11H-indolo[3,2-c]quinolines 212 via auto-tandem Pd-catalyzed intermolecular carbon–nitrogen and intramolecular carbon–carbon bond formation. The reaction was carried out by heating 4-chloroquinoline (209) (2 mmol) with a wide variety of o-chloroanilines 210 (2.4 mmol) in the presence of Pd2(dba)3 (2.5 mol%), P(t-Bu)3 (10 mol%) and K3PO4 (20 mmol) in dioxane at 125 °C for 24 h to give 11H-indolo[3,2-c]quinolines 212 in 50–82% yields, via intermediacy of 211 (Scheme 66). In the present work, the authors also reported that amine 211 is certainly an intermediate since TLC and MS analyses clearly revealed its presence during the process of the reaction and this reaction requires Pd catalysis since omitting the catalyst gave no 211 and 212.
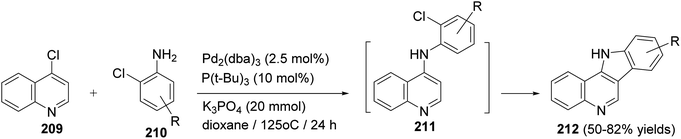 |
| | Scheme 66 Synthesis of D-ring substituted 11H-indolo[3,2-c]quinolines 212 via auto-tandem Pd-catalyzed intermolecular C–N and intramolecular C–C bond formation. | |
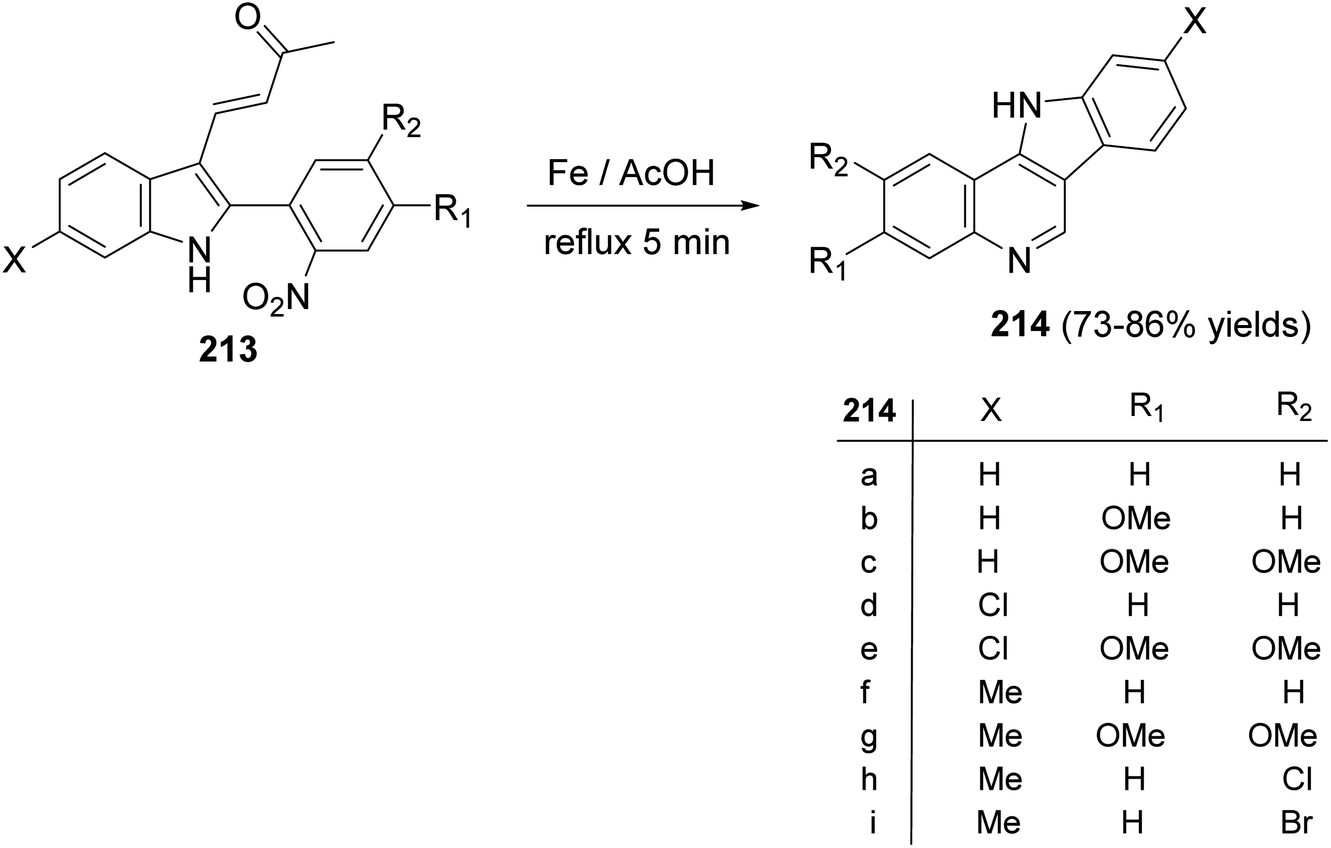
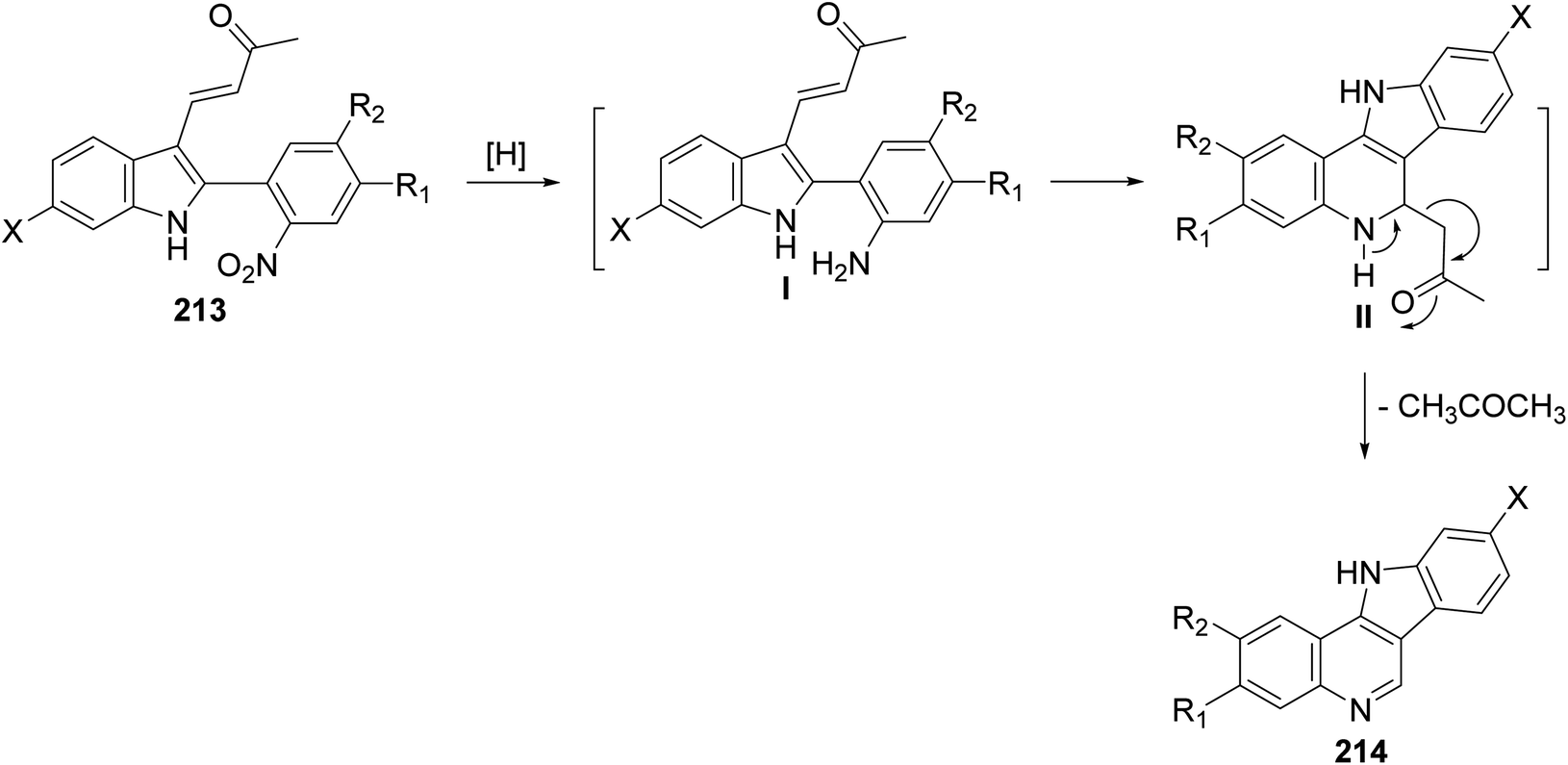
A simple and new synthesis of indolo[3,2-c]quinolines 214 via reductive cyclization of a series of 2-(2-nitrophenyl)indoles 213 was reported.94 Treatment of 213 with Fe in AcOH under reflux for 5 min gave directly 11H-indolo[3,2-c]quinolines 214, in one step, in high yields (Scheme 67). In the proposed reaction mechanism, the reduction of NO2 group gave the corresponding aniline intermediate I, which underwent intramolecular Michael addition to afford the intermediate 6-acetonyl-5,6-dihydro-11H-indolo[3,2-c]quinolines II. The aromatization of II, via elimination of one molecule of acetone, gave the desired indolo-[3,2-c]quinolines 214 (Scheme 68). The authors also reported that the reaction has a general character, electron-withdrawing halide substituents as well as electron-donating alkoxy and alkyl groups have no significant effect on the yields of products 214.
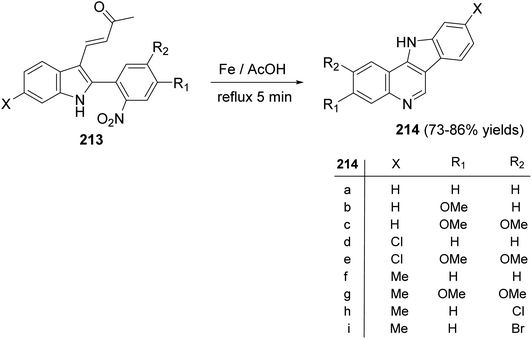 |
| | Scheme 67 Reductive cyclization of 2-(2-nitrophenyl)indoles 213 to indolo[3,2-c]quinolines 214. | |
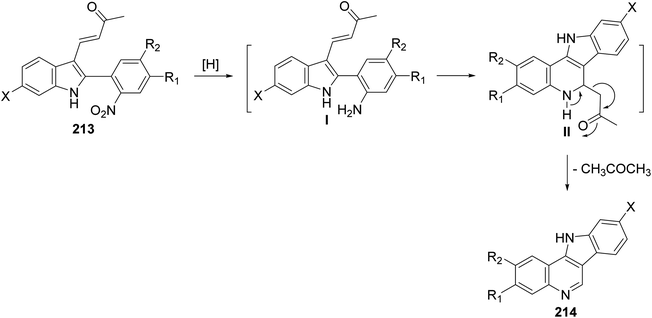 |
| | Scheme 68 The proposed mechanism of indolo[3,2-c]quinoline 214 formation. | |
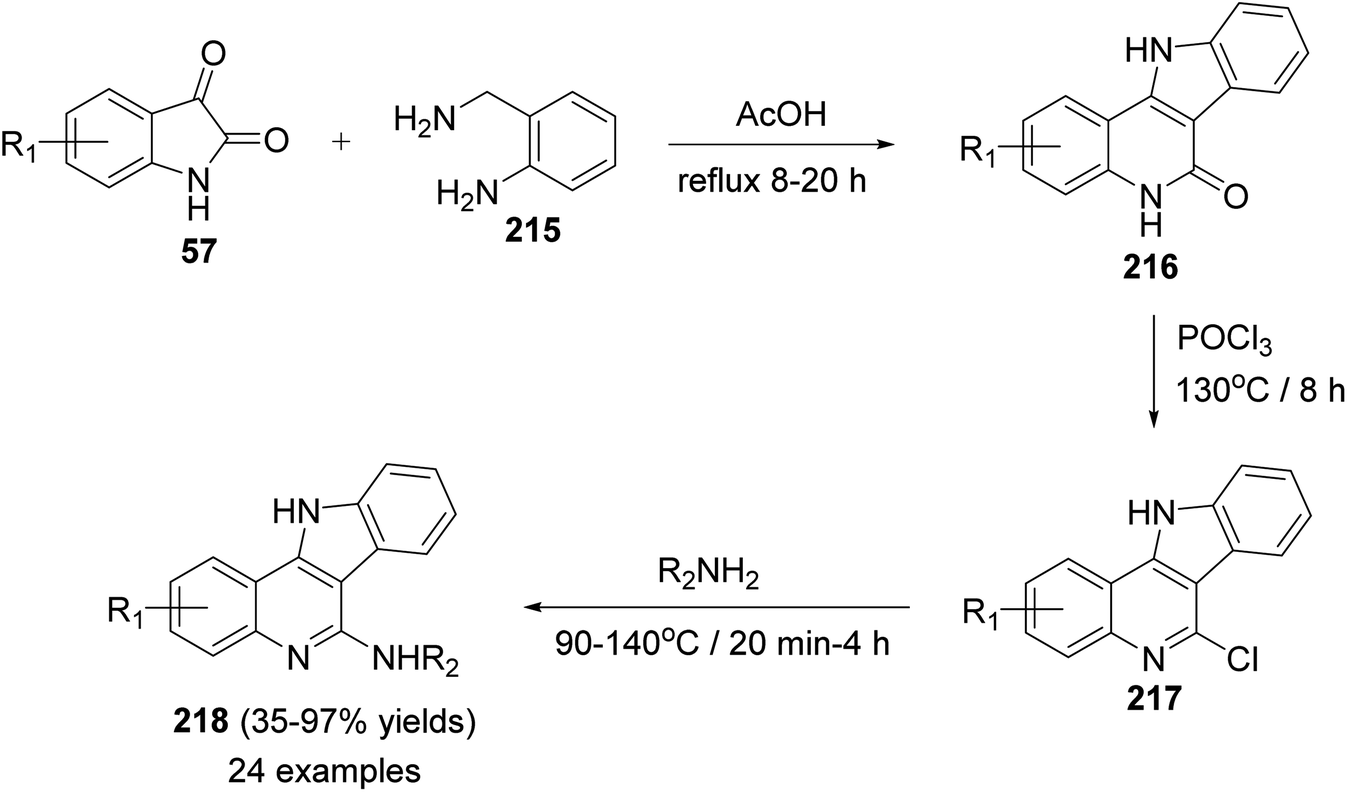
In 2014, Inokuchi et al.95 reported the synthesis of a new series of 6-amino-11H-indolo[3,2-c]quinolines 218, with different substituents on the quinoline ring, utilizing isatins 57 and 2-aminobenzylamine (215) as starting materials. Thus, the reaction of 57 with 215 in AcOH under reflux for 8–20 h provided the 6-dihydro-11H-indolo[3,2-c]quinoline-6(5H)-ones 216. Dehydrative chlorination of 216 was accomplished in POCl3 at 130 °C for 8 h to afford the corresponding 6-chloro-11H-indolo[3,2-c]quinoline 217. The 6-chloro compounds 217 when heated with the appropriate amines at 90–140 °C within 20 min to 4 h, they underwent the addition–elimination reaction of the anilines and alkyl amines to produce the new 6-amino-11H-indolo[3,2-c]quinolines 218 in 35–97% yields (Scheme 69).
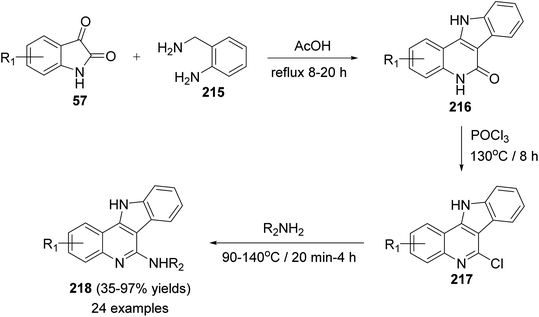 |
| | Scheme 69 Synthesis of 11H-indolo[3,2-c]quinolines 218 with amino group substituted at the C-6 position. | |
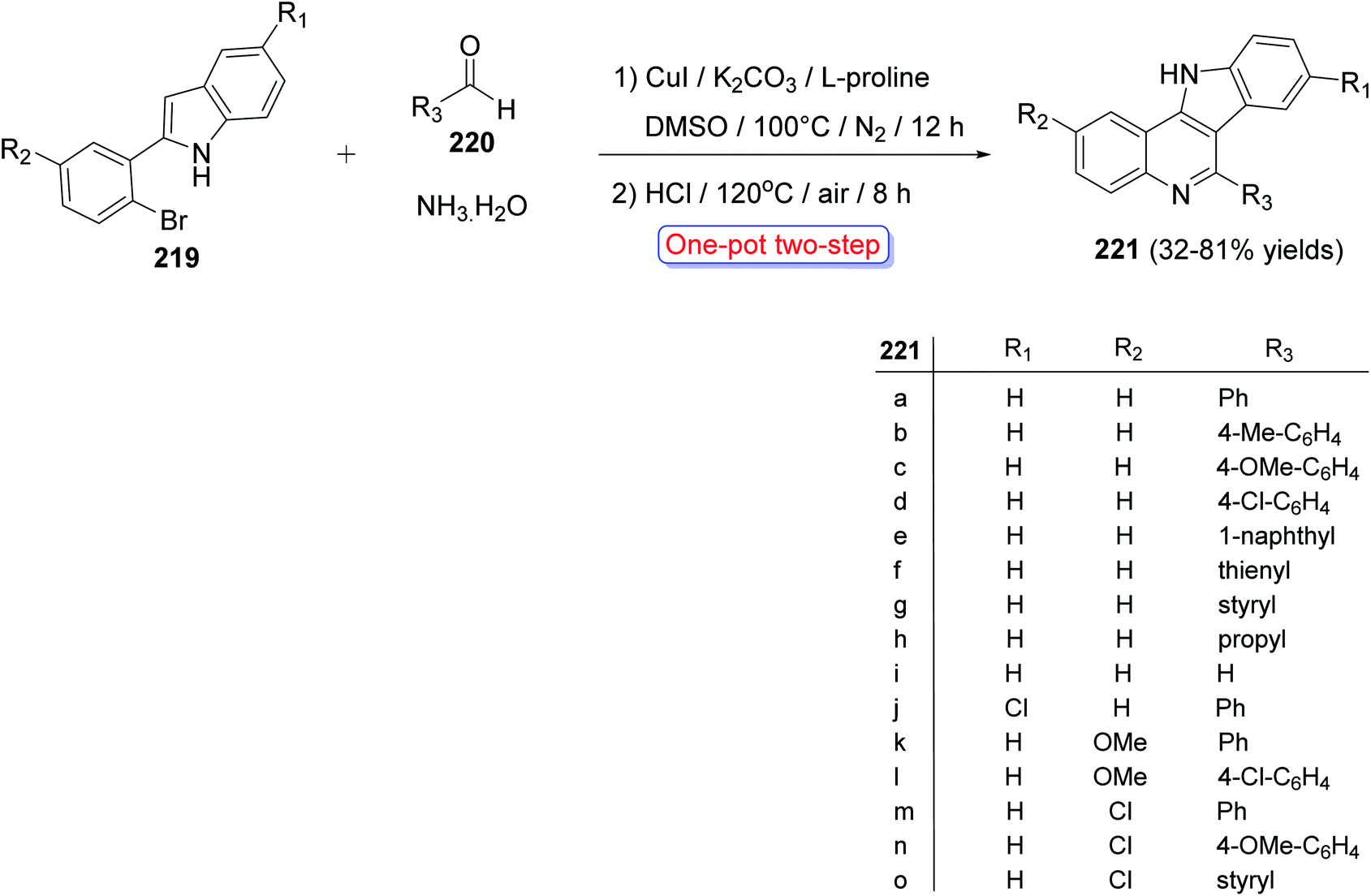
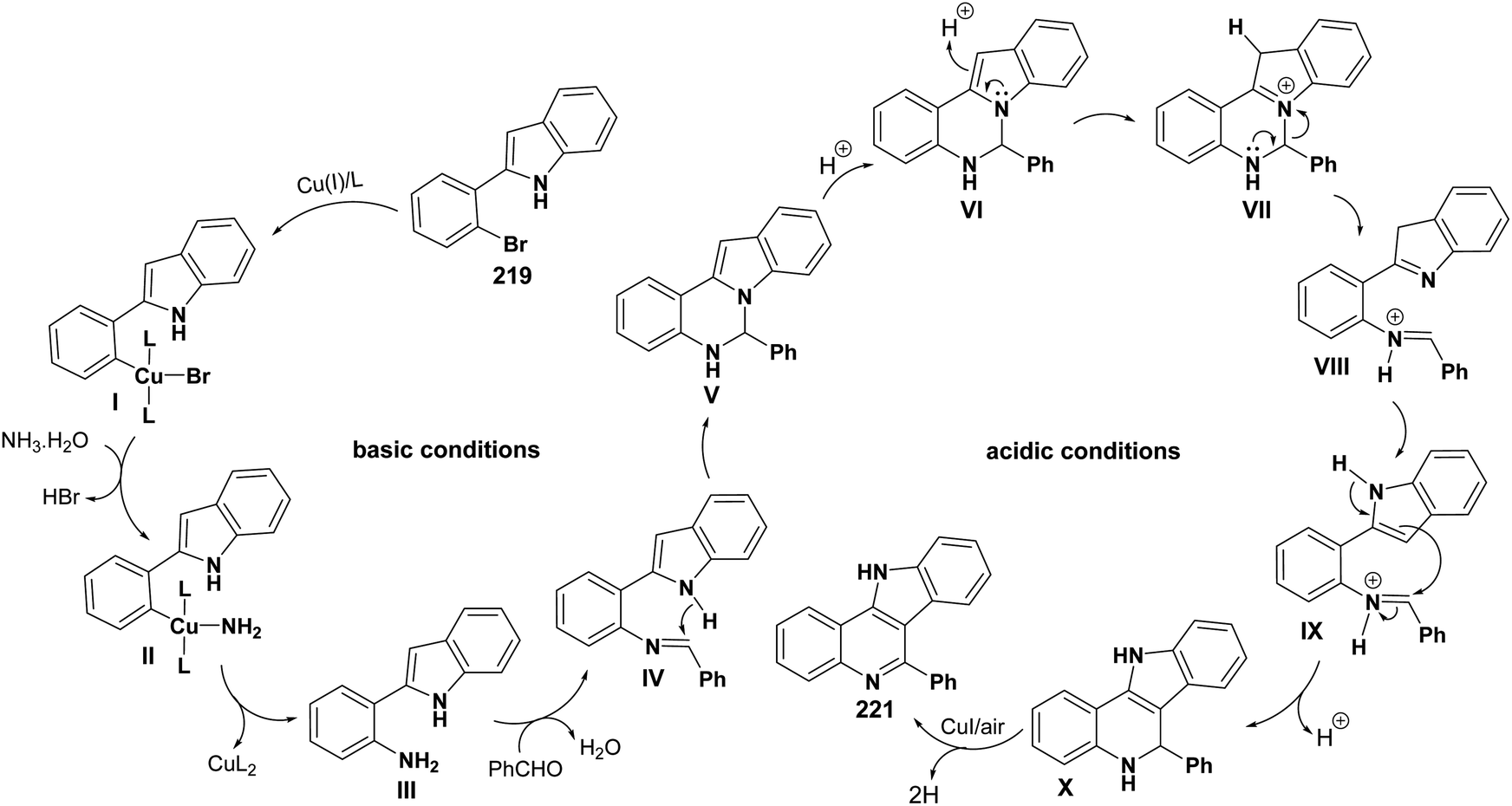
A convenient and highly selective synthesis of 11H-indolo[3,2-c]quinolines 221 through Cu-catalyzed one-pot two-step cascade reactions of 2-(2-bromoaryl)-1H-indoles 219 with aldehydes 220 and aq. NH3 was developed by Guo et al.96 in 2015. First, indoles 219 were treated with 220 and aq. NH3 in the presence of CuI, K2CO3, and L-proline under nitrogen at 100 °C for 12 h. Then, the reaction mixture was acidified with HCl and stirred at 120 °C for 8 h under air to provide the required tetracyclic product 221 in modest to good yields (32–81%) (Scheme 70). A mechanism for the Cu-catalyzed formation of 221 is outlined in Scheme 71. For aldehydes 220, it was found that alkenyl- and aryl-substituted aldehydes usually afforded the products 221 in yields higher than alkyl-substituted aldehydes. For indoles 219, different substituents (R1 and R2 group) exhibited a slight influence on the yields of the products 221.
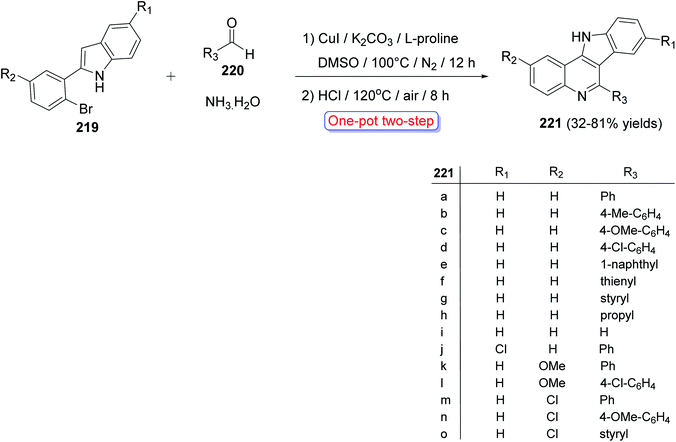 |
| | Scheme 70 One-pot two-step synthesis of 221 from 219, 220 and aq. NH3. | |
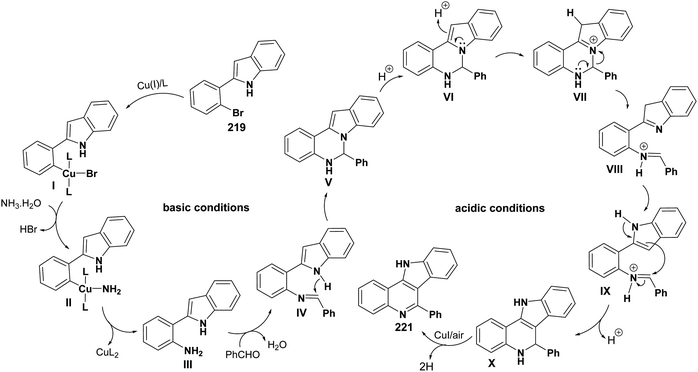 |
| | Scheme 71 Plausible mechanism for the formation of 221. | |
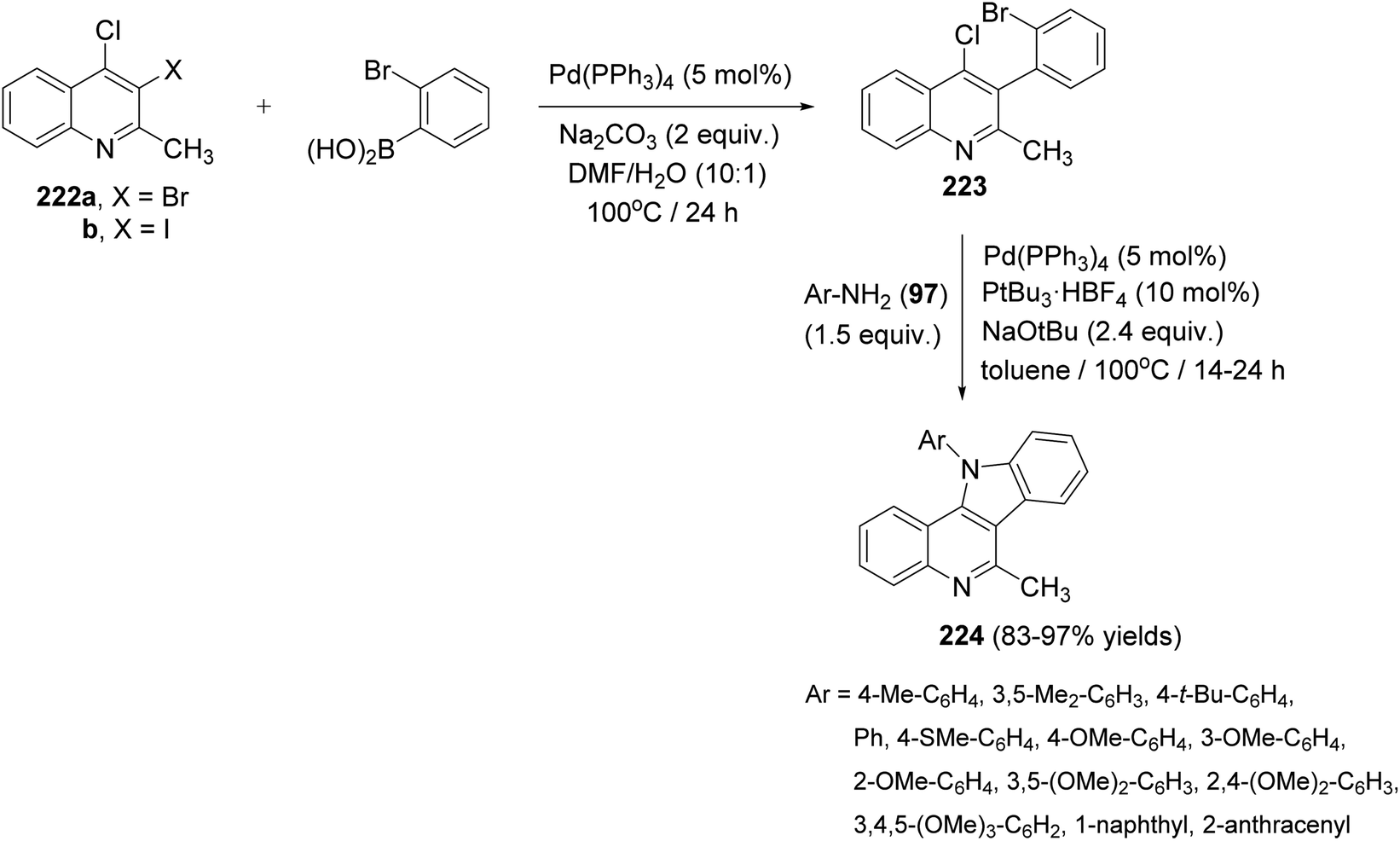
In 2017, Langer et al.97 have developed a new and convenient synthesis of 11-aryl-11H-indolo[3,2-c]quinolines 224 by chemoselective Suzuki reaction of o-dihalo-quinolines with o-bromophenylboronic acid, followed by double carbon–nitrogen coupling with primary amines. The chemoselective Suzuki reaction of 3,4-dihalo-2-methylquinolines 222 with o-bromophenylboronic acid in the presence of Pd(PPh3)4 (5 mol%) and Na2CO3 (2 equiv.) in DMF/H2O (10:1) at 100 °C for 24 h afforded 3-(2-bromophenyl)-4-chloro-2-methylquinoline (223). Heating 223 with various electron-poor, -neutral and -rich arylamines 97 (1.5 equiv.) in the presence of Pd2dba3 (5 mol%), PtBu3·HBF4 (10 mol%), and NaOtBu (2.4 equiv.) in toluene at 100 °C for 14–24 h furnished the novel tetracyclic indolo[3,2-c]-quinolines 224 in 83–97% yields (Scheme 72). In this approach, the substituents located at the 11 position are installed directly, thereby avoiding the problem of selectivity in the alkylation of the 11-unsubstituted heterocyclic compounds.
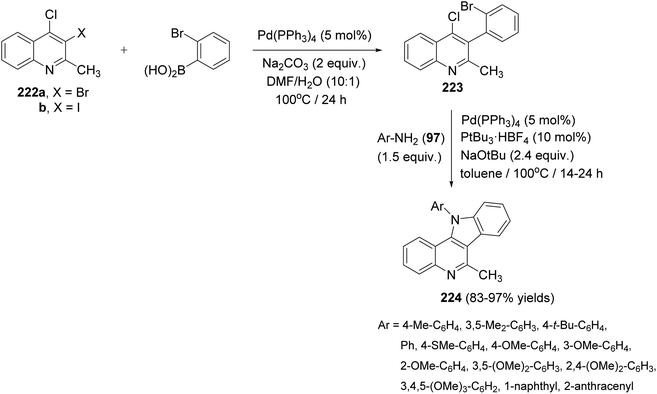 |
| | Scheme 72 A new and convenient synthesis of 11-aryl-11H-indolo[3,2-c]quinolines 224. | |
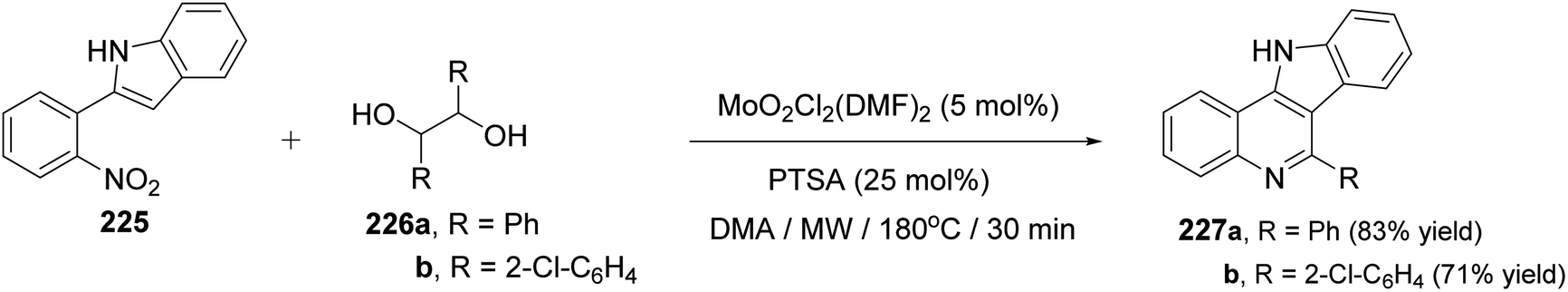
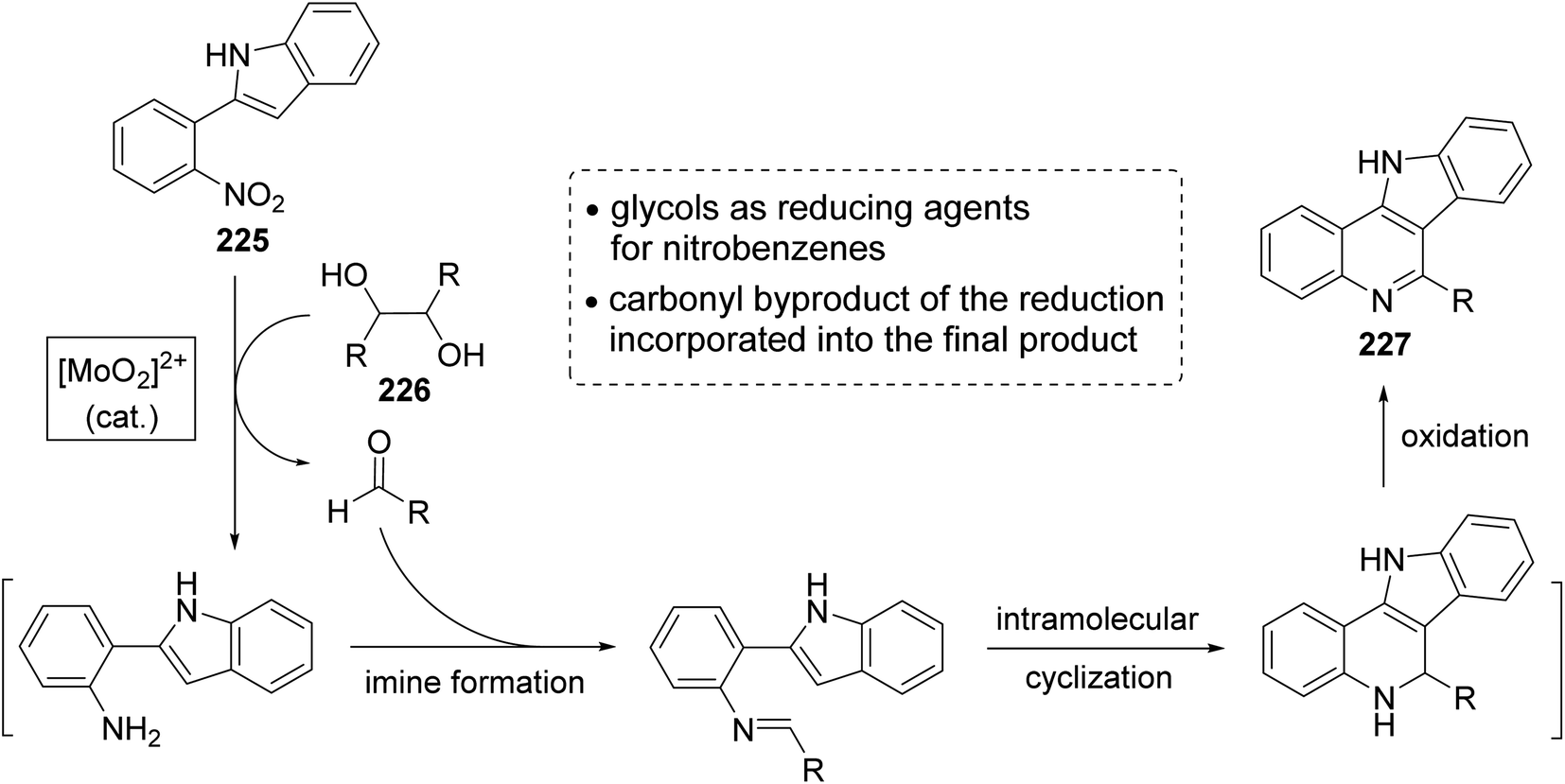
In the same year, Sanz et al.98 have reported a novel domino nitro reduction, imine formation, intramolecular cyclization, oxidation for the efficient, clean and one-pot synthesis of indolo[3,2-c]quinolines 227 from easily available nitrobenzenes as the nitrogen source and glycols as the carbonyl source and reducing agents. The process utilizes the carbonyl byproduct of the initial dioxomolybdenum(VI)-catalyzed reduction of nitrobenzenes with glycols as a reagent for the imine generation. This method represents the first sustainable domino reaction in which the waste byproduct of a reaction has been used as a reactant for the next step and included into the final product. This synthetic method proceeded by treatment 2-(2-nitrophenyl)indole (225) (1 equiv.) with secondary glycols 226 (2.2 equiv.) in the presence of a catalytic amount of a dioxomolybdenum(VI) complex and PTSA (25 mol%) in DMA as solvent and under MW irradiation at 180 °C for 30 min to give the new indolo[3,2-c]quinolines 227 in high yields (Scheme 73). A suggested mechanism for the synthesis of 227 is outlined in Scheme 74.
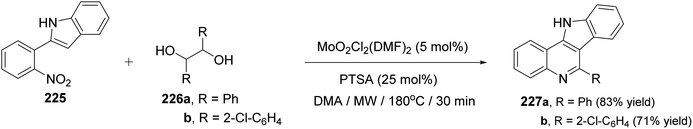 |
| | Scheme 73 Synthesis of indolo[3,2-c]quinolines 227 from 2-(2-nitrophenyl)indole (225). | |
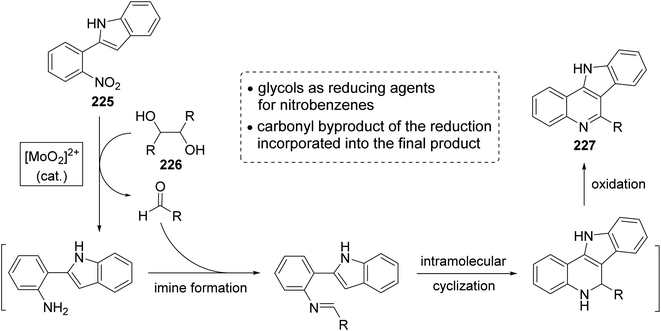 |
| | Scheme 74 A plausible mechanism for the formation of 227 from 225 and 226. | |
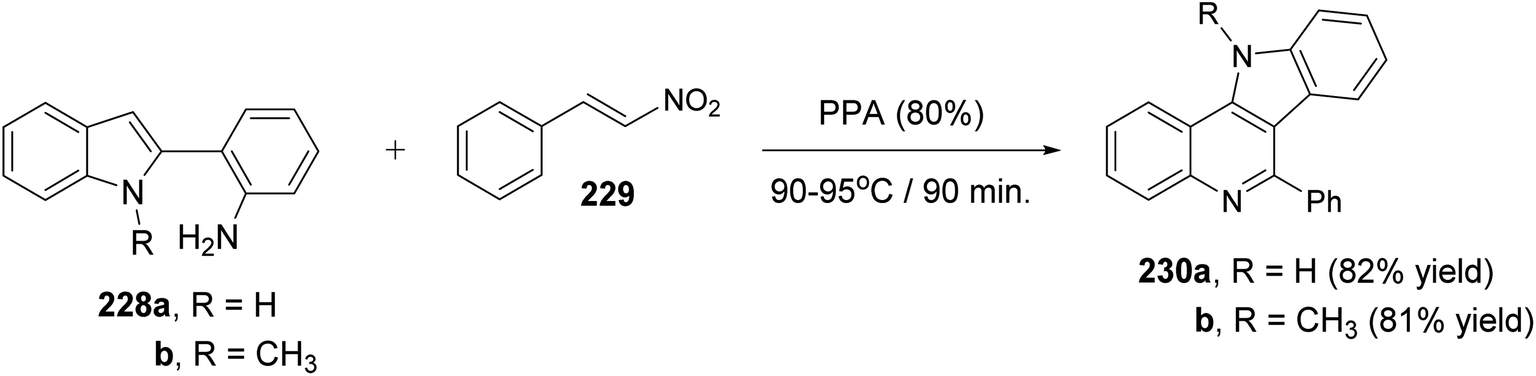
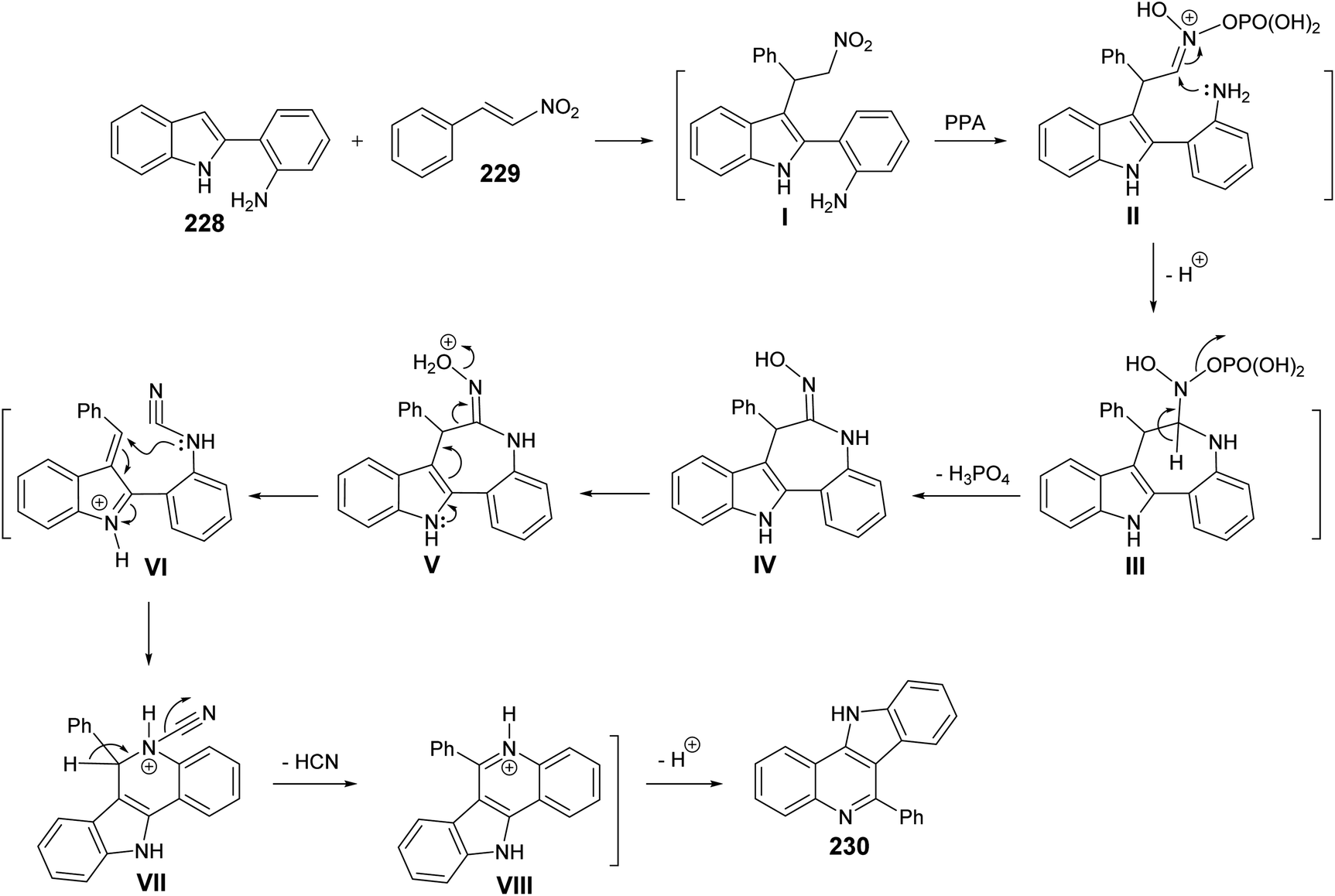
Very recently, Aksenov and his coworkers99 have reported a one-pot synthesis of indolo[3,2-c]quinolines 230 via PPA-mediated unexpected cyclization reaction between 2-(2-aminophenyl)indoles 228 and 2-nitrostyrene (229). The reaction was carried out by heating a mixture of 228 and 229 in PPA (80%) at 90–95 °C for 90 min (Scheme 75). The mechanistic pathway for the formation of 230 via cyclization of 228 with 229 is given in Scheme 76. The reaction of 228 with 229 gave the nitroalkane intermediate I. In the PPA medium, nitroalkane I is exist in the phosphorylated aci form II, with a highly electrophilic CN bond, which underwent intramolecular N-Nef reaction to provide the corresponding cyclic aminal intermediate III. Subsequent elimination of H3PO4 afforded cyclic imidoxime intermediate IV. The latter IV was protonated in acidic medium to give intermediate V, which further underwent a seven-membered ring cleavage (Werner rearrangement) to provide N-cyano-aniline VI. A subsequent 6-endo-trig cyclization of the resulting VI involving the iminium moiety of the indole to yield the indoloquinoline core in the form of N-cyano-ammonium VII. The elimination of HCN afforded 5H-indolo[3,2-c]quinolin-11-ium ion VIII, which underwent deprotonation, under basic conditions during the post-reaction work-up, to afford the desired tetracyclic indolo[3,2-c]quinolines 230.
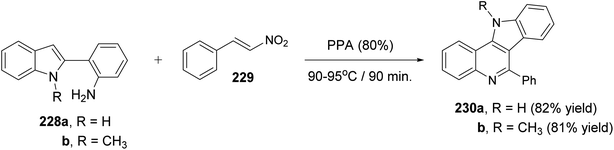 |
| | Scheme 75 Synthesis of indolo[3,2-c]quinolines 230 via PPA-mediated cyclization reaction between 228 and 229. | |
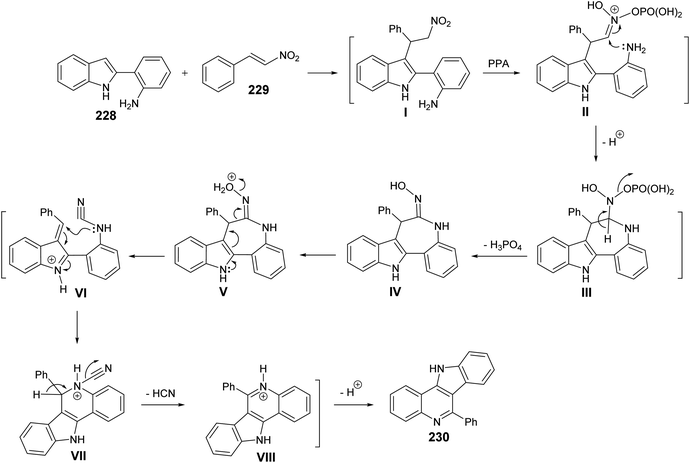 |
| | Scheme 76 A plausible mechanism for the formation of 230. | |
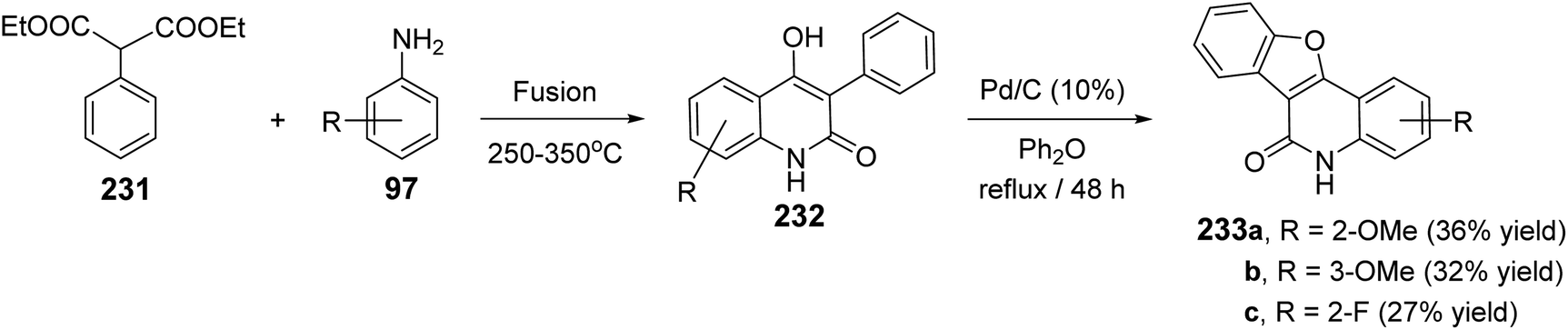
2.2.3. Benzofuro[3,2-c]quinolines. The synthesis of certain benzofuro[3,2-c]quinolin-6(5H)-ones 233 via Pd-catalyzed cyclodehydrogenation of 4-hydroxy-3-phenyl-quinolin-2(1H)-ones 232 was reported.89 Fusion of diethyl 2-phenylmalonate (231) with anilines 97 at 250–350 °C leading to the formation of 232. Refluxing 232 in diphenyl ether in the presence of a catalytic amount of Pd/C (10%) gave the respective tetracyclic products 233, in low yields (27–36%) (Scheme 77).
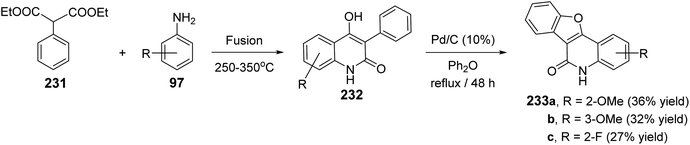 |
| | Scheme 77 Pd-catalyzed synthesis of benzofuro[3,2-c]quinolin-6(5H)-ones 233. | |
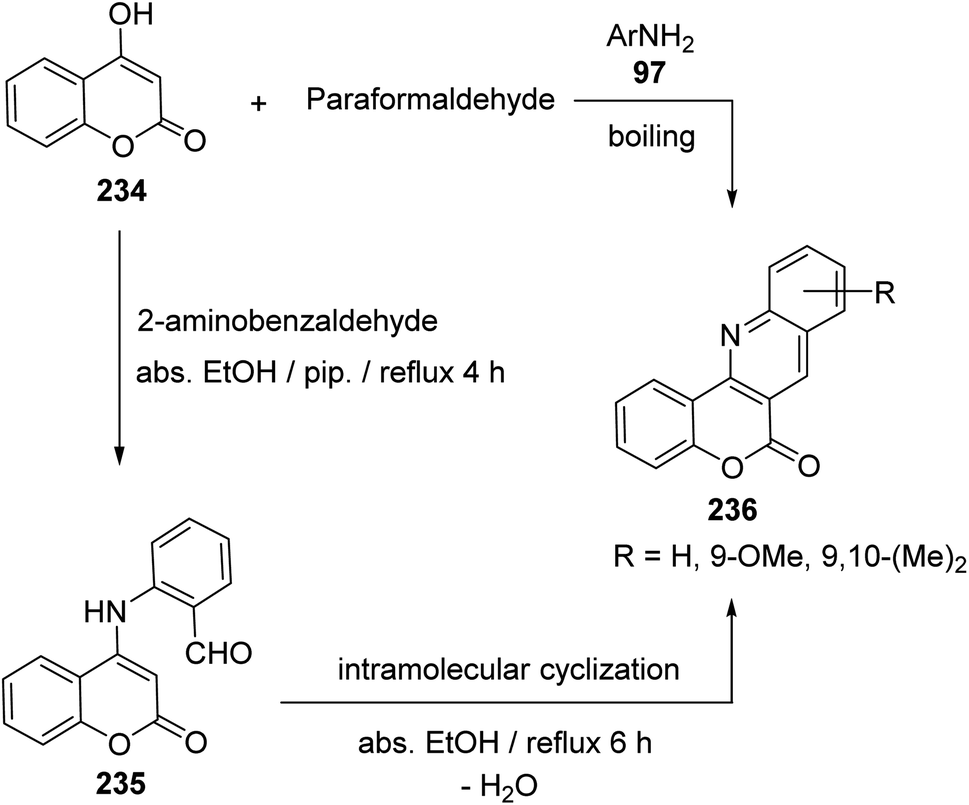
2.2.4. Chromenoquinolines. Buu-Hoi et al.100 have reported the first synthesis of 6H-chromeno[4,3-b]quinoline-6-ones 236, which are analogues of the carcinogenic dibenzacridines and benzacridines, via reaction of 4-hydroxycoumarin (234) with paraformaldehyde and primary arylamines 97 (Scheme 78). The novel synthesis of this ring system has developed by Tabaković et al.101 The reaction was carried by refluxing a 1:1 molar ratio of 234 with 2-aminobenzaldehyde in abs. EtOH in the presence of a catalytic amount of piperidine to give 2-((2-oxo-2H-chromen-4-yl)amino)benzaldehyde (235), which was converted to the tetracyclic products 236 (after refluxing 6 h) by losing a molecule of H2O via formation of intramolecular Schiff base (Scheme 78).
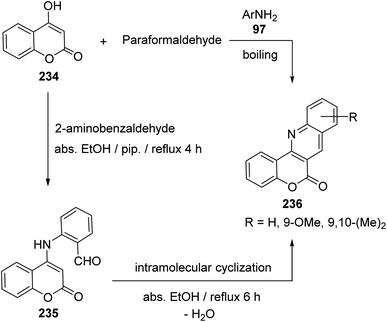 |
| | Scheme 78 Synthesis of 6H-chromeno[4,3-b]quinoline-6-ones 236. | |
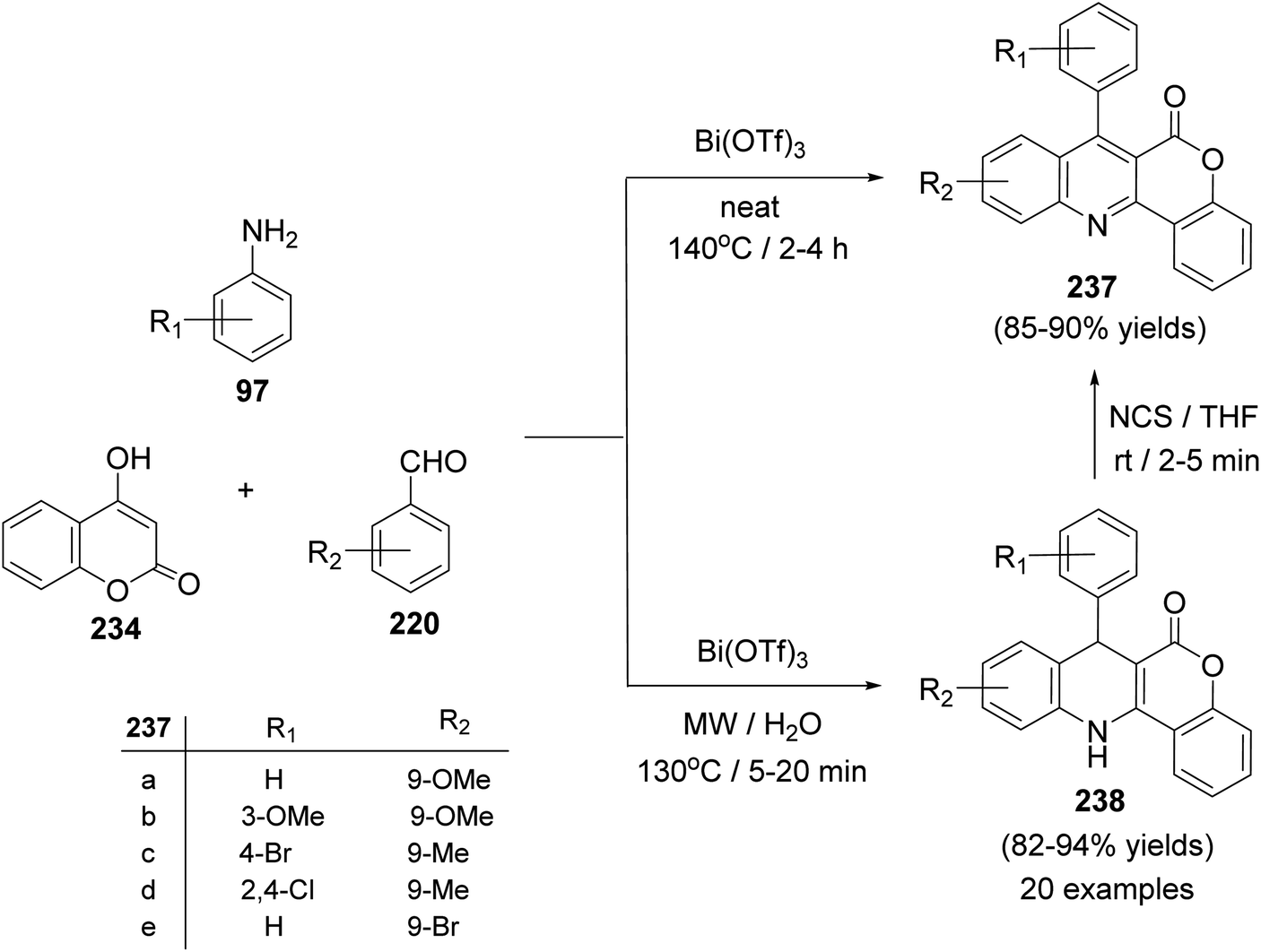
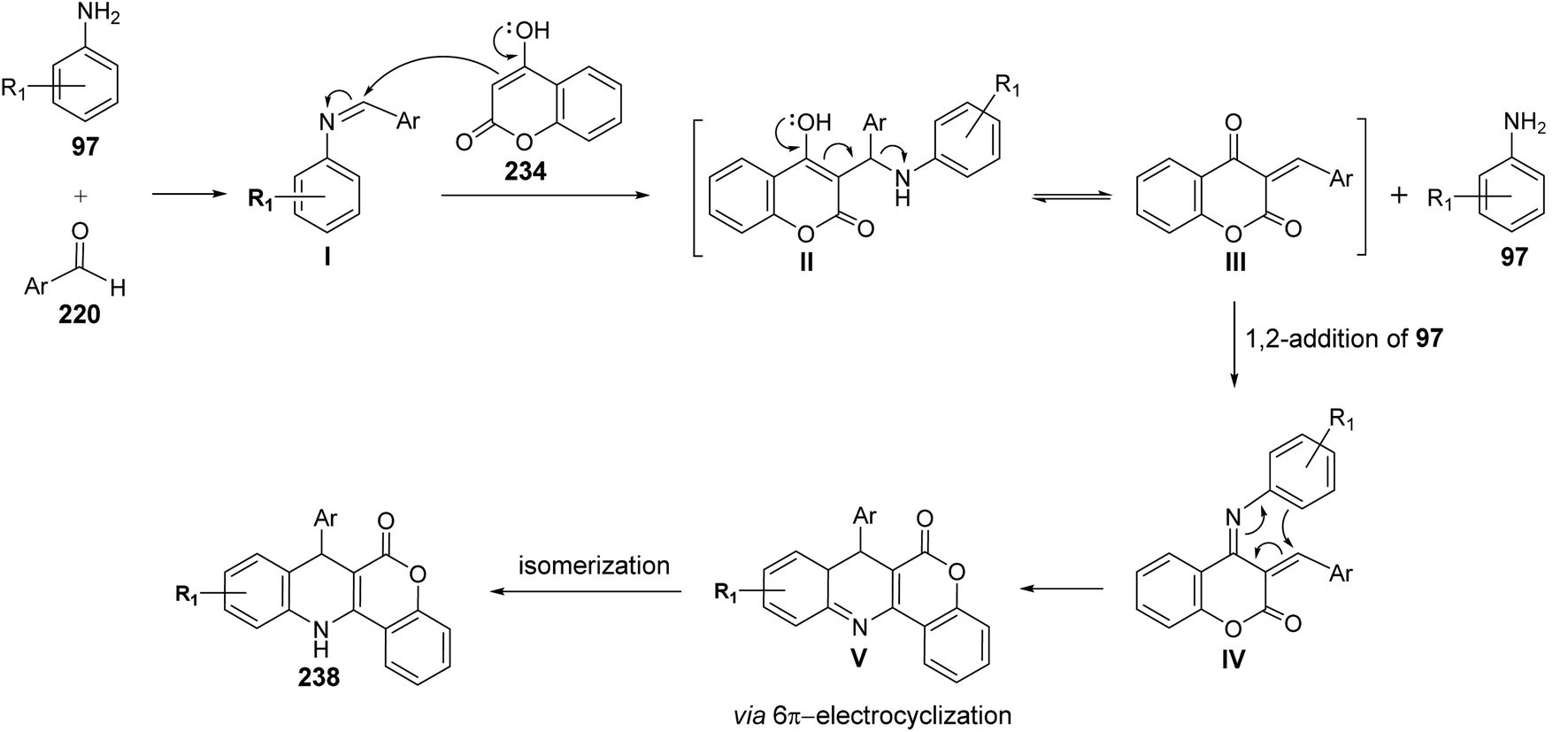
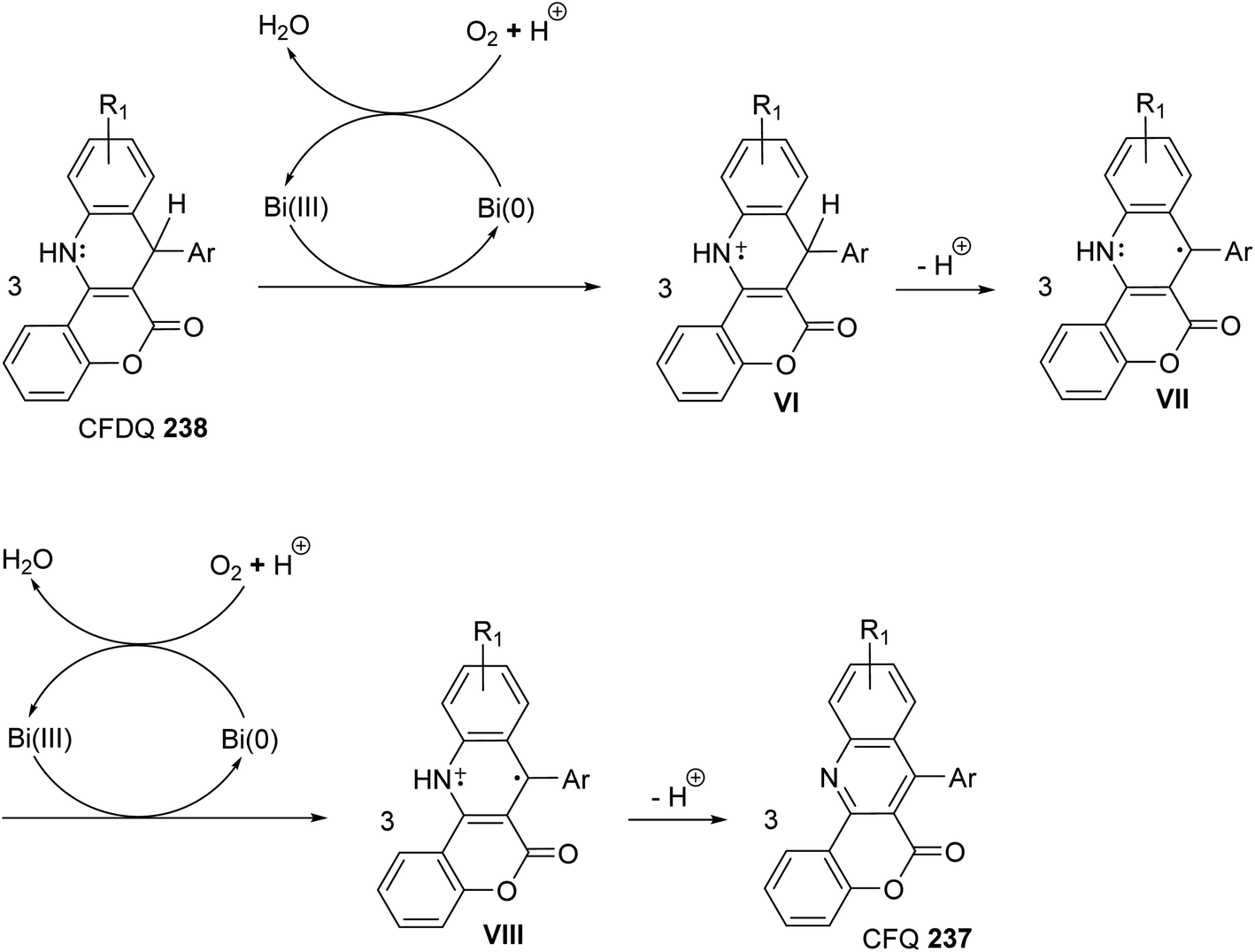
A facile one-pot, three-component reaction of 4-hydroxy-coumarin (234), aromatic amines 97 and aromatic aldehydes 220 using an environmentally benign bismuthtriflate Bi(OTf)3 as a catalyst under neat and conventional heating conditions at 140 °C produced the coumarin fused quinoline (CFQ), namely 7-aryl-6H-chromeno[4,3-b]quinoline-6-ones 237, in 85–90% yields (Scheme 79). On the other hand, microwave (MW) assisted multicomponent reactions of the same combination in the presence of Bi(OTf)3 as a catalyst in H2O at 130 °C gave coumarin fused dihydroquinolines (CFDQ) 238, in 82–94% yields (Scheme 79).102 Authors also reported an alternative and rapid route for the synthesis of CFQ 237 by stirring CFDQ 238 in the presence of N-bromosuccinamide (NBS) in THF at room temperature (Scheme 79). The plausible mechanism for the formation of CFDQ 238 is described in Scheme 80. Initially, aldehyde 220 condensed with aromatic amine 97 to form an imine I. Nucleophilic addition of 234 to intermediate I afforded an unstable intermediate II, which in equilibrium with intermediate III. Subsequent, 1,2-addition of the aromatic amines 97 to III, followed by 6π-electrocyclization and isomerisation yielded products (CFDQ) 238. The formation of 237 can be explained via the formation of 238, followed by free radical mechanism involving Bi(III)/Bi(0) as outlined in Scheme 81. The fluorescence property studies of the synthesized CFDQ 238 and CFQ 237 in different solvents showed that CFDQ 238 are more fluorescent than the CFQ 237.
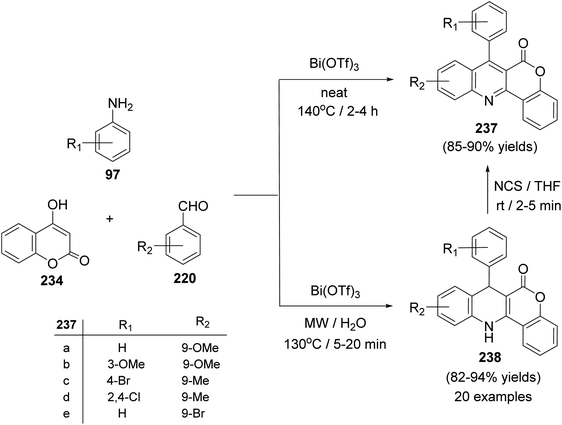 |
| | Scheme 79 Synthesis of CFQ 237 and CFDQ 238. | |
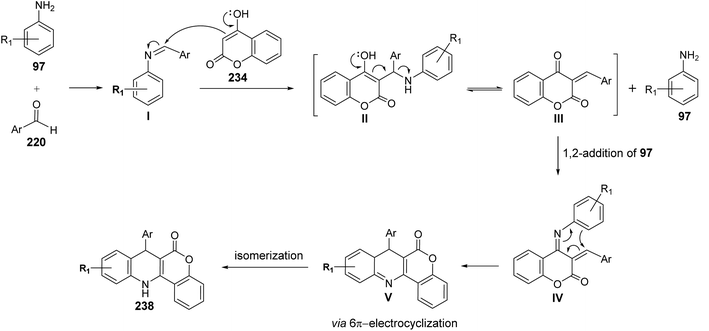 |
| | Scheme 80 Possible mechanism for the formation CFDQ 238. | |
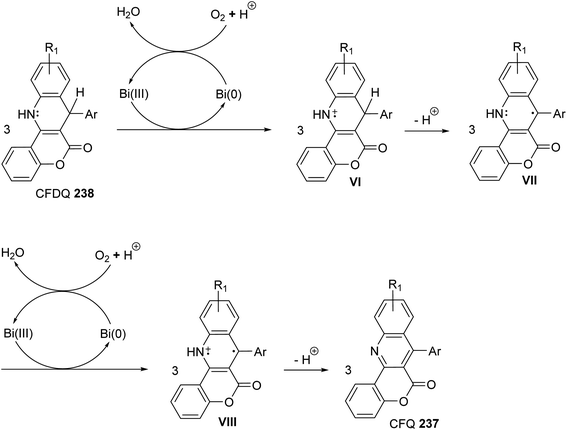 |
| | Scheme 81 Plausible mechanism for the formation CFQ 237 from CFDQ 238. | |
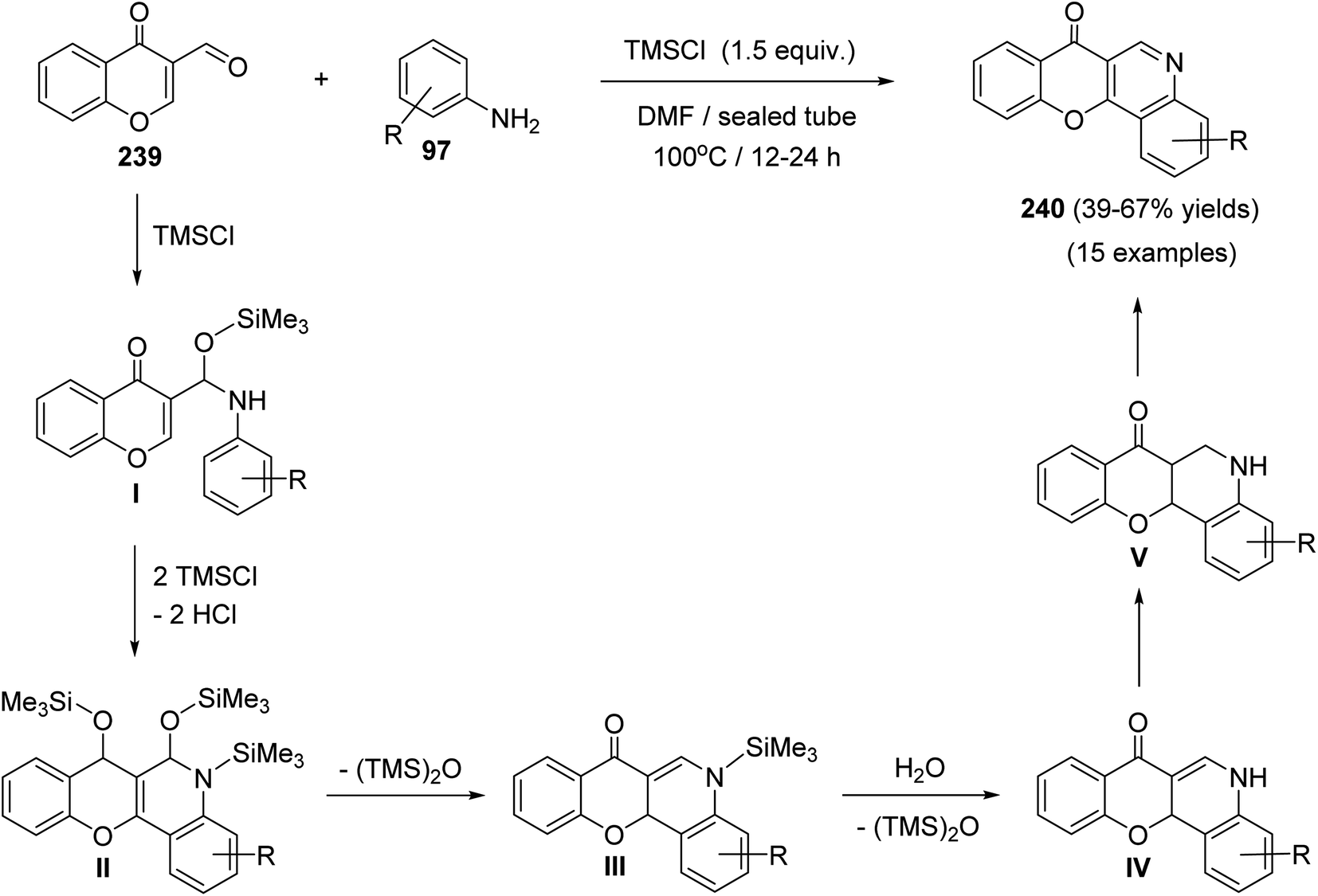
Plaskon et al.103 have described a versatile and facile method for the synthesis of 7H-chromeno[3,2-c]quinolin-7-ones 240 utilizing chlorotrimethylsilane (TMSCl) as a potential promoter and water scavenger. On heating a solution of 4-oxo-4H-chromene-3-carbaldehyde (239) and anilines 97 in the presence of TMSCl in DMF at 100 °C, the tetracyclic products 240 were obtained in 39–67% yields (Scheme 82).
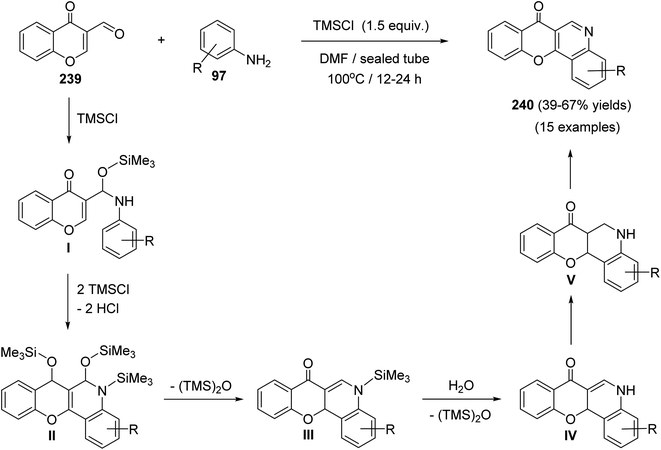 |
| | Scheme 82 TMSCl-mediated synthesis of 240 from 239. | |
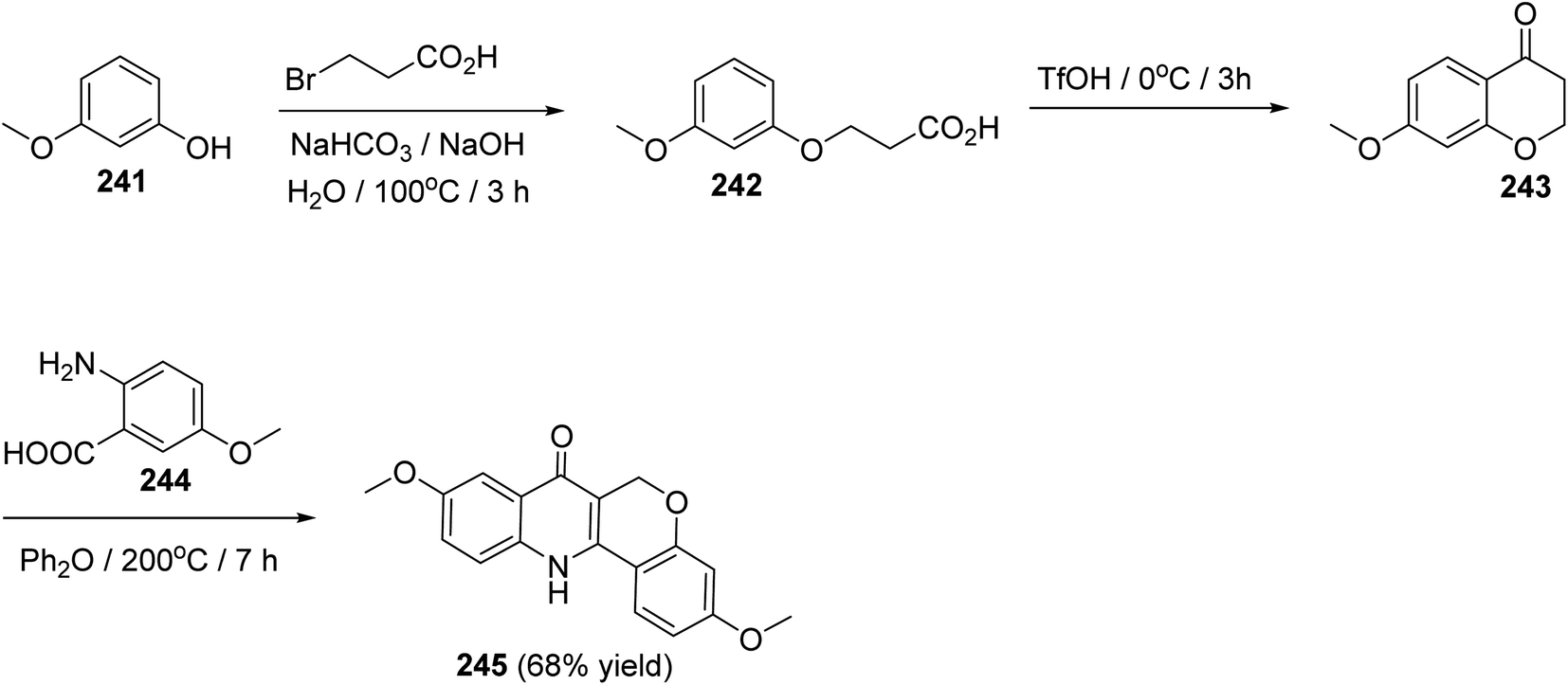
The synthesis of 3,9-dimethoxy-6,12-dihydro-6H-chromeno[4,3-b]quinoline-7-one (245), as a new class of estrogen receptor β-selective ligands, was achieved in three steps as shown in Scheme 83. Reactions were carried out by heating 3-methoxyphenol (241) with 3-bromo-propanoic acid under basic conditions at 100 °C for 3 h to give the corresponding 3-(3-methoxyphenoxy)propanoic acid (242), which underwent intramolecular Friedel–Crafts acylation when treated with TfOH at 0 °C for 3 h to provide 7-methoxychroman-4-one (243). By condensation of 243 with 5-methoxyanthranilic acid (244) in diphenyl ether at 200 °C for 7 h, the desired tetracyclic product 245 was formed in 68% yield (Scheme 83).104
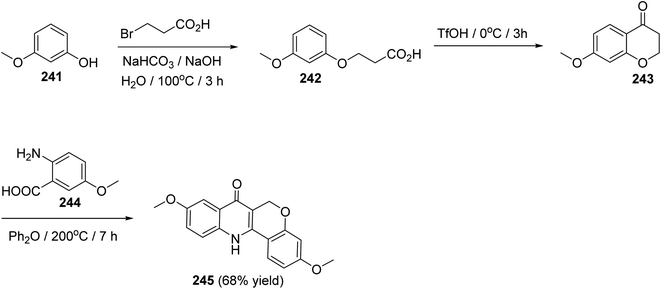 |
| | Scheme 83 Three steps synthesis of 3,9-dimethoxy-6,12-dihydro-6H-chromeno[4,3-b]-quinoline-7-one (245). | |
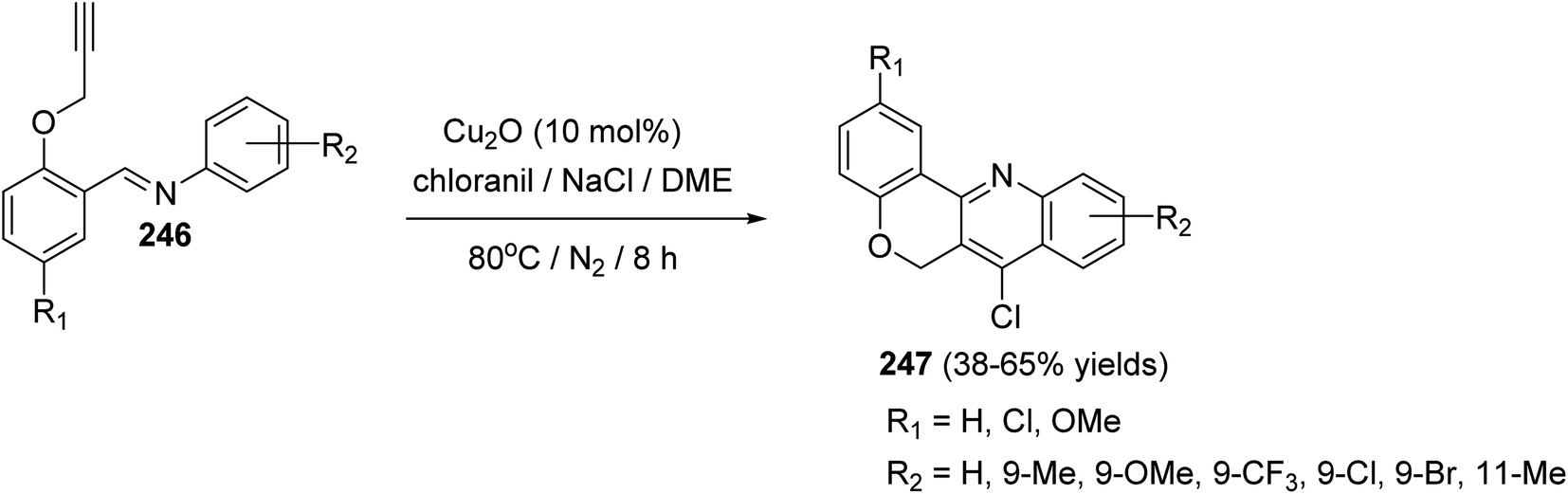
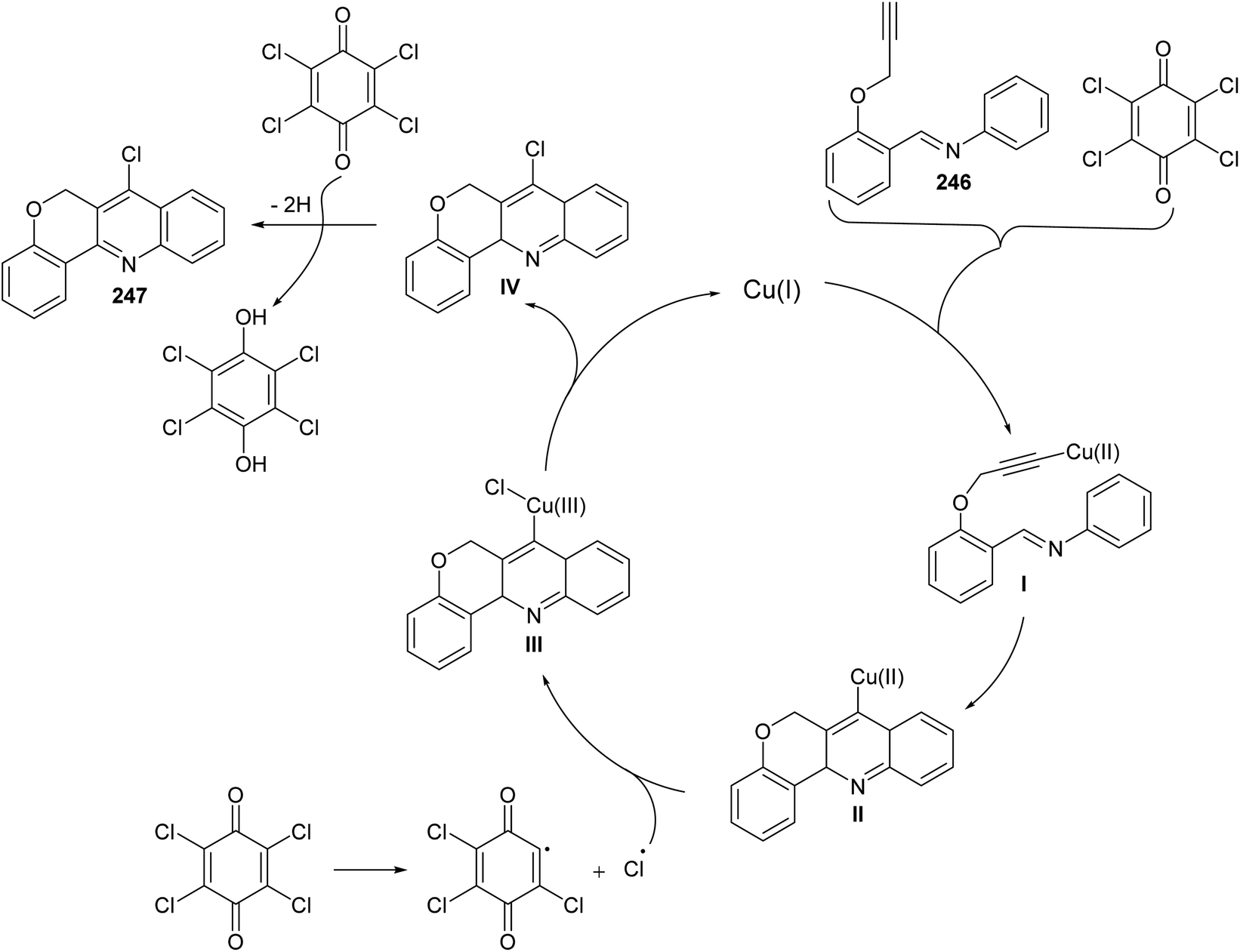
In 2016, Bao et al.105 have developed a new, efficient and convenient halogenation method to synthesize 7-halogenated chromeno[4,3-b]quinolines 247 through Cu-catalyzed cascade reactions (intramolecular aza-Diels–Alder reaction of Schiff base, followed by chlorination utilizing chloranil) (Scheme 84). The reaction was carried out by treatment Schiff base 246 (1 equiv.) with a mixture of Cu2O (10 mol%), p-chloranil (p-CHL) (2 equiv.), and NaCl (1.5 equiv.), as an additive, in dimethoxyethane (DME). The mixture was heated at 80 °C under a nitrogen atmosphere for 8 h to give the new tetracyclic product 247 in satisfactory yields (38–65%) (Scheme 84). In the proposed reaction mechanism, the reaction of Schiff base substrate 246 with a Cu(I) species in the presence of chloranil produced Cu(II) acetylide I, which undergoes intramolecular aza-Diels–Alder reaction to give intermediate II. The Cl radical originated from p-CHL reacts with intermediate II to afford intermediate III, which subsequently undergoes reductive elimination reaction to give a chlorinated intermediate IV and Cu(I) species. Oxidation of intermediate IV with p-CHL occurs to provide the desired tetracyclic product 247 (Scheme 85).
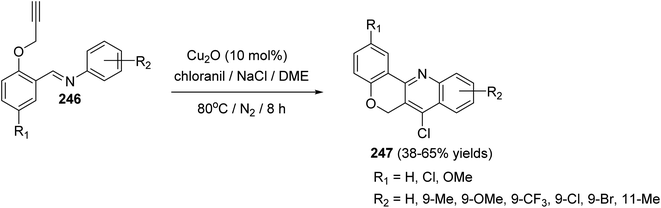 |
| | Scheme 84 Synthesis of 7-halogenated-6H-chromeno[4,3-b]quinolines 247 via Cu-catalyzed cascade reaction. | |
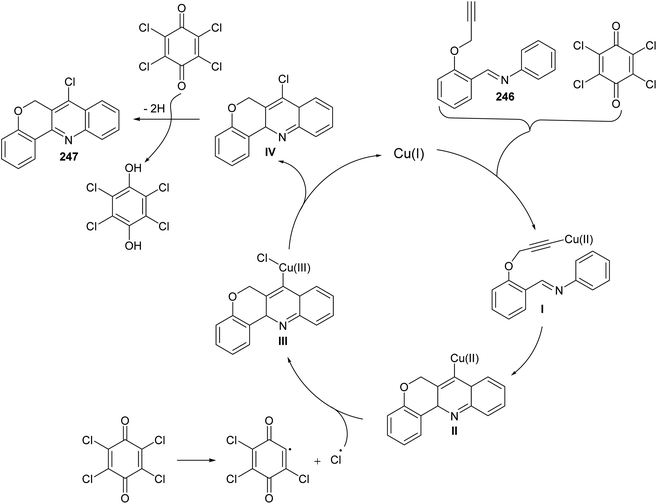 |
| | Scheme 85 A suggested mechanism for the formation of 247 from 246. | |
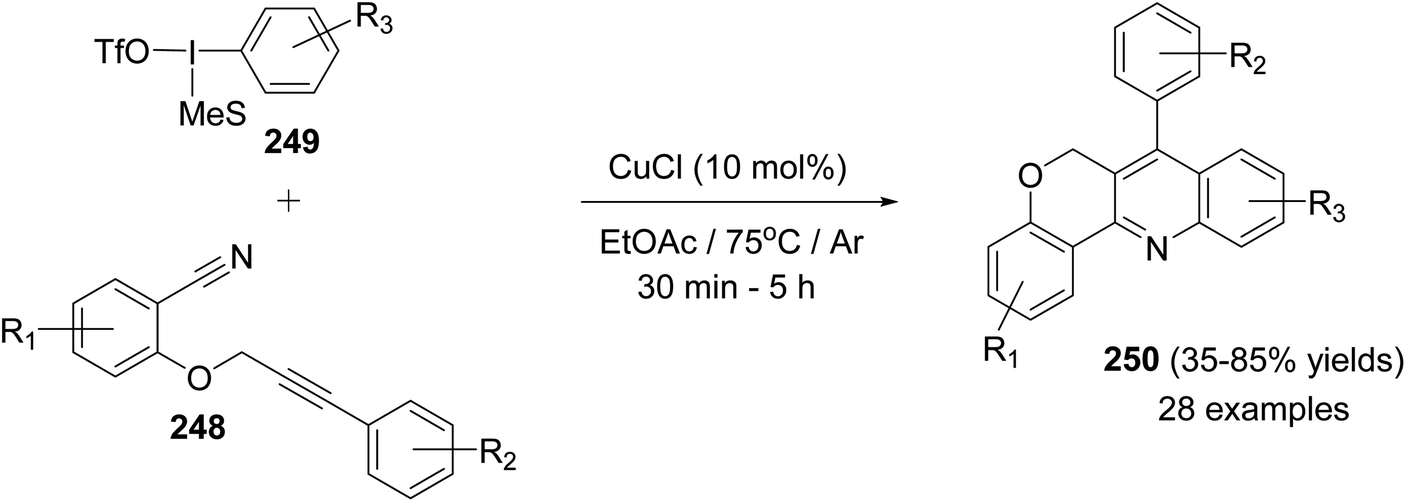
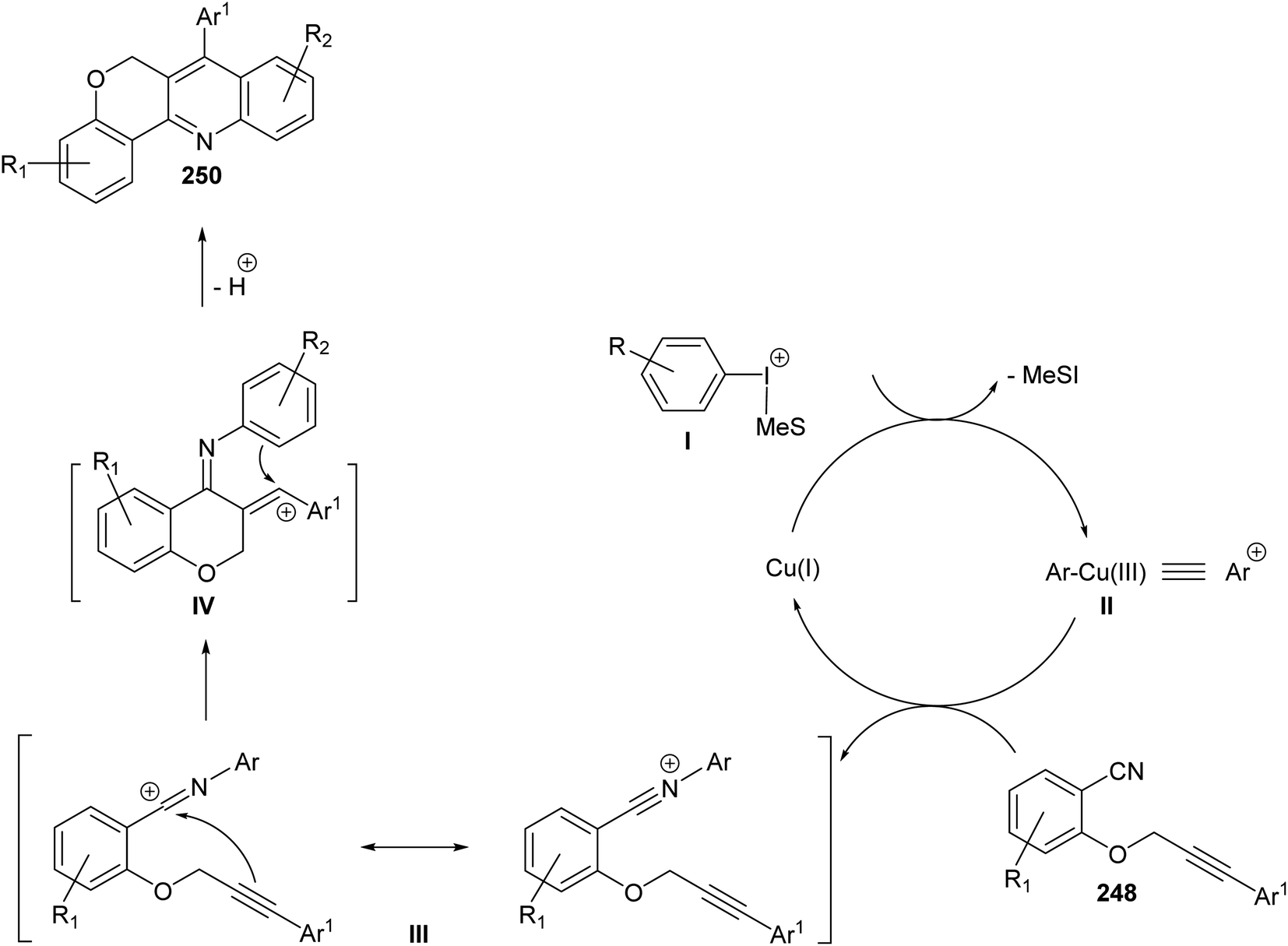
In the same year, Novák and his group106 reported the development of a novel and highly modular Cu-catalyzed oxidative transformation for the construction of chromeno-[4,3-b]quinolines 250, starting from arylpropynyloxybenzonitriles 248 and arylmesityl-iodonium triflates 249. Reaction of nitriles 248 with 249 in EtOAc in the presence of a catalytic amount of CuCl (10 mol%) at 75 °C for 30 min – 5 h afforded the polyfunctionally substituted chromeno[4,3-b]quinolines 250 in 35–85% yields (Scheme 86). A plausible mechanism was proposed (Scheme 87).
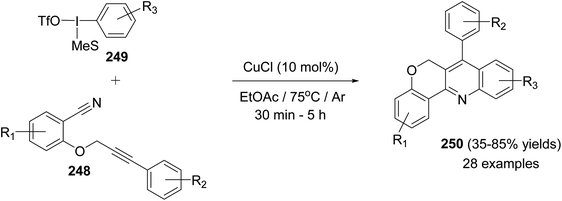 |
| | Scheme 86 Highly modular synthesis of chromeno[4,3-b]quinolines 250 via arylation-ring closure. | |
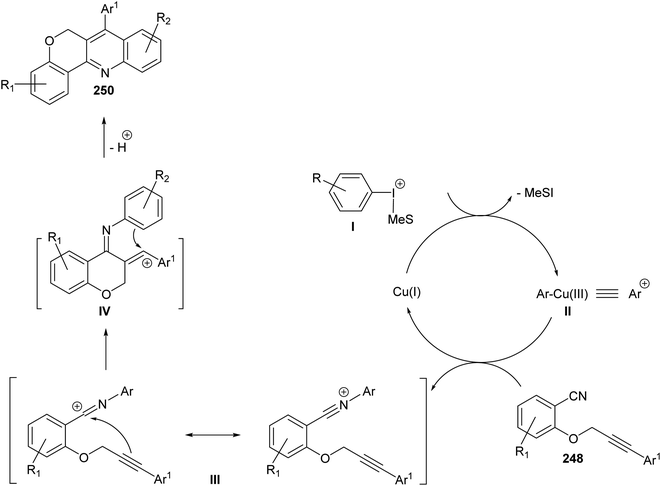 |
| | Scheme 87 A plausible mechanism for the formation of chromeno[4,3-b]quinolines 250 via arylation–cyclization reaction. | |
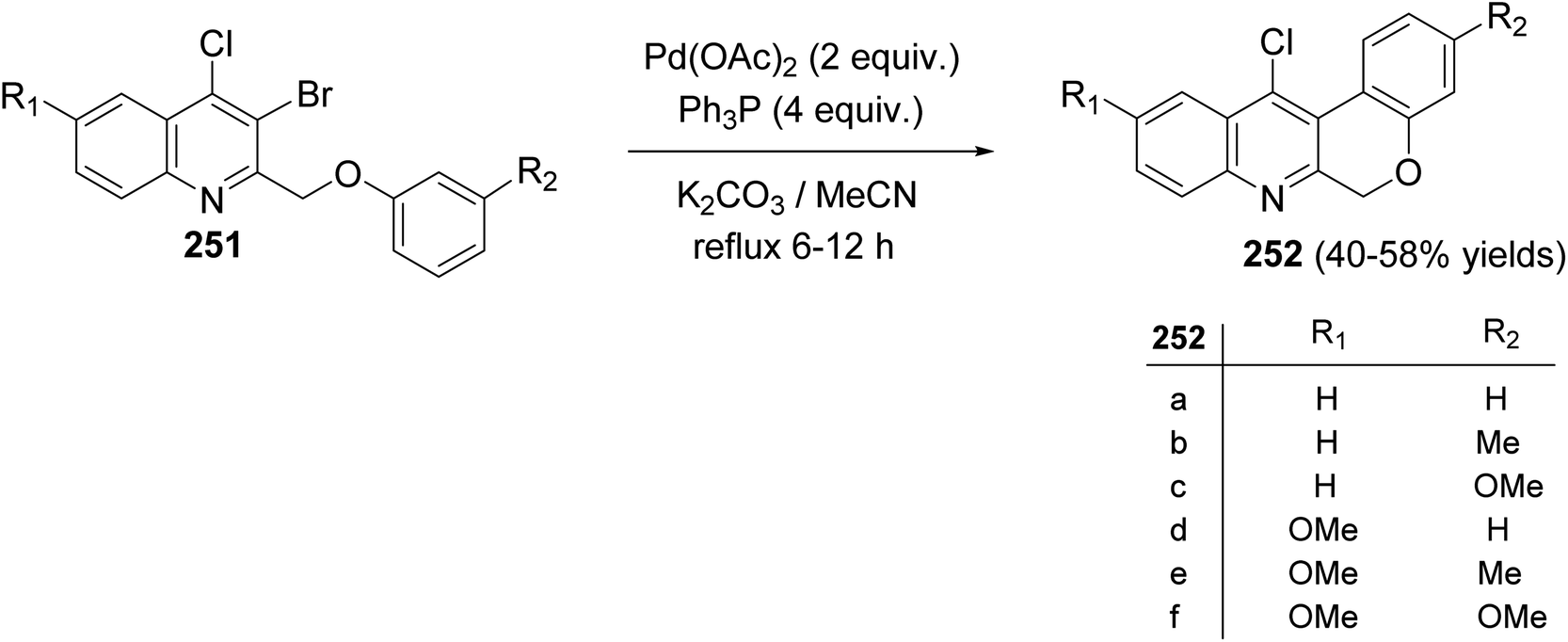
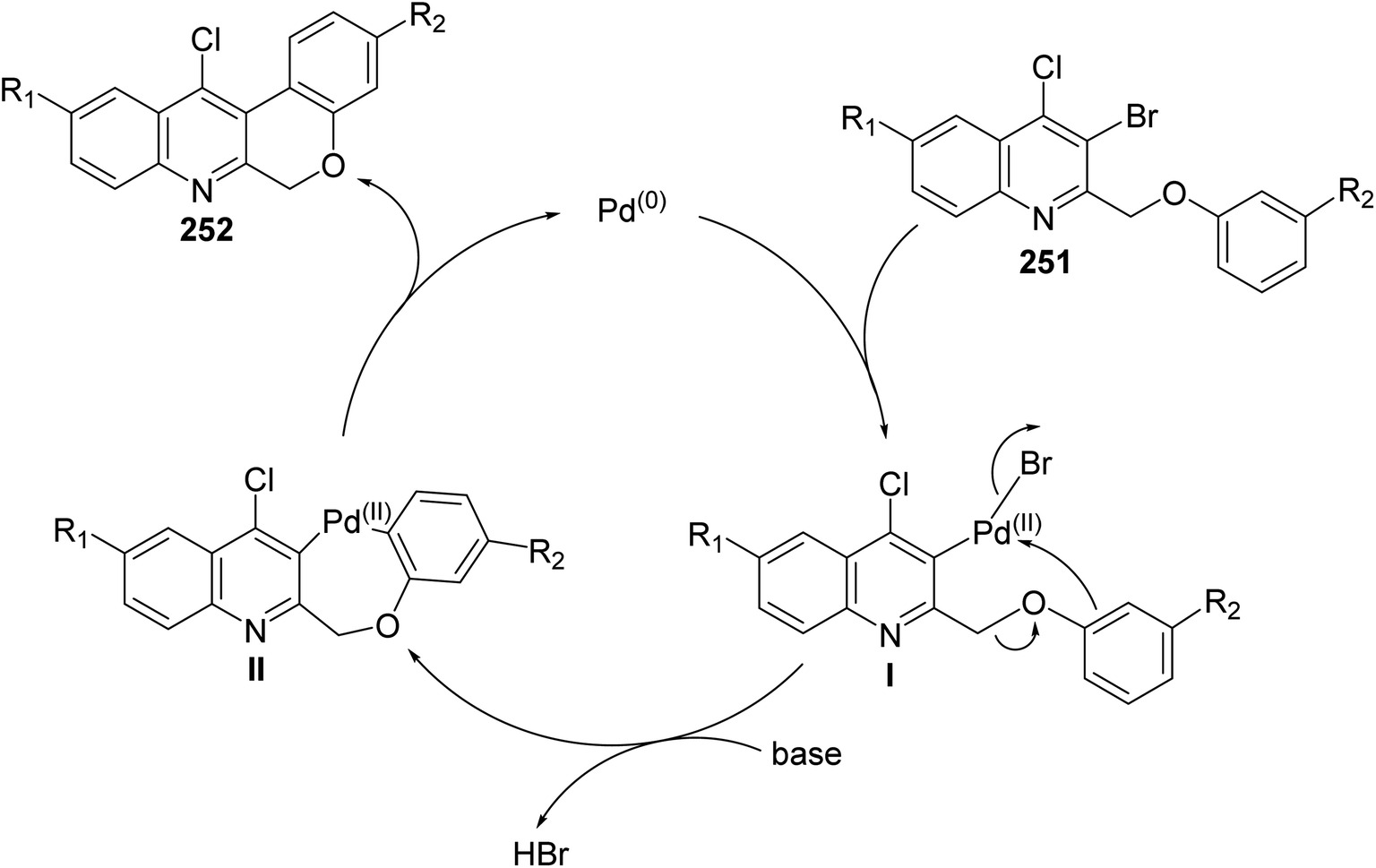
Morris and Jackson107 reported the Pd-catalyzed intramolecular cyclization of 2-[(3-substituted-phenoxy)methyl]quinolines 251 to prepare chromeno[3,4-b]quinolines 252. The cyclization was carried out, under typical Heck conditions, by refluxing a mixture of appropriate quinoline 251 (1 equiv.), Pd(OAc)2 (2 equiv.), triphenylphosphine (4 equiv.) and K2CO3 (20 equiv.) in CH3CN for 6–12 h under an atmosphere of nitrogen (Scheme 88). It was found that compound 251 containing a strong electron-withdrawing groups on the phenoxy ring retarded the cyclization and no products were obtained after heating at reflux for a long time, while electron-donating groups increased the rate of the reaction and produced the desired products 252 in reasonable yields (40–58% yields). A proposed mechanism for the formation of tetracyclic chromeno[3,4-b]quinolines 252 from 251 is shown in Scheme 89.
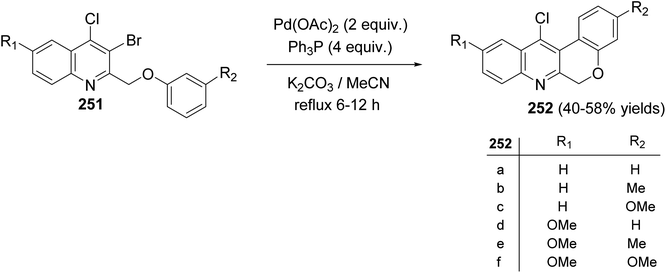 |
| | Scheme 88 Cyclization of 2-[(3-substituted-phenoxy)methyl]quinolines 251 to chromeno-[3,4-b]quinolines 252. | |
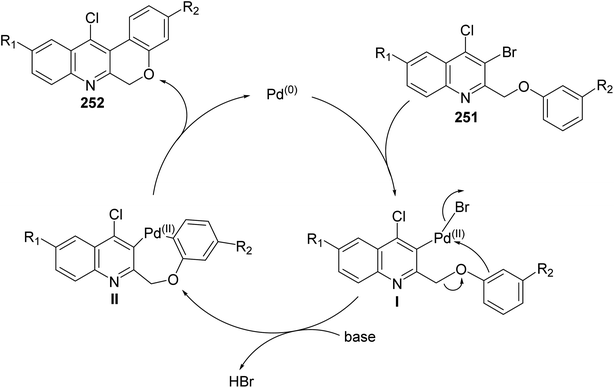 |
| | Scheme 89 Proposed mechanism for the formation of tetracyclic 252 from 251. | |
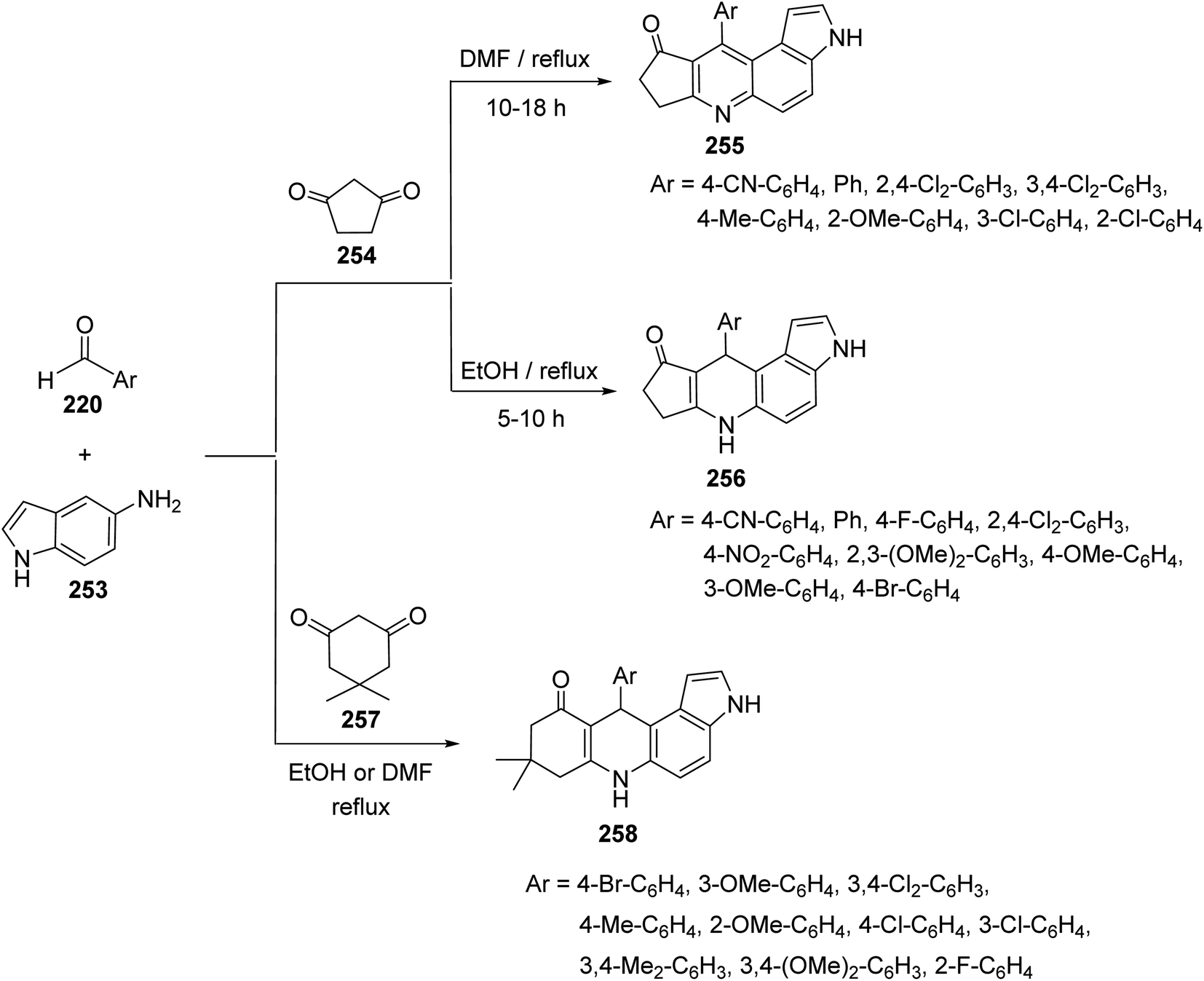
2.2.5. Cyclopenta[b]pyrrolo[3,2-f]quinolines. A combinatorial synthesis of fused heterocyclic compounds, cyclopenta[b]pyrrolo[3,2-f]quinolin-9(3H)-ones 255, 10-aryl-6,7,8,10-tetrahydrocyclopenta[b]pyrrolo[3,2-f]quinolin-9(3H)-ones 256 and 8,9-dihydro-8,8-dimethyl-11-aryl-3H-pyrrolo[3,2-a]acridin 10(6H,7H,11H)-ones 258, via three-component reaction of 1H-indol-5-amine (253), aromatic aldehydes 220 and 1,3-dicarbonyl compounds 254, 257, under catalyst-free conditions, has been reported by Zhou et al. in 2013.108 It was interesting that the designed reactions gave un-aromatized or aromatized products depending on the reaction temperature and the type of 1,3-dicarbonyl compounds. Treatment of aromatic aldehydes 220, 1H-indol-5-amine (253), and cyclopentane-1,3-dione 254 in refluxing DMF, without catalyst, resulted in the aromatized cyclopenta[b]pyrrolo[3,2-f]quinolin-9(3H)-ones 255, in high yields (83–92%). When the same reaction was carried out in refluxing EtOH, without catalysts, the un-aromatized 10-aryl-6,7,8,10-tetrahydrocyclopenta[b]pyrrolo[3,2-f]quinolin-9(3H)-ones 256 were obtained in 84–96% yields (Scheme 90). On the other hand, when 5,5-dimethylcyclohexane-1,3-dione (dimedone) (257) was subjected to react with 220 and 253, under catalyst-free conditions, the un-aromatized 11-aryl-8,9-dihydro-8,8-dimethyl-3H-pyrrolo[3,2-a]acridin-10(6H,7H,11H)-ones 258 were obtained, whether they were refluxed in DMF or EtOH (Scheme 90).
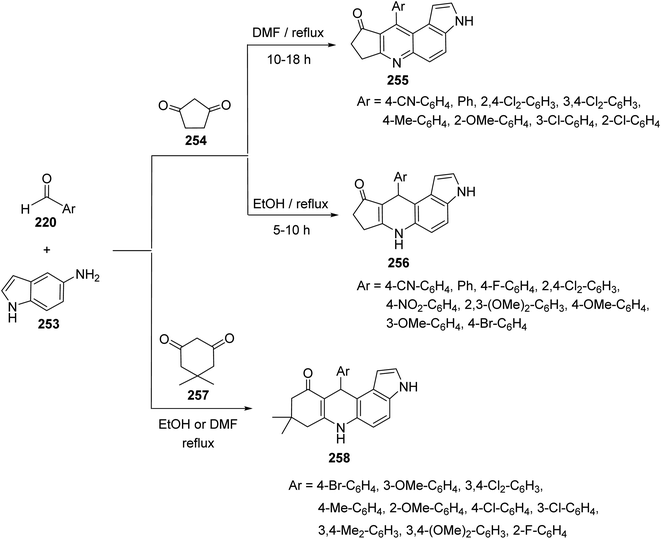 |
| | Scheme 90 One-pot synthesis of cyclopenta[b]pyrrolo[3,2-f]quinolins 255 and pyrrolo-[3,2-a]acridines 258 under catalyst-free conditions. | |
2.2.6. Benzopyranoquinolines. Meth-Cohn109 has developed an approach for the synthesis of 12-methyl-6-oxo-11,12-dihydro-6H-[2]benzopyrano[4,3-c]quinoline (262), in high yield. The reaction was accomplished by heating a mixture of N-methylformanilide (259) and homophthalic acid (260) in POCl3 at 100 °C for 10 min to give 12-methyl-6-oxo-6H-[2]benzopyrano[4,3-c]quinolinium phosphorodichloridate (261). The salt 261 showed kinetic presence for attack at the quinoline α-position. Thus, reduction of 261 with sodium borohydride in ethyl acetate at room temperature produced the desired benzopyranoquinoline 262 Scheme 91).
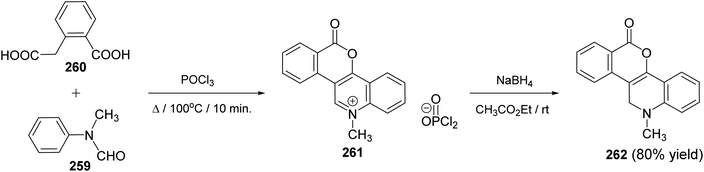 |
| | Scheme 91 Synthesis of benzopyrano[4,3-c]quinoline (262) from N-methylformanilide (259) and homophthalic acid (260). | |
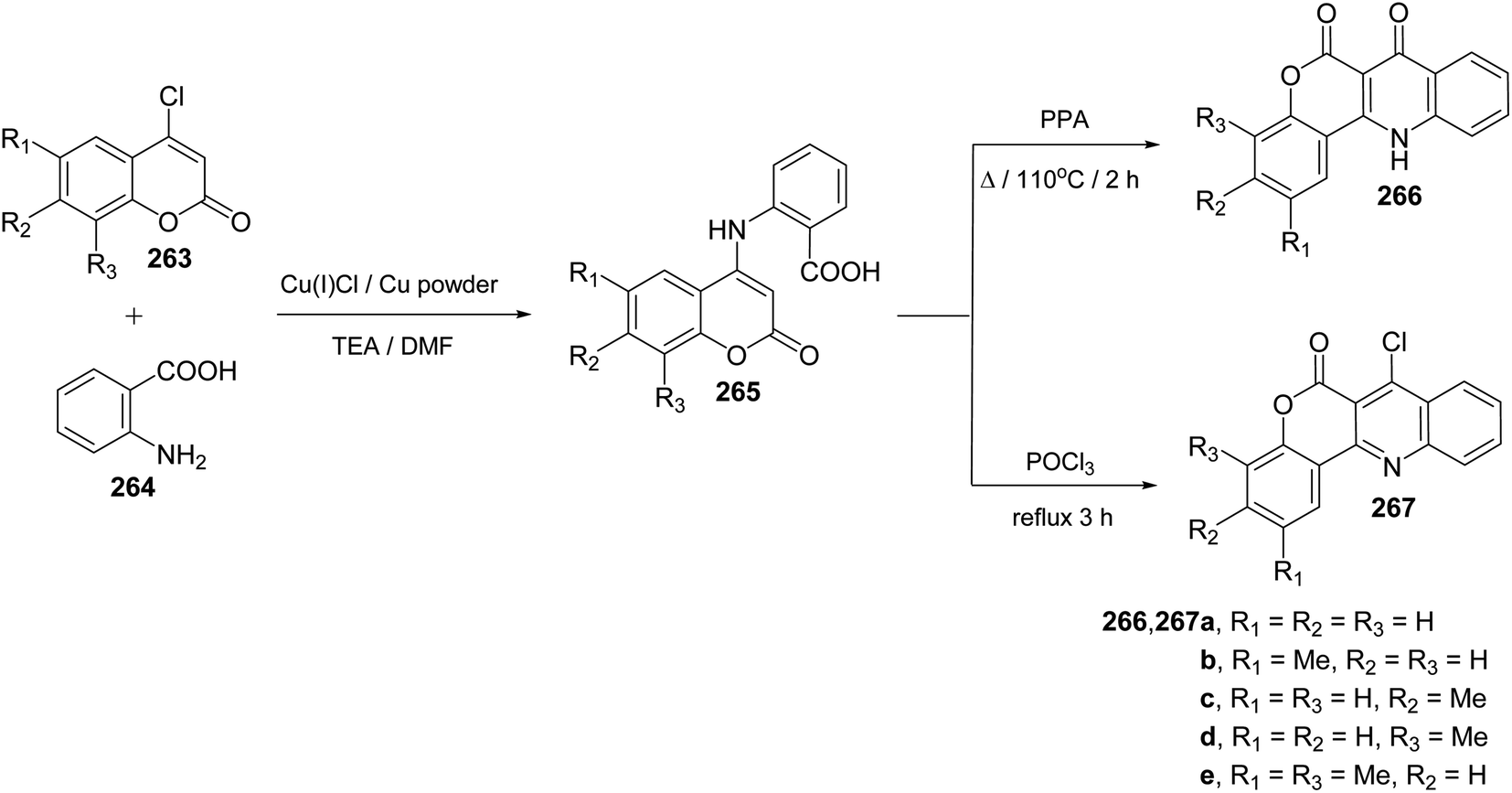
Ullmann Jordan condensation reaction of 4-chlorocoumarins 263 with anthranilic acid (264) afforded 4-(2′-carboxyphenylamino)-2H-[1]benzopyran-2-ones 265. When compounds 265 were heated with polyphosphoric acid (PPA) at 110 °C for 2 h, they underwent intramolecular cyclocondensation to furnish directly the tetracyclic ring system namely 7,12-dihydro-6H-[1]benzopyrano[4,3-b]quinoline-6,7-diones 266 in ∼90% yields (Scheme 92). On the other hand, refluxing 265 with phosphoryl chloride (POCl3) for 3 h afforded 7-chloro-benzo-pyrano[4,3-b]quinoline-6-ones 267 in ∼80% yields (Scheme 92).110
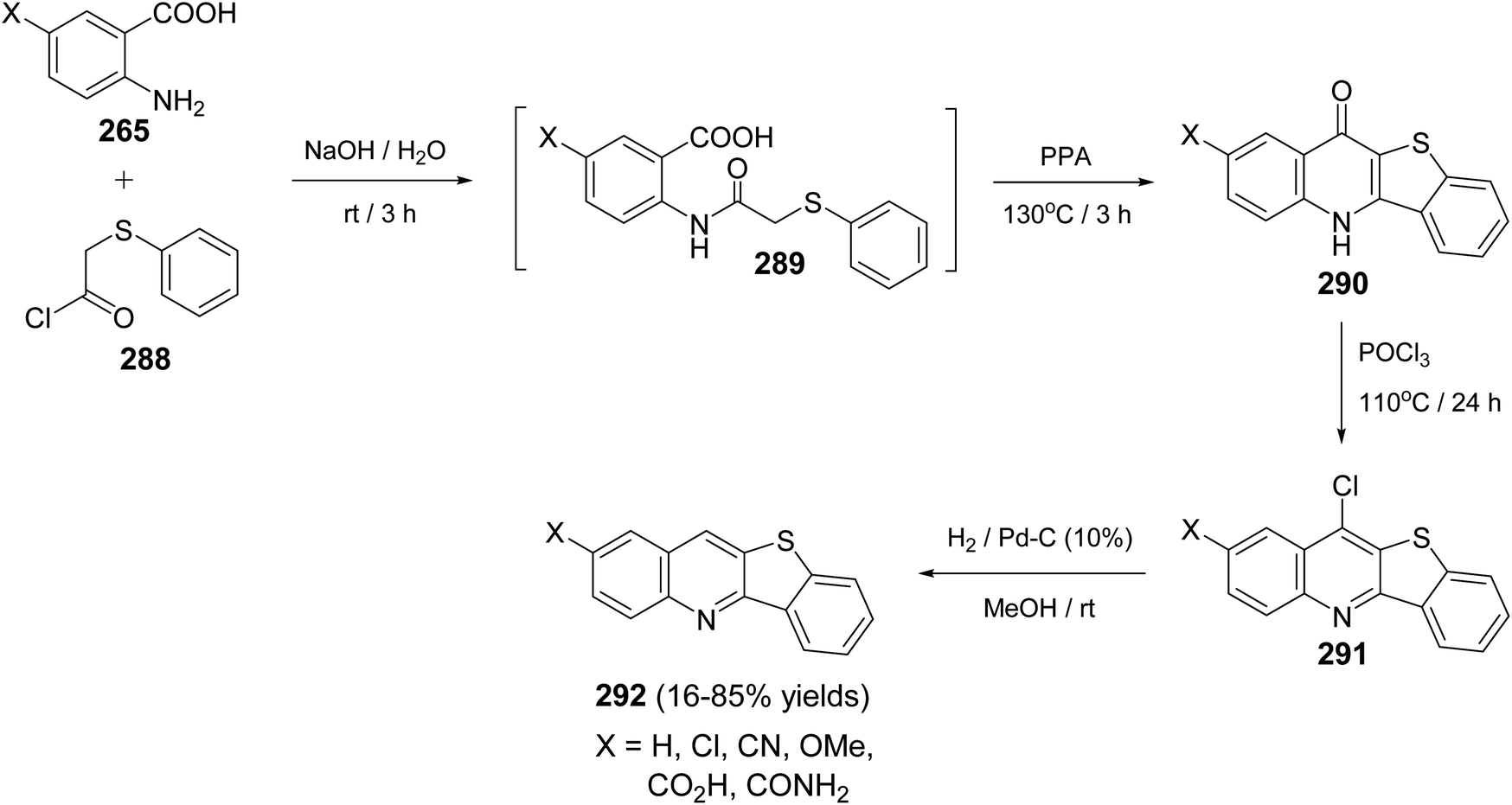
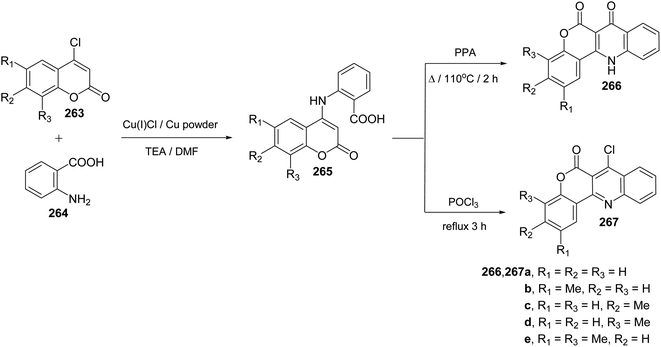 |
| | Scheme 92 Synthesis of benzopyrano[4,3-b]quinoline derivatives 266 and 267. | |
2.2.7. Benzothiopyranoquinolines. In 2003, Junjappa and his coworkers111 have reported a simple and highly efficient regioselective synthesis of new benzothiopyrano[2,3-b]quinolines 270 through TBTH/AIBN-mediated radical cyclization of the corresponding 3-(2-bromobenzoyl)-2-methylthioquinoline derivatives 268a, b. As shown in Scheme 93, the reaction proceeded by heating a solution of 268a, b (1 equiv.), TBTH (2.5 equiv.) and AIBN (catalytic) in toluene for 8–10 h under nitrogen atmosphere. The products were found to be not the tetracyclic quinolines 269, but were characterized as the novel benzothiopyrano[2,3-b]quinolines 270a, b on the basis of their spectral and analytical data. The proposed mechanistic pathway for radical cyclization of 268a, b is outlined in Scheme 93. The initially formed o-benzoyl radical 271 undergoes radical translocation by attack on the methylthio group (MeS–) to afford radical intermediate 272, which on loss of the methyl radical gives the benzothiopyranoquinolines 270a and 270b in quantitative and 98% yield, respectively.
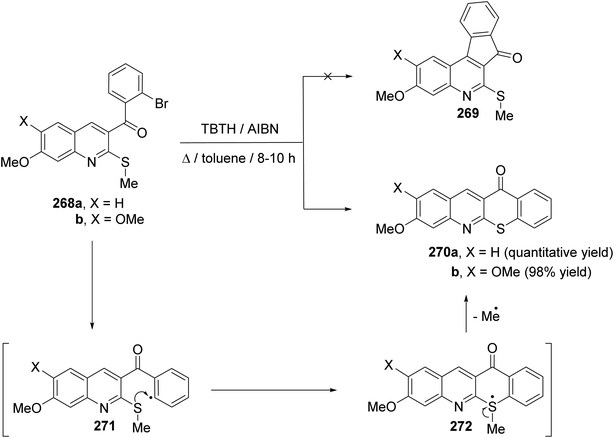 |
| | Scheme 93 A regioselective synthesis of new benzothiopyrano[2,3-b]quinolines 270. | |
2.2.8. Benzothienoquinolines. Deady et al.112 reported a one-pot, two-component synthesis of benzothieno[3,2-b]quinoline-4,11-dicarboxylic acid (275), as a new class of putative topoisomerase inhibitors, by the reaction of isatin-7-carboxylic acid (273) and benzo[b]thiophen-3(2H)-one (274). The reaction takes place at 100 °C for 8 h using NaOH (10%) as a base delivering the benzo-thieno[3,2-b]quinolines 275 in 55% yield (Scheme 94).
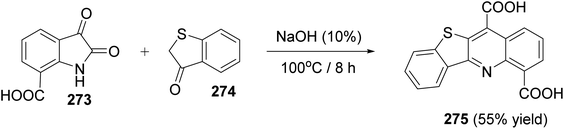 |
| | Scheme 94 A one-pot synthesis of benzothieno[3,2-b]quinoline-4,11-dicarboxylic acid (275). | |
In 2000, Rádl et al.113 developed a new procedure for the synthesis of benzothieno-[3,2-b]quinoline-11(5H)-one (280) in 88% yield, as shown in Scheme 95. Treatment of 2-mercaptobenzonitrile 276 with phenacylbromides 277a, b in DMF in the presence of K2CO3 at room temperature for 1 h gave the corresponding benzothiophenes 278, 279. When these compounds were stirred with NaH in DMF at room temperature for 2 h, they underwent intramolecular nucleophilic cyclization to provide the desired tetracyclic 280 (Scheme 95).
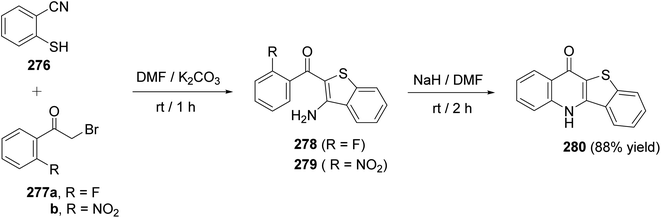 |
| | Scheme 95 A new method for the synthesis of benzothieno[3,2-b]quinoline-11(5H)-one (280). | |
In the same year, Zanardi et al.114 have devised a novel protocol for the one-pot construction of benzothieno[2,3-b]quinolines 284 based on a radical cascade reaction of the 2-(phenylalkynyl)aryl radicals 282 with some aryl isothiocyanates 283. The aryl radicals 282 were easily generated upon portionwise addition of the diazonium tetrafluoroborate 281 (1 equiv.) to a stirred pyridine solution of 283 (3 equiv.) at −10 to −20 °C. After one hour, the reaction mixture was normally subjected to chromatographic separation to give the desired benzothieno[2,3-b]quinolines 284 in 30–60% yields (Scheme 96). The reaction mechanism for the formation of 284 is outlined in Scheme 97. The aryl radicals 282 attacks the sulfur atom of the isothiocyanate 283 giving α-sulfanylimidoyl radical 285. Subsequent tandem 5-exo-dig cyclization onto the adjacent ethynyl moiety gave a linear 1-phenylvinyl radical 286. The latter intermediate 286 underwent intramolecular 6-membered cyclization onto the isothiocyanate ring to give the benzothienoquinolines 284, through the cyclohexadienyl radical 287.
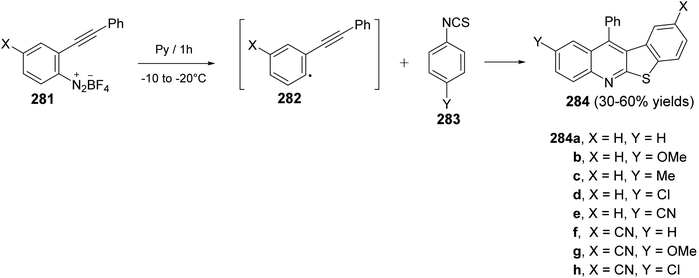 |
| | Scheme 96 Synthesis of benzothieno[2,3-b]quinolines 284 via reaction of 2-ethynylaryl radicals 282 with aryl isothiocyanates 283. | |
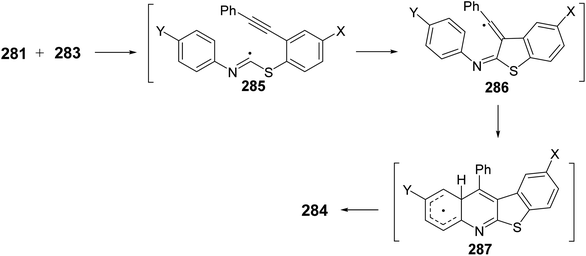 |
| | Scheme 97 Reaction mechanism for the formation of benzothieno[2,3-b]quinolines 284. | |
Zhu et al.115 in 2007, synthesized benzothieno[3,2-b]quinolines 292 by the reaction of anthranilic acid derivatives 265 with 2-(phenylthiol)acetyl chloride (288). In this reaction the substituted anthranilic acids 265 were acylated with 288 under basic conditions to give the intermediates 289. These intermediates 289 then underwent a double intramolecular cyclization reaction with polyphosphoric acid (PPA) at 130 °C for 3 h to furnish benzothieno[3,2-b]quinoline-11(5H)-ones 290. Chlorination of 290 with POCl3 at 110 °C for 24 h provided the corresponding chloro compounds 291, which hydrogenated in H2 atmosphere on Pd/C (10%) in MeOH at room temperature to give the desired tetracylic benzothieno[3,2-b]quinoline derivatives 292 in 16–85% yields (Scheme 98).
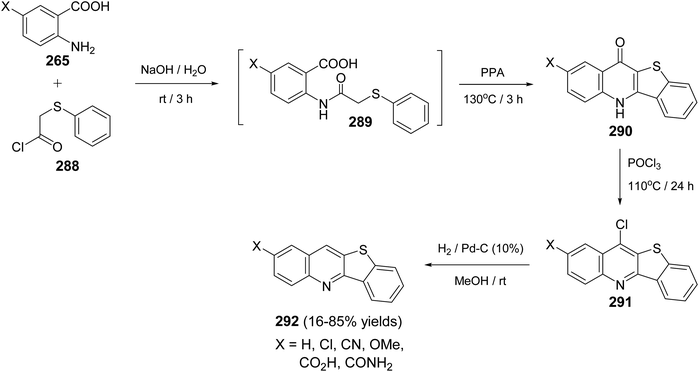 |
| | Scheme 98 Synthesis of benzothieno[3,2-b]quinolines 292 from 265 and 288. | |
Langer and his group,116 developed a highly efficient synthesis of new benzothieno-[3,2-b]quinolines 296 via the Pd-catalyzed reaction of 2-alkynyl-3-bromobenzothiophenes 293 with anilines 97. When a mixture of 293 (1 equiv.), 97 (1.3 equiv.), Pd(OAc)2 (10 mol%), Pd(t-Bu)3·HBF4 (20 mol%), KO-t-Bu (2 equiv.) and CuI (25 mol%) was heated in toluene at 105 °C for 12 h, the desired benzothieno[3,2-b]quinolines 296 were indeed produced in 60–70% yields via intermediates 294 and 295 (Scheme 99).
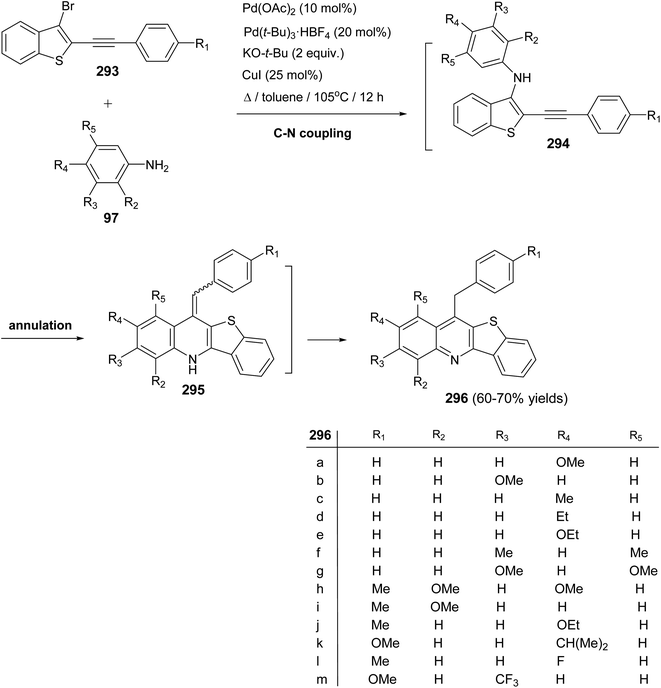 |
| | Scheme 99 A highly efficient synthesis of new benzothieno[3,2-b]quinolines 296 via the Pd-catalyzed reaction of 293 with 97. | |
A new one-pot method for the synthesis of benzothieno[3,2-b]quinoline (300) from the reaction of 3-bromobenzo[b]thiophene-2-carbaldehyde (297) with 2-aminophenylpinacol-borane (298) under Suzuki coupling conditions utilizing a stereochemically hindered ligand, 2-(cyclo-hexylphosphane)biphenyl and Ba(OH)2·8H2O, as the base, was reported by Castanheira and co-workers117 in 2014. This synthetic approach proceeded by heating a mixture of 297 (1 equiv.), 298 (1 equiv.), Pd(AcO)2 (5 mol%), 2-(cyclohexylphosphane)-biphenyl (20 mol%), Ba(OH)2·8H2O (3 equiv.) in dioxane at 100 °C for 5 h to give the tetracyclic product 300 in 30% yield (Scheme 100). The formation of 300 is assumed to proceed via a Pd-catalyzed C–N coupling followed by an intramolecular cyclization that probably occur by nucleophilic attack of the activated ortho position of the diarylamine intermediate 299 on the aldehydic carbonyl group, after deboronation (Scheme 100).
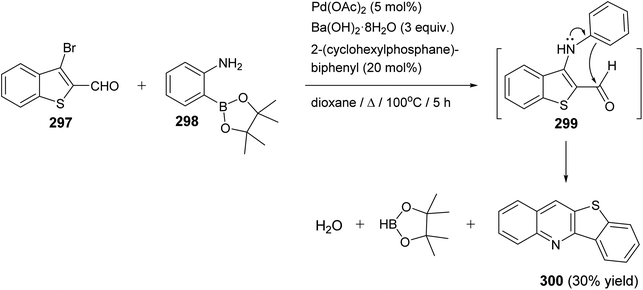 |
| | Scheme 100 One-pot synthesis of benzothieno[3,2-b]quinoline (300). | |
In 2009, Wang's group118 have developed a simple and efficient route for construction of several tetracyclic quinolines viz. benzo[f]pyrano[3,4-c]quinolines 305, benzo[f]thiopyrano[3,4-c]quinolines 306 and benzo[f]thieno[2,3-c]quinolines 307 via three-component reaction of aromatic aldehydes 220, naphthalene-2-amine (301) and heterocyclo-ketones 302–304, including tetrahydropyran-4-one 302, tetrahydrothiopyran-4-one 303 and dihydro-thiophen-3(2H)-one 304, in THF catalyzed by molecular iodine (5 mol%) at reflux temperature (Scheme 101). The mechanism involves the condensation reaction between amine 301 and aromatic aldehydes 220 to form the imine I, which reacts with iodine to produce the activated intermediate II. The enol form of cyclic ketones 302–304 attacks the latter intermediate II to give intermediate III, followed by an intramolecular cyclization to produce IV. Subsequent dehydration of IV affords dihydroquinoline V, which then undergoes air auto-oxidation to produce aromatized tetracyclic quinolines 305–307 (Scheme 102).
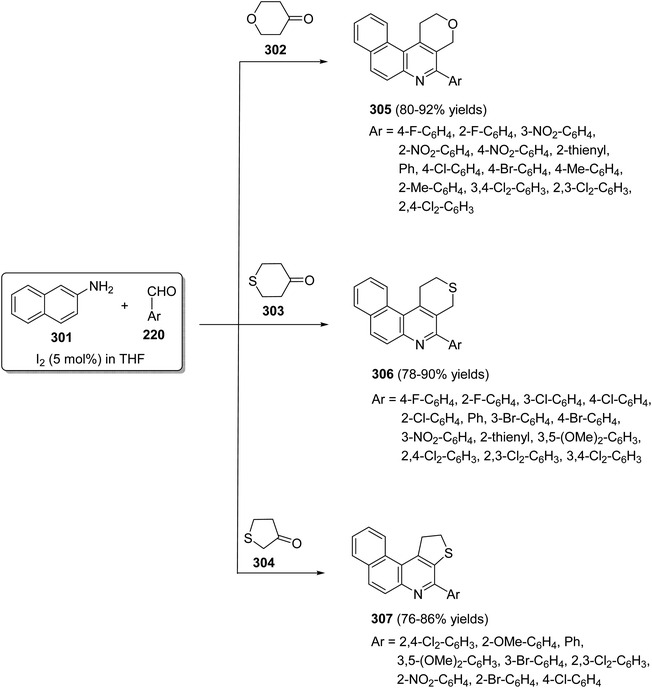 |
| | Scheme 101 Molecular iodine catalyzed synthesis of fused tetracyclic quinolines 305–307. | |
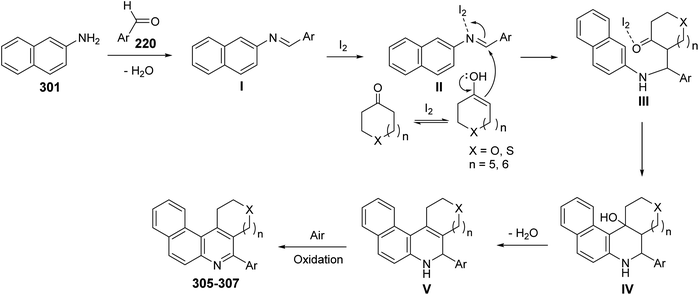 |
| | Scheme 102 A plausible mechanism for the synthesis of products 305–307. | |
2.3. Tetracyclic quinolines with three heteroatoms
2.3.1. Furothiopyrano[2,3-b]quinolines. Recently, Singh and his co-workers119 have reported a facile synthesis of tetracyclic, furothiopyrano[2,3-b]quinoline derivatives 312 via base-free I2-catalyzed cyclization reactions of 3-homoallylquinolin-2-thiones 311. The starting 3-homoallylquinoline-2-thinoes 311 were prepared, in two steps, from reaction of 2-chloroquinoline-3-carbaldehydes 308 with allyl bromides 309 in DMF (Barbies reaction), followed by treatment with Na2S in DMF at room temperature (Scheme 103). The cyclization reaction of 311 with I2 in THF at room temperature afforded the required tetracyclic quinoline, furothiopyrano[2,3-b]quinolines 312, in 80–90% yields (Scheme 103). All reactions were completed under aerobic conditions. The plausible mechanism for the formation of tetracyclic furothiopyranoquinolines 312 is shown in Scheme 104. The electrophilic addition of I2 to olefinic bond of 3-homo-allaylquinoline-2-thiones 311 leads to the formation of iodonium ion 313, which undergoes intramolecular attack by sulfide anion to give the tricyclic intermediate 314. Neighboring group participation of sulfur atom on 2-iodomethyl carbon atom in 314 gives the sulfonium salt intermediate 315. Finally, sulfonium salt ring open by the hydroxy group to give the tetracyclic ring system 312.
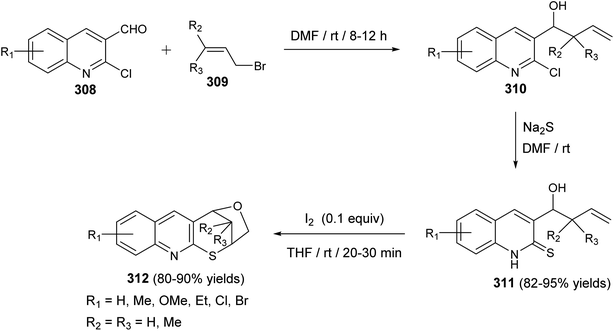 |
| | Scheme 103 Synthesis of furothiopyrano[2,3-b]quinolines 312 via iodocyclization of 3-homoallylquinolin-2-thiones 311. | |
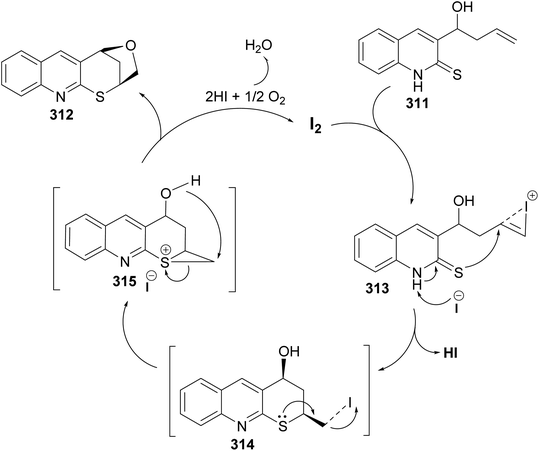 |
| | Scheme 104 Plausible mechanism for the formation of furothiopyrano[2,3-b]quinolines 312. | |
2.3.2. Quinoxalino[2,3-b]quinolines. In 2014, Che et al.120 described a rapid synthesis of fused tetracyclic quinolines, 6,11-dihydroquinoxalino[2,3-b]quinolines 319, a family of novel heterocycles with potential antitumor activity, via a sequential Ugi-variant multicomponent reaction and Pd-catalyzed bis-annulation in one-pot procedure. Reaction of cinnamic aldehydes 316, two molecules of o-iodoanilines 317 and isocyanides 318 was carried out in MeOH at room temperature in the presence of p-toluenesulfonic acid (PTSA). The reaction mixture was then treated with Pd2(dba)3, 2-dicyclohexylphosphino-2′-methylbiphenyl (Me-phos) and cesium carbonate (CsCO3) in MeCN and refluxed to produce the desired tetracyclic 6,11-dihydroquinoxalino-[2,3-b]quinolines 319, in 36–61% yields (Scheme 105). It was found that the electronic properties of substituents on the aromatic ring of cinnamic aldehyde had no effect on the reactivity, while the anilines with either electron-withdrawing or electron-donating group showed a slightly lower reactivity.
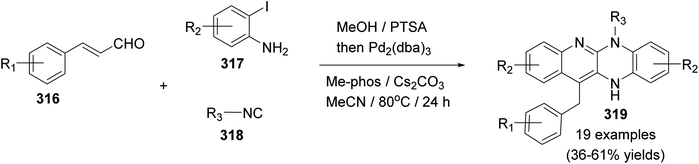 |
| | Scheme 105 Synthesis of quinolino[3,2-b]quinoxalines 319 in one-pot process. | |
2.3.3. Pyridoimidazoquinolines. Tetracyclic ring system combining an imidazo[1,2-a]pyridine skeleton condensed with a quinoline nucleus are stimulating increasing interests since they are close isosteres of a series of powerful antiproliferative compounds such as datelliptium, pazellipticine as well as ellipticinem.121
2.3.3.1. Pyrido[2′,1′:2,3]imidazo[4,5-c]quinolines. In 2015, Fan et al.121 developed a facile and unprecedented methodology for the synthesis of pyrido[2′,1′:2,3]imidazo[4,5-c]quinolines 322 via Cu-catalyzed one-pot four-component sequential reactions of 2-aminopyridine (320), 2-bromophenacyl bromide (321), aldehydes 220 and aq. NH3. The reactions were performed in three steps as shown in Scheme 106. (1) treatment of 320 with 321 in the presence of K3PO4·3H2O, as a base, in DMF at 80 °C for 4 h; (2) the reaction mixture was added with CuI, 1,10-phenanthroline (1,10-Phen) and aq. NH3 and then stirred at 80 °C for 8 h; (3) finally, the resulting mixture was added with HCl and aldehydes 220 and then stirred at 80 °C under air for 4 h to produce the desired tetracyclic 322 in 35–63% yields. It was shown that phenyl substituted aldehydes with either electron-withdrawing or electron-donating substituents on the phenyl ring underwent this cascade process easily to furnish 322 in good yields. A plausible mechanism to account for the formation of 322 is suggested in Scheme 107. The reaction is presumed to take place by an initial condensation of 320 with 321 to afford 2-(2-bromophenyl)imidazo[1,2-a]pyridine (I). The following Cu-catalyzed amination of I with NH3, as the nitrogen source, through intermediates II and III gives 2-imidazo[1,2-a]pyridine-2-yl-phenylamine (IV). Condensation of IV with aldehydes 220 affords the corresponding imine intermediate V. The subsequent intramolecular nucleophilic cyclization of V affords VI, which then undergoes an oxidative dehydrogenation under these reaction conditions to give 322 as the final products.
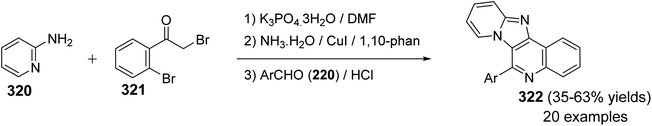 |
| | Scheme 106 Synthesis of pyrido[2′,1′:2,3]imidazo[4,5-c]quinolines 322. | |
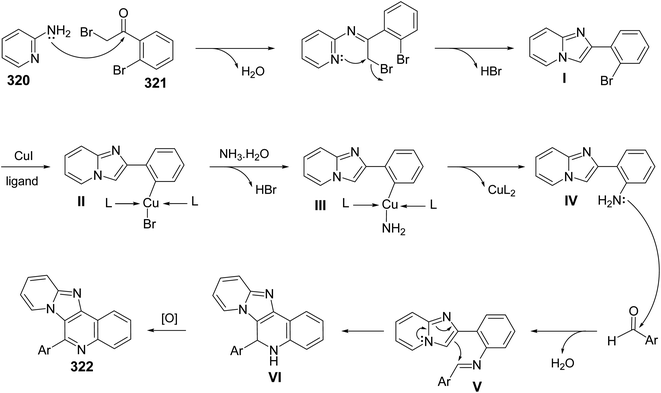 |
| | Scheme 107 Plausible mechanism for the formation of pyrido[2′,1′:2,3]imidazo[4,5-c]-quinolines 322. | |
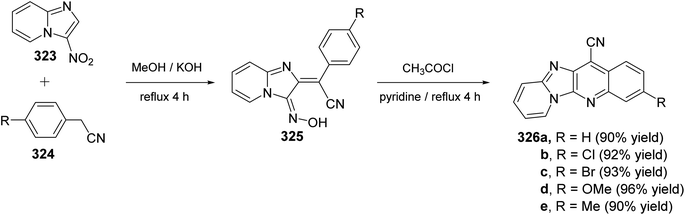 |
| | Scheme 108 Synthesis of new fluorescent tetracyclic compounds 326. | |
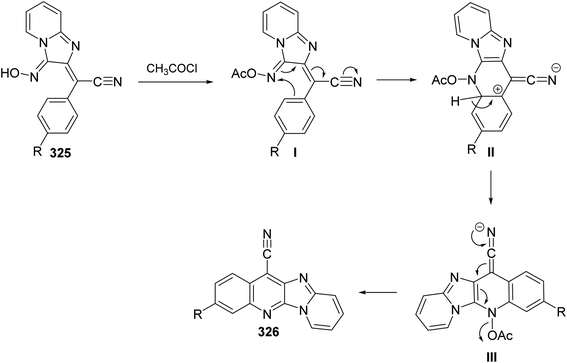 |
| | Scheme 109 Proposed mechanism for the formation of pyrido[2′,1′:2,3]imidazo[4,5-b]-quinolines 326. | |
A new strategy for the synthesis of pyrido[2′,1′:2,3]imidazo[4,5-b]quinoline (328) via a regioselective auto-tandem Pd-catalyzed inter- and intramolecular double Buchwald–Hartwig amination of 2-chloro-3-iodoquinoline (327) with aminopyridine (320) was reported.123 When a mixture of 327 (1 equiv.), 320 (1 equiv.), Pd(OAc)2 (4 mol%), XANTPHOS (4 mol%), Cs2CO3 (4 equiv.) in toluene was heated at 140 °C (pressure tube) under N2 atmosphere for 17 h, the tetracyclic 328 was obtained in 96% yield (Scheme 110).
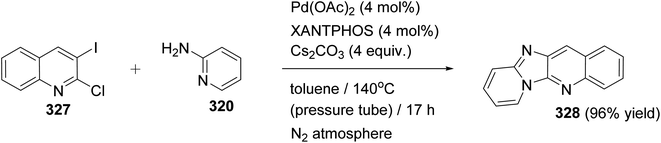 |
| | Scheme 110 Synthesis of pyrido[2′,1′:2,3]imidazo[4,5-b]quinoline (328) via auto-tandem amination on 327 with 320. | |
Guillaumet et al.124 have developed a rapid and efficient metal-free method for the synthesis of pyrido[2′,1′:2,3]imidazo[4,5-b]quinolines 330 from 2-(arylethynyl)imidazo[1,2-a]pyridin-3-amines 329 catalyzed by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). Microwave irradiation of 329 in 1,4-dioxane with a catalytic amount of DBU at 220 °C produced a series of unusual fused heterocyclic quinolines 330, in 65–97% yields (Scheme 111). A proposed reaction pathway for the formation of 330 is shown in Scheme 111. The electron rich secondary amines 329 assists in the hydroarylation of the triple bond after deprotonation by DBU. Next, aromatization produces the respective pyrido[2′,1′:2,3]imidazo[4,5-b]quinolines 330.
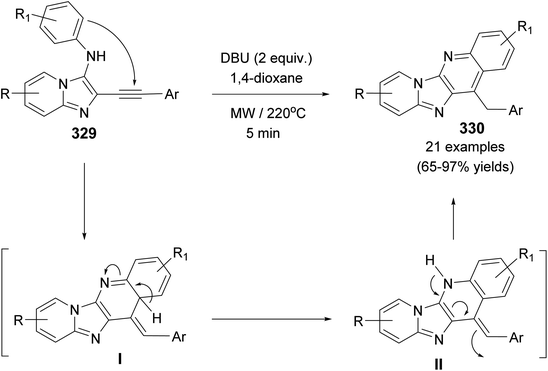 |
| | Scheme 111 DBU-catalyzed synthesis of pyrido[2′,1′:2,3]imidazo[4,5-b]quinolines 330. | |
2.3.4. Imidazo[1′,2′:1,2]pyrrolo[3,4-c]quinolines. A simple and new synthetic route for the synthesis of imidazo[1′,2′:1,2]pyrrolo-[3,4-c]quinolin-11-ones 334 based on the cascade reaction of isatins with heterocyclic ketene aminals (HKAs) has developed by Yu et al.125 in 2011. Refluxing a reaction mixture of isatins 57 and five-member heterocyclic ketene aminals (HKAs) 331 in toluene catalyzed by AcOH, forming the respective products 334 with good to excellent yields (78–94%) (Scheme 112). A suggested mechanism for the formation of 334 is shown in Scheme 112. At first, the α-C of 331 was added to the more electrophilic carbonyl center of 57 to furnish the intermediate 332. This intermediate underwent imine–enamine tautomerization, intramolecular cyclization, dehydration, and ring-opening reactions to produce the further intermediate 333. Finally, protonation of 333 followed by intramolecular cyclization resulted in the formation of the target tetracyclic 334.
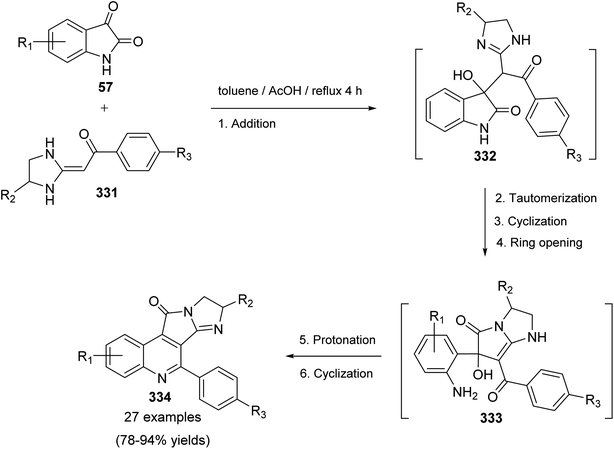 |
| | Scheme 112 Acetic acid catalyzed synthesis of imidazo[1′,2′:1,2]pyrrolo[3,4-c]quinolin-11-ones 334 via cascade of reactions. | |
2.3.5. Cyclopenta[c]pyrazolo[4,3-f]quinolines. A three-component imino Diels–Alder reaction of 1H-indazol-5-amine (335), aromatic aldehydes 220 and cyclopentanone in boiling THF catalyzed by molecular iodine led to the formation of a series of cyclopenta[c]pyrazolo[4,3-f]quinolines 336 in high yields (Scheme 113).126
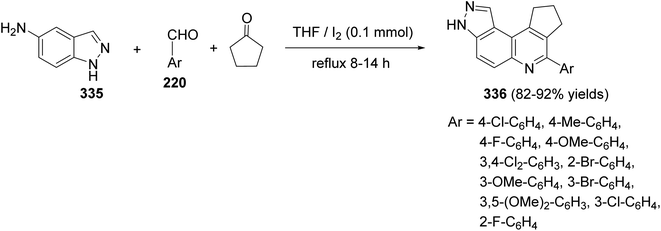 |
| | Scheme 113 Molecular I2 catalyzed synthesis of cyclopenta[c]pyrazolo[4,3-f]quinolines 336. | |
2.3.6. Benzo[h]pyrimido[4,5-b]quinolines. Guo and Yu127 reported a three component reaction between 1-naphthylamine (337), aromatic aldehydes 220 and barbituric acid (338) in ionic liquid [bmim]BF4, as a green solvent, produced a novel tetracyclic 7-aryl-11,12-dihydrobenzo[h]pyrimido[4,5-b]quinoline-8,10(7H,9H)-diones 339, in 76–92% yields (Scheme 114).
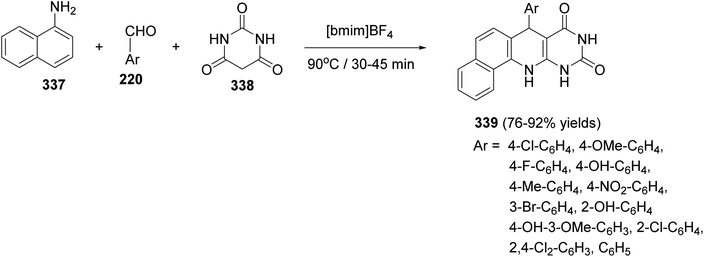 |
| | Scheme 114 Synthesis of benzo[h]pyrimido[4,5-b]quinoline-8,10-diones 339 in ionic liquid. | |
2.3.7. Benzo[g]pyrazolo[3,4-b]quinolines. In 2014, Karamthulla and coworkers128 have described a L-proline mediated synthesis of 2H-benzo[g]pyrazolo[3,4-b]quinoline-5,10(4H,11H)-diones 342, in 52–88% yields, via a three-component one-pot reaction of 3-aminopyrazoles 340, aldehydes 220 and 2-hydroxy-1,4-naphthoquinone (341) in boiling ethanol (Scheme 115). The plausible mechanism for the formation of 342 is shown in Scheme 116. The reaction proceeds via domino aldol reaction/Michael addition/intramolecular condensation/tautomerism sequence to afford the desired products 342 regioselectively.
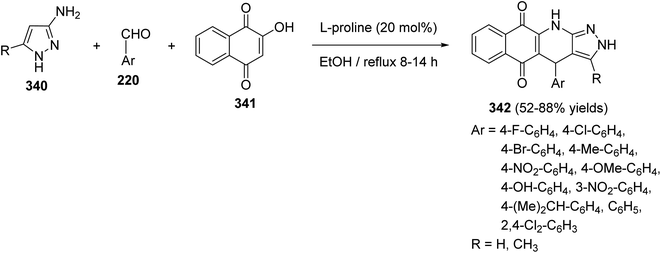 |
| | Scheme 115 L-Proline mediated synthesis of benzo[g]pyrazolo[3,4-b]quinoline-5,10-diones 342. | |
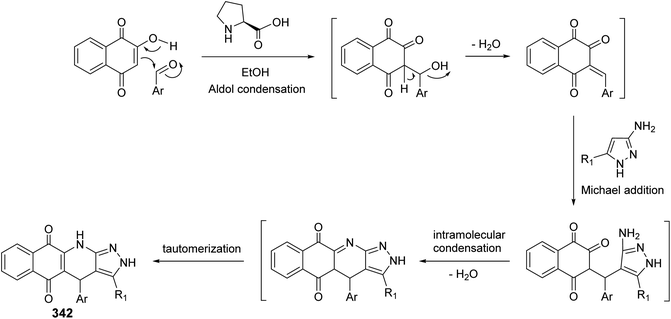 |
| | Scheme 116 Proposed mechanism for the formation of benzo[g]pyrazolo[3,4-b]quinoline-5,10-diones 342. | |
2.3.8. Thieno[3′,2′:4,5]thieno[2,3-c]quinolines. A multistep synthesis of novel thieno[3′,2′:4,5]thieno[2,3-c]quinolones 347 has been developed by Koružnjak et al.129 Refluxing a solution of 5-substituted-thiophene-3-carboxaldehyde 343 and malonic acid in pyridine gave 5-substituted 3-(3-thienyl)acrylic acids 344. When 344 was heated with SOCl2 in the presence of a catalytic amount of pyridine in chlorobenzene at 140 °C, they underwent cyclization to give the corresponding 3-chloro-thieno[2,3-b]thiophene-2-carbonyl chlorides 345. Reacting 345 with aniline derivatives 97 in refluxing toluene produced the 3-cholorothieno[2,3-b]thiophene-2-carboxamides 346, which were photochemically dehydrohalogenated at room temperature to afford the respective tetracyclic 347, in 31–76% yields (Scheme 117). The products 347 showed cytostatic activities against malignant cell lines and marked antitumor activity.
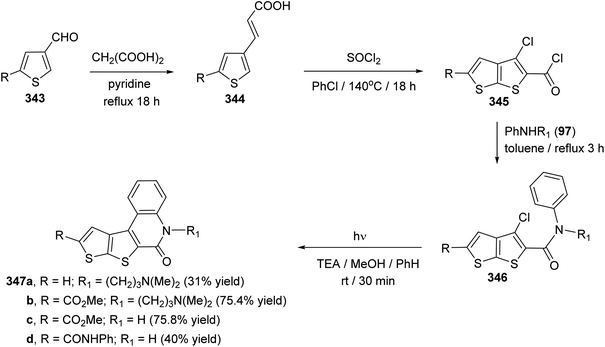 |
| | Scheme 117 A multistep synthesis of thieno[3′,2′:4,5]thieno[2,3-c]quinolones 347. | |
2.4. Tetracyclic quinolines with more than three heteroatoms
Since twenty-six years ago, Mekheimer and his group have developed different successful routes for the synthesis of a new tetracyclic systems containing the quinoline nucleus of biological importance utilizing for the first time 2,4-dichloroquinoline-3-carbonitrile (348) as a good precursor. It is well known that the chlorine atom at C-4 in 348 is more reactive than that at C-2 and is more easy to displace using nucleophiles.130–134
2.4.1. 1-Thia-3,5,6-triazaaceanthrylenes and 1-thia-3,4,5,6-tetraazaaceanthrylenes. Mekheimer et al.135 have reported the synthesis of novel tetracyclic ethyl 5-alkyl-5H-1-thia-3,5,6-triazaaceanthrylene-2-carboxylates and ethyl 5-alkyl-5H-1-thia-3,4,5,6-tetraaza-aceanthrylene-2-carboxylates using ethyl 3-amino-4-chloro-thieno[3,2-c]quinoline-2-carboxylate (350) as starting material, which was prepared from reaction of 348 with ethyl 2-mercaptoacetate (349) in DMF in the presence of TEA at room temperature in 98% yield (Scheme 118). Refluxing 350 in an excess of primary alkylamines furnished 351 in 70–75% yields, which were treated with an excess of TEO at reflux temperature to afford the angularly annulated tetracyclic ethyl 5-alkyl-5H-1-thia-3,5,6-triazaaceanthrylene-2-carboxylates 352 in 72–88% yields (Scheme 118). In contrast to the other amines, when 350 was reacted with excess of benzylamine, thieno[3,2-c]quinoline derivative 356 was obtained, as a viscous oil, which was refluxed with TEO to give 1-thia-3,5,6-triazaaceanthrylenes 357 in 64% yield (Scheme 118). Refluxing 351 with ethyl chloroformate and phenyl isothiocyanate furnished the new tetracyclic 353 and 354 in 86–96% and 78–83% yields, respectively. On the other hand, diazotization of 351 with NaNO2 in H2SO4 (70%) at −5 °C gave the novel ethyl 5-alkyl-5H-1-thia-3,4,5,6-tetraazaaceanthrylene-2-carboxylates 355 in 73–89% yields (Scheme 118).
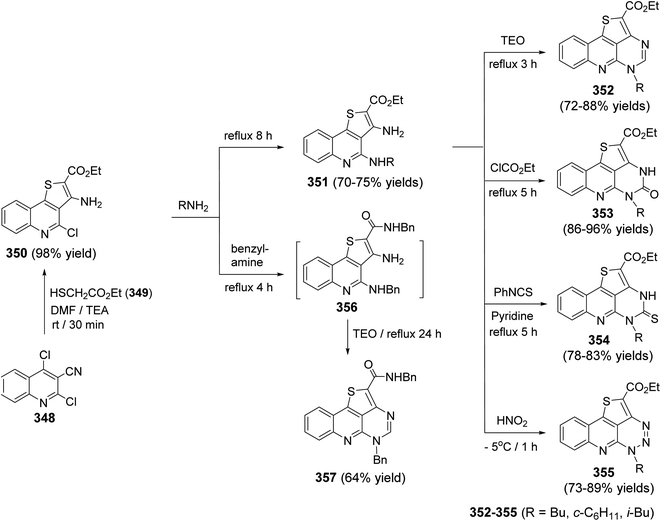 |
| | Scheme 118 Synthesis of ethyl 5-alkyl-5H-1-thia-3,5,6-triazaaceanthrylenes 352–354, 357 and ethyl 5-alkyl-5H-1-thia-3,4,5,6-tetraazaaceanthrylenes 355. | |
2.4.2. 1,2,3,5,6-Pentaazaaceanthrylenes. In 2003, Mekheimer et al.136 described the synthesis of new tetracyclic ring systems combining both a pyrimidine and a pyrazoloquinoline moieties starting from 3-amino-4-chloro-1-phenyl-1H-pyrazolo[4,3-c]quinoline (358), which was obtained by reaction of 348 with phenylhydrazine.136 Reaction of 358 with phenyl isothiocyanate in absolute pyridine under reflux conditions led to the formation of 1,5-diphenyl-1,2,3,5,6-pentaazaaceanthrylene-4(3H)-thione (359) in 68% yield. When 358 was reacted with primary aliphatic amines in DMF at reflux temperature, the corresponding 3,4-diaminopyrazolo[4,3-c]quinolines 360 were obtained in 79–85% yields, which refluxed with an excess of TEO to afford the novel 1,2,3,5,6-pentaazaaceanthrylenes 361 in 80–92% yields (Scheme 119). Also, compounds 361 were prepared by an alternative route. As an example, reacting 358 with TEO under reflux conditions gave the intermediate 4-chloro-3-(ethoxymethyleneamino)-1-phenyl-pyrazolo-[4,3-c]quinoline (362), which then refluxed, in situ, with iso-butylamine in DMF to furnish the desired tetracyclic 361 (Scheme 119).
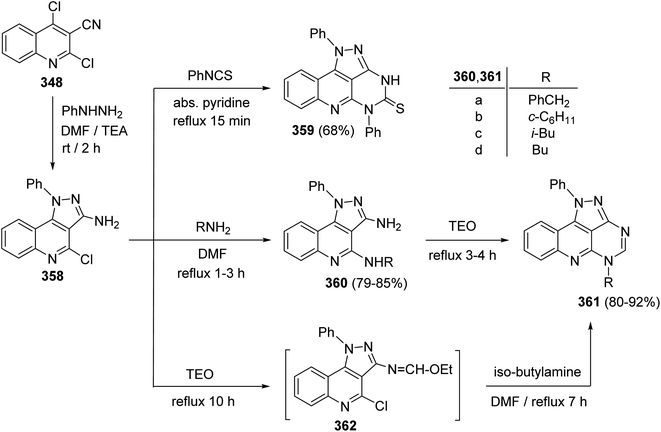 |
| | Scheme 119 Synthesis of 1,2,3,5,6-pentaazaaceanthrylenes 359 and 361. | |
2.4.3. 1,2,3,4,5,6-Hexaazaaceanthrylenes and 5,7,8,10a,11-pentaazabenzo[a]fluorenes ring systems. The synthesis of new 5-alkyl-1,5-dihydro-1,2,3,4,5,6-hexaazaaceanthrylenes 366 was developed by Mekheimer137 in 2001. Reactions were carried out by heating the key 3-amino-4-chloro-1H-pyrazolo[4,3-c]quinoline (363) with an excess of the primary alkylamines at reflux temperature to afford the corresponding 4-alkylamino-3-amino-1H-pyrazolo[4,3-c]-quinolines 364, in 82–86% yields, which were transformed into the tetracyclic 5-alkyl-1,5-dihydro-1,2,3,4,5,6-hexaazaaceanthrylenes 366, when treated with NaNO2 in H2SO4 (70%) at −5 °C. The yields of the last step are 66–87% (Scheme 120). The author discarded the other possible structure 365 based on the spectral data.
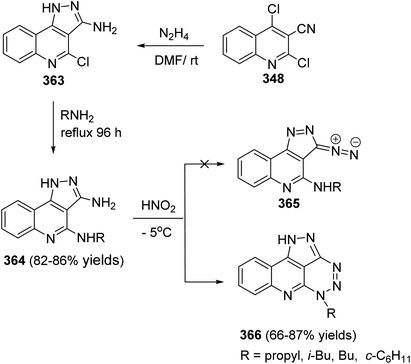 |
| | Scheme 120 Synthesis of 1,2,3,4,5,6-hexaazaaceanthrylenes 366 from 363. | |
On the other hand, reacting compound 363 with arylamines 97 in DMF under reflux conditions afforded the corresponding 4-arylamino-pyrazolo[4,3-c]quinolines 367 in 80–88% yields. Diazotisation of the amines 367 under ordinary conditions gave the 4-arylamino-3-diazo-1H-pyrazolo[4,3-c]quinolines 369 in 82–94% yields. Treatment the 3-diazo compounds 369 with ethyl acetoacetate (370) in absolute EtOH at room temperature gave the intermediate 371, which then underwent intramolecular cyclo-condensation to afford directly the novel tetracyclic ring system 372 in 70–83% yields, for which the alternative isomeric structure 373 is theoretically possible (Scheme 121).137
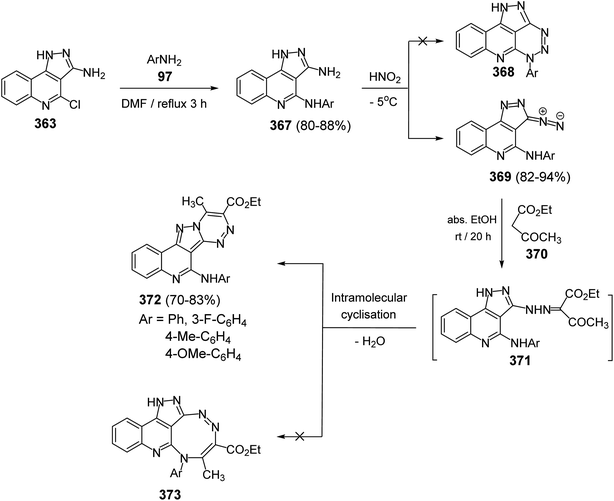 |
| | Scheme 121 Synthesis of 5,7,8,10a,11-pentaazabenzo[a]fluorenes 372. | |
2.4.4. 5-Thia-1,2,3,6-tetraazaacephenanthrylenes and 5-thia-1,3,6-triazaacephenan-thrylenes. Mekheimer et al.138 have reported a new protocol for the construction of novel tetracyclic pyrimidothienoquinolines and 1,2,3-triazinothienoquinolines through 4-alkyl-amino-2-chloro-quinoline-3-carbonitriles 374. By heating a mixture of 374 with ethyl 2-mercaptoacetate (349) and excess of sodium ethoxide in absolute EtOH at reflux temperature, 3-aminothieno[2,3-b]quinolines 375 were formed in 70–94% yields (Scheme 122). Diazotization of 375 resulted in the formation of 5-thia-1,2,3,6-tetraazaacephenanthryl-enes 376 in 67–75% yields (Scheme 122). However, when 375 were refluxed with TEO, 5-thia-1,3,6-triazaacephenanthrylenes 377 were indeed obtained in 65–81% yields (Scheme 122).138 On the other hand, reaction of 375 with cyclohexylamine in DMF under reflux conditions afforded the corresponding amides 378, which in treatment with an excess of TEO did not afford the perianellated tetracyclic system 379, but furnished instead the unexpected linear isomeric pyrimidothienoquinoline derivatives 380 in good yields (Scheme 122).138
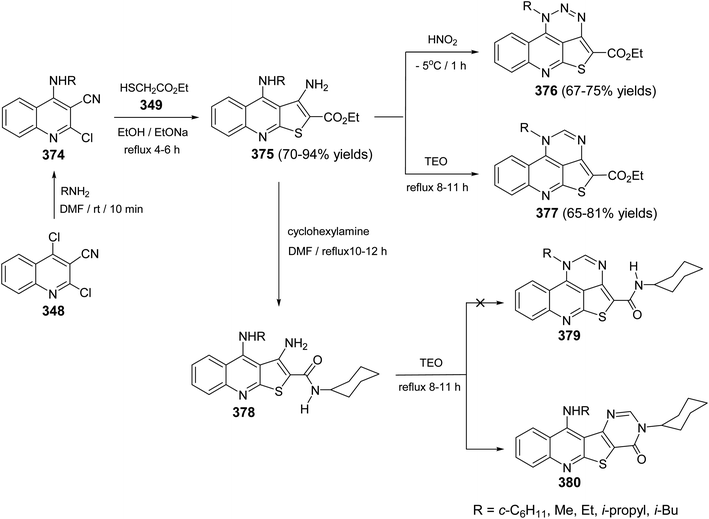 |
| | Scheme 122 Synthesis of novel 1H-5-thia-1,2,3,6-tetraazaacephenanthrylenes 376, 1H-5-thia-1,3,6-triazaacephenanthrylenes 377 and linear pyrimidothienoquinolines 380. | |
2.4.5. 1,4,5,6,6a-Pentaazabenzo[a]indacenes, 1,3,5,6-tetraazaaceanthrylenes and 5,7,9,11 tetraazabenzo[a]fluorenes. The reaction of 348 with methyl glycinate hydrochloride (381) was investigated by Mekheimer139 to synthesize the new 1,4,5,6,6a-pentaazabenzo[a]indacenes 387. The reaction was carried out by treatment 348 with an excess of 381 in DMF in the presence of TEA at room temperature to afford one of the two isomeric structures 382 or 383 (Scheme 123). Spectral data could not unequivocally differentiate these isomers. To chemically verify the structure of 383, tetrazolo[1,5-a]quinolines 385 was prepared, in 98% yield, by heating 383 with sodium azide in DMF at 70–75 °C (Scheme 123). The azidoquinolines 384 was ruled out based on the IR spectrum which has no azido band. When 383 was heated with sodium methoxide in MeOH at reflux temperature, it underwent intramolecular cyclization to provide methyl 3-amino-4-chloro-1H-pyrrolo[3,2-c]quinoline-2-carboxylate (386) in 85% yield. Refluxing 386 with sodium azide in DMF afforded the novel tetracyclic 1,4,5,6,6a-pentaaza-benzo[a]indacenes 387 in 96% yield. Alternatively, compound 387 could also be obtained by refluxing 385 with sodium methoxide in MeOH (Scheme 123).
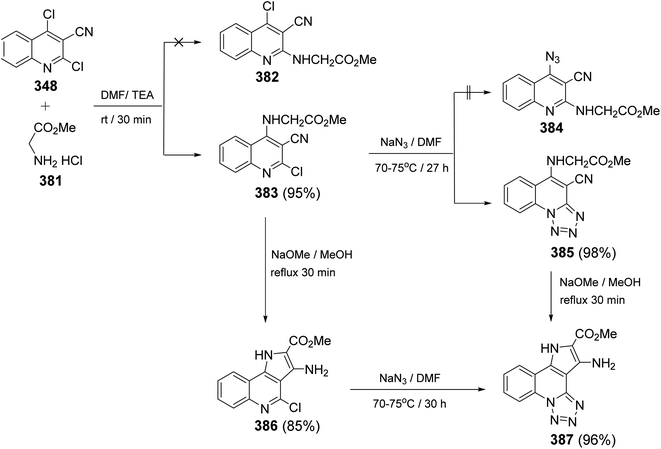 |
| | Scheme 123 Synthesis of methyl 3-amino-1,4,5,6,6a-pentaazabenzo[a]indacene-2-carboxylate (387). | |
On the other hand, when a solution of 386 in dry DMF was treated with ethyl iodide in the presence of anhydrous K2CO3 at 55–60 °C, the methyl 3-amino-4-chloro-1-ethyl-pyrrolo-[3,2-c]quinoline-2-carboxylate (388), major product, was obtained in 61% yield. In addition to 388, minor amounts of 390 was also obtained. Reaction of 388 and 390 with sodium azide, under similar reaction conditions as described before for the synthesis of 387, yielded the novel tetracyclic systems 389 and its analog 391, respectively, in quantitative yields (Scheme 124). Alternatively, compound 391 could also be obtained by heating a solution of 389 in dry DMF with ethyl iodide at 55–60 °C (Scheme 124).139
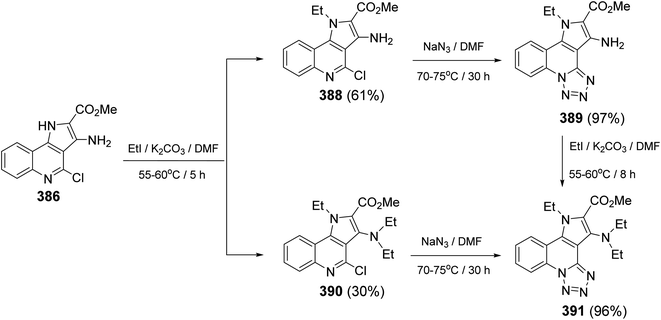 |
| | Scheme 124 Synthesis of new 1,4,5,6,6a-pentaazabenzo[a]indacenes 389 and 391. | |
Also, Mekheimer and his co-workers139 have reported the synthesis of novel tetracyclic systems incorporating a pyrimidine nucleus in addition to the pyrroloquinoline moiety utilizing the N-ethylpyrrolo[3,2-c]quinoline derivative 388 as starting material. Thus, reaction of 388 with isothiocyanates in dry pyridine under reflux conditions gave the corresponding novel methyl 1-ethyl-5-substituted-4-thioxo-3(4H)-1,3,5,6-tetraazaaceanthrylene-2-carboxylates 392 in 63–80% yields. On the other hand, treatment of 388 (1 equiv.) with morpholine (10 equiv.) in boiling absolute EtOH yielded the corresponding pyrrolo[3,2-c]quinoline derivative 393a, in 79% yield, which reacted with isothiocyanates, under the same conditions described above, to yield the interesting 5,7,9,11-tetraazabenzo[a]fluorenes 394, in 55–81% yields, as new model systems for pyrrolopyrimidoquinolines (Scheme 125). Furthermore, when 388 was heated in an excess of butylamine at reflux temperature, the versatile intermediate pyrrolo[3,2-c]quinolines 393b was formed. Refluxing 393b with ethyl chloroformate yielded the new methyl 5-butyl-1-ethyl-4-oxo-3(4H)-1,3,5,6-tetraazaaceanthrylene-2-carboxylate (395) in 63% yield. Meanwhile, refluxing 393b with TEO afforded the methyl 5-butyl-1-ethyl-1,3,5,6-tetraazaaceanthrylene-2-carboxylate (396) in 70% yield (Scheme 126).
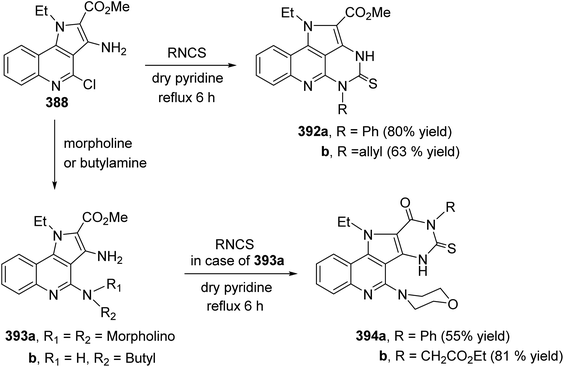 |
| | Scheme 125 Synthesis of methyl 1-ethyl-4-thioxo-3(4H)-1,3,5,6-tetraazaaceanthrylene-2-carboxylates 392 and 5,7,9,11-tetraazabenzo[a]fluorenes 394. | |
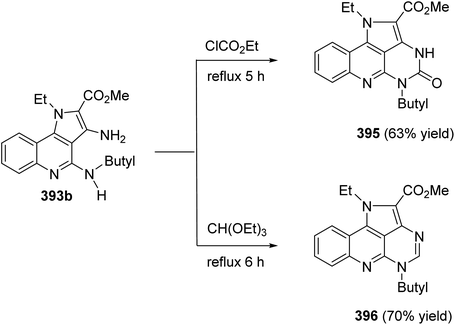 |
| | Scheme 126 Synthesis of methyl 5-butyl-1-ethyl-4-oxo-3(4H)-1,3,5,6-tetraazaaceanthrylene-2-carboxylate (395) and methyl 5-butyl-1-ethyl-1,3,5,6-tetraazaaceanthrylene-2-carboxylate (396). | |
2.4.6. 1,2,3,4,5,6-Hexaazaacephenanthrylenes ring systems. Refluxing 4-amino-2-chloro-quinoline-3-carbonitriles 397 with an excess of hydrazine hydrate (80%) afforded 3-amino-4-arylamino-1H-pyrazolo[3,4-b]quinolines 398 in 58–67% yields. Diazotization of 398 yielded the new tetracyclic 1-aryl-1,5-dihydro-1,2,3,4,5,6-hexaazaacephenanthrylenes 399, in 67–76% yields (Scheme 127). The other possible structure 400 was readily ruled out for the reaction products on the basis of spectral data.140
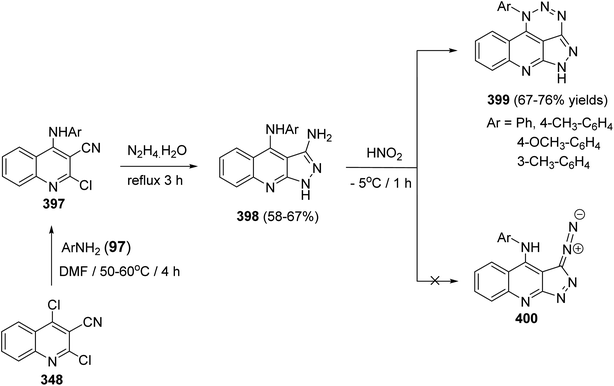 |
| | Scheme 127 Synthesis of 1-aryl-1,2,3,4,5,6-hexaazaacephenanthrylenes 399 from 348. | |
2.4.7. Isoxazolo[3′,4′:4,5]pyrrolo(or thieno)[2,3-c]quinolines. An efficient approach was developed by Mekheimer and co-workers141 to synthesis the new tetracyclic isoxazolo[3′,4′:4,5]pyrrolo(or thieno)[3,2-c]quinolines 403. By refluxing of 350 and 386 with primary aliphatic amines in DMF, the corresponding amines 351, 401 were obtained, which were transformed into the 3-azido-pyrrolo(or thieno)quinolines 402 in 74–93% yields, when treated with sodium nitrite in H2SO4 (70%) at −5 °C, followed by reaction of the non-isolated pyrrolo(or thieno)quinoline diazonium sulfate with an aqueous solution of NaN3 at −5 °C. Conversion of azides 402 into the angular tetracyclic isoxazolo[3′,4′:4,5]pyrrolo(or thieno)[2,3-c]quinolines 403 was performed by refluxing in bromo-benzene (Scheme 128).
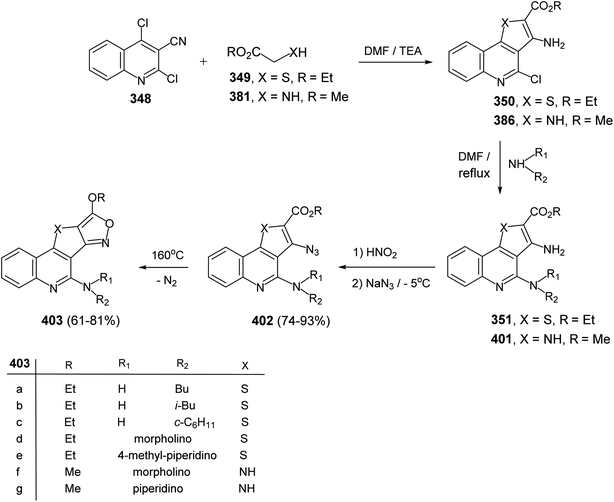 |
| | Scheme 128 Synthesis of isoxazolo[3′,4′:4,5]pyrrolo(or thieno)[3,2-c]quinolines 403. | |
2.4.8. [1,2,4]Triazino[4′,3′:1,5]pyrazolo[4,3-c]quinolines. In 2017, Mekheimer et al.142 have described the first synthesis of new four heterocyclic ring systems 10-amino-6,9-disubstituted-[1,2,4]triazino[4′,3′:1,5]pyrazolo[4,3-c]quinoline derivatives. Reacting 348 with cyanoacetic acid hydrazide (404) in refluxing MeOH in the presence of Et3N gave the unexpected 3-amino-4-chloro-1H-pyrazolo[4,3-c]quinoline (363) (Scheme 129). The mechanism for the formation of 363 is shown in Scheme 130. Heating 363 with cyclic secondary amines (piperidine, morpholine and 1-methyl-piperazine) in DMF at reflux temperature furnished the corresponding 4-amino-pyrazolo[4,3-c]quinolines 405 (Scheme 129).
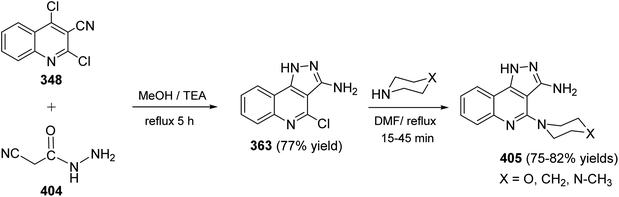 |
| | Scheme 129 Synthesis of 3-amino-1H-pyrazolo[4,3-c]quinoline derivatives 405. | |
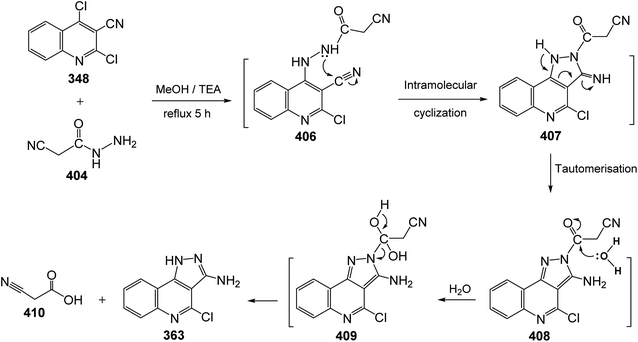 |
| | Scheme 130 Reaction mechanism for the formation of 3-amino-4-chloro-1H-pyrazolo-[4,3-c]quinoline (363). | |
Then, compounds 363 and 405 were utilized as precursors for the synthesis of the new tetracyclic systems 413. Diazotization of 363 and 405 yielded the corresponding diazonium salts 411, which were then subjected to couple with different active methylene nitriles 412, in aqueous EtOH containing sodium acetate, to give the novel perianellated tetracyclic ring system 10-amino-6,9-disubstituted-[1,2,4]triazino[4′,3′:1,5]pyrazolo[4,3-c]quinolines 413 (Scheme 131).142 The structures of all the newly synthesized compounds were unambiguously confirmed by spectroscopic and analytical techniques. Furthermore, X-ray crystallographic analysis on compound 413c was performed to determine the absolute configuration of the products 413. The probable mechanism leading to the formation of 9-substituted-[1,2,4]triazino[4′,3′:1,5]pyrazolo[4,3-c]quinolines 413 is outlined in Scheme 132.
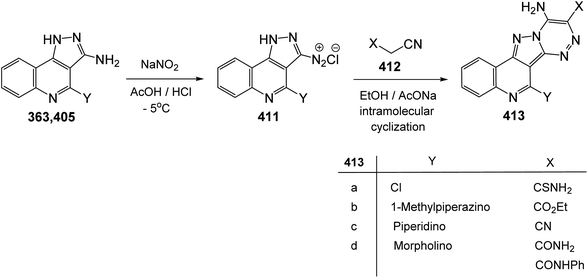 |
| | Scheme 131 Coupling reaction of 411 with active methylenes 412. | |
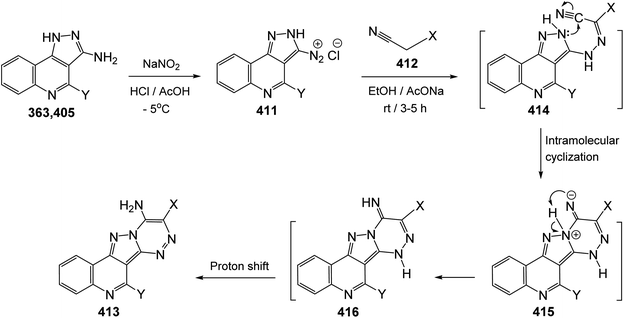 |
| | Scheme 132 Reaction mechanism for the formation of 9-substituted-[1,2,4]triazino[4′,3′:1,5]-pyrazolo[4,3-c]quinolines 413. | |
2.4.9. Benzo[c]pyrimido[4,5,6-ij][2,7]naphthyridines. In 2018, Mekheimer and his group143 have developed unprecedented synthesis of benzo[c]pyrimido[4,5,6-ij][2,7]naphthyridines with structural analogy to pyrido[4,3,2-mn]-acridines present in the marine tetracyclic pyridoacridine alkaloids. Reaction of 348 with an equimolecular amount of cyanoacetamides 417 in absolute MeOH containing a catalytic amount of K2CO3 at room temperature gave the new benzo[c][2,7]naphthyridines 418, only in one step (Scheme 133). When 418 were reacted with phenyl isothiocyanate in refluxing dry pyridine, the corresponding novel 3-alkyl-2-oxo-6-phenyl-5-thioxo-3,4,5,6-tetrahydro-2H-benzo[c]pyrimido[4,5,6-ij][2,7]naphthyridine-1-carbonitriles 420 were indeed formed in good yields, via intermediacy of 419 (Scheme 134).
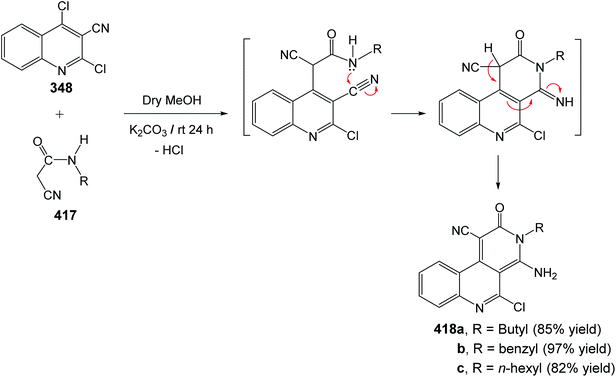 |
| | Scheme 133 Synthesis of 3-alkyl-4-amino-5-chloro-2-oxo-2,3-dihydrobenzo[c][2,7]naphthy-ridine-1-carbonitriles 418. | |
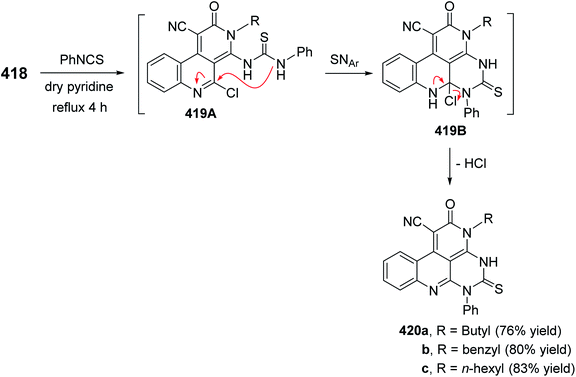 |
| | Scheme 134 Synthesis of 3-alkyl-2-oxo-6-phenyl-5-thioxo-3,4,5,6-tetrahydro-2H-benzo[c]-pyrimido[4,5,6-ij][2,7]naphthyridine-1-carbonitriles 420. | |
When 421, obtained by reacting 418 (R = butyl) with various alkyl amines, were heated with acetic anhydride under reflux conditions, the novel 3,6-dialkyl-5-methyl-2-oxo-3,6-dihydro-2H-benzo[c]pyrimido[4,5,6-ij][2,7]naphthyridine-1-carbonitriles 424 were isolated in very good yields (Scheme 135).143
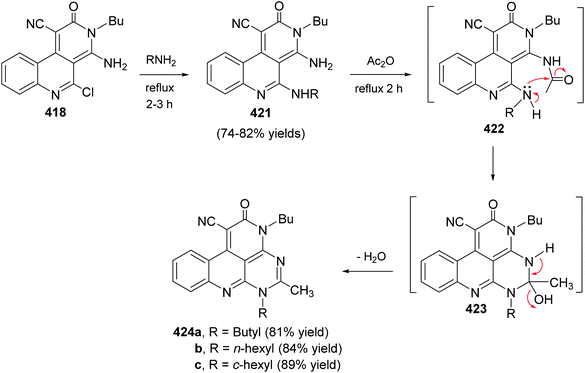 |
| | Scheme 135 Synthesis of 3,6-dialkyl-5-methyl-2-oxo-3,6-dihydro-2H-benzo[c]pyrimido-[4,5,6-ij][2,7]naphthyridine-1-carbonitriles 424. | |
Moreover, refluxing 421 with TEO gave the new 3,6-dialkyl-2-oxo-3,6-dihydro-2H-benzo[c]pyrimido[4,5,6-ij][2,7]naphthyridine-1-carbonitriles 425 in 80–86% yields (Scheme 136). On the other hand, treatment of 421 with NaNO2 in H2SO4 (70%) at −5 °C yielded the previously unreported 3,6-dialkyl-2-oxo-3,6-dihydro-2H-benzo[c][1,2,3]triazino[4,5,6-ij]-[2,7]naphthyridine-1-carbonitriles 427 in excellent yields (Scheme 136).143
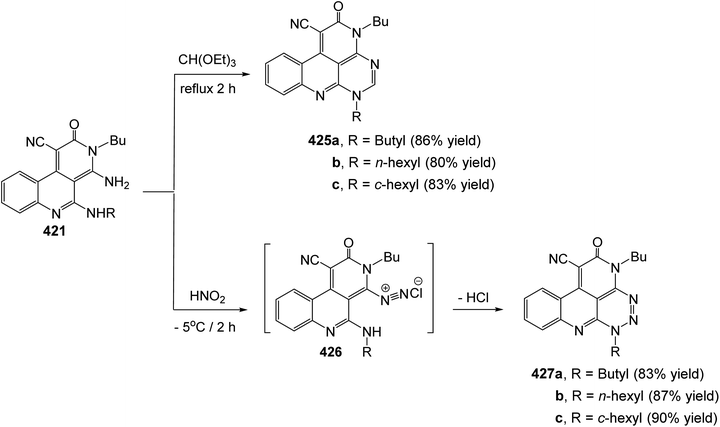 |
| | Scheme 136 Synthesis of benzo[c]pyrimido[4,5,6-ij][2,7]naphthyridine-1-carbonitriles 425 and benzo[c][1,2,3]triazino[4,5,6-ij][2,7]naphthyridine-1-carbonitriles 427. | |
2.4.10. Pyrimido[1′,2′:1,5]pyrazolo[4,3-c]quinolines. The synthesis of tetracyclic pyrimidopyrazoloquinolines 428 has described by Hassan and his group.144 On reacting tetracyanoethylene (TCNE) (2 equiv.) (as π-acceptor) with pyrazolo[4,3-c]quinolines 363, 364137 (1 equiv.) (as donor) in DMF furnished a green colour, which rapidly disappeared to give the tetracyclic products, namely, 8-amino-6-substituted-pyrimido[1′,2′:1,5]pyrazolo[4,3-c]quinoline-9,10-dicarbonitriles 428, in 51–55% yields. This behavior can be explained as due to initial formation of unstable charge-transfer complexes between TCNE and pyrazoloquinolines 363, 364 followed by completion of electron transfer from 363, 364 to TCNE leading to the formation of TCNE anion radical in contact with pyrazoloquinoline cation radical I. These radicals combine to furnish the adduct II. Elimination of one molecule of HCN gives III which undergoes intramolecular cyclization via attack of the amino group on the CN one to afford the pyrimidopyrazoloquinolines 428 (Scheme 137).
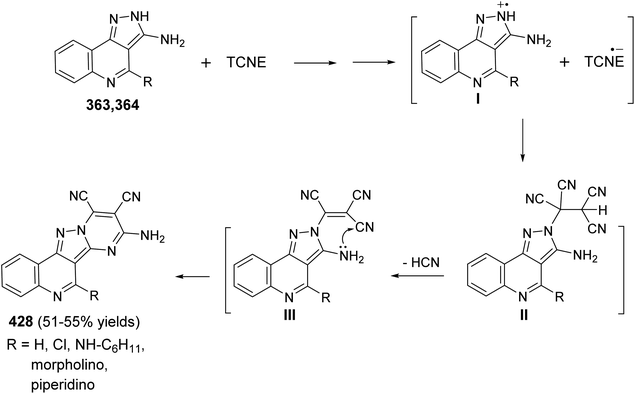 |
| | Scheme 137 Synthesis of 8-amino-6-substituted-pyrimido[1′,2′:1,5]pyrazolo[4,3-c]quinoline-9,10-dicarbonitriles 428. | |
Conflicts of interest
There are no conflicts to declare.
Notes and references
- S. Mukherjee and M. Pal, Drug Discov. Today, 2013, 18, 389 CrossRef CAS PubMed.
- S. Mukherjee and M. Pal, Curr. Med. Chem., 2013, 20, 4386 CrossRef CAS PubMed.
- P. Zajdel, A. Partyka, K. Marciniec, A. J. Bojarski, M. Pawlowski and A. Wesolowska, Future Med. Chem., 2014, 6, 57 CrossRef CAS PubMed.
- S. Bawa, S. Kumar, S. Drabu and R. Kumar, J. Pharm. BioAllied Sci., 2010, 2, 64 CrossRef CAS PubMed.
- B. Gryzło and K. Kulig, Mini-Rev. Med. Chem., 2014, 14, 332 CrossRef PubMed.
- K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245 CrossRef CAS PubMed.
- A. P. Gorka, A. d. Dios and P. D. Roepe, J. Med. Chem., 2013, 56, 5231 CrossRef CAS PubMed.
- S. Bongarzone and M. L. Bolognesi, Expet Opin. Drug Discov., 2011, 6, 251 CrossRef CAS PubMed.
- K. A. Reynolds, W. A. Loughlin and D. J. Young, Mini-Rev. Med. Chem., 2013, 13, 730 CrossRef CAS PubMed.
- A. Lilienkampf, J. Mao, B. Wan, Y. Wang, S. G. Franzblau and A. P. Kozikowski, J. Med. Chem., 2009, 52, 2109 CrossRef CAS PubMed.
- R. S. Keri and S. A. Patil, Biomed. Pharmacother., 2014, 68, 1161 CrossRef CAS PubMed.
- S. Singh, G. Kaur, V. Mangla and M. K. Gupta, J. Enzyme Inhib. Med. Chem., 2014, 25032745 Search PubMed.
- G. R. Proctor and A. L. Harvey, Curr. Med. Chem., 2000, 7, 295 CrossRef CAS PubMed.
- R. Musiol, Curr. Pharm. Des., 2013, 19, 1835 CrossRef CAS PubMed.
- J. Polanski, H. Niedbala, R. Musiol, B. Podeszwa, D. Tabak, A. Palka, A. Mencel, J. Finster, J. F. Mouscadet and M. L. Bret, Lett. Drug Des. Discov., 2006, 3, 175 CrossRef CAS.
- J. Polanski, H. Niedbala, R. Musiol, B. Podeszwa, D. Tabak, A. Palka, A. Mencel, J. F. Mouscadet and M. Le Bret, Lett. Drug Des. Discov., 2007, 4, 99 CrossRef CAS.
- D. Dubé, M. Blouin, C. Brideau, C. C. Chan, S. Desmarais, D. Ethier, J. P. Falgueyret, R. W. Friesen, M. Girard, Y. Girard, J. Guay, D. Riendeau, P. Tagari and R. N. Young, Bioorg. Med. Chem. Lett., 1998, 8, 1255 CrossRef.
- J. Jiang, M. Hoang, J. R. Young, D. Chaung, R. Eid, C. Turner, P. Lin, X. Tong, J. Wang, C. Tan, S. Feighner, O. Palyha, D. L. Hreniuk, J. Pan, A. W. Sailer, D. J. MacNeil, A. Howard, L. Shearman, S. Stribling, R. Camacho, A. Strack, L. H. Van der Ploeg, M. T. Goulet and R. J. DeVita, Bioorg. Med. Chem. Lett., 2006, 16, 5270 CrossRef CAS PubMed.
- J. Jiang, P. Lin, M. Hoang, L. Chang, C. Tan, S. Feighner, O. C. Palyha, D. L. Hreniuk, J. Pan, A. W. Sailer, N. R. Morin, D. J. MacNeil, A. D. Howard, L. H. Y. Van der Ploeg, M. T. Goulet and R. J. DeVita, Bioorg. Med. Chem. Lett., 2006, 16, 5275 CrossRef CAS PubMed.
- T. Ulven, P. B. Little, J.-M. Receveur, T. M. Frimurer, ø. Rist, P. K. Nørregaard and T. Högberg, Bioorg. Med. Chem. Lett., 2006, 16, 1070 CrossRef CAS PubMed.
- T. Ulven, T. M. Frimurer, J.-M. Receveur, P. B. Little, ø. Rist, P. K. Nørregaard and T. Högberg, J. Med. Chem., 2005, 48, 5684 CrossRef CAS PubMed.
- R. Arienzo, D. E. Clark, S. Cramp, S. Daly, H. J. Dyke, P. Lockey, D. Norman, A. G. Roach, K. Stuttle, M. Tomlinson, M. Wong and S. P. Wren, Bioorg. Med. Chem. Lett., 2004, 14, 4099 CrossRef CAS PubMed.
- M. Orhan Püsküllü, B. Tekiner and S. Suzen, Mini-Rev. Med. Chem., 2013, 13, 365 Search PubMed.
- G. Dzimbeg, B. Zorc, M. Kralj, K. Ester, K. Pavelić, G. Andrei, R. Snoeck, J. Balzarini, E. De Clercq and M. Mintas, Eur. J. Med. Chem., 2008, 43, 1180 CrossRef CAS PubMed.
- R. Musiol, M. Serda, S. Hensel-Bielowka and J. Polanski, Curr. Med. Chem., 2010, 17, 1960 CrossRef CAS PubMed.
- C. M. M. Gómez, V. V. Kouznetsov, M. A. Sortino, S. L. Alvarez and S. A. Zacchino, Bioorg. Med. Chem., 2008, 16, 7908 CrossRef PubMed.
- D. H. Boschelli, Y. D. Wang, F. Ye, B. Wu, N. Zhang, M. Dutia, D. W. Powell, A. Wissner, J. M. Weber and F. Boschelli, J. Med. Chem., 2001, 44, 822 CrossRef CAS PubMed.
- N. Muruganantham, R. Sivakumar, N. Anbalagan, V. Gunasekaran and J. T. Leonard, Biol. Pharm. Bull., 2004, 27, 1683 CrossRef CAS PubMed.
- S. Eswaran, A. S. Adhikari and N. S. Shetty, Eur. J. Med. Chem., 2009, 44, 4637 CrossRef CAS PubMed.
- M. Kidwai, R. K. Bhushan, P. Sapra, K. R. Saxena and R. Gupta, Bioorg. Med. Chem., 2000, 8, 69 CrossRef CAS PubMed.
- N. M. Shah, M. P. Patel and R. G. Patel, Eur. J. Med. Chem., 2012, 54, 239 CrossRef CAS PubMed.
- A. Mahamoud, J. Chevalier, A. Davin-Regli, J. Barbe and J.-M. Pages, Curr. Drug Targets, 2006, 7, 843 CrossRef CAS PubMed.
- M. P. Maguire, K. R. Sheets, K. McVety, A. P. Spada and A. Zilberstein, J. Med. Chem., 1994, 37, 2129 CrossRef CAS PubMed.
- N. Costedoat-Chalumeau, B. Dunogué, N. Morel, V. Le Guern and G. Guettrot-Imbert, Presse Med., 2014, 43, 167 CrossRef PubMed.
- I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, J. Org. Chem., 1994, 59, 2623 CrossRef CAS.
- S. Bongarzone and M. L. Bolognesi, Expet Opin. Drug Discov., 2011, 6, 251 CrossRef CAS PubMed.
- A. Mahamoud, J. Chevalier, A. Davin-Regli, J. Barbe and J. M. Pagès, Curr. Drug Targets, 2006, 7, 843 CrossRef CAS PubMed.
- A. Shah, V. C. Diculescu, R. Qureshi and A. M. Oliveira-Brett, Bioelectrochem, 2010, 79, 173 CrossRef CAS PubMed.
- K. Cimanga, T. De Bruyne, L. Pieters, J. Totte, L. Tona, K. Kambu, D. Vanden Berghe and A. J. Vlietinck, Phytomedicine, 1998, 5, 209 CrossRef CAS PubMed.
- Z.-W. Mei, L. Wang, W.-J. Lu, C.-Q. Pang, T. Maeda, W. Peng, M. Kaiser, I. E. El Sayed and T. Inokuchi, J. Med. Chem., 2013, 56, 1431 CrossRef CAS PubMed.
- P. Grellier, L. Ramiaramanana, V. Millerioux, E. Deharo, J. Schrével, F. Frappier, F. Trigalo, B. Bodo and J. L. Pousset, Phytother Res., 1996, 10, 317 CrossRef CAS.
- E. Shaban, M. Świtalska, L. Wang, N. Wang, F. Xiu, I. Hayashi, T. A. Ngoc, S. Nagae, S. El-Ghlban, S. Shimoda, A. El Gokha, I. El Sayed, J. Wietrzyk and T. Inokuchi, Molecules, 2017, 22, 1954 CrossRef PubMed.
- L. Wang, M. Świtalska, Z. W. Mei, W. J. Lu, Y. Takahara, X. W. Feng, I. E. T. El-Sayed, J. Wietrzyk and T. Inokuchi, Bioorg. Med. Chem., 2012, 20, 4820 CrossRef CAS PubMed.
- I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, J. Org. Chem., 1994, 59, 2623 CrossRef CAS.
- C. W. Wright, J. Addae-Kyereme, A. G. Breen, J. E. Brown, M. F. Cox, S. L. Croft, Y. Gökçek, H. Kendrick, R. M. Phillips and P. L. Pollet, J. Med. Chem., 2001, 44, 3187 CrossRef CAS PubMed.
- P. Grellier, L. Ramiaramanana, V. Millerioux, E. Deharo, J. Schrével, F. Frappier, F. Trigalo, B. Bodo and J.-L. Pousset, Phytother Res., 1996, 10, 317 CrossRef CAS.
- Z. Sui, J. Altom, V. N. Nguyen, J. Fernandez, J. I. Bernstein, J. J. Hiliard, J. F. Barrett, B. L. Podlogar and K. A. Ohemengy, Bioorg. Med. Chem., 1998, 6, 735 CrossRef CAS PubMed.
- Y. Ishihara, Y. Kiyota and G. Goto, Chem. Pharm. Bull., 1990, 38, 3024 CrossRef CAS PubMed.
- P. Kumar, C. U. Dinesh and B. Pandey, Tetrahedron Lett., 1994, 35, 9229 CrossRef CAS.
- G. Kim and G. Keum, Heterocycles, 1997, 45, 1979 CrossRef CAS.
- P. Pigeon and B. Decroix, Synth. Commun., 1998, 28, 2507 CrossRef CAS.
- D.-B. Reuschling and F. Kröhnke, Chem. Ber., 1971, 104, 2103 CrossRef CAS.
- H. Z. Alkhathlan, M. A. Al-Jaradah, K. A. Al-Farhan and A. A. Mousa, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 373 CrossRef CAS.
- Y. Zhou, L. Qian and W. Zhang, Synlett, 2009, 843 CAS.
- M. Janjic, R. Prebil, U. Groselj, D. Kralj, C. Malavasic, A. Golobic, K. Stare, G. Dahmann, B. Stanovnik and J. Svete, Helv. Chim. Acta, 2011, 94, 1703 CrossRef CAS.
- I. M. Sakhautdinov, I. R. Batyrshin, N. A. Sergeeva, F. Z. Galin and M. S. Yunusov, Russ. J. Org. Chem., 2012, 48, 788 CrossRef CAS.
- B. Xu, Z. Cheng and L. Fu, Tetrahedron Lett., 2014, 55, 7194 CrossRef CAS.
- K. Mishra, A. K. Pandey, J. B. Singh and R. M. Singh, Org. Biomol. Chem., 2016, 14, 6328 RSC.
- L. W. Deady, J. Desneves, A. J. Kaye, M. Thompson, G. J. Finlay, B. C. Baguley and W. A. Denny, Bioorg. Med. Chem., 1999, 7, 2801 CrossRef CAS PubMed.
- C.-H. Tseng, Y.-L. Chen, P.-J. Lu, C.-N. Yangd and C.-C. Tzenga, Bioorg. Med. Chem., 2008, 16, 3153 CrossRef CAS PubMed.
- T.-C. Chen, D.-S. Yu, S.-J. Chen, C.-L. Chen, C.-C. Lee, Y.-Y. Hsieh, L.-C. Chang, J.-H. Guh, J.-J. Lin and H.-S. Huang, Arabian J. Chem., 2019, 12, 4348 CrossRef CAS.
- Y.-L. Chen, H.-M. Hung, C.-M. Lu, K.-C. Li and C.-C. Tzeng, Bioorg. Med. Chem., 2004, 12, 6539 CrossRef CAS PubMed.
- G. A. Salman, S. Janke, N. N. Pham, P. Ehlers and P. Langer, Tetrahedron, 2018, 74, 1024 CrossRef CAS.
-
(a) W. Peczynska-Czoch, F. Pognan, E. Kaczmarek and J. Boratyńiski, J. Med. Chem., 1994, 37, 3503 CrossRef CAS PubMed;
(b) L. Kaczmarek, W. Peczynska-Czoch, J. Osiadacz, M. Mordarski, W. A. Sokalski, J. Boratynski, E. Marcinkowska, H. Glazman-Kusnierczyk and C. Radzikowski, Bioorg. Med. Chem., 1999, 7, 2457 CrossRef CAS PubMed.
-
(a) W.-J. Lu, M. Świtalska, L. Wang, M. Yonezawa, I. E.-T. El-Sayed, J. Wietrzyk and T. Inokuchi, Med. Chem. Res., 2013, 22, 4492 CrossRef CAS;
(b) W.-J. Lu, K. J. Wicht, L. Wang, K. Imai, Z.-W. Mei, M. Kaiser, I. E.-T. El-Sayed, T. J. Egan and T. Inokuchi, Eur. J. Med. Chem., 2013, 64, 498 CrossRef CAS;
(c) L. Wang, W.-J. Lu, T. Odawara, R. Misumi, Z.-W. Mei, W. Peng, I. E. El-Sayed and T. Inokuchi, J. Heterocycl. Chem., 2014, 51, 1106 CrossRef CAS;
(d) M. Okada, Z.-W. Mei, Md. I. Hossain, L. Wang, T. Tominaga, T. Takebayashi, M. Murakami, M. Yasuda, T. Shigehiro, T. Kasai, A. Mizutani, H. Murakami, I. E.-T. El Sayed, S. Dan, T. Yamori, M. Seno and T. Inokuchi, Med. Chem. Res., 2016, 25, 879 CrossRef CAS.
- S. Ali, Y.-X. Li, S. Anwar, F. Yang, Z.-S. Chen and Y.-M. Liang, J. Org. Chem., 2012, 77, 424 CrossRef CAS PubMed.
- R. Sunke, V. Kumar, M. A. Ashfaq, S. Yellanki, R. Medisetti, P. Kulkarni, E. V. Venkat Shivaji Ramarao, N. Z. Ehteshamc and M. Pal, RSC Adv., 2015, 5, 44722 RSC.
- R. Ghorbani-Vaghei and S. M. Malaekehpoor, Tetrahedron Lett., 2012, 53, 4751 CrossRef CAS.
- C. Shi, Q. Zhang and K. K. Wang, J. Org. Chem., 1999, 64, 925 CrossRef CAS PubMed.
- H. Takeda, T. Ishida and Y. Takemoto, Chem. Lett., 2009, 38, 772 CrossRef CAS.
- W. Ali, A. Dahiya, R. Pandey, T. Alam and B. K. Patel, J. Org. Chem., 2017, 82, 2089 CrossRef CAS PubMed.
- S. Kundal, B. Chakraborty, K. Paul and U. Jana, Org. Biomol. Chem., 2019, 17, 2321 RSC.
- P. M. Fresneda, P. Molina and S. Delgado, Tetrahedron, 2001, 57, 6197 CrossRef CAS.
- G. S. M. Sundaram, C. Venkatesh, U. K. Syam Kumar, H. Ila and H. Junjappa, J. Org. Chem., 2004, 69, 5760 CrossRef CAS PubMed.
- O. Amiri-Attou, Th. Terme and P. Vanelle, Synlett, 2005, 3047 CAS.
- P. T. Parvatkar and S. G. Tilve, Tetrahedron Lett., 2011, 52, 6594 CrossRef CAS.
- P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, Tetrahedron Lett., 2007, 48, 7870 CrossRef CAS.
- P. T. Parvatkar, P. S. Parameswaran and S. G. Tilve, J. Org. Chem., 2009, 74, 8369 CrossRef CAS PubMed.
- M. J. Haddadin, R. M. Bou Zerdan, M. J. Kurth and J. C. Fettinger, Org. Lett., 2010, 12, 5502 CrossRef CAS PubMed.
- A. Khorshidi and Kh. Tabatabaeian, J. Mol. Catal. A: Chem., 2011, 344, 128 CrossRef CAS.
- M. K. Vecchione, A. X. Sun and D. Seidel, Chem. Sci., 2011, 2, 2178 RSC.
- H. K. Kadam, P. T. Parvatkar and S. G. Tilve, Synthesis, 2012, 44, 1339 CrossRef CAS.
- H. K. Kadam and S. G. Tilve, J. Heterocycl. Chem., 2016, 53, 2066 CrossRef CAS.
- Z. Yan, C. Wan, J. Wan and Z. Wang, Org. Biomol. Chem., 2016, 14, 4405 RSC.
- L. Shi and B. Wang, Org. Lett., 2016, 18, 2820 CrossRef CAS PubMed.
- L. Fan, M. Liu, Y. Ye and G. Yin, Org. Lett., 2017, 19, 186 CrossRef CAS PubMed.
- C. Challa, J. Ravindran, M. M. Konai, S. Varughese, J. Jacob, B. S. D. Kumar, J. Haldar and R. S. Lankalapalli, ACS Omega, 2017, 2, 5187 CrossRef CAS PubMed.
- P. T. Parvatkar, P. S. Parameswaran, D. Bandyopadhyay, S. Mukherjee and B. K. Banik, Tetrahedron Lett., 2017, 58, 2948 CrossRef CAS.
- Y.-L. Chen, C.-H. Chung, I.-L. Chen, P.-H. Chen and H.-Y. Jeng, Bioorg. Med. Chem., 2002, 10, 2705 CrossRef CAS PubMed.
- P. Molina, M. Alajarin and A. Vidal, Tetrahedron, 1990, 46, 1063 CrossRef CAS.
- S. Cacchi, G. Fabrizi, P. Pace and F. Marinelli, Synlett, 1999, 620 CrossRef CAS.
- K. Kobayashi, Y. Izumi, K. Hayashi, O. Morikawa and H. Konishi, Bull. Chem. Soc. Jpn., 2005, 78, 2171 CrossRef CAS.
- C. Meyers, G. Rombouts, K. T. J. Loones, A. Coelho and B. U. W. Maes, Adv. Synth. Catal., 2008, 350, 465 CrossRef CAS.
- M. G. Uchuskin, A. S. Pilipenko, O. V. Serdyuk, I. V. Trushkovc and A. V. Butin, Org. Biomol. Chem., 2012, 10, 7262 RSC.
- N. Wang, M. Switalska, M.-Y. Wu, K. Imai, T. A. Ngoc, C.-Q. Pang, L. Wang, J. Wietrzyk and T. Inokuchi, Eur. J. Med. Chem., 2014, 78, 314 CrossRef CAS PubMed.
- S. Guo, L. Tao, W. Zhang, X. Zhang and X. Fan, J. Org. Chem., 2015, 80, 10955 CrossRef CAS PubMed.
- N. N. Pham, S. Janke, G. A. Salman, T. T. Dang, T. S. Le, A. Spannenberg, P. Ehlers and P. Langer, Eur. J. Org. Chem., 2017, 5554 CrossRef CAS.
- R. Rubio-Presa, M. R. Pedrosa, M. A. Fernández-Rodríguez, F. J. Arnáiz and R. Sanz, Org. Lett., 2017, 19, 5470 CrossRef CAS PubMed.
- A. V. Aksenov, D. A. Aksenov, G. D. Griaznov, N. A. Aksenov, L. G. Voskressensky and M. Rubin, Org. Biomol. Chem., 2018, 16, 4325 RSC.
- N. P. Buu-Hoi, M. Mangane and P. Jacquignon, J. Chem. Soc. C, 1966, 50 RSC.
- K. Tabaković, I. Tabaković, M. Trkovnik, A. Jurić and N. Trinajstić, J. Heterocycl. Chem., 1980, 17, 801 CrossRef.
- M. N. Khan, S. Pal, S. Karamthulla and L. H. Choudhury, New J. Chem., 2014, 38, 4722 RSC.
- A. S. Plaskon, S. V. Ryabukhin, D. M. Volochnyuk, K. S. Gavrilenko, A. N. Shivanyuk and A. A. Tolmachev, J. Org. Chem., 2008, 73, 6010 CrossRef CAS PubMed.
- A. T. Vu, A. N. Campbell, H. A. Harris, R. J. Unwalla, E. S. Manas and R. E. Mewshaw, Bioorg. Med. Chem. Lett., 2007, 17, 4053 CrossRef CAS PubMed.
- X. Yu, J. Wang, Z. Xu, Y. Yamamoto and M. Bao, Org. Lett., 2016, 18, 2491 CrossRef CAS.
- K. Aradi, P. Bombicz and Z. Novák, J. Org. Chem., 2016, 81, 920 CrossRef CAS.
- A. L. C. Morris and Y. A. Jackson, Synthesis, 2011, 229 CAS.
- Y.-J. Zhou, D.-S. Chen, Y.-L. Li, Y. Liu and X.-S. Wang, ACS Comb. Sci., 2013, 15, 498 CrossRef CAS PubMed.
- O. Meth-Cohn, Synthesis, 1986, 76 CrossRef CAS.
- V. V. Mulwad and B. S. Mahaddalkar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1999, 38, 29 Search PubMed.
- P. K. Mahata, C. Venkatesh, U. K. Syam Kumar, H. Ila and H. Junjappa, J. Org. Chem., 2003, 68, 3966 CrossRef CAS PubMed.
- L. W. Deady, A. J. Kaye, G. J. Finlay, B. C. Baguley and W. A. Denny, J. Med. Chem., 1997, 40, 2040 CrossRef CAS PubMed.
- S. Rádl, P. Konvicka and P. Váchal, J. Heterocycl. Chem., 2000, 37, 855 CrossRef.
- L. Benati, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo and G. Zanardi, J. Org. Chem., 2000, 65, 8669 CrossRef CAS PubMed.
- X. Y. Zhu, L. G. Mardenborough, S. Li, A. Khan, W. Zhang, P. Fan, M. Jacob, S. Khan, L. Walkerb and S. Y. Ablordeppey, Bioorg. Med.
Chem., 2007, 15, 686 CrossRef CAS PubMed.
- G. A. Salman, M. Hussain, V. Iaroshenko, A. Villinger and P. Langer, Adv. Synth. Catal., 2011, 353, 331 CrossRef CAS.
- A. R. O. Rodrigues, M. S. D. Carvalhoa, J. A. V. Cardoso, R. C. Calhelha, M.-J. R. P. Queiroz, P. J. G. Coutinho and E. M. S. Castanheira, J. Photochem. Photobiol., A, 2014, 294, 20 CrossRef CAS.
- X.-S. Wang, Q. Li, J.-R. Wu and S.-J. Tu, J. Comb. Chem., 2009, 11, 433 CrossRef CAS PubMed.
- M. Asthana, N. Sharma, R. Kumar, J. B. Singh and R. M. Singh, Tetrahedron Lett., 2014, 55, 4378 CrossRef CAS.
- C. Che, B. Yang, X. Jiang, T. Shao, Z. Yu, C. Tao, S. Li and S. Lin, J. Org. Chem., 2014, 79, 436 CrossRef CAS PubMed.
- X.-S. Fan, J. Zhang, B. Li and X.-Y. Zhang, Chem.–Asian J., 2015, 10, 1281 CrossRef CAS PubMed.
- M. Rahimizadeh, M. Pordel, M. Bakavoli and H. Eshghi, Dyes Pigm., 2010, 86, 266 CrossRef CAS.
- K. T. J. Loones, B. U. W. Maes and R. A. Dommisse, Tetrahedron, 2007, 63, 8954 CrossRef CAS.
- M. Arnould, M.-A. Hiebel, S. Massip, J. M. Leger, C. Jarry, S. Berteina-Raboin and G. Guillaumet, Chem.–Eur. J., 2013, 19, 12249 CrossRef CAS PubMed.
- F. Yu, S. Yan, L. Hu, Y. Wang and J. Lin, Org. Lett., 2011, 13, 4782 CrossRef CAS PubMed.
- W. Wang, H. Jiang, M.-M. Zhang and X.-S. Wang, J. Heterocycl. Chem., 2014, 51, 830 CrossRef CAS.
- H. Y. Guo and Y. Yu, Chin. Chem. Lett., 2010, 21, 1435 CrossRef CAS.
- S. Karamthulla, S. Pal, T. Parvin and L. H. Choudhury, RSC Adv., 2014, 4, 15319 RSC.
- J. D. Koružnjak, N. Slade, B. Zamola, K. Pavelic and G. Karminski-Zamola, Chem. Pharm. Bull., 2002, 50, 656 CrossRef PubMed.
- W. Steinschifter and W. Stadlhauer, J. Prakt. Chem., 1994, 336, 311 CrossRef CAS.
- A. Albert, Chemie der Heterocyclen, Verlag Chernie. Weinheim, 1962 Search PubMed.
- H. Lettau, Chcmie der Heterocyclen, S. Hirzel-Verlag, Leipzig, 1981 Search PubMed.
- G. Illuminati, Adv. Heterocycl. Chem., 1964, 3, 285 CrossRef CAS.
- M. L. Bclli, G. Illuminati and G. Marino, Tetrahedron, 1963, 19, 345 CrossRef.
- R. A. Mekheimer, K. U. Sadek, H. A. Abd El-Nabi, A. A.-H. Mohamed, E. A. Ebraheem and M. B. Smith, J. Heterocycl. Chem., 2005, 42, 567 CrossRef CAS.
- R. A. Mekheimer, E. K. Ahmed, H. A. El-Faham, L. H. Kamel and D. Doepp, J. Chem. Res., 2003, 388 CrossRef.
- R. A. Mekheimer, Synth. Commun., 2001, 31, 1971 CrossRef CAS.
- R. A. Mekheimer, E. Kh. Ahmed, H. A. El-Fahham and L. H. Kamel, Synthesis, 2001, 97 CrossRef CAS.
- R. A. Mekheimer, Synthesis, 2000, 2078 CrossRef CAS.
- R. A. Mekheimer, J. Chem. Soc., Perkin Trans. 1, 1999, 2183 RSC.
- R. A. Mekheimer, A. M. Abdel Hameed and K. U. Sadek, Arkivoc, 2008, xvi, 144 Search PubMed.
- R. A. Mekheimer, M. A. Al-Sheikh, H. Y. Medrasi and Gh. A. Bahatheg, Synth. Commun., 2017, 47, 1052 CrossRef CAS.
- R. A. Mekheimer, M. A. Al-Sheikh, H. Y. Medrasi and Gh. A. Bahatheg, Mol. Divers., 2018, 22, 159 CrossRef CAS PubMed.
- A. A. Hassan, R. Mekheimer and N. K. Mohamed, Pharmazie, 1997, 52, 589 CAS.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mariam A. Al-Sheikh
b,
Hanadi Y. Medrasi
b and
Kamal U. Sadek
a
*a,
Mariam A. Al-Sheikh
b,
Hanadi Y. Medrasi
b and
Kamal U. Sadek
a