
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceH2O-prompted CO2 capture on metal silicates in situ generated from SBA-15†
Meijun Li *b,
Mengkun Tianc,
Hao Chena,
Shannon Mark Mahurinb,
Zili Wub and
Sheng Dai*ab
*b,
Mengkun Tianc,
Hao Chena,
Shannon Mark Mahurinb,
Zili Wub and
Sheng Dai*ab
aDepartment of Chemistry, University of Tennessee, 1420 Circle Drive, Knoxville, TN 38996, USA
bChemical Sciences Division, Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN 37831, USA. E-mail: lim4@ornl.gov; dais@ornl.gov
cDepartment of Material Science and Engineer, University of Tennessee, 1420 Circle Drive, Knoxville, TN 38996, USA
First published on 4th August 2020
Abstract
A series of metal silicates, NaMSi10Ox (M = Cu, Mn and Ni), were prepared by in situ doping of metals into mesoporous SBA-15 under a hydrothermal process, displaying a continuous framework of SiO4 structure with a narrow pore size distribution. These metal silicate materials were tested for CO2 adsorption behavior in the absence and presence of water. The results exhibited that the effect of H2O on the CO2 capture capability of metal silicates depends on the types of metal inserted into SBA-15. Compared to the dry condition, H2O addition enhances CO2 uptake dramatically for NaCuSi10Ox by 25%, and slightly for NaNiSi10Ox (∼10%), whereas little effect is shown on NaMnSi10Ox. The metal silicate materials are stable after adsorption of CO2 under wet conditions, which is benefited from their synthesis method, hydrothermal conditions. The improvement of CO2 uptake on metal silicates by H2O is attributed to the competitive and synergistic adsorption mechanism on the basis of IR investigations, where initially adsorbed H2O acts as a promoter for further CO2 capture through a hydration reaction, i.e., formation of bicarbonate and carbonates on the surface of the samples. These observations provide new possibilities for the design and synthesis of porous metal silicate materials for CO2 capture under practical conditions where moisture is present.
Introduction
The significantly increased concentration of carbon dioxide in the atmosphere is the major contributor to global warming, and CO2 is mainly generated from processes such as vehicles, refineries, and thermal power stations and the combustion of natural gas.1–3 Therefore, developing technologies for reducing the amount of carbon dioxide emission has been attracting wide attention throughout the industrial and academic communities4,5 owing to the environmental sustainability. There are many techniques available to remove CO2 with different advantages and limitations. To date, the most effective way is carbon capture and storage (CCS) based on solid adsorbents.6,7Currently, a wide range of materials with fine structural and chemical properties have been investigated as possible CO2 captors at low (303–353 K) or high (T > 473 K) temperatures, such as various activated carbons,8–10 zeolites,11–13 silicates,14–16 metal oxides,17–19 metal–organic frameworks (MOFs)20–25 and zirconates/aluminates,26–29 each presenting some advantages and disadvantages. Solid sorbents impregnated or grafted with polyamines and related groups are widely researched materials, which combine the advantages of both the amine group and solid materials to maximize the sorption properties. These functionalized solids not only present higher CO2 selectivity and adsorption capacity, but also possess lower temperatures and less energy input for regeneration. For example, a capture capacity of 12 mmol g−1 is reported for cationic polymerization of oxazolines on mesoporous silica under simulated flue gas conditions.30 However, the thermal stability of the amine group employed should be considered carefully for long term implementation, which is particularly important when the adsorbent is subject to steam stripping and/or high temperature regeneration. In addition, MOFs20–25 have been extensively investigated in carbon storage/separation due to their enriched morphological texture, chemical tunability, highly available porous surface, aromaticity and densely coordinated unsaturated metal sites. However, most MOF adsorbents exhibit decreased CO2 uptake capacities under wet conditions. Many efforts have been dedicated to improving the water resistance properties of MOFs. Recently, Ibarra et al.31 reported the progress in CO2 capture under humid conditions in MOFs in their review paper, which includes an emerging technology of enhancement of CO2 capture by confining water within the pores of MOFs.
Alkaline ceramics, such as alkali-based silicates, zirconates or aluminates, have attracted much attention for CO2 capture because this type of material exhibits high CO2 uptake, thermal stability, low cost, repeated regenerability and tolerance against water, and have been used in recent attempts towards tailoring materials for selective CO2 capture.32 Typically, CO2 capture on alkali-based silicates presents a double-step sorption mechanism, with an initial chemical sorption of CO2 to form carbonates on the surface and further reaction with the alkaline atom, which accordingly depends on different factors such as the chemical composition, the structure, different microstructural factors and the presence of water.33–36 Therefore, CO2 capture can be significantly affected by the tunability of their compositions and structure, such as replacement of alkali elements by transition metals.
Notably, CO2 capture on metal silicates/aluminates has received little attention,37,38 and it has been only recently shown that microporous copper silicate synthesized from sodium silicate and copper sulfate has a stable CO2 uptake of 156 cm3 cm−3 in the presence of water vapor.38 It is well known that most flue gas streams under consideration for carbon capture are highly humid, which typically causes the reduction of CO2 uptake capabilities, or leads to absorbent decomposition in some cases. Therefore, the development of efficient, stable and sustainable absorbents based on more available and low-cost materials, such as transition metal salts, could favor the technologies of the CO2 capture processes in the presence of H2O. Herein, we develop a new synthetic method for metal (Cu, Ni and Mn) silicate sorbents derived by in situ doping of metals into mesoporous SBA-15 through a hydrothermal synthesis. The synthesized metal silicates, exhibiting a quartz crystallized structure with narrow pore size distribution, are found to possess comparable or even increased CO2 sorption capacity in the presence of water vapor. Based on in situ FTIR study, a competitive/synergistic adsorption model is proposed for CO2 adsorption with H2O addition on the metal silicate materials.
Experimental
Materials
The following raw materials were used as provided: copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, Acros Organics); manganese(II) nitrate tetrahydrate (Mn(NO3)2·4H2O, Sigma-Aldrich); nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, Sigma-Aldrich); sodium carbonate anhydrous (Na2CO3, Fisher Chemicals); tetraethyl orthosilicate (TEOS, Acros Organics); hydrochloric acid (HCl, 37%, Fisher Chemicals).Sample preparation
Ordered mesoporous SBA-15 was synthesized by a hydrothermal method as reported in the literature39 with some modifications. In a typical synthesis, 2.8 mmol P123 and 2.9 mol HCl (83 mL) were added into DI water (28 mol) in a Nalgene PP bottle (1000 mL), followed by stirring for 3 hours until P123 was fully dissolved and the solution was clear. Tetraethyl orthosilicate (TEOS, 0.17 mol) was then added to this solution and stirred for another 30 min. The bottle was then capped and kept in an oven at 373 K for 24 hours. The reaction mixture was cooled down and then filtered. The white powder was dried at 333 K for 24 h, followed by air calcination at 823 K for 4 hours with a heating rate of 1 K min−1 and air flow rate of ∼120 mL min−1.The metal silicates were prepared through a hydrothermal method in an autoclave. The metal salts (0.5 mmol) (Cu(NO3)2·3H2O, Ni(NO3)2·6H2O or Mn(NO3)2), Na2CO3 (2.5 mmol), and SBA-15 (2 mmol) were loaded into a PTFE-lined stainless-steel autoclave along with 5 mL of deionized water. The autoclave was sealed and heated to 463 K for 48 hours and then cooled to room temperature. The products were washed with deionized water and ethanol to remove excess starting materials. The obtained metal silicates were dried at 373 K for 12 hours. The actual metal and silicon content in the materials was analyzed by ICP. It was found that the actual ratio of metal to silicon (∼1/10) was lower than the ratio of metal to silicon in the starting solution. Therefore, the samples are denoted as NaMSi10Ox (M = Cu, Ni and Mn).
Characterization
The scanning electron microscope (SEM) imaging was performed on a Zeiss Auriga microscope with an electron beam operation of 5 keV, which is a dual beam FIB (Focused Ion Beam) with a field emission electron column for high resolution electron imaging and a Canion Ga+ column for precision ion beam milling. Elemental distributions of the samples were characterized by energy dispersive X-ray spectroscopy (EDS) performed in a Zeiss EVO MA15 scanning electron microscope (SEM) working at 20 kV. The resolution of this microscope is about 5 nm. The EDS spectra were analyzed by the Bruker QUANTAX software.Nitrogen adsorption isotherms were measured at 77 K using a Gemini VII 2390 Surface Area Analyzer (Micrometrics Instrument Corp.). Prior to the adsorption measurements, the metal silicate samples were degassed under flowing nitrogen for 2 h at 453 K to remove any residual water and surface species. The surface area was calculated using the Brunauer–Emmett–Teller (BET) method in the relative pressure range of 0.05 to 0.25. The total pore volume (Vt) was estimated from the N2 adsorption isotherm at a relative pressure P/P0 = 0.97. The pore size distributions (PSDs) were calculated using the Barrett–Joyner–Halenda (BJH) algorithm for cylindrical pores according to the improved Kruk–Jaroniec–Sayari (KJS) method.
X-ray diffraction (XRD) patterns were collected on a PANalytical powder diffractometer using CuKα radiation. The working voltage was 40 kV, and the current was 40 mA. The intensity data were collected at room temperature in the 2θ range of 0°–4° and 15°–70°.
CO2 sorption measurements were carried out at 303 and 273 K using a Gemini VII 2390 Surface Area Analyzer (Micrometrics Instrument Corp.). Prior to the CO2 adsorption measurements, all metal silicate samples were treated under flowing nitrogen for 5 h at 453 K.
Adsorption of dry CO2, N2/H2O, and CO2/H2O was performed using a thermogravimetric analysis (TGA) device (Model Seiko 6300 TG/DTA, RT Instruments, Inc.) at 323 K. The experiments were performed using distilled water and two different carrier gases: bubbling nitrogen (N2) or carbon dioxide (CO2) through distilled water at room temperature to get saturated water vapor pressure. The relative humidity of pristine flue gas varies from case to case, but is normally at low levels. Therefore, a relative humidity of ∼25% (saturated water vapor pressure at room temperature) was chosen as a representative case for investigation. The gas flow rate used in all of the experiments was 60 mL min−1. The net CO2 adsorption capacity in the presence of H2O was calculated by subtracting the N2/H2O adsorption value from the CO2/H2O adsorption value.
In situ IR study of dry CO2 and CO2/H2O adsorption on the metal silicate samples was conducted using a Thermo Nicolet Nexus 670 spectrometer in diffuse reflectance mode (DRIFTS) while the exiting stream was analyzed by an OmniStar GSD-301 (Pfeiffer-Balzer) quadrupole mass spectrometer (QMS). The samples were pretreated in a DRIFTS cell (HC-900, Pike Technologies, nominal cell volume of 6 cm3) at 453 K in helium flow (25 mL min−1) for 1.5 h before cooling down to 298 K for CO2 adsorption. 2% CO2/He (25 mL min−1) was flowed over the samples at room temperature for 30 min before switching to helium purging for another 30 min. Time-resolved IR spectra were collected during the adsorption process and the subsequent thermal desorption process.
Results and discussion
The successful synthesis of the metal silicates with the SBA-15 doped by different types of metals is confirmed by XRD, N2 isothermal adsorption and SEM characterization. The small angle XRD patterns of the samples (Fig. S1†) indicate that SBA-15 is successfully produced, characterized by three well-resolved diffraction peaks at 0.86, 1.49, and 1.70. These typical peaks disappeared with the introduction of metal salt under hydrothermal conditions, which results in the appearance of different diffraction peaks at a higher angle range (Fig. 1). Fig. 1 shows the XRD profiles of the synthesized metal silicates and pristine SBA-15 from 15° to 70°. The pristine SBA-15 only exhibits a broad diffraction peak at higher angle diffraction (Fig. 1A-a). All synthesized metal silicate samples present distinct diffraction peaks characteristic of quartz (PDF #: 83-0539) as seen in Fig. 1A-b–d, regardless of the metal type. This suggests that the formed metal (Cu, Ni and Mn) silicate materials have a continuous framework of SiO4 (tetrahedrally coordinated) structures. The XRD patterns do not show any evidence of metal oxides forming for CuO, NiO and MnO. From the enlarged portion (the bottom figure, Fig. 1B), it can be seen that the characteristic diffraction peak at around 26.6 degrees of all metal doped samples is slightly shifted to the higher angle side relative to the pure quartz phase (PDF #: 83-0539). The observed key features for metal silicate materials, peak shift and absence of dopant oxide XRD peaks, evidently confirm the successfully incorporation of metals (Cu, Ni and Mn) into the synthesized silicates with quartz crystalline phase. To further confirm the location of metals in the synthesized silicates, EDS mapping acquired by SEM (Fig. 2) shows that O, Si, Na and the metal (Cu, Ni and Mn) elements are uniformly distributed at the micrometer scale, and the EDS intensity of Si to Cu (Mn and Ni) is similar, although the ratio of Si/metal is 10 (ICP results). Combined with the XRD results, it's highly possible that part of the introduced metal is incorporated into the structure of the silicate, while the rest of the metal is homogeneously dispersed on the surface of the silicates. Apparently, Na ions are present over the surface of the synthesized silicates. Fig. 3 gives the surface morphology and structure of the prepared metal silicates and pristine SBA-15 examined by SEM. SBA-15 shows a smooth surface and twisted belt morphology with clearly observable porous structure. The synthesized NaCuSi10Ox, NaNiSi10Ox and NaMnSi10Ox exhibited rod, ball and cotton-like structures, respectively. All three metal silicate samples show rough surface characteristics and irregular porous structures, which indicates the structural changes of the synthesized metal silicates from pristine SBA-15. This is in agreement with the results from XRD and the N2 adsorption isotherms as described below.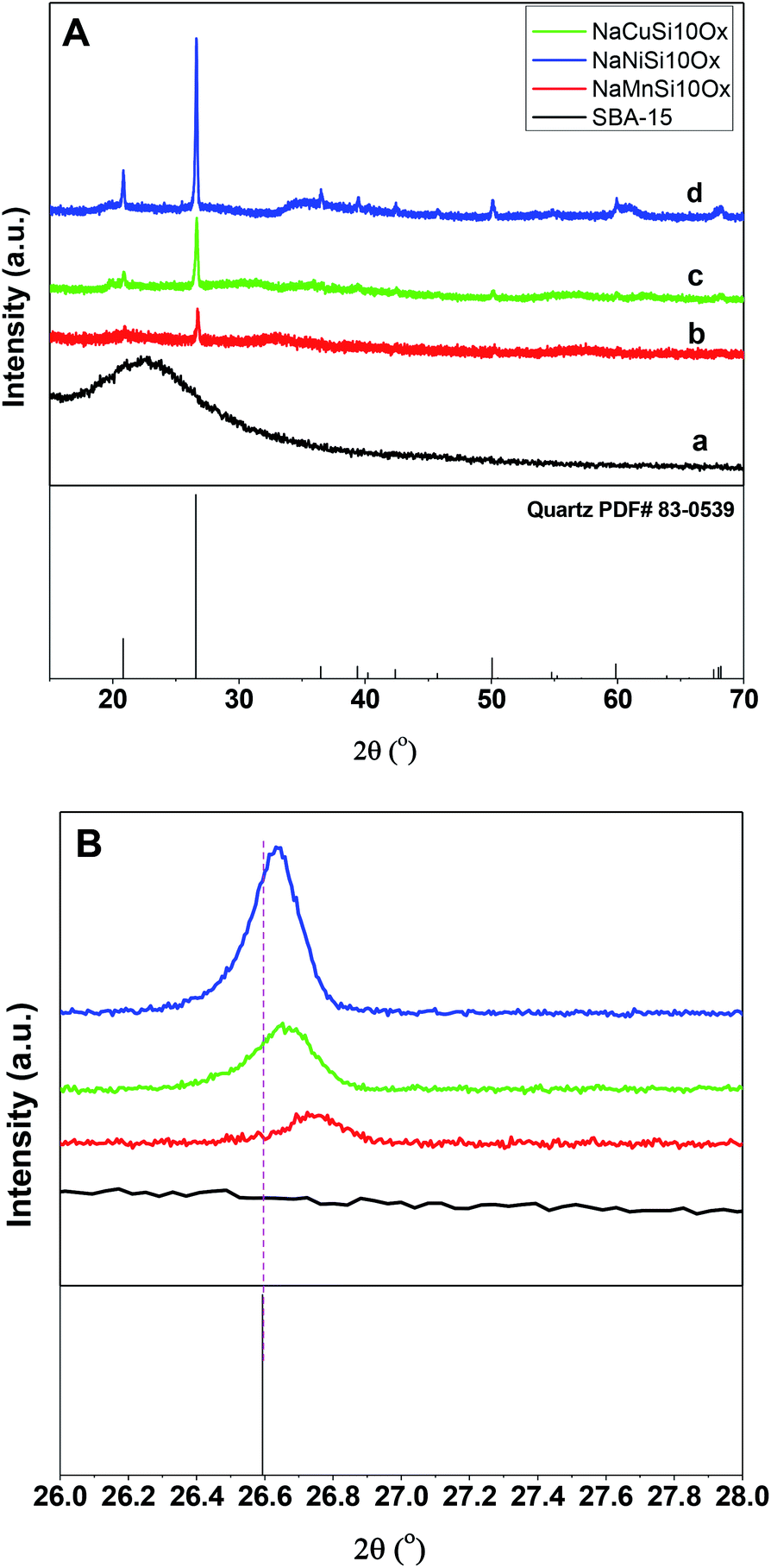 | ||
| Fig. 1 The XRD patterns (A) and the enlarged XRD patterns (B) of the as synthesized metal silicates and pristine SBA-15 materials. | ||
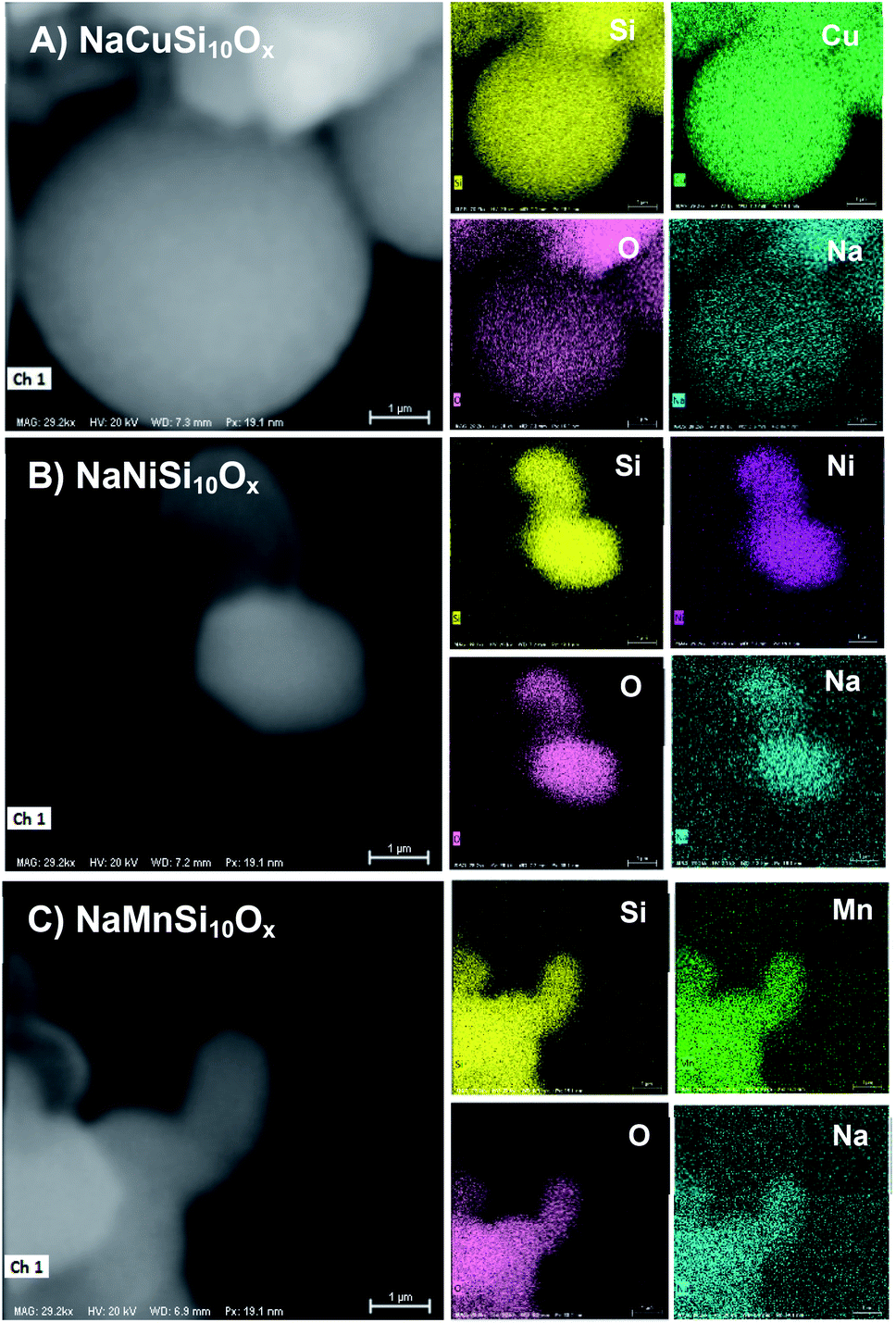 | ||
| Fig. 2 The SEM-EDS mappings of (A) NaCuSi10Ox, (B) NaNiSi10Ox and (C) NaMnSi10Ox. | ||
 | ||
| Fig. 3 The SEM images of (A) NaCuSi10Ox, (B) NaNiSi10Ox, (C) NaMnSi10Ox and (D) SBA-15. | ||
The porous structures of the metal silicates and pristine SBA-15 were characterized by nitrogen adsorption at 77 K. Fig. 4 gives a comparison of the N2 adsorption–desorption isotherms and the corresponding pore size distribution for these materials. SBA-15 displays a typical type IV isotherm, which indicates the presence of a mesoporous structure. The N2 adsorption–desorption isotherms for the metal silicate samples can also be classified as type IV isotherms according to the IUPAC classification (Fig. 4A). However, the volume of adsorbed N2 and the areas of the hysteresis loops for the metal silicates are much smaller compared with those for the pristine SBA-15. This can be ascribed to the crystallization process of the metal silicates, where the degradation and reconstruction of the framework of pristine SBA-15 occur with the incorporation of metals.40 The pore size (Fig. 4B) of the synthesized metal silicates is decreased to 1.5–5 nm, compared with the pristine SBA-15 silica which has a mesopore size distribution range of 6–10 nm. In particular, the pore size for copper silicate narrows down to 1.5–2.5 nm, characteristic of super-microporous materials,41 though the BJH model might not be accurate for analyzing microporosity. The pore size distribution for nickel and manganese silicates covers two ranges, 1.5–2.5 and 3–4.5 nm for manganese silicate, and 1.5–2.5 and 3–5 nm for nickel silicate. The calculated textural properties of SBA-15 silica and the synthesized metal silicates are summarized in Table 1. Compared with pristine SBA-15, the surface area and pore volume decrease for all of the synthesized metal silicate samples. Again, this may be due to the framework reconstruction and crystallization caused by the incorporation of metals into the pristine SBA-15, in accordance with the XRD results. In brief, the metal silicates with micro–mesoporous composites and quartz structures are successfully made by reconstruction of the pristine SBA-15 framework with in situ introduction of metals during the hydrothermal synthesis process.
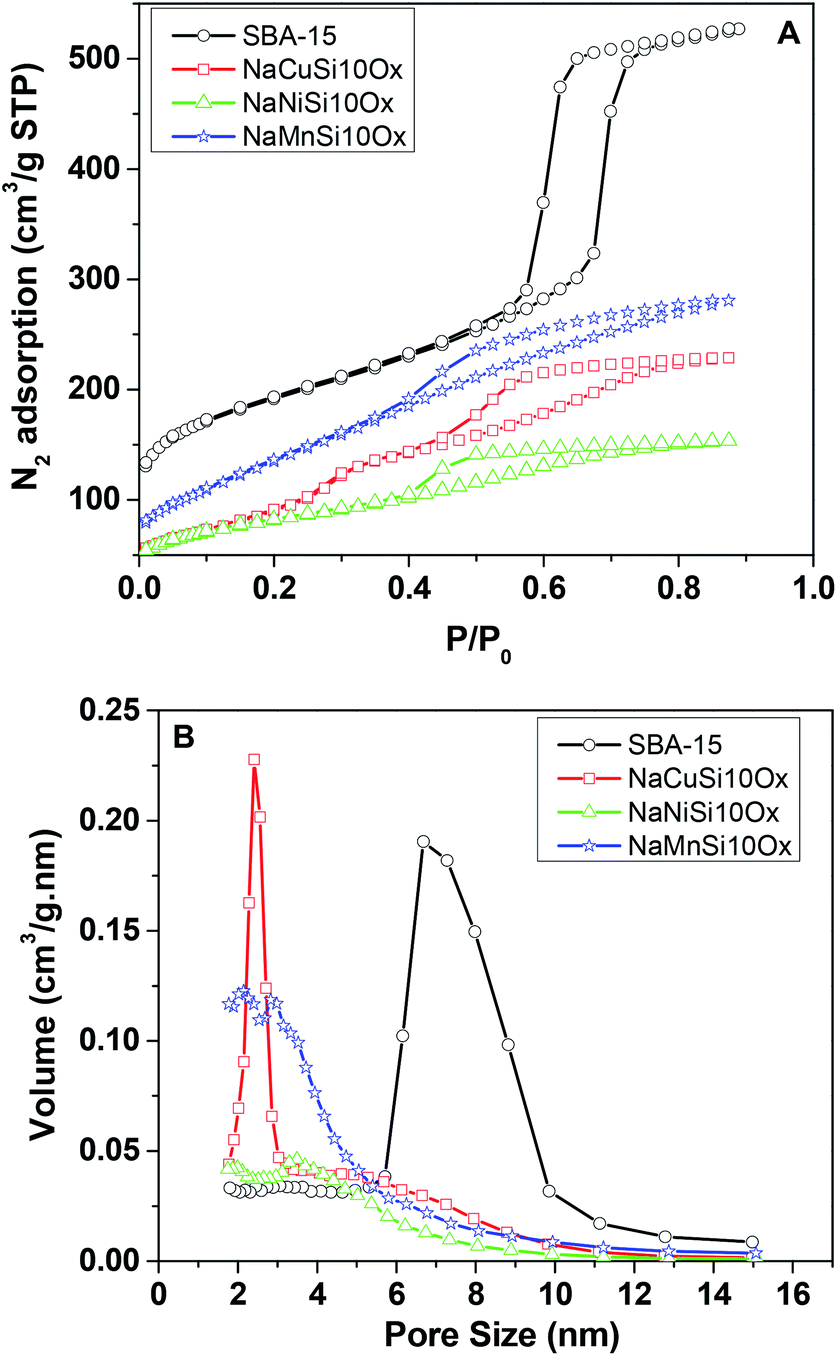 | ||
| Fig. 4 N2 adsorption isotherms (A) and corresponding pore size distributions (B) for the metal silicates and SBA-15. | ||
| Samples | Actual ratio of metal to silicon | Surface area (m2 g−1) | Vt (cm3 g−1) | CO2 adsorption at 273 K (mmol g−1) | CO2 adsorption at 303 K (mmol g−1) |
|---|---|---|---|---|---|
| NaCuSi10Ox | 10.7 | 445 | 0.46 | 1.57 | 1.18 |
| NaNiSi10Ox | 11.9 | 314 | 0.28 | 1.00 | 0.80 |
| NaMnSi10Ox | 11.6 | 551 | 0.53 | 1.75 | 1.28 |
| SBA-15 | 0 | 652 | 0.83 | 0.85 | 0.62 |
The CO2 adsorption capacities of all samples were measured at two temperatures (273 and 303 K) and the results are shown in Fig. 5 and Table 1. For the pristine SBA-15, the CO2 adsorption capacities at 273 K and 303 K are 0.85 and 0.62 mmol g−1 at 1 bar, respectively. The CO2 total capacities on the metal silicates at both temperatures are higher than that on SBA-15, though their surface area and pore volume are lower than those of pristine SBA-15. This can be attributed to the formation of a crystallized metal silicate structure and the incorporation of metals, i.e., CO2 adsorption on the metal silicates is impacted by both the textural parameters (surface area and pore volume) and the chemical properties (the type of metal). As can be seen in Fig. 5, the CO2 capacities differ slightly for the metal silicate samples at both temperatures: ∼1.75 mmol g−1 @ 273 K (∼1.18 mmol g−1 @ 303 K) for NaMnSi10Ox, ∼1.57 mmol g−1 @ 273 K (∼1.02 mmol g−1 @ 303 K) for NaCuSi10Ox and ∼1.00 mmol g−1 @ 273 K (∼0.81 mmol g−1 @ 303 K) for NaNiSi10Ox, following the trend of decreasing surface area and pore volume of these samples. To clearly show the effect of metals in the framework/over the silicate surface on CO2 adsorption behavior, the CO2 capacities were normalized to the total surface area, as presented in Fig. 6. NaCuSi10Ox has higher CO2 capacity than NaMnSi10Ox and NaNiSi10Ox at 273 K with the latter two giving similar capacities of CO2 uptake. This suggests that the type of metal in the silicate plays a role in the CO2 adsorption behavior, but not significantly. The significant increase in CO2 adsorption capacity of the metal silicates compared to the pristine SBA-15 demonstrates a stronger effect from the texture parameters. Sorbent materials for CO2 capture from humid flue gases are urgently required due to the increased cost for prior dehydration of the gas stream. So TGA experiments were carried out to further analyze and evaluate the potential of the synthesized metal silicates for CO2 capture under humidified conditions. To test the influence of water vapor, the pretreated metal silicate samples were exposed to dry CO2, humidified CO2 and humidified N2 for 10 hours at 323 K. The net CO2 adsorption capacity in the presence of H2O is calculated by subtracting the N2/H2O adsorption value from the CO2/H2O adsorption value, and breakthrough curves with time are sketched in Fig. 7. It is found that the presence of water induces a longer breakthrough time for CO2 capture compared with dry gases, which implies the existence of competitive adsorption between H2O and CO2 molecules, i.e., the CO2 and H2O possibly adsorb on the same surface sites of the metal silicate samples. Competitive adsorption is a general phenomenon for multicomponent involved systems. Interestingly, compared with the dry CO2 case, H2O addition clearly enhances the CO2 uptake amount for the NaCuSi10Ox and NaNiSi10Ox samples at a longer adsorption time, and has almost no effect on the NaMnSi10Ox sample as shown in Fig. 7A–C. This indicates that adsorbed H2O could act as a helper for further CO2 capture, possibly through hydrogen bonding or a hydration reaction (see below). In the presence of H2O, the quantity of CO2 adsorbed by the NaCuSi10Ox and NaNiSi10Ox samples is 0.76 and 0.46 mmol g−1 at 450 min, respectively, which represents 27% and 10% improvement relative to the dry gas stream conditions. The NaMnSi10Ox sample gives 0.61 mmol g−1 CO2 capacity, which is similar to that (0.62 mmol g−1) under dry gas conditions. Obviously, NaCuSi10Ox is the best adsorbent among the three metal silicates under wet conditions, followed by the NaMnSi10Ox and NaNiSi10Ox, despite the differences in surface area and pore volume for all materials (Table 1). Different from the dry conditions, where the CO2 uptake mainly relies on the texture parameters of the metal silicate materials, the effect of H2O addition on the CO2 uptake is more dependent on the type of metal in the metal silicate than on the texture parameters. It is worth mentioning that all samples are highly stable after CO2 capture in wet conditions, which is evident from the characteristic XRD pattern (Fig. S2†). To better understand how the presence of water vapor affects the CO2 capture on the metal silicates, FT-IR was employed to investigate the adsorption of dry and humidified CO2 on all samples. Fig. 8 compares the IR spectra taken as a function of time after the NaCuSi10Ox sample was exposed to CO2 in the absence and presence of water. As shown in Fig. 8A and B, upon initial CO2 adsorption (less than 2 min), the most intense bands appear at 2354, 2342, 1675, 1631 and 1360 cm−1. The band at 2354 cm−1 with a shoulder at 2342 cm−1 is attributed to physisorbed CO2, while the IR features in the range of 1700 to 1200 cm−1 are characteristic of carbonates and bicarbonates.42–45 Generally, there are two types of basic sites on inorganic oxide materials, basic hydroxyl groups and basic surface oxygen (normally designated as a Lewis base) that can interact with the acidic CO2. Therefore, CO2 adsorption on these two types of basic sites can result in bicarbonate (with OH groups) and various carbonate species (with surface O). The bands at 1675 and 1360 cm−1 are assigned to carbonates, and the band at 1631 cm−1 reflects the presence of bicarbonates.43–45 It is noted that the initial adsorption bands are similar in frequency for CO2 adsorption under dry and humid conditions, indicating that similar surface species are formed with/without H2O addition. However, these IR features evolve differently as a function of adsorption time in the absence and presence of water. Under dry conditions (Fig. 8A), the overall types of surface adsorbed CO2, carbonate and bicarbonate species do not vary as a function of adsorption time, but the relative intensity (amount) continuously increases for the same type of surface species. Under wet conditions (Fig. 8B), in addition to the initial bands at 1675, 1630 and 1360 cm−1, there are two new bands appearing at 1550 and 1390 cm−1 and the band at 1631 cm−1 is relatively intensified with increased adsorption time (3 to 30 min). The relative enhancement of the 1631 cm−1 band and the appearance of the band at 1390 cm−1 are caused by newly developed bicarbonate (acid carbonate) species produced from further reaction of adsorbed CO2 and H2O.43,45,46 Adsorbed water can also contribute through its bending mode to the 1631 cm−1 band.
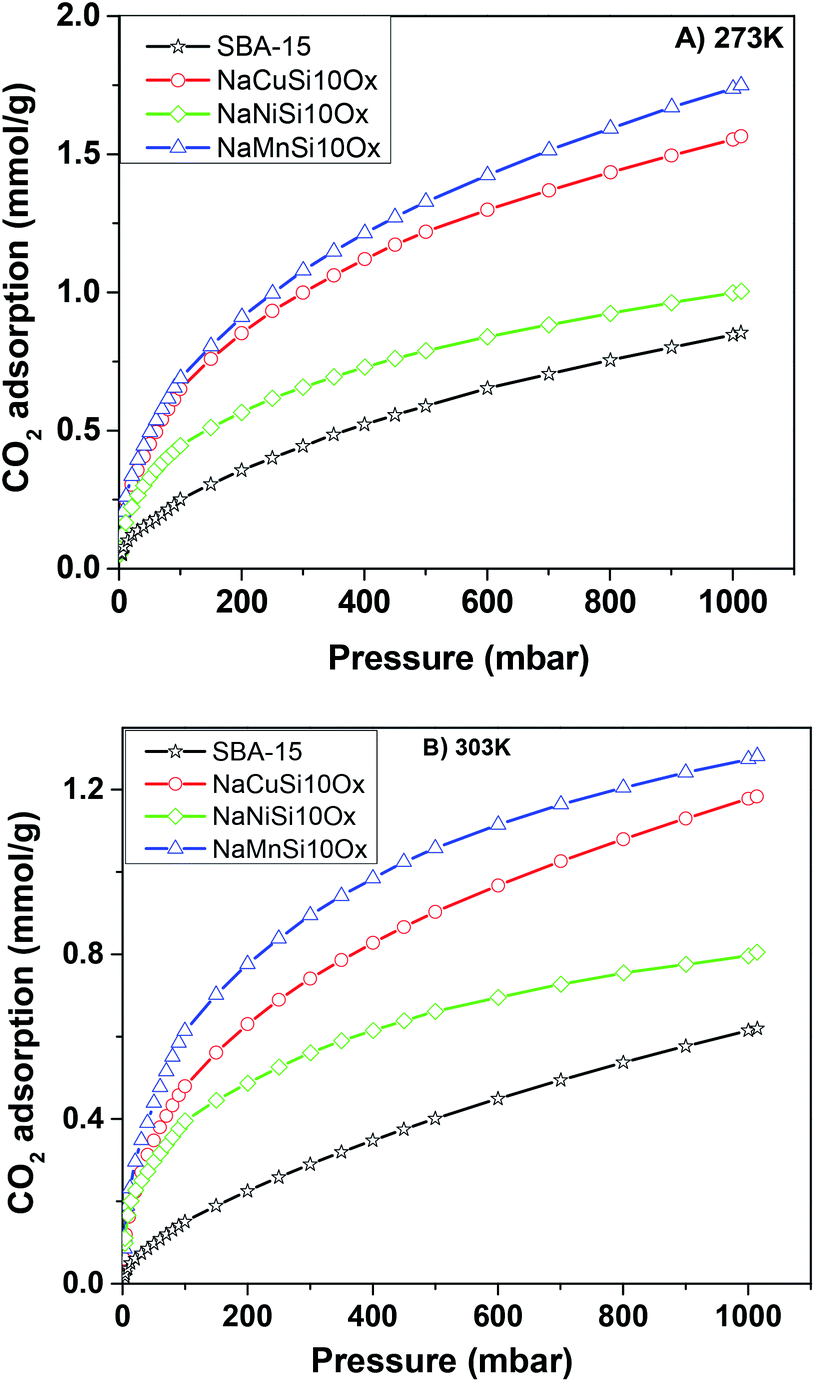 | ||
| Fig. 5 The CO2 adsorption isotherms for the metal silicate and pristine SBA-15 samples up to a maximum pressure of 1 bar at (A) 273 K and (B) 303 K. | ||
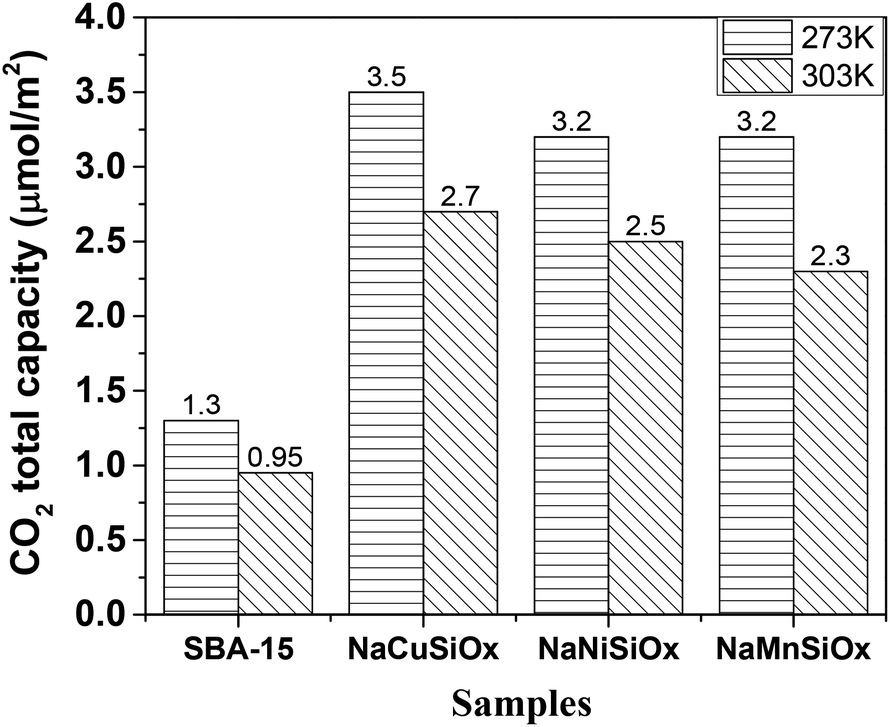 | ||
| Fig. 6 CO2 total capacity of metal silicates and SBA-15 samples normalized to the surface area. | ||
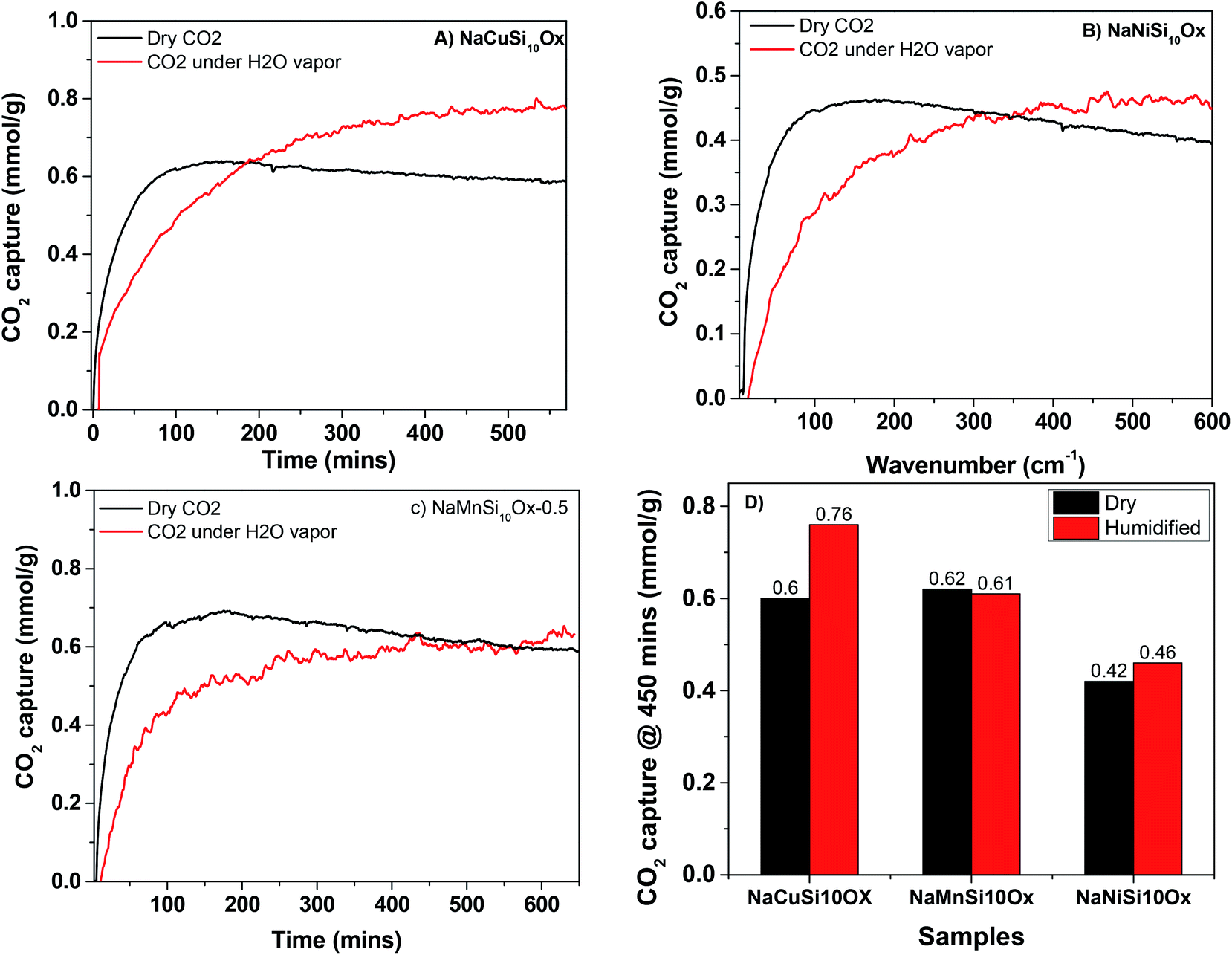 | ||
| Fig. 7 CO2 adsorption on metal silicates in the absence and presence of water vapor as a function of time, (A) NaCuSi10Ox, (B) NaNiSi10Ox and (C) NaMnSi10Ox, and (D) the CO2 uptakes on the metal silicates at 450 min in the absence and presence of water vapor. | ||
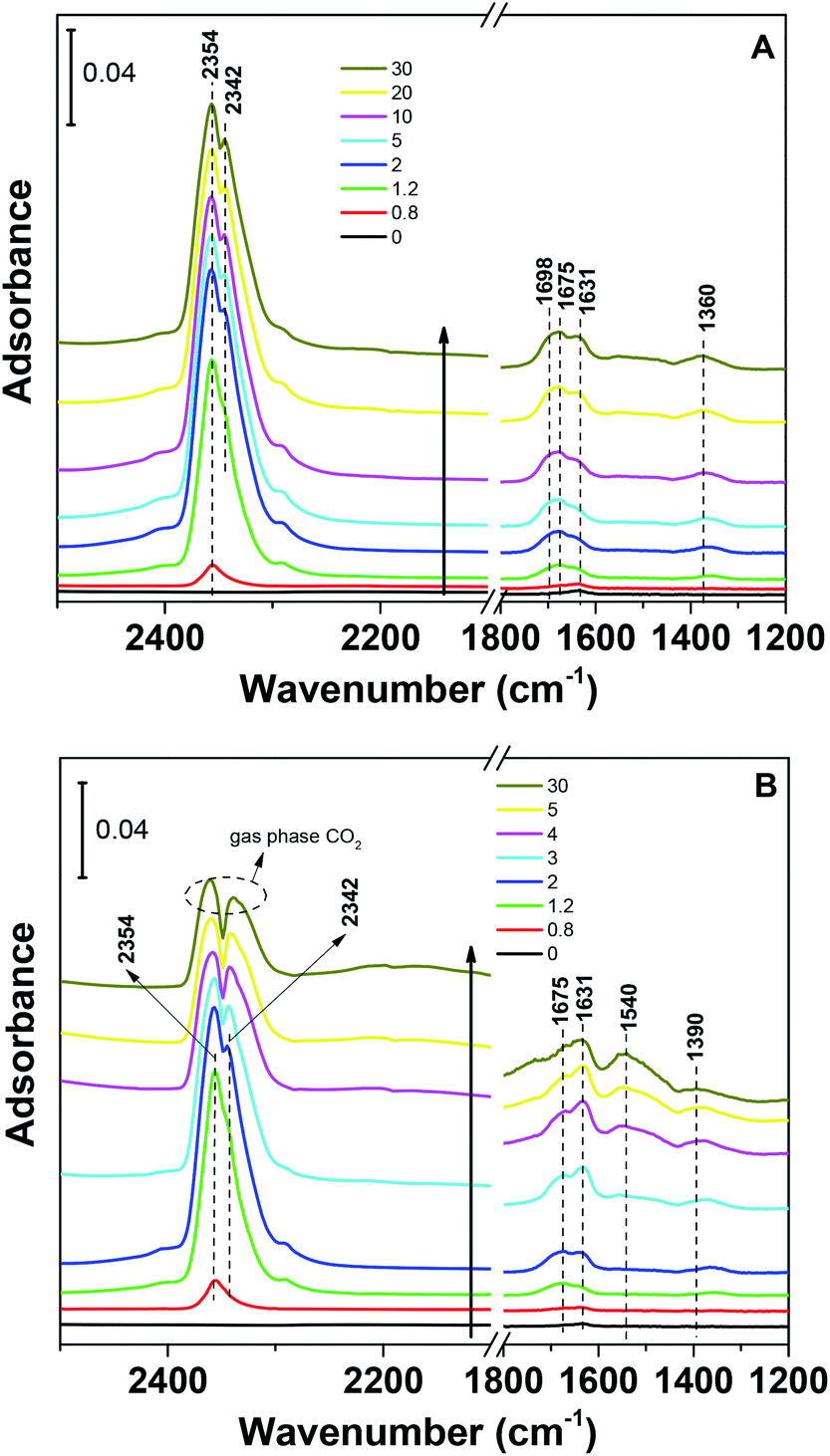 | ||
| Fig. 8 IR spectra from CO2 adsorption at room temperature on NaCuSi10Ox for 30 min: (A) in the absence of water vapor, and (B) in the presence of water vapor. | ||
The band at 1550 cm−1 is possibly due to unidentate carbonate, which is generated by the H2O interacting with bidentate carbonate as evidenced from the disappearance of the band at 1698 cm−1.46 These observations confirmed that H2O addition is able to react with adsorbed CO2 species to form bicarbonate and unidentate carbonate, and thus leads to the improvement of the CO2 uptake on the sample. It was also demonstrated in a number of studies that CO2 capture on silicates can be enhanced at low temperature by water vapor addition,47–49 producing acid carbonate (MHCO3) species instead of carbonate (Na2CO3) species. Those reports are in agreement with our IR observations on the NaCuSi10Ox sample in the presence of water.
It is worthwhile to mention that the initial bands at 2354 and 2342 cm−1 (shoulder) due to physisorbed CO2 are replaced with those from gas phase CO2 as a function of adsorption time in the presence of H2O, which occurs simultaneously with the evolution of the carbonate species. This seems to suggest that H2O also competes with physisorbed CO2 for similar adsorption sites on the NaCuSi10Ox sample, where H2O eventually dominates the shared sites. These IR results are in agreement with the TGA observations.
To investigate the stability of the various carbonate species formed on the NaCuSi10Ox sample, IR spectroscopy was used to follow desorption of the surface species formed from CO2 adsorption in flowing He gas. The IR spectra during desorption at room temperature are shown in Fig. 9, for both dry and wet conditions. Prolonged purging (30 min in flowing He) at room temperature does not noticeably change the IR features of the carbonates and bicarbonates under both dry and wet conditions. The IR bands corresponding to physisorbed CO2 species decline in intensity with increased purging time but are still observable even after purging in He for 30 min for the case of CO2 adsorption in the absence of H2O (Fig. 9A). In contrast, the IR bands due to gas phase CO2 readily vanish upon He purging to 2 min for the case of CO2 adsorption in the presence of H2O (Fig. 9B). This indicates that the water addition is beneficial to the removal of physisorbed CO2 species. Further heating as shown in Fig. S3A and B† gradually diminishes the IR features of carbonates and bicarbonates, and eventually the IR bands in the region of 1700–1200 cm−1 disappear when the temperature increases up to 453 K. There is no apparent difference in the IR feature evolution of the various carbonate and bicarbonate species as a function of temperature in the absence and presence of water.
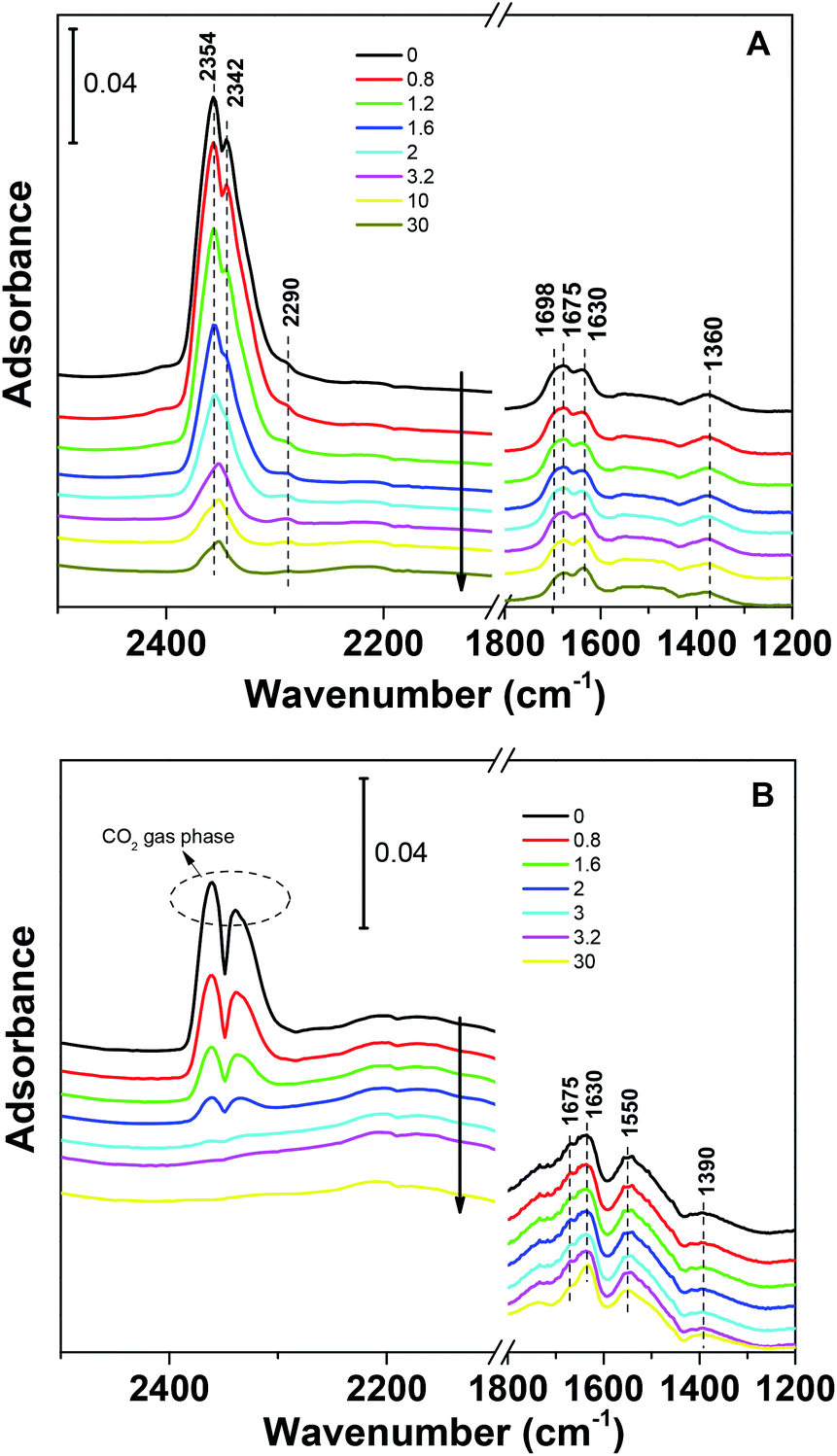 | ||
| Fig. 9 IR spectra during CO2 desorption on NaCuSi10Ox as a function of He purging time (30 min) for (A) dry and (B) wet conditions, respectively. | ||
IR experiments for CO2 adsorption and desorption on NaNiSi10Ox and NaMnSi10Ox in the absence and presence of water were also conducted and the results are shown in Fig. S3 and S4.† One similar observation is that in the presence of water, IR bands corresponding to the physisorbed CO2 species are replaced by CO2 gas phase features with increased adsorption time for all three samples, implying the competitive adsorption between CO2 and H2O on the metal silicates. However, the contrast in IR features due to carbonates between the dry and wet CO2 gases is much less evident on the NaNiSi10Ox and NaMnSi10Ox samples than on the NaCuSi10Ox sample, especially on NaMnSi10Ox. Meanwhile, for the NaNiSi10Ox sample, the bicarbonate feature at 1633 cm−1 slightly increases in intensity with adsorption time in the presence of H2O, and the weak bands ascribed to bicarbonates at 1493 and 1390 cm−1 are only observed upon prolonged adsorption time (20–30 min). For NaMnSi10Ox, the change in IR features in the carbonates region is barely noticeable with wet CO2 gas adsorption compared with the dry CO2 case. Upon desorption at room temperature, the IR features due to the physisorbed CO2 species under dry conditions decrease dramatically in intensity, and thus disappear with prolonged He purging in 5 min (Fig. S3B and S4B†) for both NaCuSi10Ox and NaMnSi10Ox. This suggests that interaction of CO2 with the two silicates (Ni, Mn) is much weaker than that with NaCuSi10Ox, where physisorbed CO2 species still persist after He purging for 30 min at room temperature.
The bonding strength of the chemisorbed CO2, i.e., carbonate species, on these metal silicates is comparable according to the MS profiles of CO2 evolved during the IR-TPD process (Fig. S5†) under both dry and wet conditions. However, it shows that in general the carbonate species formed in the presence of H2O require higher temperature to desorb (up to 573 K) than those formed in the absence of H2O (up to 400 K). The higher the desorption temperature, the stronger the interaction is for CO2 with the materials. So the MS results during CO2-TPD signify that H2O addition does reinforce the stabilization of the carbonate (bicarbonate) species on the metal silicates, consistent with the IR observation of these new species with the addition of water.
On the basis of the IR results, it is reasonable to postulate that CO2 and H2O exhibit a competitive and cooperative adsorption model on the metal silicate samples, especially for NaCuSiOx and NaNiSiOx, a similar behavior to that reported for CO2 and H2O coadsorbed on activated alumina.50 It can be concluded that the H2O and CO2 molecules competitively co-adsorbed on the same sites of the metal silicate surface initially. Depending on the adsorption strength of CO2 vs. H2O, a stronger adsorption strength of CO2, for example on NaCuSi10Ox, may permit its co-adsorption and synergistic interaction with H2O to produce new adsorbed bicarbonate and carbonate species and thus results in enhanced CO2 capture capacity. In contrast, the presence of H2O has little effect on NaMnSi10Ox which has a weaker interaction with CO2. The results indicate that the three synthesized metal silicate materials could potentially be applied as CO2 capture sorbents in humidified conditions with better/comparable CO2 uptakes compared with dry conditions.
Conclusions
CO2 capture under dry and wet conditions has been evaluated on metal silicate materials (NaCuSi10Ox, NaNiSi10Ox and NaMnSi10Ox) at low temperatures (273–323 K). These metal silicate samples were directly derived by incorporation of metals into SBA-15 through a hydrothermal method. All of the metal silicates exhibit greatly higher CO2 capture capability than the pristine SBA-15. CO2 capture capability strongly depends on the textural parameters, such as surface area, of the metal silicates in the absence of H2O. H2O addition is able to facilitate the CO2 uptake on the metal silicates, even though competitive adsorption is present between CO2 and H2O on the samples. The enhancement in CO2 capture depends on the type of metal introduced in/on the silicates. Metal silicates such as NaCuSi10Ox with strongest adsorption strength of CO2 show the greatest increase in CO2 capture capacity, due to the competitive and synergistic interaction between CO2 and H2O on the same surface sites in forming new surface bicarbonate and carbonate species. On the basis of the TGA and IR results, the presence of water not only improves the CO2 capture performance but also facilitates the removal of physisorbed CO2 during the desorption process. These findings provide insights for designing new sustainable adsorbents based on metal silicate sorbents derived from SBA-15, of which the structure and the CO2 sorption performance can be modified by controllable incorporation of different types of metals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Center for Understanding and Control of Acid Gas-Induced Evolution of Materials for Energy (UNCAGE-ME), an Energy Frontier Research Center funded by U.S. Department of Energy, Office of Science, Basic Energy Sciences. Part of the work, including the IR and Raman studies, was conducted at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility.References
- D. A. Yang, H. Y. Cho, J. Kim, S. T. Yang and W. S. Ahn, Energy Environ. Sci., 2012, 5, 6465–6473 RSC.
- W. Liu, D. King, J. Liu, B. Johnson, Y. Wang and Z. G. Yang, JOM, 2009, 61, 36–44 CrossRef CAS.
- J. Goldemberg, Science, 2007, 315, 808–810 CrossRef CAS PubMed.
- M. Wang, A. Lawal, P. Stephenson, J. Sidders and C. Ramshaw, Chem. Eng. Res. Des., 2011, 89, 1609–1624 CrossRef CAS.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9–20 CrossRef CAS.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- J. Y. Wang, L. Huang, R. Y. Yang, Z. Zhang, J. W. Wu, Y. S. Gao, Q. Wang, D. O'Hare and Z. Y. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- J. Y. Yu, M. Y. Guo, F. Muhammad, A. F. Wang, F. Zhang, Q. Li and G. S. Zhu, Carbon, 2014, 69, 502–514 CrossRef CAS.
- K. Huang, S. H. Chai, R. T. Mayes, G. M. Veith, K. L. Browning, M. A. Sakwa-Novak, M. E. Potter, C. W. Jones, Y. T. Wu and S. Dai, Chem. Commun., 2015, 51, 17261–17264 RSC.
- M. J. Li, K. Huang, J. A. Schott, Z. L. Wu and S. Dai, Microporous Mesoporous Mater., 2017, 249, 34–41 CrossRef CAS.
- R. V. Siriwardane, M. S. Shen, E. P. Fisher and J. A. Poston, Energy Fuels, 2001, 15, 279–284 CrossRef CAS.
- J. Shang, G. Li, R. Singh, Q. F. Gu, K. M. Nairn, T. J. Bastow, N. Medhekar, C. M. Doherty, A. J. Hill, J. Z. Liu and P. A. Webley, J. Am. Chem. Soc., 2012, 134, 19246–19253 CrossRef CAS PubMed.
- T. D. Pham, Q. L. Liu and R. F. Lobo, Langmuir, 2013, 29, 832–839 CrossRef CAS PubMed.
- R. B. Khomane, B. K. Sharma, S. Saha and B. D. Kulkarni, Chem. Eng. Sci., 2006, 61, 3415–3418 CrossRef CAS.
- A. Sanna and M. M. Maroto-Valer, Ind. Eng. Chem. Res., 2016, 55, 4080–4088 CrossRef CAS.
- M. T. Rodriguez and H. Pfeiffer, Thermochim. Acta, 2008, 473, 92–95 CrossRef CAS.
- H. A. Mosqueda, C. Vazquez, P. Bosch and H. Pfeiffer, Chem. Mater., 2006, 18, 2307–2310 CrossRef CAS.
- M. J. Li, U. Tumuluri, Z. L. Wu and S. Dai, Chemsuschem, 2015, 8, 3651–3660 CrossRef CAS PubMed.
- J. Baltrusaitis and V. H. Grassian, J. Phys. Chem. B, 2005, 109, 12227–12230 CrossRef CAS PubMed.
- Z. J. Zhang, Y. G. Zhao, Q. H. Gong, Z. Li and J. Li, Chem. Commun., 2013, 49, 653–661 RSC.
- Y. Y. Liu, Z. Y. U. Wang and H. C. Zhou, Greenhouse Gases: Sci. Technol., 2012, 2, 239–259 CrossRef CAS.
- X. Peng, X. Cheng and D. P. Cao, J. Mater. Chem., 2011, 21, 11259–11270 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runčevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef CAS PubMed.
- S. Mukherjee, N. Sikdar, D. O'Nolan, D. M. Franz, V. Gascón, A. Kumar, N. Kumar, H. S. Scott, D. G. Madden, P. E. Kruger, B. Space and M. J. Zaworotko, Sci. Adv., 2019, 5, eaax9171 CrossRef CAS PubMed.
- Q. Xiao, X. D. Tang, Y. F. Liu, Y. J. Zhong and W. D. Zhu, Chem. Eng. J., 2011, 174, 231–235 CrossRef CAS.
- X. S. Yin, Q. H. Zhang and J. G. Yu, Inorg. Chem., 2011, 50, 2844–2850 CrossRef CAS PubMed.
- T. Avalos-Rendon, J. Casa-Madrid and H. Pfeiffer, J. Phys. Chem. A, 2009, 113, 6919–6923 CrossRef CAS PubMed.
- S.-Z. Kang, T. Wu, X. Li and J. Mu, Mater. Lett., 2010, 64, 1404–1406 CrossRef CAS.
- G. Qi, L. Fu and E. P. Giannelis, Nat. Commun., 2014, 5, 5796 CrossRef CAS PubMed.
- E. González-Zamora and I. A. Ibarra, Mater. Chem. Front., 2017, 1, 1471–1484 RSC.
- M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 RSC.
- S. Shan, Q. Jia, L. Jiang and Y. Wang, in Advanced Materials Research, ed. Y. Zhang, 2011, vol. 213, pp. 515–518 Search PubMed.
- Q. Zhang, D. Han, Y. Liu, Q. Ye and Z. Zhu, AIChE J., 2013, 59, 901–911 CrossRef.
- G. G. Santillan-Reyes and H. Pfeiffer, Int. J. Greenhouse Gas Control, 2011, 5, 1624–1629 CrossRef CAS.
- Y. Duan and K. Parlinski, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 104113–104123 CrossRef.
- K. S. Ganesh Tilekar, K. Kale, R. Raskar and A. Gaikwad, Front. Chem. Sci. Eng., 2011, 5, 15 Search PubMed.
- S. J. Datta, C. Khumnoon, Z. H. Lee, W. K. Moon, S. Docao, T. H. Nguyen, I. C. Hwang, D. Moon, P. Oleynikov, O. Terasaki and K. B. Yoon, Science, 2015, 350, 302–306 CrossRef CAS PubMed.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- H. F. Yin, Z. Ma, H. G. Zhu, M. F. Chi and S. Dai, Appl. Catal., A, 2010, 386, 147–156 CrossRef CAS.
- W. A. Steele, G. Zgrablich and W. Rudzinski, Equilibria and Dynamics of gas adsorption on heterogeneous solid surfaces, Study in Surf. Sci. Catal., 1997 Search PubMed.
- A. M. Turek, I. E. Wachs and E. Decanio, J. Phys. Chem., 1992, 96, 5000–5007 CrossRef CAS.
- I. M. Hill, S. Hanspal, Z. D. Young and R. J. Davis, J. Phys. Chem. C, 2015, 119, 9186–9197 CrossRef CAS.
- Z. H. Cheng, A. Yasukawa, K. Kandori and T. Ishikawa, Langmuir, 1998, 14, 6681–6686 CrossRef CAS.
- K. Bhattacharyya, W. Q. Wu, E. Weitz, B. K. Vijayan and K. A. Gray, Molecules, 2015, 20, 15469–15487 CrossRef CAS PubMed.
- J. V. Evans and T. L. Whateley, Trans. Faraday Soc., 1967, 63, 2769–2777 RSC.
- T. L. Avalos-Rendon and H. Pfeiffer, Energy Fuels, 2012, 26, 3110–3114 CrossRef CAS.
- J. S. Falcone, J. L. Bass, P. H. Krumrine, K. Brensinger and E. R. Schenk, J. Phys. Chem. A, 2010, 114, 2438–2446 CrossRef CAS PubMed.
- J. Ortiz-Landeros, C. Gomez-Yanez and H. Pfeiffer, J. Solid State Chem., 2011, 184, 2257–2262 CrossRef CAS.
- G. Li, P. Xiao and P. Webley, Langmuir, 2009, 25, 10666–10675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional temperature programmed desorption IR data. See DOI: 10.1039/d0ra02736g |
| This journal is © The Royal Society of Chemistry 2020 |