
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dendrimer crown-ether tethered multi-wall carbon nanotubes support methyltrioxorhenium in the selective oxidation of olefins to epoxides†
Bruno Mattia Bizzarri *a,
Angelica Fanellia,
Lorenzo Bottaa,
Claudia Sadunb,
Lorenzo Gontranic,
Francesco Ferellade,
Marcello Crucianellid and
Raffaele Saladino*a
*a,
Angelica Fanellia,
Lorenzo Bottaa,
Claudia Sadunb,
Lorenzo Gontranic,
Francesco Ferellade,
Marcello Crucianellid and
Raffaele Saladino*a
aDipartimento di Scienze Biologiche ed Ecologiche, Università della Tuscia, Via S. Camillo de Lellis, 01100 Viterbo, Italy. E-mail: bm.bizzari@unitus.it; saladino@unitus.it
bDipartimento di Chimica, La Sapienza Università di Roma, Piazzale Aldo Moro 5, 00185 Roma, Italy
cDipartimento di Ingegneria Industriale, Università di Roma Tor Vergata, Viale degli Ingegneri, 00133 Roma, Italy
dDipartimento di Scienze Fisiche e Chimiche, Università dell'Aquila, Via Vetoio, 67100 L'Aquila, Italy
eLaboratori Nazionali del Gransasso, INFN S.S. 17/bis km 18 + 910, I-67010 Assergi, AQ, Italy
First published on 1st May 2020
Abstract
Benzo-15-crown-5 ether supported on multi-wall carbon nanotubes (MWCNTs) by tethered poly(amidoamine) (PAMAM) dendrimers efficiently coordinated methyltrioxorhenium in the selective oxidation of olefins to epoxides. Environmentally friendly hydrogen peroxide was used as a primary oxidant. Up to first and second generation dendrimer aggregates were prepared by applying a divergent PAMAM methodology. FT-IR, XRD and ICP-MS analyses confirmed the effective coordination of methyltrioxorhenium by the benzo-15-crown-5 ether moiety after immobilization on MWCNTs. The novel catalysts converted olefins to the corresponding epoxides in high yield without the use of Lewis base additives, or anhydrous hydrogen peroxide, the catalyst being stable for more than six oxidative runs. In the absence of the PAMAM structure, the synthesis of diols largely prevailed.
Introduction
Multi-wall carbon nanotubes (MWCNTs) are efficient supports for the immobilization of reactive organometallic species thanks to their high surface area to volume ratio value, chemical stability, and unique redox, rheological and structural properties.1,2 They show surface defect sites for the anchoring of spacer chains and selective recognition sites, as for example for crown ethers.3 Different applications of MWCNTs-crown ether functionalized systems are reported,4 including dispersive micro-solid phase extraction procedures,5 exchange reaction in quasi-solid-state processes,6 absorption of metals and organic pollutants,7 and ion-detection.8 In these latter cases, MWCNT-crown ether tethered poly(amidoamine) (PAMAM) dendrimers received increasing attention.9 These supramolecular aggregates are characterized by recognition sites on both the dendrimer core and the end-termination of the system.10,11 The divergent methodology for the preparation of PAMAM dendrimers encompasses the grafting of 1,3-diamino propane intermediates with methyl acrylate and ethylenediamine, followed by linkage of the crown ether moiety. The chemical complexity of PAMAM dendrimers (number of dendrimer side-chains and type of ramification) depends on the successive methyl acrylate/ethylenediamine treatments, and controls the distribution of the recognition sites, the leaching of the active species,12–14 and the dispersibility of MWCNTs in organic solvents.15Crown ethers are coordination sites for methyltrioxorhenium (MTO),16,17 a potent catalyst for the activation of hydrogen peroxide (H2O2) by formation of two reactive intermediates, namely monoperoxo [MeRe(O2)O2] and bis(peroxo) [MeRe(O2)O2] η2-metal complexes.18 The chemistry of MTO is largely reviewed,19 including σ C–H oxygen atom insertion20 and oxy-functionalization,21 oxidative nucleophilic substitution,22 oxidative desulfurization (ODS),23 synthesis of quinone derivatives,24 Baeyer–Villiger rearrangement,25 metathesis,26 methanolysis of glycals,27 and epoxidation of olefins in traditional28 and non-conventional solvents, such as chiral aliphatic amines,24 fluorinated solvent29 and ionic liquids.30 The formation of diols is the major drawback in the epoxidation of olefins, as a consequence of the acidic property and high affinity of Re(VII) for the oxygen atom. Lewis bases such as pyridine, pyridine derivatives,31,32 and bipyridines,33,34 have been used as additives to inhibit this side-reaction, their efficacy depending from the stability of the corresponding MTO complex. As an alternative, anhydrous H2O2 and urea-H2O2 adduct (UHP) have been also applied.35 The immobilization of MTO on solid supports can solve the problem of selectivity, stability and recyclability of the catalyst. Immobilization procedures have been reported including organic polymers bearing Lewis bases, such as chitosan,36 poly(4-vinylpyridine),37 poly(4-vinylpyridine-N-oxide),38 and MTO/nitrogen ligand complexes,39 and in alternative different inorganic supports.40–42 More recently, MWCNTs wrapped on poly(4-vinylpyridine)/MTO showed high reactivity in the desulfurization of synthetic diesel fuel.43 Lewis-base sites generally enhance the formation of epoxides,44,45 but drawbacks relative to toxicity of the Lewis base additives (e.g. pyridine and pyridine derivatives), partial selectivity, low reactivity and leaching of rhenium have yet to be completely solved.
We report here the preparation and structural characterization of novel heterogeneous catalysts based on the immobilization of MTO on MWCNTs functionalized with benzo-15-crown-5 ether (B15C5) in the presence of tethered PAMAM dendrimers. Up to first and second generation of B15C5 PAMAM aggregates were obtained by applying the divergent methodology. The variety of catalysts was increased by the use of alkyl-diamino linkers of various length (from C-2 to C-6 atoms, respectively). FT-IR, ICP-MS and XRD analyses confirmed the coordination of MTO by the B15C5 moiety after immobilization on MWCNTs. The novel catalysts were applied in the epoxidation of a large panel of olefins using H2O2 (35% water solution) as green oxidant in the absence of Lewis bases additives to yield epoxides in high yield and selectivity. As a general trend, alkyl-diamino linkers with longer carbon atom chain afforded the most reactive systems in PAMAM aggregates. Diols were obtained as the main reaction products in the absence of the dendrimer structure, highlighting the role of PAMAM in the selectivity of the reaction.
Results and discussion
Preparation of MWCNTs-B15C5/MTO catalysts IV A–D
MWCNTs-B15C5/MTO catalysts IV A–D were prepared by immobilization of MTO on previously functionalized MWCNTs-B15C5 support. Briefly, MWCNTs I were treated with different alkyl-diamino linkers, ethylene diamine (EDA), propylene diamine (PDA), cadaverine (CV), and hexamethylene diamine (HAD) (from C-2 to C-6 atoms, respectively), in the presence of N,N-di-isopropylcarbodiimide (DIC), 1-hydroxybenzotriazole (HOBt), and N,N-diisopropyl ethylamine (DIPEA) in DMF at 25 °C for 24 h, to yield intermediates II A–D.46 The coupling procedure was repeated with 4′-carboxy-benzo-15-crown-5 (B15C5) as electrophilic reagent, to afford MWCNTs-B15C5 III A–D (Scheme 1). The FT-IR analysis of intermediates II A–D and III A–D showed the shift of the peak at 1730 cm−1 (stretching vibration of the carboxylic group in native MWCNTs I) at 1637 cm−1 (SI#1, Fig. S1 and S2†), confirming the formation of the amide bond.47 Moreover, intermediates III A–D showed the characteristic peaks at 1120 cm−1 and 1210 cm−1, corresponding to C–O stretching vibrational mode of B15C5 (SI#1, Fig. S2†).8 MWCNTs-B15C5 III A–D were successively treated with MTO in THF under gentle stirring at 25 °C for 6 h, to afford MWCNTs-B15C5/MTO catalysts IV A–D.43 Catalysts IV A–D showed the typical Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrational mode at 953 cm−1 (SI#1, Fig. S3†). Scanning Electron Microscopy (SEM) analysis of IV A–D (Fig. 1) confirmed the integrity of MWCNTs after the functionalization procedure. Powder X-ray analysis (XRD) was applied to evaluate the coordination geometry of MTO in representative intermediate III D and catalyst IV D.48–50 The X-ray data of III D and IV D were treated by the difference method of neutron diffraction and isotopic substitution (NDIS)37,51–53 in order to solve the contemporary presence of highly structured MWCNTs and amorphous B15C5 (generally indicated as support in the next discussion). Assuming that the general structure of III D was not significantly altered during the MTO loading, structural data for MTO/B15C15 complex were obtained by subtracting the radial distribution D(r) function of III D to that of IV D (Fig. 2, panels A and B) (see SI#2† for a detailed description of the difference method technique).54 The structure and distribution functions of III D and IV D are reported in SI#2 (Fig. S5 and S6,† respectively).
O stretching vibrational mode at 953 cm−1 (SI#1, Fig. S3†). Scanning Electron Microscopy (SEM) analysis of IV A–D (Fig. 1) confirmed the integrity of MWCNTs after the functionalization procedure. Powder X-ray analysis (XRD) was applied to evaluate the coordination geometry of MTO in representative intermediate III D and catalyst IV D.48–50 The X-ray data of III D and IV D were treated by the difference method of neutron diffraction and isotopic substitution (NDIS)37,51–53 in order to solve the contemporary presence of highly structured MWCNTs and amorphous B15C5 (generally indicated as support in the next discussion). Assuming that the general structure of III D was not significantly altered during the MTO loading, structural data for MTO/B15C15 complex were obtained by subtracting the radial distribution D(r) function of III D to that of IV D (Fig. 2, panels A and B) (see SI#2† for a detailed description of the difference method technique).54 The structure and distribution functions of III D and IV D are reported in SI#2 (Fig. S5 and S6,† respectively).
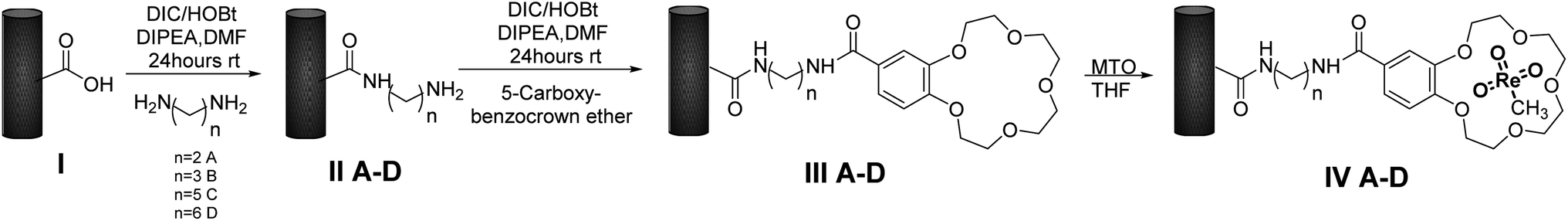 | ||
| Scheme 1 Synthesis of catalysts IV A–D. | ||
 | ||
| Fig. 1 SEM images of catalyst IV A–D. The structure of MWCNTs is not modified after the functionalization procedure with B15C5. | ||
 | ||
| Fig. 2 Red line represents the theoretical model. Panel (A) weighted experimental radial distribution function D(r) of III D. Panel (B) experimental radial distribution function D(r) of IV D. Panel (C) difference curve for the two radial distribution functions, D(r)[IV D] − D(r)[III D] [–○–]. Panel (D) balls and sticks view of the complex between MTO and B15C5. | ||
Note that the oscillation pattern in the D(r) function of the two samples remain almost the same, confirming the hypothesis that the MTO loading didn't alter the structural order of the support. The D(r) function showed three peaks, set at about 1.80, 2.45, and 3.00 Å, respectively (Fig. 2, panel C). The first coordination sphere of the Re atom consists of six interactions assuming a quite octahedral conformation around the metal. The first peak was fitted by three metal–oxygen interaction lengths of exactly 1.80 Å and one metal–carbon interaction of 1.75 Å, respectively. The second peak at 2.45 Å is due to the other two interactions Re–O necessary to complete the octahedral coordination sphere. These interactions are also responsible for the peak at 3.00 Å (mutual interactions). The carbon atoms of the crown ether moiety might also contribute to this peak. The successive signals in the D(r) function were not further considered, since they derived from long-range MTO-MWCNTs interactions. In Fig. 2 (panel C) the function is compared with the theoretical peak shape function calculated for the octahedral model represented in Fig. 2 (panel D). The Re–O and Re–C bond distance values are quite in accord with the mean values previously reported for MTO/Ln complexes.55 As shown by the model (Fig. 2, panel D), the Re atom is coordinated by two adjacent oxygen atoms of the B15C5 ring. The model refers to the couple of oxygen atoms bonded to the aromatic ring, but the possibility that another couple of adjacent oxygen atoms is involved in the interaction with the metal cannot be completely ruled-out.
Moreover, the first Re coordination sphere analysis does not allow to determine the relative position of the methyl group with respect to the crown-ether oxygen atoms, due to the small electron differences between this group and the oxygen ones.
The distances between Re atom and ligands in the octahedral arrangement are reported in Table 1.
Preparation of MWCNTs-B15C5/PAMAM/MTO catalysts V E–H
We prepared first and second-generation dendrimer catalysts V E–F and V G–H, respectively. As a general procedure, intermediates II A and II D were treated with methyl acrylate in MeOH at 25 °C for three days, followed by addition of alkyl-diamino linkers EDA and HAD, to yield III E–F, respectively (Scheme 2, pathway A). The reaction of III E–F with B15C5 in the presence of DIC, HOBT, and DIPEA, in DMF at 25 °C for 24 h, afforded IV E–F. These latter intermediates were further converted to first-generation MWCNTs-B15C5/PAMAM/MTO catalysts V E–F by treatment with MTO in THF at 25 °C. In a similar way, two successive treatments of II A and II D with methyl acrylate and alkyl-diamino linkers afforded III G–H (Scheme 2, pathway B), successively converted into IV G–H under previously reported experimental conditions.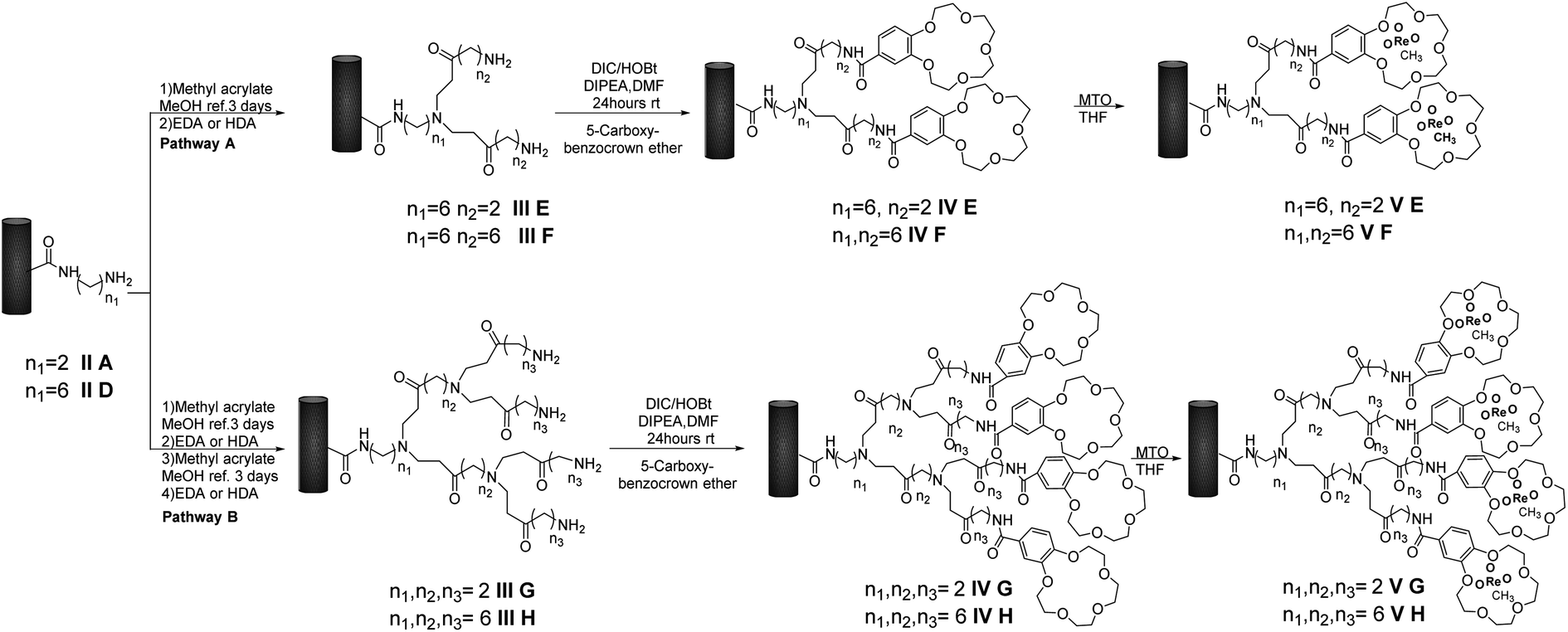 | ||
| Scheme 2 Synthesis of MWCNTs-B15C5/PAMAM/MTO catalysts V E–H. Pathway A: preparation of the first-generation of dendrimer hybrids V E–F. Pathway B: preparation of the second-generation of dendrimer hybrids V G–H. | ||
Treatment of IV G–H with MTO in THF at 25 °C yielded the second-generation MWCNTs-B15C5/PAMAM/MTO catalysts IV G–H. FT-IR analyses of catalysts V E–H are reported in SI#1 (Fig. S4†).
Irrespective from the sample, the characteristic ReO stretching vibrational mode at 950 cm−1, and the CONH stretching vibrational mode at 1630 cm−1, were always observed, besides to B15C5 C–O stretching signals at 1120, 1210, and 1250 cm−1, respectively. The integrity of MWCNTs in catalysts V E–H after the PAMAM functionalization was confirmed by SEM analysis (Fig. 3). Moreover, some sections of the MWCNTs in catalysts V F and V H, as representative examples, showed the presence of the PAMAM coating in TEM images (SI#6†). The loading factor of catalysts IV A–D and V E–H was evaluated by Inductively Coupled Plasma Mass-Spectrometry (ICP-MS) analysis (Table 2, entries 1–8). Catalysts V E–F showed loading factors of the same order of magnitude than IV A–D (Table 2, entries 5–6 versus entries 1–4). As a general trend, a slight decrease of the loading factor was observed for catalysts V G–H (Table 2, entries 7–8). Note that, among PAMAM catalysts, the higher value of the loading factor was obtained in the presence of the longest linker in terminal position (Table 2, entry 6 versus entry 5, and entry 8 versus entry 7).
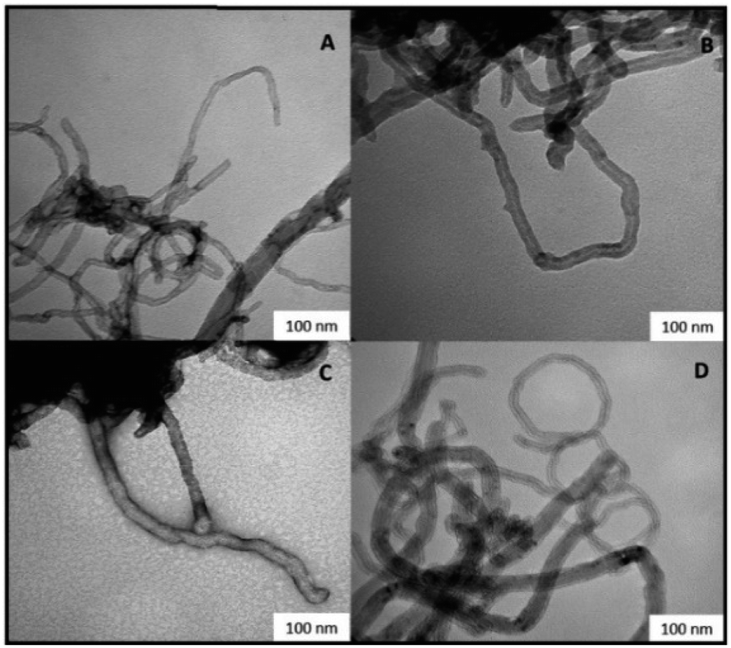 | ||
| Fig. 3 Scanning electron microscopy images of catalysts V E–H. Panel (A) catalyst V E. Panel (B) catalyst V F. Panel (C) catalyst V G. Panel (D) catalyst V H. | ||
| Entry | Catalyst | Rea (%) | Loading factorb |
|---|---|---|---|
| a Values are expresses as wt/% with respect to the total weight of the sample. Measurements were repeated in triplicate.b Amount of mg of MTO per gram of support.c The n value represents the number of carbon atoms in the linker as reported in Scheme 2. | |||
| 1 | IV A (n = 2)c | 29.9 ± 0.3 | 1.6 |
| 2 | IV B (n = 3) | 16.4 ± 0.3 | 0.9 |
| 3 | IV C (n = 5) | 30.6 ± 0.4 | 1.7 |
| 4 | IV D (n = 6) | 8.3 ± 0.3 | 0.5 |
| 5 | V E (n1 = 6, n2 = 2) | 22.3 ± 0.5 | 1.2 |
| 6 | V F (n1 = 6, n2 = 6) | 26.4 ± 0.4 | 1.4 |
| 7 | V G (n1 = n2 = n3 = 2) | 3.7 ± 0.2 | 0.2 |
| 8 | V H (n1 = n2 = n3 = 6) | 4.1 ± 0.3 | 0.3 |
Examples of the decreased accessibility to coordinative sites into higher generation dendrimer systems have been reported as a consequence of conformational changes, steric hindrance and encapsulation processes.56
Epoxidation of olefins with catalysts IV A–D and V E–H
Catalysts IV A–D and V E–H have been evaluated for the epoxidation of a large panel of aliphatic and aromatic olefins, including 1-hexene 1, 1-octene 2, trans-3-octene 3, vinyl-cyclohexane 4, styrene 5, cis-stilbene 6, and allylbenzene 7. The oxidation was performed by treating the selected olefin (0.05 mmol) with H2O2 (35% water solution, 0.5 mmol) and the appropriate amount of catalyst (containing 0.0025 mmol of MTO) at 25 °C, in the absence of Lewis base additives as resumed in Scheme 3.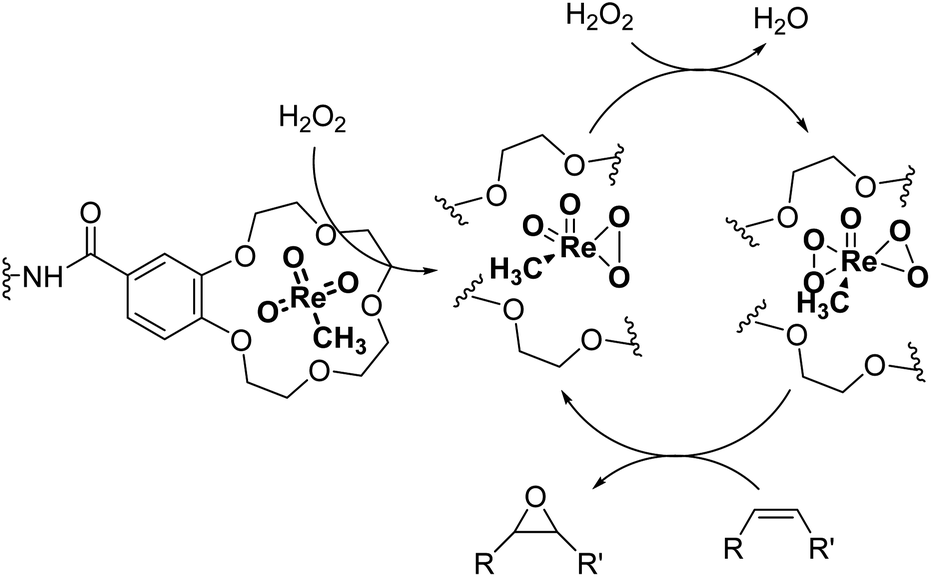 | ||
| Scheme 3 Schematic representation for the MTO/H2O2 epoxidation of olefins. The reaction proceeds through the formation of monoperoxo [MeRe(O2)O2] and bis(peroxo) [MeRe(O2)O2] η2-metal complexes.18 | ||
The reaction products have been detected by gas-chromatography associated to mass-spectrometry (GC-MS) and nuclear magnetic resonance (NMR) analysis after chromatographic purification (SI#3 and SI#4,† respectively). The oxidation with MTO alone was used as reference. Irrespective from the experimental conditions, the oxidation of 1 with MTO and catalysts IV A–D afforded the diol 9 as the only recovered product (Table 3, entries 1–5), IV B being the most efficient catalyst (Table 3, entry 3) The corresponding epoxide was not detected in the reaction mixture, confirming the detrimental effect of the Lewis acidity of MTO in the ring-opening of the oxirane ring, even in the presence of B15C5. As a general trend, the catalytic activity was improved by increasing the number of carbon atoms in the alkyl-diamino spacer, as in the case of IV C–D (Table 3, entries 4 and 5), the lowest reactivity being observed with IV A, characterized by a linker with only two carbon atoms. MWCNTs-B15C5/PAMAM/MTO catalysts V E–H showed a different selectivity in the oxidation of compound 1, the epoxide 8 being obtained as the only recovered product in quantitative yield and high conversion of the substrate (Table 3, entries 6–9). In particular, the second-generation of PAMAM catalysts V G–H was more reactive than the first-generation counterpart V E–F (Table 3, entries 6–7 versus entries 8–9). It is reasonable to suggest that the high selectivity toward the synthesis of the epoxide in the oxidation of 1 with PAMAM catalysts may be associated to the occurrence of additional interactions between MTO and nitrogen atoms in the dendrimer structure, acting as internal Lewis base sites. In accordance with this hypothesis, the vibrational mode at 492 cm−1, corresponding to stretching of the Re–N group, was detected for V G, as representative example, by Far Infrared analysis (FIR) (SI#5;† MTO/1,2-cyclohexanediamine complex was used as reference). The ligand properties of tertiary amine atoms in PAMAM dendrimer are reported.57 For example, octahedral Co(II)-PAMAM dendrimer complexes showed a slight distorted octahedral coordination of the metal atom by the polymeric matrix.58,59 Moreover, atomistic molecular dynamics (MD) simulations on ethylenediamine (EDA) PAMAM dendrimer wrapped on MWCNTs (up to 11th generation) showed the high mobility and the low distance occurring between tertiary nitrogen atoms and the terminal amino groups of the structure, where the metal coordinative sites are expected to be located.58 The oxidation of aliphatic olefins 1-octene 2, trans-3-octene 3 and vinyl-cyclohexane 4 proceeded in a similar way. The epoxides 10, 12 and 14 were obtained in quantitative yield in the presence of catalysts V E–H (Table 3, entries 15–18, 24–27, and 33–36, respectively), while diols 11, 13 and 15 were isolated as the main recovered products with MTO alone and catalysts IV A–D (Table 3). In this latter case, only small amounts of epoxides were detected.
| Entry | Substrate | Catalyst | Product | Conversion (%) | Yield of epoxideb (%) | Yield of diolb (%) | TONc |
|---|---|---|---|---|---|---|---|
| a The reaction was performed by treating the selected olefin (0.5 mmol) with a stoichiometric amount of H2O2 (35%, water solution) in the presence of the appropriate catalyst (0.5 mol/% with respect to loaded MTO) in CH2Cl2 (5.0 mL) at 25 °C for 5 h. Experiments were performed in triplicate. A: ethylene diamine NH2(CH2)2NH2. B: propylene diamine NH2(CH2)3NH2. C: cadaverine NH2(CH2)5NH2. D: hexamethylene diamine NH2(CH2)6NH2.b The product yield is calculated with respect to converted substrate. N.d. not determined.c Turn over number expressed as the ratio between the mmol of epoxide versus the mmol of MTO in the heterogeneous catalyst. | |||||||
| 1 | 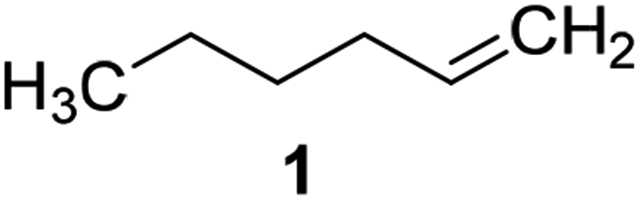 |
MTO |  |
94 | n.d.c | >98 | |
| 2 | IV A | 16 | n.d. | >98 | — | ||
| 3 | IV B | 84 | n.d. | >98 | — | ||
| 4 | IV C | 68 | n.d. | >98 | — | ||
| 5 | IV D | 58 | n.d | >98 | — | ||
| 6 | V E | 89 | >98 | n.d. | 178 | ||
| 7 | V F | 91 | >98 | n.d. | 182 | ||
| 8 | V G | >98 | >98 | n.d. | 200 | ||
| 9 | V H | >98 | >98 | n.d. | 200 | ||
| 10 | 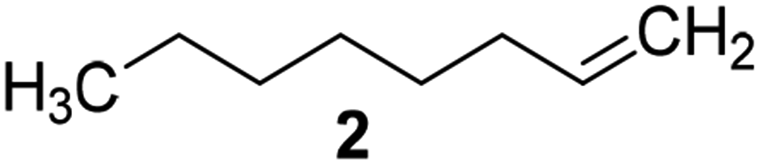 |
MTO |  |
80 | n.d | >98 | — |
| 11 | IV A | 12 | n.d | >98 | — | ||
| 12 | IV B | 83 | 4 | 96 | 8 | ||
| 13 | IV C | 64 | 11 | 87 | 22 | ||
| 14 | IV D | 58 | n.d. | n.d. | — | ||
| 15 | V E | 80 | >98 | n.d. | 160 | ||
| 16 | V F | 77 | >98 | n.d. | 154 | ||
| 17 | V G | >98 | >98 | n.d. | 200 | ||
| 18 | V H | >98 | >98 | n.d. | 200 | ||
| 19 |  |
MTO |  |
84 | n.d. | >98 | 0 |
| 20 | IV A | 18 | 3 | 97 | 6 | ||
| 21 | IV B | 79 | 12 | 85 | 24 | ||
| 22 | IV C | 69 | 12 | 88 | 24 | ||
| 23 | IV D | 62 | >98 | n.d | 124 | ||
| 24 | V E | 85 | >98 | n.d | 170 | ||
| 25 | V F | 89 | >98 | n.d | 178 | ||
| 26 | V G | >98 | >98 | n.d | 200 | ||
| 27 | V H | >98 | >98 | n.d | 200 | ||
| 28 | 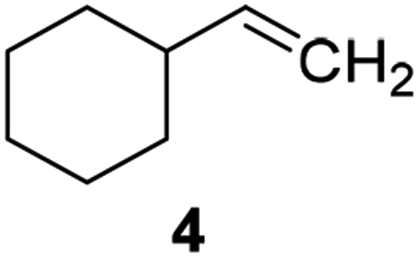 |
MTO |  |
88 | n.d. | >98 | — |
| 29 | IV A | 16 | n.d.b | >98 | — | ||
| 30 | IV B | 83 | 14 | 85 | 28 | ||
| 31 | IV C | 64 | 3 | 97 | 6 | ||
| 32 | IV D | 58 | n.d.b | >98 | — | ||
| 33 | V E | 93 | >98 | n.d | 186 | ||
| 34 | V F | 90 | >98 | n.d | 180 | ||
| 35 | V G | >98 | >98 | n.d | 200 | ||
| 36 | V H | >98 | >98 | n.d | 200 | ||
| 37 |  |
MTO |  |
70 | n.d.b | >98 | — |
| 38 | IV A | 31 | n.d.b | >98 | — | ||
| 39 | IV B | 48 | 5 | 90 | 10 | ||
| 40 | IV C | 36 | 8 | 92 | 16 | ||
| 41 | IV D | 29 | 11 | 89 | 22 | ||
| 42 | V E | 64 | >98 | n.d | 128 | ||
| 43 | V F | 67 | >98 | n.d | 134 | ||
| 44 | V G | 72 | >98 | n.d | 144 | ||
| 45 | V H | 74 | >98 | n.d | 148 | ||
| 46 |  |
MTO | 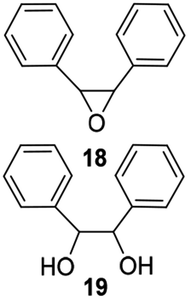 |
80 | n.d.b | >98 | — |
| 47 | IV A | 18 | n.d.b | >98 | — | ||
| 48 | IV B | 35 | 5 | 95 | 10 | ||
| 49 | IV C | 36 | 7 | 93 | 14 | ||
| 50 | IV D | 34 | 4 | 96 | 8 | ||
| 51 | V E | 65 | >98 | n.d | 130 | ||
| 52 | V F | 61 | >98 | n.d | 122 | ||
| 53 | V G | 74 | >98 | n.d | 148 | ||
| 54 | V H | 77 | >98 | n.d | 154 | ||
| 55 | 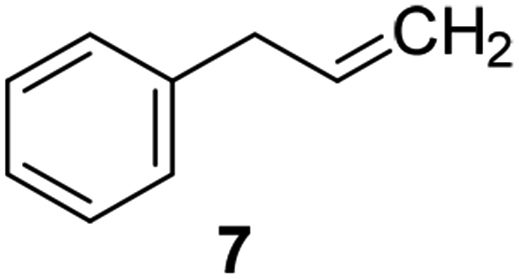 |
MTO | 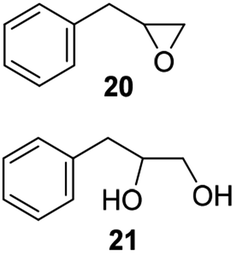 |
69 | n.d.b | >98 | — |
| 56 | IV A | 21 | n.d.b | >98 | — | ||
| 57 | IV B | 40 | 10 | 90 | 20 | ||
| 58 | IV C | 37 | 7 | 93 | 14 | ||
| 59 | IV D | 39 | 9 | 91 | 18 | ||
| 60 | V E | 52 | >98 | n.d | 104 | ||
| 61 | V F | 48 | >98 | n.d | 96 | ||
| 62 | V G | 62 | >98 | n.d | 124 | ||
| 63 | V H | 89 | >98 | n.d | 178 | ||
The second generation of PAMAM catalysts was slightly more reactive than the first-generation counterpart in the conversion of substrate. Catalysts V E–H efficiently catalyzed the epoxidation of low reactive aromatic olefins, styrene 5, cis-stilbene 6, and allylbenzene 7, affording the corresponding epoxides 16, 18, and 20, as the only recovered products in quantitative yield, from acceptable (52%) to high (89%) conversion of substrate (Table 3, entries 42–45, 51–54, 60–63, respectively). Instead, diols 17, 19, and 21, largely prevailed in the oxidation performed with MTO and catalysts IV A–D. The recyclability of catalysts V F and V H in the oxidation of compound 1 was studied as representative example (Table 4). After the first run, the catalyst was recovered by filtration, washed with CH2Cl2, and used without any further purification. Catalysts V F and V H showed a slight decrease in the conversion of substrate (Table 4, entries 2–6 and entries 7–12, respectively) yielding the epoxide 8 in quantitative yield for at least six successive reactions. In accordance with these data, a low decrease of the loading factor of the catalyst was observed by ICP-MS analysis of the sample at the end of any run (Table 4).
| Run | Substrate | Catalyst | Conversion (%) | Yield of epoxidea (%) | Reb (%) |
|---|---|---|---|---|---|
| a The reactions were performed by treating the olefin 1 (0.5 mmol) with H2O2 (0.5 mmol) and the appropriate quantity of MTO catalyst (containing 0.0025 mmol of active rhenium species) calculated with respect to the MTO loading value. After each reaction, the catalyst was recovered by filtration, washed with CH2Cl2, and used without any further purification for the successive run.b Values are expresses as wt/% with respect to the total weight of the sample. Measurements were repeated in triplicate. | |||||
| 1 | 1 | V F | 91 | >98 | 26.4 ± 0.4 |
| 2 | 90 | >98 | 26.1 ± 0.3 | ||
| 3 | 90 | >98 | 26.0 ± 0.2 | ||
| 4 | 85 | >98 | 24.3 ± 0.4 | ||
| 5 | 84 | >98 | 23.9 ± 0.3 | ||
| 6 | 82 | >98 | 21.6 ± 0.2 | ||
| 7 | V H | >98 | >98 | 4.1 ± 0.3 | |
| 8 | >98 | >98 | 4.0 ± 0.2 | ||
| 9 | >98 | >98 | 4.0 ± 0.2 | ||
| 10 | 97 | >98 | 4.0 ± 0.3 | ||
| 11 | 95 | >98 | 3.5 ± 0.2 | ||
| 12 | 92 | >98 | 3.3 ± 0.1 | ||
Experimental section
Materials
1-Hexene 1, 1-octene 2, trans-3-octene 3, vinyl-cyclohexane 4, styrene 5, cis-stilbene 6, allylbenzene 7, organic solvents, MTO, methyl acrylate, ethylene diamine (EDA), propylene diamine (PDA), cadaverine (CV), hexamethylene diamine (HDA), diisopropylcarbodiimide (DIC), hydroxybenzotriazole (HOBt), N,N-diisopropyl ethylamine (DIPEA), 4′-carboxy-benzo-15-crown-5, and GH polypro membrane filters were purchased from Sigma-Aldrich-Merck (Germany) and used without further purification. Gas chromatography associated to mass spectrometry (GC-MS) was recorded on a Varian 410 GC-320 MS (Palo Alto, CA, USA) using a VF-5 ms column (30 m, 0.25 mm, 0.25 μm), and an electron beam of 70 eV. All experiments were done in triplicate. Ultrapure HNO3 and HCl obtained from a sub-boiling system (DuoPUR, Milestone, Bergamo, Italy) and ultrapure 18.2 MΩ water from a Milli-Q (Millipore, Burlington, MA, USA) were used for the sample dissolution. 1H NMR and 13C NMR spectra were recorded on a Bruker (400 MHz) spectrometer.Preparation of oxidized MWCNTs I
In a round-bottomed flask, MWCNTs (1.0 g) and sulphonitric mixture [H2SO4–HNO3 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 500 mL] were kept under magnetic stirring for 4.0 h at 25 °C, sonicated for 1 h and heated for 12 h at 40 °C. The reaction mixture was cooled down to 25 °C and the suspension was washed with cold H2O (400 mL). The supernatant was removed by centrifugation (4000 × g rpm, 30 min). The residue was washed with deionized H2O (200 mL) and filtered using GH polypro membrane filters 0.2 (μm). The resulting MWCNTs I were dried under reduced pressure and used without further purification.
:1), 500 mL] were kept under magnetic stirring for 4.0 h at 25 °C, sonicated for 1 h and heated for 12 h at 40 °C. The reaction mixture was cooled down to 25 °C and the suspension was washed with cold H2O (400 mL). The supernatant was removed by centrifugation (4000 × g rpm, 30 min). The residue was washed with deionized H2O (200 mL) and filtered using GH polypro membrane filters 0.2 (μm). The resulting MWCNTs I were dried under reduced pressure and used without further purification.
Preparation of MWCNTs-B15C5/MTO catalysts IV A–D
MWCNTs I (400 mg) were suspended in dimethyl formamide (500 mL) and treated with N,N-diisopropylcarbodiimide (DIC; 250 mg, 2.0 mmol), hydroxybenzotriazole (HOBt; 270 mg, 2.0 mmol), and N,N-diisopropyl ethylamine (DIPEA; 700 μL, 4.0 mmol) in a round-bottomed flask with an egg-shaped magnetic stirring bar for 15 min at 25 °C. Thereafter, the appropriate diamine (ethylene diamine for II A, propylene diamine for II B, cadaverine for II C, and hexamethylene diamine for II D; 2.0 mmol) was added to the solution under gentle stirring for 12 h at 30 °C. The resulting solution was washed with DMF (5 × 5.0 mL), centrifugated (4000 × g rpm, 20 min), and filtered using GH polypro membrane filters (0.2 μm). The resulting intermediates II A–D were dried under reduced pressure. Successively, 4′-carboxy-benzo-15-crown-5 (1.5 g, 6.0 mmol) was dissolved in DMF (250 mL), followed by treatment with DIC (790 mg, 6.0 mmol) and DIPEA (2.1 mL, 12 mmol), in a 500 mL round-bottomed flask under stirring for 4 h at 25 °C. Thereafter II A–D (200 mg) were added to the solution and the mixture stirred for 8 h at 30 °C. The resulting intermediates III A–D were washed with DMF and H2O, centrifugated (4000 × g rpm, 20 min), and filtered using GH polypro membrane filters (0.2 μm). Intermediates III A–D (125 mg) were suspended in THF (50 mL) in a round-bottomed flask, and MTO (25 mg) in THF (25 mL) was added dropwise. After 1 h the suspension was concentrated under reduced pressure, washed with THF (5 × 5.0 mL) and filtered using GH polypro membrane filters (0.2 μm) to afford MTO catalysts IV A–D.Preparation of MWCNTs-B15C5/PAMAM/MTO catalysts V E–H
The grafting of MWCNTs I with PAMAM was performed by applying a slightly modified procedure from the literature.8 Methyl acrylate (10 mL) was added to a suspension of the appropriate intermediate II D in methanol (10 mL), and the resulting mixture was stirred at 80 °C for 3 days. Thereafter, ethylene diamine (EDA, 0.15 mol, 400 mg), and in alternative, hexamethylene diamine (HAD, 0.15 mol, 775 mg), was added to the suspension in order to prepare the first-generation PAMAM intermediates III E–F, respectively. Two successive treatments of II A and II D with methyl acrylate and the appropriate alkyl-diamino linker afforded III G–H, respectively. Next, the appropriate PAMAM intermediate III E–H (100 mg) in DMF (50 mL) was added to a mixture of 4′-carboxybenzo-15-crown-5 (2.0 mmol, 624 mg), DIC (2.0 mmol, 406 mg), and HOBt (2.0 mmol, 270 mg) in dry CH2Cl2 (20 mL). The reaction mixture was stirred for 48 h at room temperature and then filtered on GH polypro membrane filters (0.2 μm) to afford PAMAM intermediates IV E–H, respectively. The black solid was washed several times with DMF (3 × 2.0 mL) and CH2Cl2 (5 × 10 mL) and dried under vacuum. PAMAM intermediates IV E–H (100 mg) were than suspended in THF (25 mL) and a solution of MTO (25 mg) in THF (25 mL) was added dropwise. After 1 h the suspension was concentrated under reduced pressure, washed with THF (5 × 5.0 mL) and filtered using GH polypro membrane filters (0.2 μm) to afford catalysts V E–H.FT-IR analysis
FT-IR analyses were performed, at room temperature, with a PerkinElmer Spectrum One and a BRUKER VERTEX 70V spectrometers, respectively, both equipped with an ATR unit. Mid Infrared spectra were recorded by averaging 32 scans, with a resolution of 4 cm−1. In the case of FIR (Far Infrared) configuration, spectra were registered within the (600–50 cm−1) interval.Scanning electron microscopy (SEM) and transmission electron microscopy (TEM)
For the scanning electron microscopy (SEM), the sample suspensions (50 L) were let to adsorb onto carbon tape attached to aluminum stubs and air dried at 25 °C. The observation was made by a JEOL JSM 6010LA electron microscope (Waltham, MA, USA) using Scanning Electron (SE) and Back Scattered Electrons (BSE) detectors. Energy Dispersive Spectroscopy (EDS) analysis was carried out to reveal the chemical elements. Transmission electron microscopy (TEM) analysis was carried out by JEM-1200 EXII Transmission Electron Microscope. For this aim one drop of nanocomposite was put on copper lace coated with carbon and dried at 25 °C.XRD analysis
The X-ray diffraction data were collected by a Bruker D8 Advance with DaVinci design located at CNIS – La Sapienza University of Rome diffractometer with a molybdenum source. The angle dispersive instrument is equipped with a Mo Kα X-ray tube (λ = 0.7107 Å), whose radiation was focused onto the sample with Göbel mirrors. The 2θ angle range available was 2.75–142° with a step of 0.25° within Bragg–Brentano para-focusing geometry. The scattered intensity was collected with the Lynxeye XE Energy-Dispersive 1-D detector.Inductively coupled plasma mass-spectrometry (ICP-MS) analysis
The samples were weighed (from 1.0 mg to 10 mg) and transferred in fluorinated ethylene propylene (FEP) vials, previously washed to avoid any kind of external contamination. Regia solution was chosen for the mineralization. In particular, 750 μL of HCl and 150 μL HNO3 were added and the solution was heated to 80 °C for 3 hours. The volume was adjusted to 5.0 mL and then diluted another 10 times before the ICP-MS analysis. The analysis was performed with ultrahigh vacuum PHI 1257 system and an Agilent 7500 ICP-MS instrument under clean room ISO6 (Santa Clara, CA, USA), respectively.Oxidation of olefins 1–7
A 5.0 mL flask equipped with a magnetic bar was charged with CH2Cl2 (5.0 mL), the olefins 1–7 (0.5 mmol), the appropriate amount of selected MTO catalyst (0.0025 mmol of MTO calculated with respect to the MTO loading value) and H2O2 (35% water solution, 50 μL, 0.5 mmol). The two-phase mixture was stirred vigorously at room temperature for 5 h. The progress of the reaction was monitored at half-hour intervals by gas chromatography associated to mass spectrometry (GC-MS) analysis of small aliquots of the organic phase. The conversion of olefins and yield of corresponding products were determined by GC-MS using n-octane as internal standard and by comparison with commercially available standards. GC-MS was recorded on a Varian 410 GC-320 MS (Palo Alto, CA, USA) using a VF-5 ms column (30 m, 0.25 mm, 0.25 m), and an electron beam of 70 eV. Helium was used as the carrier gas at a constant flow of 1.5 mL min−1. The GC oven temperature was started at 50 °C (1 min hold) ramped at 20 °C min−1 to 115 °C (10 min hold), then ramped 5 °C min−1 to 280 °C (10 min hold).Conclusions
MTO was efficiently supported on B15C5 and PAMAM functionalized MWCNTs. The novel catalysts showed a different selectivity in the oxidation of olefins depending on the chemical complexity of the system. Epoxides were synthesized in quantitative yield and high conversion of substrate in the presence of PAMAM catalysts, while diols largely prevailed when B15C5 was directly linked to the surface of MWCNTs. XRD analysis showed that the Re atom was coordinated by an octahedral structure through interaction with a couple of oxygen atoms of the B15C5 ring. The high selectivity observed in the synthesis of epoxides with PAMAM catalysts was probably due to the Lewis base property of the nitrogen atoms present in the dendrimer aggregate in buffering the Lewis acidic character of Re(VII) atom. The reactivity of the dendrimer-based catalysts increased by increasing the order of complexity of the structure, the PAMAM of second-generation affording higher yield of epoxide with respect to the first-generation counterpart. In the PAMAM series, terminal alkyl-diamino spacer bearing carbon atoms (cadaverine) afforded the most reactive catalysts. Note that PAMAM catalysts V E–H afforded styrene epoxide in yield higher than poly(4-vinylpyridine) and microencapsulated polystyrene MTO based systems,37 highlighting the beneficial role of the dendrimer structure in the selectivity of the reaction. In the absence of PAMAM, the Lewis acidity of MTO was responsible for the ring-opening of epoxides to diols. In this latter case, the reactivity decreased in the presence of alkyl-diamino spacer bearing more than three carbon atoms (propyl diamine). Finally, catalyst V H retained the catalytic activity for at least six successive runs without any appreciable leaching of the metal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MIUR Ministero dell'Istruzione, dell'Università della Ricerca Italiano, progetto PRIN 2017-ORIGINALE CHEMIAE in Antiviral Strategy – Origin and Modernization of Multi-Component Chemistry as a Source of Innovative Broad Spectrum Antiviral Strategy, cod. 2017BMK8JR is acknowledged.Notes and references
- L. Botta, B. M. Bizzarri, M. Crucianelli and R. Saladino, J. Mater. Chem. B, 2017, 5, 6490–6510 RSC.
- V. Dhand, W. R. Lee, S.-J. Park, K. Y. Rhee and G. Mittal, J. Ind. Eng. Chem., 2014, 21, 11–25 Search PubMed.
- A. S. Attiyat, G. D. Christian, C. V. Cason and R. A. Bartsch, Electroanalysis, 1992, 1, 51–56 CrossRef.
- J. P. Camarena, H. Espinoza-Gómez, R. Somanathan, H. Tiznado, E. Vélez-López, R. Romero-Rivera, M. A. Martínez-López, M. Avalos-Borjaa, G. Alonso-Nuñez and E. Rogel-Hernández, J. Nanosci. Nanotechnol., 2011, 11, 5539–5545 CrossRef CAS PubMed.
- L. J. Du, Y. H. Hu, Q. Y. Wang, Q. D. Zhang, Y. B. Chen, L. Q. Peng, S. L. Pan, Q. Li and J. Cao, Food Chem., 2018, 262, 118–128 CrossRef CAS PubMed.
- L.-Y. Lin, K.-C. Ho, C.-Y. Liu, K.-F. Lin, R. Vittal, C.-G. Wu, K.-C. Huang, C.-Y. Chen and Y. H. Chang, J. Mater. Chem., 2011, 21, 18467 RSC.
- L. Danyang, N. Lanli, D. Yimin, Z. Jiaqi, C. Tianxiao and Z. Yi, J. Mater. Sci.: Mater. Electron., 2018, 30, 1161–1174 CrossRef.
- F. Xavier Rius, P. Blondeau, G. Kerric, E. J. Parra and G. A. Crespo, J. Mater. Chem., 2012, 22, 16611 RSC.
- M. A. Herrero, F. M. Toma, K. T. Al-Jamal, K. Kostarelos, A. Bianco, T. Da Ros, F. Bano, L. Casalis, G. Scoles and M. Prato, J. Am. Chem. Soc., 2009, 131, 9843–9848 CrossRef CAS PubMed.
- D. Astruc, D. Wang, C. Deraedt, L. Liang, R. Ciganda and J. Ruiz, Synthesis, 2016, 47, 2017–2031 CrossRef.
- A. M. Caminade, A. Ouali, R. Laurent, C. O. Turrin and J. P. Majoral, Coord. Chem. Rev., 2018, 305, 478–497 Search PubMed.
- Y. P. Sun, W. Huang, Y. Lin, K. Fu, A. Kitaygorodskiy, L. A. Riddle, Y. J. Yu and D. L. Carroll, Chem. Mater., 2001, 13, 2864–2869 CrossRef CAS.
- M. Holzinger, J. Abraham, P. Whelan, R. Graupner, L. Ley, F. Hennrich, M. Kappes and A. Hirsch, J. Am. Chem. Soc., 2003, 125, 8566–8580 CrossRef CAS PubMed.
- E. L. Diz, C. Ehli, D. M. Guldi, M. Prato, S. Campidelli, C. Sooambar, E. L. Diz, C. Ehli, D. M. Guldi and M. Prato, J. Am. Chem. Soc., 2006, 128, 12544–12552 CrossRef PubMed.
- A. Desmecht, T. Steenhaut, F. Pennetreau, S. Hermans and O. Riant, Chem.–Eur. J., 2018, 24, 12992–13001 CrossRef CAS PubMed.
- M. J. Li, C. C. Ko, G. P. Duan, N. Zhu and V. W. W. Yam, Organometallics, 2007, 26, 6091–6098 CrossRef CAS.
- X. Zhang, Z. Zhou, S. Ma and C. Shu, Solvent Extr. Ion Exch., 1993, 11, 585–601 CrossRef CAS.
- I. R. Beattie and P. J. Jones, Inorg. Chem., 1979, 18, 2318–2319 CrossRef CAS.
- M. Crucianelli, R. Saladino and F. De Angelis, ChemSusChem, 2010, 3, 524–540 CrossRef CAS PubMed.
- G. Bianchini, M. Crucianelli, C. Crestini and R. Saladino, Top. Catal., 2006, 40, 221–227 CrossRef CAS.
- R. Saladino, V. Neri, P. Checconi, I. Celestino, L. Nencioni, A. Palamara and M. Crucianelli, Chemistry, 2013, 19, 2392–2404 CrossRef CAS PubMed.
- L. Botta, S. Filippi, B. M. Bizzarri, R. Meschini, M. Caputo, L. Proietti-De-Santis, C. Iside, A. Nebbioso, G. Gualandi and R. Saladino, Bioorg. Med. Chem. Lett., 2019, 29, 78–82 CrossRef CAS PubMed.
- A. Di Giuseppe, M. Crucianelli, F. De Angelis, C. Crestini and R. Saladino, Appl. Catal., B, 2009, 89, 239–245 CrossRef CAS.
- R. Saladino, V. Neri, A. Farina, C. Crestini, L. Nencioni and A. T. Palamara, Adv. Synth. Catal., 2008, 350, 321–331 CrossRef CAS.
- R. Bernini, E. Mincione, M. Cortese, G. Aliotta, A. Oliva and R. Saladino, Tetrahedron Lett., 2001, 42, 5401–5404 CrossRef CAS.
- A. M. J. Rost, H. Schneider, J. P. Zoller, W. A. Herrmann and F. E. Kühn, J. Organomet. Chem., 2005, 690, 4712–4718 CrossRef CAS.
- G. Soldaini, A. Goti, C. Crestini, R. Saladino, M. Crucianelli and F. Cardona, J. Mol. Catal. A: Chem., 2008, 284, 108–115 CrossRef.
- R. Saladino, V. Neri, A. R. Pelliccia and E. Mincione, Tetrahedron, 2003, 59, 7403–7408 CrossRef CAS.
- R. Saladino, M. C. Ginnasi, D. Collalto, R. Bernini and C. Crestini, Adv. Synth. Catal., 2010, 352, 1284–1290 CrossRef CAS.
- R. Saladino, R. Bernini, V. Neri and C. Crestini, Appl. Catal., A, 2009, 360, 171–176 CrossRef CAS.
- C. C. Romão, F. E. Kühn and W. A. Herrmann, Chem. Rev., 1997, 97, 3197–3246 CrossRef PubMed.
- S. M. Nabavizadeh, Inorg. Chem., 2003, 42, 4204–4208 CrossRef CAS PubMed.
- P. Ferreira, W. Xue, Ä. Bencze, E. Herdtweck, F. E. Ku and R. V June, Inorg. Chem., 2001, 40, 5834–5841 CrossRef CAS PubMed.
- F. E. Kühn, A. M. Santos and W. A. Herrmann, Dalton Trans., 2005, 2483–2489 RSC.
- W. Adam and C. M. Mitchell, Angew. Chem., Int. Ed., 1996, 35, 533–535 CrossRef CAS.
- A. Di Giuseppe, M. Crucianelli, M. Passacantando, S. Nisi and R. Saladino, J. Catal., 2010, 276, 412–422 CrossRef CAS.
- R. Saladino, V. Neri, A. Rita Pelliccia, R. Caminiti and C. Sadun, J. Org. Chem., 2002, 67, 1323–1332 CrossRef CAS PubMed.
- R. Saladino, V. Neri, E. Mincione and P. Filippone, Tetrahedron, 2002, 58, 8493–8500 CrossRef CAS.
- R. Saladino, E. Mincione, O. A. Attanasi and P. Filippone, Pure Appl. Chem., 2003, 75, 265–272 CAS.
- R. Buffon, A. Auroux, F. Lefebvre, M. Leconte, A. Choplin, J. Basset and W. A. Herrmann, J. Mol. Catal., 1992, 76, 287–295 CrossRef.
- A. Sakthivel, G. Raudaschl-Sieber and F. Kühn, Dalton Trans., 2006, 468–472 RSC.
- A. Fong, S. L. Scott, B. Peters, L. Delevoye, R. M. Gauvin, K. C. Szeto, A. Gallo, M. Taoufik and J. Rieb, J. Am. Chem. Soc., 2016, 138, 12935–12947 CrossRef PubMed.
- D. Piccinino, I. Abdalghani, G. Botta, M. Crucianelli, M. Passacantando, M. L. Di Vacri and R. Saladino, Appl. Catal., B, 2017, 200, 392–401 CrossRef CAS.
- S. M. Nabavizadeh and M. Rashidi, J. Am. Chem. Soc., 2006, 128, 351–357 CrossRef CAS PubMed.
- M. Zhou, M. Liu, L. Huang, J. Zhang, J. Wang, X. Li, F. Kühn and S. Zang, Green Chem., 2015, 17, 1186–1193 RSC.
- I. Abdalghani, S. Nisi, R. Saladino, M. Passacantando, B. Bizzarri, M. Ferrante, M. Crucianelli, L. Botta and A. Taddei, Nanomaterials, 2018, 8, 516 CrossRef PubMed.
- E. J. Parra, P. Blondeau, G. A. Crespoa and F. Xavier Riusa, Chem. Commun., 2011, 47, 2438–2440 RSC.
- D. Atzei, D. De Filippo, A. Rossi, R. Caminiti and C. Sadun, Spectrochim. Acta, 1995, 51, 11 CrossRef.
- D. Atzei, D. De Filippo, A. Rossi, R. Caminiti and C. Sadun, Inorg. Chim. Acta, 1996, 248, 203 CrossRef CAS.
- D. Atzei, C. Sadun and L. Pandolfi, Spectrochim. Acta, 2000, 56, 531 CrossRef CAS.
- R. H. Tromp and G. W. J. Neilson, Phys. Chem., 1996, 100, 7380–7383 CrossRef CAS.
- R. Caminiti, M. Carbone, S. Panero and C. Sadun, J. Phys. Chem., 1999, 103, 10348–10355 CrossRef CAS.
- D. Atzei, T. Ferri, C. Sadun, P. Sangiorgi and R. Caminiti, J. Am. Chem. Soc., 2001, 123, 2552–2558 CrossRef CAS PubMed.
- K. Jurkiewicz, M. Pawlyta and A. Burian, Carbon, 2018, 4, 68 CAS.
- J. Takacs, P. Kiprof, J. Riede and W. A. Herrmann, Organometallics, 1990, 9, 782–787 CrossRef CAS.
- R. M. Crooks, B. I. Lemon, L. Sun, L. K. Yeung and M. Zhao, Dendrimers III, Springer Berlin Heidelberg, Berlin, Heidelberg, 2001 Search PubMed.
- A. S. Ertürk, M. Ulvi Gürbüz, M. Tülü and A. Bozdoğan, Maced. J. Chem. Chem. Eng., 2016, 35, 263–270 CrossRef.
- L. Banci, A. Bencini, C. Benelli, D. Gatteschi and C. Zanchini, Structures versus Special Properties, Springer Berlin Heidelberg, Berlin, Heidelberg, 1982 Search PubMed.
- V. Velachi, D. Pramanik, A. Sood and P. Maiti, Soft Matter, 2013, 9, 1372 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SI#1 FT-IR of intermediates II A–D, III A–D and catalysts IV A–D and V E–H; SI#2: difference method of neutron diffraction and isotopic substitution SI#3: mass spectrometry profiles of compounds 1–21; SI#4: nuclear magnetic resonance spectra of compounds 1–21, SI#5 far IR of IV D and V G compared to the reference MTO-1,2-cyclohexanediamine complex and SI#6 transmission electron microscopy (TEM) of V F and V H. See DOI: 10.1039/d0ra02785e |
| This journal is © The Royal Society of Chemistry 2020 |