
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSc3N@Ih-C80 based donor–acceptor conjugate: role of thiophene spacer in promoting ultrafast excited state charge separation†
Rubén Caballero a,
Luis David Serviána,
Habtom B. Gobezec,
Olivia Fernandez-Delgadob,
Luis Echegoyen*b,
Francis D'Souza*c and
Fernando Langa*a
a,
Luis David Serviána,
Habtom B. Gobezec,
Olivia Fernandez-Delgadob,
Luis Echegoyen*b,
Francis D'Souza*c and
Fernando Langa*a
aInstituto de Nanociencia Nanotecnología y Materiales Moleculares (INAMOL), Universidad de Castilla-La Mancha, Campus de la Fábrica de Armas, 45071 Toledo, Spain. E-mail: fernando.langa@uclm.es
bDepartment of Chemistry and Biochemistry, University of Texas at El Paso, 500 W University Avenue, El Paso, Texas 79968, USA. E-mail: luis.echegoyen@utep.edu
cDepartment of Chemistry, University of North Texas, 1155 Union Circle, Denton, Texas 305070, USA. E-mail: Francis.dsouza@unt.edu
First published on 27th May 2020
Abstract
Light induced charge separation in a newly synthesized triphenylamine–thiophene-Sc3N@Ih-C80 donor–acceptor conjugate and its C60 analog, triphenylamine–thiophene-C60 conjugate is reported, and the significance of the thiophene spacer in promoting electron transfer events is unraveled.
Endohedral metallofullerenes (EMF) are compounds of great interest due to their fascinating structure and unique electronic properties1–4 which are different from those of empty fullerenes.5,6 While the early efforts were dedicated to their electronic structure, their chemical functionalization has aroused significant interest in recent years,7–9 not only to utilize their interesting properties but also to prepare new compounds with potential applications in photovoltaics,10,11 biomedical12–15 and materials science applications.16,17 Due to their higher lowest unoccupied molecular orbital (LUMO) compared to those of empty fullerene cages, EMFs are excellent acceptor materials in organic photovoltaics, capable of increasing the open circuit voltage of the devices.10 To this end, a few covalent donor–acceptor systems comprising EMFs have been prepared,8,18–20 which showed the higher stability of the radical-ion pair in comparison with analogous empty fullerenes.21–31
Up until now, several methods have been developed to functionalize EMFs such as Diels–Alder,7,32,33 Bingel–Hirsch reaction,34–37 cycloaddition with carbenes or benzynes etc.38–41 Among these, 1,3-dipolar cycloaddition of azomethine ylides is perhaps the most used to functionalize EMFs,38–42 although other dipoles as nitrile imines43 have been used as well. On the other hand, nitrile oxides react with alkenes to afford 2-isoxazolines and can be prepared from aldoximes by treatment with a chlorinated agent and a weak base, generally (except for hindered nitrile oxides) prepared in situ to circumvent dimerization.44 Cycloadditions of nitrile oxides on fullerenes,45 carbon nanotubes46,47 or graphene48 are known but only one example of the cycloaddition of a stable nitrile oxide to Sc3N@Ih-C80 has been described49 in an elegant study showing that the isoxazoline ring is fused to a [5,6]-bond junction in the endohedral cage.
Here, we describe the synthesis and photophysical study employing femtosecond transient absorption (fs-TA) spectroscopy, of a donor–acceptor conjugate formed by a triphenylamine moiety as a donor group and Sc3N@Ih-isoxazoline-C80 as the acceptor, linked by a thiophene spacer (Fig. 1). The analogous C60 derivative was also prepared for comparison. Here we show that the central thiophene ring promotes π-conjugation between the donor and acceptor entities thus facilitating charge separation processes in these conjugates, useful for optoelectronic applications.
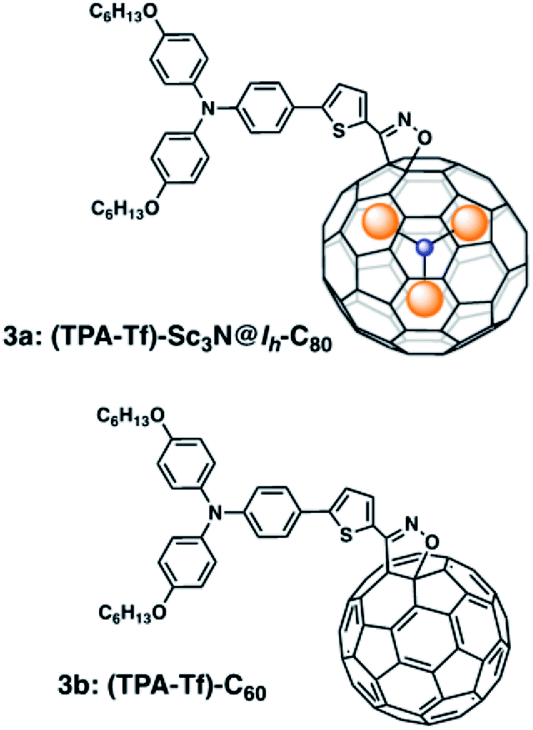 | ||
| Fig. 1 Structure of the newly synthesized triphenylamine–thiophene-Sc3N@Ih-C80 endohedral fullerene conjugate, (3a) and its C60 analog derivative (3b). | ||
Scheme 1 shows the synthetic approach for both donor–acceptor conjugates. Reaction of aldehyde 150 with hydroxylamine hydrochloride in ethanol/pyridine afforded oxime 2, as a mixture of Z–E isomers, in 77% yield. Then, 2 was reacted with N-chlorosuccinimide (NCS) and after 30 min a solution of Sc3N@Ih-C80 in 1,2-dichlorobenzene (o-DCB) and triethylamine was added and stirred for 72 h at room temperature. The progress of the reaction was followed by HPLC, which showed that a new single peak appears as that for Sc3N@Ih-C80 disappears. After purification by column chromatography, followed by preparative HPLC, conjugate 3a was obtained in 25% yield. A similar procedure was followed to prepare the C60-based compound 3b (see the Experimental section in the ESI for details†). Compound 3a was characterized by means of MALDI-TOF mass analysis showing the molecular ion peak at 1677.28 amu. The 1H NMR spectrum contains all the expected signals of the addend. The thiophene H-atoms appear at 7.96 and 7.24 ppm, deshielded compared to those in the oxime due to the presence of the fullerene cage. Note that this signal is broad as a consequence of the hindered rotation. In the 13C NMR spectrum, the most relevant signals are those of the sp3 C-atoms of the functionalized fullerene cage at 97.7 and 70.3 ppm. Similar features are observed in the spectrum of the C60-based compound 3b. Finally, the appearance of a broad UV-Vis absorption around 700 nm and the lack of absorption in the 800 nm region, expected for the [6,6]-isomer, confirmed the formation of the [5,6]-isomer; a result that agrees well with the previously described examples of 1,3-dipolar cycloadditions on Sc3N@Ih-C80.49 Interestingly, 3a undergoes a fast retrocycloaddition when refluxed in o-DCB while 3b is stable under these conditions.
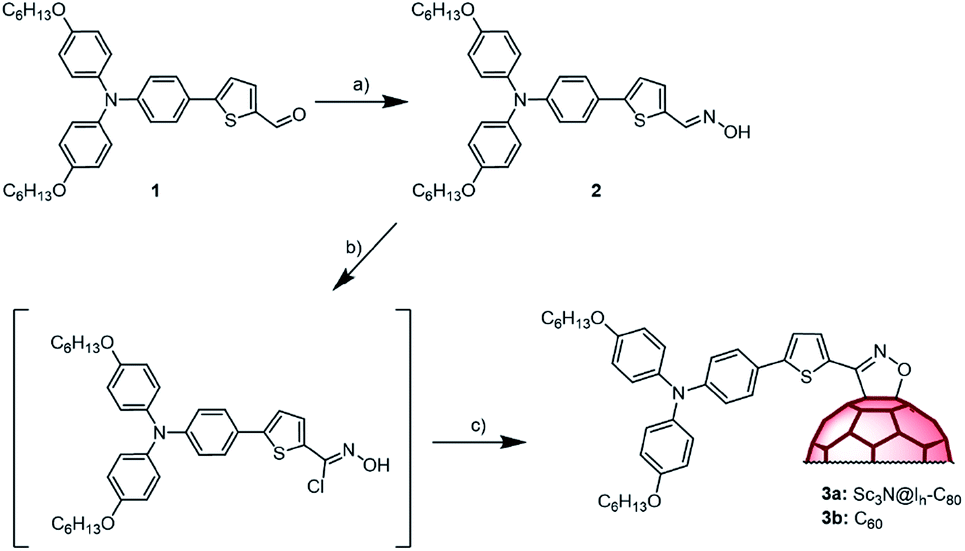 | ||
| Scheme 1 Reagents and conditions: (a) hydroxylamine hydrochloride, pyridine; (b) NCS; pyridine (c) Et3N, Sc3N@Ih-C80 or C60, r.t. | ||
The absorption spectrum of 3a and 3b in dichloromethane together with the parent oxime 2 are shown in Fig. 2a. Oxime 2 exhibits a maximum at 390 nm which is red-shifted to 412 nm and 407 nm in cycloadducts 3a and 3b, respectively, indicating a stronger influence of the Sc3N@Ih-C80 cage when compared to C60 over the electronic properties of the organic addend. In the case of 3a, a band around 690 nm is observed and the spectrum lacked the typical band around 800 nm ascribed to [6,6]-EMF adducts;51 again confirming the [5,6]-structure for 3a, as discussed above.
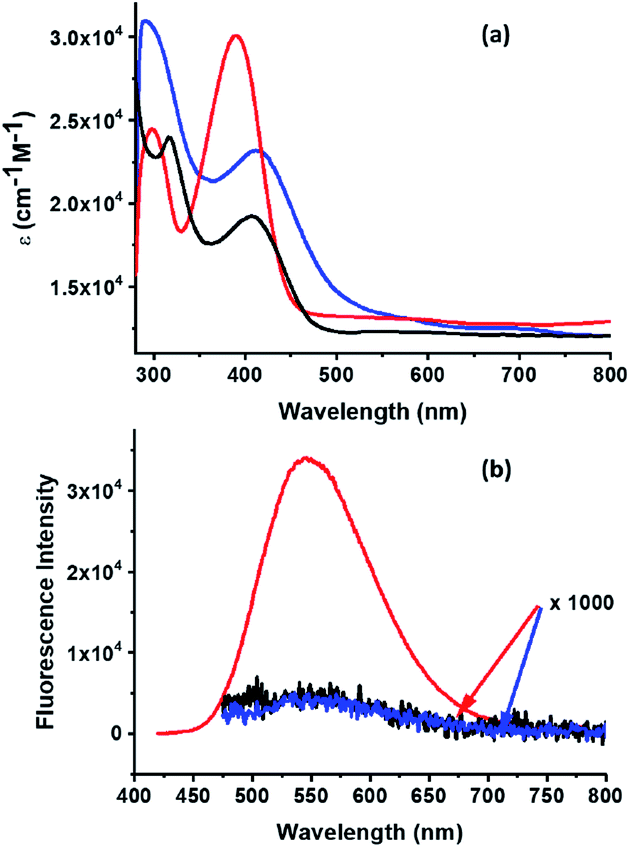 | ||
| Fig. 2 (a) Absorption and (b) fluorescence spectra of 10−6 M solutions of 2 (red), 3a (black) and 3b (blue) in CH2Cl2. | ||
The photophysical properties of 3a and 3b were first studied in dichloromethane by steady-state fluorescence spectroscopy with a 407 nm excitation wavelength (λex), which mostly excites the TPA moiety. The model oxime 2 showed strong fluorescence with a maximum at 544 nm. The fluorescence of this chromophore was fully quenched upon connection to the fullerene cages for both 3a and 3b (Fig. 2b). It is possible that photoinduced charge separation is the reason for the observed fluorescence quenching (vide infra).
The electrochemical properties of 3a and 3b, oxime 2 and pristine Sc3N@Ih-C80 and C60 were investigated by cyclic voltammetry (CV) and Osteryoung square wave voltammetry (OSWV) and the results are summarized in Table 1. Representative CVs for 3a and 3b are shown in Fig. 3a. In the anodic region, the first oxidation potentials of 3a and 3b, attributed to the alkoxy-TPA-thiophenyl moiety, are positively shifted by 80 mV and 110 mV, respectively, compared to that for oxime 2 (isostructural with the organic addend of 3a–b), suggesting the existence of some electronic interactions with the respective fullerene cage. In the cathodic region, the first reduction potential of 3a is positively shifted by 270 mV when compared to that of pristine Sc3N@Ih-C80. This behavior is similar to what was previously observed for an isoxazoline Sc3N@Ih-C80 derivative, confirming the strong influence that functionalization has on the electrochemical properties of the EMF cage.52 However, the first reduction of 3b and C60 showed similar values (−1.05 V and −1.03 V, respectively) as described in other donor-isoxazolinofullerene derivatives.44 This behavior is different from that for C60 derived pyrrolidinofullerenes, where the first reduction is about 120 mV more negative than for pristine C60 as a result of the saturation of one of the double bonds.
| ERed5 | ERed4 | ERed3 | ERed2 | ERed1 | EOx1 | EOx2 | EOx3 | |
|---|---|---|---|---|---|---|---|---|
a Determined by OSWV using o-DCB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile (4:1) as a solvent. A Ag/AgNO3 (0.01 M) electrode was used as a reference and checked against the Fc/Fc+ couple on a glassy carbon electrode, Pt was used as counter electrode at 20 °C and 0.1 M tetrabutylammonium hexafluorophosphate as supporting electrolyte with scan rate = 100 mV s−1.b Non-reversible process.c Value corresponding to the overlap of two reduction processes. :acetonitrile (4:1) as a solvent. A Ag/AgNO3 (0.01 M) electrode was used as a reference and checked against the Fc/Fc+ couple on a glassy carbon electrode, Pt was used as counter electrode at 20 °C and 0.1 M tetrabutylammonium hexafluorophosphate as supporting electrolyte with scan rate = 100 mV s−1.b Non-reversible process.c Value corresponding to the overlap of two reduction processes. |
||||||||
| 3a | — | −2.23c | −1.50c | −0.93 | 0.29 | 0.53b | 0.80b | |
| Sc3N@C80 | −2.35 | −2.23 | −1.72 | −1.59 | −1.20b | 0.69 | — | — |
| 3b | −2.39 | −1.97 | −1.82b | −1.50 | −1.05 | 0.32 | 0.87b | — |
| C60 | — | −2.36 | −1.88 | −1.42 | −1.03 | — | — | — |
| 2 | — | — | — | — | — | 0.21 | 0.68b | 0.79b |
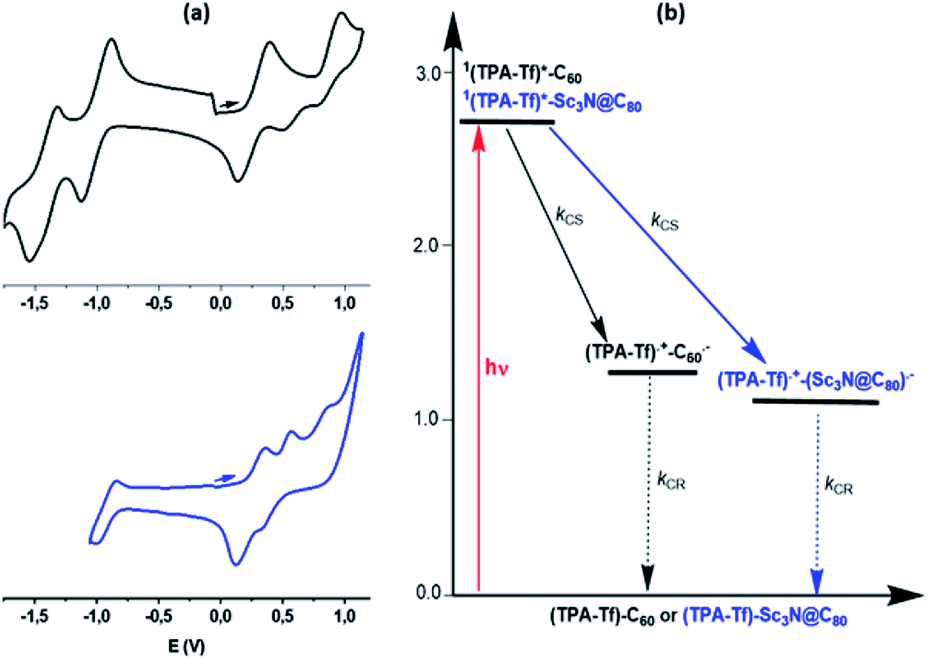 | ||
| Fig. 3 (a) Cyclic voltammograms of 3b (black) and 3a (blue) in o-DCB:acetonitrile (4:1) (b) energy level diagram showing the different photochemical events occurring for 3a and 3b due to TPA excitation. Energies of different states were evaluated from spectral and electrochemical studies. Solid arrows indicate major photo-processes, dashed arrow indicates minor photo-processes. CS = charge separation, CR = charge recombination. | ||
From the optical absorption and emission, electrochemical redox potential and structural parameters, Gibbs free-energy changes associated with charge separation (CS) and charge recombination (CR) were estimated according to eqn (i) and (ii)53
| −ΔGCR = Eox − Ered + ΔGS | (i) |
| −ΔGCS = ΔE0–0 − (−ΔGCR) | (ii) |
The energy level diagram shown in Fig. 3b was constructed to visualize the possible photochemical events. Excitation of TPA in the dyads leads to a population of its lowest excited singlet state, 1TPA-Tf*. The 1TPA-Tf* state can undergo a thermodynamically allowed CS to the appended fullerene. The fluorescence spectra in Fig. 2b reveals that attaching Sc3N@Ih-C80 or C60 to TPA via the thiophene spacer leads to quantitative quenching and no new peaks due to fullerene emission suggesting no energy transfer. The data suggest that CS is the most likely quenching mechanism. The high ΔGCS and ΔGCR values and the spatial closeness between the donor and acceptor entities along with the strong electronic coupling by through the thiophene spacer are anticipated to accelerate both the CS and CR processes. Further, fs-TA studies were performed in solvents of different polarity to clarify these predictions. Prior to this, in an effort to spectrally characterize electron transfer products, TPA-Tf (compound 2) was chemically oxidized using NOBF4 in benzonitrile, as shown in Fig. S11.† Increased additions of the oxidizing agent decreased the intensity of the main peak at 390 nm and resulted in the appearance of new peaks at 375, 486, 564, 731 and 967 nm, corresponding to (TPA-Tf)+˙. Spectral characteristics of Sc3N@Ih-C80−˙ and C60−˙ with peaks around 1000 nm have been reported earlier.54,55
Fs-TA studies were performed in three solvents of varying polarity. Fig. 4 shows fs-TA spectra of 2, 3a and 3b at the indicated delay times in o-DCB while those recorded in toluene and benzonitrile are shown in Fig. S12 and S13,† respectively. In all these solvents, immediately after excitation, the instanteneously formed 12* (1TPA-Tf*) revealed a negative peak in the 450–550 nm region due to contributions of ground state bleaching (GSB) and stimulated emission (SE). Positive peaks in the 600 and 1240 nm region due to excited state absorption (ESA) were also observed. With time, the near-IR peak revealed a blue-shift, likely due to solvation or vibrational cooling,56,57 and at longer delay times, the recovery and decay of negative and positive peaks was associated with emergence of new peaks in the 735 and 1000 nm range likely due to 32*, formed via intersystem crossing (ISC).
 | ||
| Fig. 4 Fs-TA spectra at the indicated delay times of (a) 2 (λex = 390 nm), (b) 3a (λex = 500 nm), and (c) 3b (λex = 390 nm) in o-DCB. The DAS are shown in the right hand panels. | ||
For 3a and 3b ultrafast electron transfer events were observed. As shown in Fig. 4b and c, the recovery and decay of peaks corresponding to 1(TPA-Tf)* were rapid with new peaks developing within a few picoseconds characteristic of (TPA-Tf)+˙-Sc3N@Ih-C80−˙ in the case of 3a and (TPA-Tf)+˙-C60−˙ in the case of 3b. These are signature peaks of Sc3N@Ih-C80−˙ and C60−˙ in the near-IR region and (TPA-Tf)+˙ in the visible region. Further, decay associated spectra (DAS) were generated from global analysis to evaluate time constants for CS and CR, as shown in the right-hand panels of Fig. 4. Changing the solvent revealed solvent polarity dependent electron transfer kinetics, showing that polar solvents facilitated the charge separation process. The measured time constants and rates are given in Table 2.
| Compound | Solvent | τCS, ps | kCS, s−1 | τCR, ps | kCR, s−1 | Ref. |
|---|---|---|---|---|---|---|
| 3a | Toluene | 4.4 | 2.23 × 1011 | 65.7 | 1.50 × 1010 | t.w. |
| o-DCB | 1.0 | 10.0 × 1011 | 5.6 | 1.78 × 1011 | t.w. | |
| PhCN | 1.02 | 9.80 × 1011 | 4.3 | 2.32 × 1011 | t.w. | |
| 3b | Toluene | 4.2 | 2.38 × 1011 | 732 | 1.36 × 109 | t.w. |
| o-DCB | 7.0 | 1.43 × 1011 | 18 | 5.56 × 1010 | t.w. | |
| PhCN | 2.9 | 3.44 × 1011 | 6.3 | 1.59 × 1011 | t.w. | |
| TPA-Sc3N@C80 | THF | — | 3.4 × 1010 | 1.7 × 109 | 54 | |
| PhCN | — | — | — | 4.5 × 108 | 54 | |
| TPA-C60 | THF | — | 7.6 × 1010 | — | 8.0 × 109 | 54 |
| PhCN | — | — | — | 6.5 × 109 | 54 |
The data presented in Table 2 reveals the following: (i) the kCS values are generally higher for 3a compared to 3b which could be attributed to the free-energy changes (see Fig. 3b) where facile charge separation for 3a over 3b was noted. (ii) Although some charge stabilization is observed in nonpolar toluene, in general kCR values are also much faster on the order of 1010 to 1011 s−1. (iii) A comparison of electron transfer rates between the present system and the previously reported TPA-fullerene conjugate54 where charge stabilization was observed to some extent suggests that the thiophene entity enhances the π-conjugation between the donor and acceptor entities in 3a and 3b, thus facilitating electron transfer events in both directions.
Conclusions
In summary, new donor–acceptor conjugates, TPA-Tf-Sc3N@Ih-C80, and its C60 analog, TPA-Tf-C60, were synthesized by a 1,3-dipolar cycloaddition reaction of nitrile oxides. The thiophene unit promotes the π-conjugation between the donor and acceptor entities. An energy level diagram constructed using spectral and electrochemical data revealed an exothermic excited state charge separation, more so for the TPA-Tf-Sc3N@Ih-C80 conjugate. Photoexcitation of the TPA-Tf entity in the conjugates promoted charge separation within a few picoseconds, attributed to the π-linker, thiophene, connecting the donor and acceptor units, accesible redox potentials and spatial closeness of the donor and acceptor entities of the conjugates. Further studies to fully explore the potential of the current synthetic methodology to functionalize endohedral fullerenes for photochemical and optoelectronic applications are underway in our laboratories.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
Support from MINECO (Spain) (grant CTQ2016-79189-R to FL), the Junta de Comunidades de Castilla-la Mancha and European Social Fund (grant SBPLY/17/180501/000254), the US National Science Foundation (CHE-18001317 to L. E. and CHE-1401188 to F. D.), and The Robert A. Welch Foundation (for an endowed chair to L. E. (grant AH-0033)) is gratefully acknowledged.Notes and references
- N. Shigeru, K. Kaoru and A. Takeshi, Bull. Chem. Soc. Jpn., 1996, 69, 2131–2142 CrossRef.
- X. Lu, L. Feng, T. Akasaka and S. Nagase, Chem. Soc. Rev., 2012, 41, 7723–7760 RSC.
- T. Akasaka and X. Lu, Chem. Rec., 2012, 12, 256–269 CrossRef CAS PubMed.
- X. Lu, T. Akasaka and S. Nagase, Chem. Commun., 2011, 47, 5942–5957 RSC.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS.
- L. Dunsch and S. Yang, Small, 2007, 3, 1298–1320 CrossRef CAS.
- P. Jin, Y. Li, S. Magagula and Z. Chen, Coord. Chem. Rev., 2019, 388, 406–439 CrossRef CAS.
- Y. Chai, X. Liu, B. Wu, L. Liu, Z. Wang, Y. Weng and C. Wang, J. Am. Chem. Soc., 2020, 142, 4411–4418 CrossRef CAS.
- X. Lu, L. Bao, T. Akasaka and S. Nagase, Chem. Commun., 2014, 50, 14701–14715 RSC.
- R. B. Ross, C. M. Cardona, D. M. Guldi, S. G. Sankaranarayanan, M. O. Reese, N. Kopidakis, J. Peet, B. Walker, G. C. Bazan, E. Van Keuren, B. C. Holloway and M. Drees, Nat. Mater., 2009, 8, 208–212 CrossRef CAS PubMed.
- J. R. Pinzón, M. E. Plonska-Brzezinska, C. M. Cardona, A. J. Athans, S. S. Gayathri, D. M. Guldi, M. Á. Herranz, N. Martín, T. Torres and L. Echegoyen, Angew. Chem., Int. Ed., 2008, 47, 4173–4176 CrossRef PubMed.
- S. Yang, T. Wei and F. Jin, Chem. Soc. Rev., 2017, 46, 5005–5058 RSC.
- J. W. Meng, D. L. Wang, P. C. Wang, L. Jia, C. Chen and X.-J. Liang, J. Nanosci. Nanotechnol., 2010, 10, 8610–8616 CrossRef CAS PubMed.
- T. Li and H. C. Dorn, Small, 2017, 13, 1603152 CrossRef.
- R. D. Bolskar, in Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes, ed. F. Cataldo and T. Da Ros, Springer Netherlands, Dordrecht, 2008, pp. 157–180 Search PubMed.
- X. Lu, T. Akasaka and S. Nagase, Acc. Chem. Res., 2013, 46, 1627–1635 CrossRef CAS.
- M. N. Chaur, F. Melin, A. L. Ortiz and L. Echegoyen, Angew. Chem., Int. Ed., 2009, 48, 7514–7538 CrossRef CAS PubMed.
- B. Liu, H. Fang, X. Li, W. Cai, L. Bao, M. Rudolf, F. Plass, L. Fan, X. Lu and D. M. Guldi, Chem.–Eur. J., 2015, 21, 746–752 CrossRef CAS PubMed.
- J. R. Pinzón, C. M. Cardona, M. Á. Herranz, M. E. Plonska-Brzezinska, A. Palkar, A. J. Athans, N. Martín, A. Rodríguez-Fortea, J. M. Poblet, G. Bottari, T. Torres, S. S. Gayathri, D. M. Guldi and L. Echegoyen, Chem.–Eur. J., 2009, 15, 864–877 CrossRef.
- Y. Takano, M. Á. Herranz, N. Martín, S. G. Radhakrishnan, D. M. Guldi, T. Tsuchiya, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2010, 132, 8048–8055 CrossRef CAS PubMed.
- M. Rudolf, L. Feng, Z. Slanina, W. Wang, S. Nagase, T. Akasaka and D. M. Guldi, Nanoscale, 2016, 8, 13257–13262 RSC.
- D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22–36 RSC.
- H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 CrossRef CAS PubMed.
- D. M. Guldi, M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1997, 119, 974–980 CrossRef CAS.
- N. Martín, L. Sánchez, M. Á. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015–1024 CrossRef.
- D. M. Guldi, A. Gouloumis, P. Vázquez and T. Torres, Chem. Commun., 2002, 2056–2057, 10.1039/b205620h.
- N. Martín, L. Sánchez, M. A. Herranz and D. M. Guldi, J. Phys. Chem. A, 2000, 104, 4648–4657 CrossRef.
- S. Fukuzumi, K. Ohkubo, H. Imahori and D. M. Guldi, Chem.–Eur. J., 2003, 9, 1585–1593 CrossRef CAS PubMed.
- D. M. Guldi, I. Zilbermann, A. Gouloumis, P. Vázquez and T. Torres, J. Phys. Chem. B, 2004, 108, 18485–18494 CrossRef CAS.
- D. I. Schuster, P. Cheng, P. D. Jarowski, D. M. Guldi, C. Luo, L. Echegoyen, S. Pyo, A. R. Holzwarth, S. E. Braslavsky, R. M. Williams and G. Klihm, J. Am. Chem. Soc., 2004, 126, 7257–7270 CrossRef CAS PubMed.
- D. I. Schuster, K. Li, D. M. Guldi, A. Palkar, L. Echegoyen, C. Stanisky, R. J. Cross, M. Niemi, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2007, 129, 15973–15982 CrossRef CAS PubMed.
- S. Sato, Y. Maeda, J.-D. Guo, M. Yamada, N. Mizorogi, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2013, 135, 5582–5587 CrossRef CAS.
- S. Osuna, M. Swart and M. Solà, J. Am. Chem. Soc., 2009, 131, 129–139 CrossRef CAS PubMed.
- N. Alegret, A. Rodríguez-Fortea and J. M. Poblet, Chem.–Eur. J., 2013, 19, 5061–5069 CrossRef CAS PubMed.
- M. Garcia-Borràs, M. R. Cerón, S. Osuna, M. Izquierdo, J. M. Luis, L. Echegoyen and M. Solà, Angew. Chem., Int. Ed., 2016, 55, 2374–2377 CrossRef PubMed.
- J. R. Pinzón, T. Zuo and L. Echegoyen, Chem.–Eur. J., 2010, 16, 4864–4869 CrossRef PubMed.
- N. Alegret, M. N. Chaur, E. Santos, A. Rodríguez-Fortea, L. Echegoyen and J. M. Poblet, J. Org. Chem., 2010, 75, 8299–8302 CrossRef CAS PubMed.
- B. Cao, T. Wakahara, Y. Maeda, A. Han, T. Akasaka, T. Kato, K. Kobayashi and S. Nagase, Chem.–Eur. J., 2004, 10, 716–720 CrossRef CAS.
- Y. Takano, Z. Slanina, J. Mateos, T. Tsuchiya, H. Kurihara, F. Uhlik, M. Á. Herranz, N. Martín, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2014, 136, 17537–17546 CrossRef CAS PubMed.
- Y. Maeda, M. Kimura, C. Ueda, M. Yamada, T. Kikuchi, M. Suzuki, W.-W. Wang, N. Mizorogi, N. Karousis, N. Tagmatarchis, T. Hasegawa, M. M. Olmstead, A. L. Balch, S. Nagase and T. Akasaka, Chem. Commun., 2014, 50, 12552–12555 RSC.
- T. Cai, L. Xu, M. R. Anderson, Z. Ge, T. Zuo, X. Wang, M. M. Olmstead, A. L. Balch, H. W. Gibson and H. C. Dorn, J. Am. Chem. Soc., 2006, 128, 8581–8589 CrossRef CAS.
- T. Cai, Z. Ge, E. B. Iezzi, T. E. Glass, K. Harich, H. W. Gibson and H. C. Dorn, Chem. Commun., 2005, 3594–3596, 10.1039/b503333k.
- C. Ma, B. Xu, G. Xie, J. He, X. Zhou, B. Peng, L. Jiang, B. Xu, W. Tian, Z. Chi, S. Liu, Y. Zhang and J. Xu, Chem. Commun., 2014, 50, 7374–7377 RSC.
- K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863–910 CrossRef CAS.
- F. Langa, P. de la Cruz, E. Espíldora, A. González-Cortés, A. de la Hoz and V. López-Arza, J. Org. Chem., 2000, 65, 8675–8684 CrossRef CAS.
- M. Alvaro, P. Atienzar, P. de la Cruz, J. L. Delgado, V. Troiani, H. Garcia, F. Langa, A. Palkar and L. Echegoyen, J. Am. Chem. Soc., 2006, 128, 6626–6635 CrossRef CAS PubMed.
- M. Vizuete, M. J. Gómez-Escalonilla, J. L. G. Fierro, P. Atienzar, H. García and F. Langa, ChemPhysChem, 2014, 15, 100–108 CrossRef CAS.
- H. Uceta, M. Vizuete, J. R. Carrillo, M. Barrejón, J. L. G. Fierro, M. P. Prieto and F. Langa, Chem.–Eur. J., 2019, 25, 14644–14650 CrossRef CAS.
- L. Bao, M. Chen, W. Shen, C. Pan, K. B. Ghiassi, M. M. Olmstead, A. L. Balch, T. Akasaka and X. Lu, Inorg. Chem., 2016, 55, 4075–4077 CrossRef CAS PubMed.
- A. Aljarilla, C. Herrero-Ponce, P. Atienzar, S. Arrechea, P. d. l. Cruz, F. Langa and H. García, Tetrahedron Lett., 2013, 69, 6875–6883 CrossRef CAS.
- N. Martín, M. Altable, S. Filippone, A. Martín-Domenech, R. Martínez-Álvarez, M. Suarez, M. E. Plonska-Brzezinska, O. Lukoyanova and L. Echegoyen, J. Org. Chem., 2007, 72, 3840–3846 CrossRef.
- C. Shu, T. Cai, L. Xu, T. Zuo, J. Reid, K. Harich, H. C. Dorn and H. W. Gibson, J. Am. Chem. Soc., 2007, 129, 15710–15717 CrossRef CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- J. R. Pinzón, D. C. Gasca, S. G. Sankaranarayanan, G. Bottari, T. Torres, D. M. Guldi and L. Echegoyen, J. Am. Chem. Soc., 2009, 131, 7727–7734 CrossRef PubMed.
- C. B. KC, G. N. Lim and F. D'Souza, Nanoscale, 2015, 7, 6813–6826 RSC.
- J. Moreno, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, S. Hecht and S. A. Kovalenko, J. Chem. Phys., 2015, 143, 024311 CrossRef CAS PubMed.
- M. Fakis, P. Hrobarik, O. Yushchenko, I. Sigmundova, M. Koch, A. Rosspeintner, E. Stathatos and E. Vauthey, J. Phys. Chem. C, 2014, 118, 28509–28519 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and experimental details, additional fs-TA spectral data. See DOI: 10.1039/d0ra04379f |
| This journal is © The Royal Society of Chemistry 2020 |