
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRsmG forms stable complexes with premature small subunit rRNA during bacterial ribosome biogenesis†
Sudeshi M. Abedeera ,
Caitlin M. Hawkins‡
and
Sanjaya C. Abeysirigunawardena*
,
Caitlin M. Hawkins‡
and
Sanjaya C. Abeysirigunawardena*
Department of Chemistry and Biochemistry, Kent State University, Kent, Ohio 44242, USA. E-mail: sabeysir@kent.edu
First published on 11th June 2020
Abstract
The ribosome is the ribonucleoprotein machine that carries out protein biosynthesis in all forms of life. Perfect synchronization between ribosomal RNA (rRNA) transcription, folding, post-transcriptional modification, maturation, and assembly of r-proteins is essential for the rapid formation of structurally and functionally accurate ribosomes. Many RNA nucleotide modification enzymes may function as assembly factors that oversee the accuracy of ribosome assembly. The protein RsmG is a methyltransferase enzyme that is responsible for N7 methylation in G527 of 16S rRNA. Here we illustrate the ability of RsmG to bind various premature small subunit ribosomal RNAs with contrasting affinities. Protein RsmG binds with approximately 15-times higher affinity to premature 16S rRNA with the full leader sequence compared to that of mature 16S rRNA. Various r-proteins which bind to the 5′-domain influence RsmG binding. The observed binding cooperativity between RsmG and r-proteins is sensitive to the maturation status of premature small subunit rRNA. However, neither the maturation of 16S rRNA nor the presence of various r-proteins significantly influence the methylation activity of RsmG. The capability of RsmG to bind to premature small subunit rRNA and alter its binding preference to various RNA–protein complexes based on the maturation of rRNA indicates its ability to influence ribosome assembly.
1 Introduction
Ribosomes are ribonucleoprotein complexes that are responsible for protein biosynthesis in all kingdoms of life. Assembly of functionally accurate ribosomes from rRNAs and more than 50 proteins is critical for cell survival. The 30S ribosomal subunit can be assembled in vitro by the hierarchical addition of 21 ribosomal proteins to 16S rRNA.1,2 Unlike during in vitro assembly of 30S ribosomes, perfect coordination between rRNA transcription and folding, precursor rRNA maturation, post-transcriptional modification of rRNA, and binding of r-proteins is required during the synthesis of new ribosomes in vivo.Several rRNA modification enzymes can act as ribosome assembly factors that are essential to control flux through various parallel assembly pathways during in vivo ribosome biogenesis. Binding of the rRNA modification enzyme may preferentially stabilize several assembly intermediates, thus providing a thermodynamic advantage for the loading of r-proteins. On the other hand, binding antagonism between rRNA modification enzymes and r-proteins may prevent premature binding of r-proteins. For example, rRNA modification enzyme KsgA functions as an assembly factor that controls rRNA maturation and subsequent translation initiation. In addition, the role played by KsgA as an assembly factor is far more important for ribosome biogenesis than its methyltransferase activity.3–5 Similarly, several enzymes associated with ribosome assembly such as ATPases and methyltransferases function as assembly factors during ribosome biogenesis in which their enzymatic activity serves only as a checkmark for the successful completion of their job as an assembly factor.6,7
Here we study the ability of ribosomal small subunit methyltransferase G (RsmG) enzyme to bind to E. coli small subunit precursor rRNAs. Protein RsmG (or GidB) that is part of a SAM-dependent methyltransferase superfamily is observed in both Gram-positive and Gram-negative bacteria. RsmG binds to 30S 5′-domain and methylates N7 of G527 that is located at the highly conserved 530 loop of 16S helix 18 (Fig. 1a, c and d).8 Spontaneous streptomycin-resistant mutations were observed in both RsmG and uS12 after incubation of wildtype S. coelicolor A3(2) with various concentrations of streptomycin.9,10 Chromosomal deletion of RsmG resulted in the loss of methylation as well as low-level streptomycin resistance, although bacterial growth is unaffected. Generation of spontaneous compensatory mutations in ribosomal protein uS12 in response to mutations in RsmG suggests a link between rRNA methylation and ribosome assembly.11 Ribosome structure near 16S helix 2 and helix 18 pseudoknots are significantly different for streptomycin-resistant ribosomes compared to their wildtype counterparts,12 perhaps due to the emergence of streptomycin resistant rRNA mutations near 30S pseudoknots12–14 and in proteins9,10 those bind in their vicinity. These results suggest a strong correlation between rRNA methylation, ribosome assembly, and structural integrity near rRNA pseudoknots. The objective of this research is to investigate the relationship between RsmG binding, rRNA maturation, and ribosome assembly and RsmG binding thermodynamics underlying such correlations.
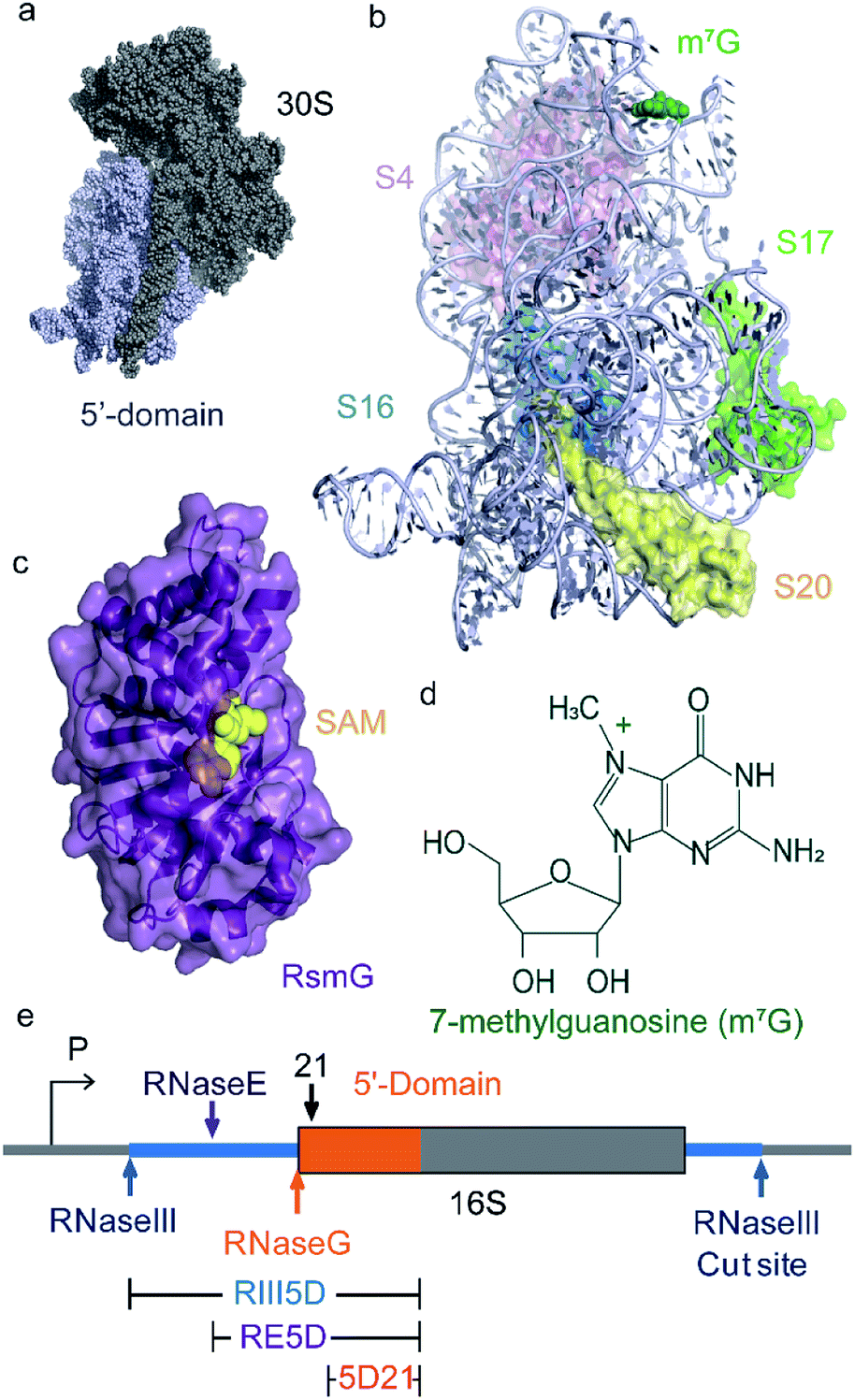 | ||
| Fig. 1 RsmG binds to 30S 5′-domain and methylates N7 of G527 at 16S helix 18. (a) The crystal structure of the 30S (small) subunit of the E. coli ribosome. The 5′-domain is shown in light blue spheres. (b) The ribbon structure of the 5′-domain with the primary (uS4, uS17, bS20) and secondary (bS16) assembly proteins. RsmG is responsible for the 7-methylguanosine (m7G) modification shown in green spheres. (PDB ID 2I2P). (c) The X-ray crystal structure of T. thermophilus (PDB ID: 3G8B) RsmG (purple ribbon) with SAM (yellow spheres) bound at its active site. (d) The chemical structure of 7-methylguanosine (m7G). (e) 17S rRNA is transcribed from the P1 or P2 promoters of the rrn operon as shown. The 5′-domain RNA is shown in orange. Cut sites of RNases that process 5′-leader and 3′-trailer sequences are indicated with arrows. The different rRNA constructs (mature and precursor) used in this study are also indicated. | ||
2 Materials and methods
2.1 Synthesis and purification of 5′-domain rRNAs
Three different E. coli 16S 5′-domain RNAs with varying lengths of 5′-leader were used in FRET-based binding assays. These three RNAs included a mature 5′-domain rRNA construct (5D21; nts. 21–556 E. coli numbering) and two 16S precursor rRNAs starting from RNaseIII (RIII5D; nts. −115–556) and RNaseE (RE5D; nts. −66–556) cut sites. The pseudoknot mutant (PK-Mut: G505A, C507U) for 5′-domain RNA was obtained by site-directed mutagenesis (Q5 site-directed mutagenesis kit, NEB). All 5′-domain RNA constructs are extended at the 3′-end to base pair with a fluorophore-labelled complementary DNA oligomer, as previously described by Abeysirigunawardena and Woodson 2016. Extended 5′-domain RNA constructs were in vitro transcribed with T7 RNA polymerase using PCR products generated using p17S plasmid (based on rrnC operon). RNAs were purified by 4% denaturing PAGE using standard protocols as described previously. Phenol–chloroform extraction was performed on all RNAs followed by ethanol precipitation prior to their use. RNA precipitates were resuspended in appropriate buffers and their concentrations were determined using A260 (absorbance at 260 nm) measured using Nanodrop spectrophotometer and their respective extinction coefficients.2.2 Purification and labeling of RsmG protein
Overexpression vector for E. coli RsmG (RsmG_pET-24b(+)) was purchased from GenScript. Double mutant RsmG (RsmG: C172S, A14C) with a single solvent accessible Cys residue was obtained by site-directed mutagenesis (Q5 site-directed mutagenesis kit, NEB). This mutant gave the best FRET signal and labeling efficiency out of many tested. RsmG was overexpressed in BL21(DE3) cells and purified by Ni-NTA column (AKTA start, GE Healthcare). Purified proteins were dialyzed thrice in 80 mM HEPES-K pH 7.6, 1 M KCl, 1 mM TCEP, overnight and stored at −80 °C in 500 μL aliquots. The concentration of the protein was determined using the absorbance at 280 nm (ε280 = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 450 M−1 cm−1). Protein RsmG: C172S, A14C (16.8 μM, 500 μL) in 80 mM HEPES-K pH 7.6, 1 M KCl, 1 mM TCEP was pre-incubated at 20 °C for 30 min. A six-fold molar excess of Cy5 maleimide mono-reactive dye (GE Healthcare) was added and incubated at 20 °C for 2 hours. The labeling reaction was then quenched by adding 10 mL of 20 mM Tris–HCl pH 7.5, 6 M urea and 6 mM BME. Unreacted dye was removed by dialysis (20 mM HEPES-K pH 7.6, 1 M KCl, 1 mM TCEP with three buffer exchanges) followed by affinity chromatography described earlier. Isolated Cy5-labeled RsmG: C172S, A14C was then dialyzed against storage buffer (80 mM HEPES-K pH 7.6, 1 M KCl, 1 mM TCEP, three buffer exchanges) and stored at −80 °C in 50 μL aliquots, in a light–tight container. The labeling efficiency was determined to be almost 100% by SDS-PAGE. Concentration of the labeled protein was determined using the absorbance at 650 nm (ε650 = 250000 M−1 cm−1) measured using Nanodrop spectrophotometer.
450 M−1 cm−1). Protein RsmG: C172S, A14C (16.8 μM, 500 μL) in 80 mM HEPES-K pH 7.6, 1 M KCl, 1 mM TCEP was pre-incubated at 20 °C for 30 min. A six-fold molar excess of Cy5 maleimide mono-reactive dye (GE Healthcare) was added and incubated at 20 °C for 2 hours. The labeling reaction was then quenched by adding 10 mL of 20 mM Tris–HCl pH 7.5, 6 M urea and 6 mM BME. Unreacted dye was removed by dialysis (20 mM HEPES-K pH 7.6, 1 M KCl, 1 mM TCEP with three buffer exchanges) followed by affinity chromatography described earlier. Isolated Cy5-labeled RsmG: C172S, A14C was then dialyzed against storage buffer (80 mM HEPES-K pH 7.6, 1 M KCl, 1 mM TCEP, three buffer exchanges) and stored at −80 °C in 50 μL aliquots, in a light–tight container. The labeling efficiency was determined to be almost 100% by SDS-PAGE. Concentration of the labeled protein was determined using the absorbance at 650 nm (ε650 = 250000 M−1 cm−1) measured using Nanodrop spectrophotometer.
2.3 Purification of ribosomal proteins
Proteins uS4, uS17, bS16 and bS20 from pET-24b derivatives were overexpressed in BL21(DE3) cells and purified by cation exchange chromatography (UNO™ S6, BioRad) as previously described in Culver and Noller 1999. Purified proteins were dialyzed thrice in 80 mM HEPES-K pH 7.6, 1 M KCl, 6 mM BME, overnight and stored at −80 °C in 50 μL aliquots.2.4 FRET-based RsmG titrations
Extended 5′-domain RNAs (10 nM) was incubated with Cy3-babeled DNA oligomer (4 nM; 100% labeling efficiency) in HK buffer (80 mM HEPES-K pH 7.6 and 330 mM KCl) at 70 °C for 5 min and then, cooled at 25 °C for 5 min. The annealed RNAs (10 nM) were then folded in HKM buffer (80 mM HEPES-K pH 7.6, 330 mM KCl, 4–20 mM MgCl2) at 37 °C for 15 min. For FRET titrations in the presence of r-proteins, a three-fold molar excess of uS4, uS17 or combination of uS4, bS16, and bS20 (15 nM) was added to the mixture and incubated at 37 °C for another 15 min. Labeled RNA was titrated with Cy5-labeled RsmG in 2 nM increments, at 30 °C. The fluorescence emission (550–700 nm) was measured upon excitation at 532 nm using PTI QuantaMaster 400 fluorometer (Horiba Scientific). Fluorescence intensities at 570 nm (donor) and 670 nm (acceptor) were used to calculate the FRET efficiency (EFRET) values at each RsmG concentration (EFRET = I670/(I570 + I670)). Final EFRET values are the average of three independent titrations. The change in FRET efficiency vs. RsmG–Cy5 concentration with volume correction was fit to the quadratic equation (eqn (1)) using Origin software, to determine the Kd values for binding between RsmG and each 5′-domain RNA construct. In eqn (1), F is the change in fluorescence intensity per 1 nM of RsmG.| EFRET = F × ((Kd + [RNA] + [RsmG]) − sqrt ((Kd + [RNA] + [RsmG])2 − 4 × [RNA] × [RsmG]))/2 | (1) |
The ΔGlink for binding of RsmG and respective r-protein(s) to various mature (5D21) and precursor (RE5D and RIII5D) 16S rRNA constructs was calculated using eqn (2), in which the Kd of RsmG-5′-domain rRNA complex in the absence and presence of r-protein(s) for a given 5′-domain rRNA construct are given as K1 and K2, respectively.
|
ΔGlink = RTln(K2/K1)
| (2) |
Errors for ΔGlink was obtained by propagation of errors for K1 and K2.
2.5 Filter binding assay
Two 5′-domain RNAs (5D21; nts. 21–556 and RIII5D; nts. −115–556) and three leader fragments of bacterial small subunit rRNA (RIII-RE; nts. −115, −66, RIII-RG; nts. −115–1, and RE-RG; nts. −66–1) were used in filter binding assays. All of these RNAs were in vitro transcribed using α-32P-ATP, low-A NTP mix, and T7 RNA polymerase following standard protocols (Cazenave and Uhlenbeck 1994).15 Following the transcription reaction (at 37 °C for 1 hour), reaction mixtures were passed through TE-10 columns to remove free nucleotides. 5′-Domain RNAs (500–1000 counts) were pre-folded in HKM20 (80 mM HEPES-K pH 7.6, 330 mM KCl, 20 mM MgCl2) for 15 minutes followed by the incubation at 37 °C with protein RsmG (0–1 μM) in the same HKM20 buffer for another 15 minutes. For 5′-leader RNA fragments, RNAs were incubated with varying concentrations of RsmG in HK50 buffer (80 mM HEPES-K pH 7.6 and 50 mM KCl) at 30 °C for 15 minutes followed by the incubation with RsmG (0–5 μM) for another 15 minutes. The RNA–protein mixture (10 μL) was then spotted onto nitrocellulose membrane on top of a Nylon membrane pre-soaked in binding buffer (20 mM Tris–HCl pH 7.5, 100 mM CH3COOK, 200 mM KCl, 2.5 mM MgCl2, and 1 mM DTT) and vacuum-dried. The spotted samples were washed twice with 20 μL each from 1 × HK50 buffer. The two membranes were vacuum dried and exposed to an intensifying screen. Radiograph of the membranes were obtained using phosphorimager (Typhoon FLA 9500). Intensities of RNA–protein complexes and free RNAs on nitrocellulose and Nylon membranes, respectively, were measured using ImageJ. Fraction of RNA–protein complexes (FB) was plotted against [RsmG]. Non-linear fitting of these titration curves was performed using eqn (1) to obtain Kds for various RsmG–RNA complexes.2.6 Activity assay for RsmG
Both mature and precursor RNAs (30 pmol) were pre-folded (incubation at 37 °C for 15 minutes) in 50 μL HKM buffer (80 mM HEPES-K pH 7.6, 330 mM KCl, 4–20 mM MgCl2). Where appropriate, 1.5-fold molar excess of r-protein(s) were added to pre-folded RNAs and incubated at 37 °C for another 15 minutes. For methylation reaction of RNA by RsmG, 5′-domain RNA (30 pmol) was incubated with 1.5-fold molar excess of RsmG in the presence of S-adenosyl methionine (1 mM) in HKM buffer at 37 °C for 1 hour. Phenol/chloroform extraction of methylated RNAs were performed followed by ethanol precipitation. The RNA pellet was then treated with NaBH4 (10 μL 1 M Tris–HCl pH 8.3 and 10 μL 8 mg mL−1 NaBH4) and incubated on ice for 10 min in the dark followed by ethanol precipitation. Aniline (10 μL aniline, 60 μL acetic acid and 930 μL deionized water) was added to the RNA pellet and incubated at 60 °C in dark for 10 min. The reaction mixture was then ethanol precipitated, and the pellet was dissolved in 20 μL of deionized water. A primer extension assay was performed using radiolabeled (32P) DNA primer (p540, Powers and Noller 1995)16 and Superscript III Reverse Transcriptase enzyme (Invitrogen) following the manufacturer's standard protocol. Each sample (20 μL) was mixed with 20 μL of loading buffer (50% glycerol, 2% xylene cyanol and 2% bromophenol blue) and 2 μL of the mixture was loaded onto 8% denaturing PAGE gel followed by electrophoresis for 1–2 hours. Gels were exposed to intensifying screens and autoradiographs were recorded using Typhoon FLA 9500. Intensities of the stop signal at G527 position was measured with ImageJ software. The intensity values were normalized to a reference band (A532) to obtain relative RsmG activity.2.7 Electrophoretic mobility shift assay (EMSA) for DNA oligomer binding
Radiolabeled SA5 DNA oligomer (1 nM, 10 μL) was incubated with 0–50 nM of each modified 5′-domain RNAs in HK buffer (80 mM HEPES-K pH 7.6 and 330 mM KCl) at 70 °C for 5 min followed by incubation at 25 °C for 5 min. Each sample was then mixed with 10 μL of loading buffer (50% glycerol, 2% xylene cyanol and 2% bromophenol blue) and loaded (5 μL) onto 8% native PAGE gel. RNA–DNA complexes were electrophoresed for 1 hour at 15 W per gel. The gel was exposed to an intensifying screen and an autoradiograph was recorded using Typhoon FLA 9500. Gel bands (free DNA and RNA–DNA hybrid) in autoradiographs were quantified using ImageJ. Fraction of RNA–DNA complex were calculated. Kds for RNA–DNA complexes were determined using non-linear curve fitting (eqn (1)) in Origin.3 Results and discussion
3.1 RsmG binds tightly to 30S 5′-domain rRNAs with 5′-leader sequences
Nucleotide modifications in rRNA are added sequentially from 5′- to 3′-end in the same direction as rRNA transcription.17,18 Folding of rRNA, rRNA modifications, and ribosome assembly probably co-occur with its transcription. Transcription of the bacterial rRNA starts from two promoter sites (P1 or P2) located in the rrn operon. The primary rRNA transcript is processed by RNaseIII to produce precursor small (17S) and large (25S) subunit rRNAs and a tRNA.19,20 Precursor small subunit rRNA (17S), is further processed by a series of RNases, as shown in Fig. 1e to produce mature 16S rRNA.21,22 Nucleotide modifications in the 30S 5′-domain are likely to occur before rRNA is matured, and before many late-binding r-proteins are bound to rRNA. We hypothesized that RsmG binds preferably to precursor rRNA compared to its mature counterparts. To test the hypothesis that RsmG can bind to premature rRNA, we performed a FRET-based RsmG titration assay using both mature and unprocessed E. coli 16S 5′-domain rRNA constructs.The mature 5′-domain rRNA construct (5D21) contained the 16S rRNA sequence from nucleotide 21 to nucleotide 556 (E. coli numbering) whereas, unprocessed precursor 5′-domain rRNA constructs started at positions −66 (RE5D) and −115 (RIII5D) of the 5′-leader sequence (Fig. 1e). These 5′-domain rRNA constructs were fluorescently labeled with Cyanine 3 (Cy3) at their 3′-ends by annealing a complementary Cy3-labeled DNA oligomer to the 3′-extension on rRNA as previously described (Abeysirigunawardena and Woodson 2016).23 EMSAs were performed to confirm the ability of DNA oligomer to bind to the 3′-extension on each rRNA construct used in this study (Fig. S1†). RsmG was Cyanine 5 (Cy5)-labeled at its only Cysteine residue at position 14. Approximately 0.1 FRET efficiency was observed for rRNA–RsmG complexes. However, no significant decrease in donor fluorescence was observed when unlabeled protein was added to Cy3-labeled rRNA. In addition, no significant FRET signal was observed between RsmG and the free DNA primer. The FRET between 5DCy3 and RsmGCy5 was used to determine equilibrium dissociation constants (Kd) for various 5′-domain rRNA–RsmG complexes (Fig. 2a–c).
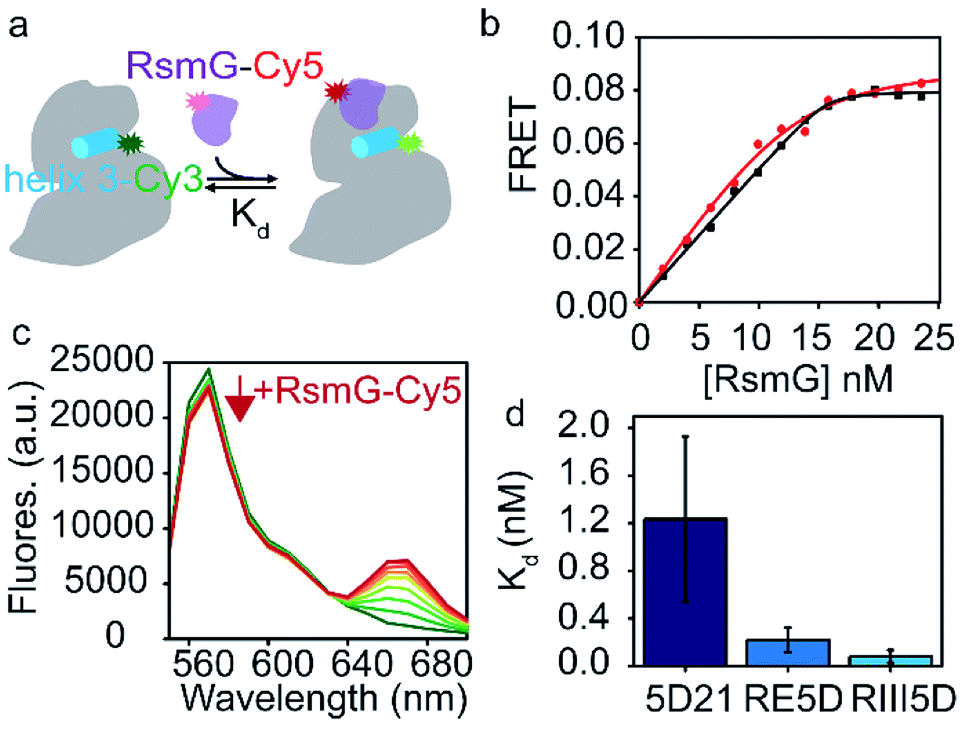 | ||
| Fig. 2 Unprocessed precursor 16S rRNAs are thermodynamically preferred by RsmG. (a) FRET-based assay used to determine equilibrium dissociation constants (Kd) for different rRNA–RsmG complexes. RsmG is fluorescently labeled with Cyanine 5 (Cy5) at its N-terminus, while rRNA is Cyanine 3 (Cy3)-labelled at the end of helix 3 as shown. (b) FRET vs. [RsmG–Cy5] titration curves for RIII5D (black) and 5D21 (red) 16S rRNA constructs. The curves represent the least square fit to quadratic equation. (c) Fluorescence spectra obtained for 5D21-Cy3 RNA at 0–25 nM RsmG–Cy5 concentrations in HKM20 buffer (80 mM HEPES pH 7.6, 330 mM KCl, 20 mM MgCl2) before acceptor direct excitation corrections are shown. Donor fluorescence (570 nm) decrease with each addition of RsmG–Cy5 is indicated by the red arrow. (d) Kds obtained for RsmG complexes with mature (5D21) and premature (RE5D and RIII5D) 16S rRNA constructs at 20 mM [Mg2+] (HKM20 buffer) are shown. | ||
Our data shows a gradual decrease in affinity as the rRNA matures from unprocessed precursor (RIII5D) to mature 5′-domain rRNA (5D21) (Fig. 2d). The affinities are significantly different between RIII5D (Kd = 0.08 ± 0.05 nM) and 5D21 (Kd = 1.2 ± 0.7 nM). Despite the high error for the Kd of 5D21, the affinity of the processed RNA was found to be decreased by 15-fold in comparison to RIII5D. Furthermore, filter binding assays performed with RIII5D, and 5D21 illustrated a similar trend in affinities for RsmG (Fig. S2†). The higher affinities of RsmG towards unprocessed precursor constructs of 16S rRNA suggest a thermodynamic preference of RsmG towards unprocessed precursor 16S rRNA, that can presumably be attributed to the formation of additional RNA–protein contacts with the leader sequences of unprocessed precursor rRNA, especially in the region between RNaseIII and RNaseE cut sites. Patches of positively charged amino acids away from the RsmG active site9,24 may interact with the 5′-leader causing the observed increase in the stability of these RsmG complexes with premature rRNA.
3.2 RsmG forms additional contacts with different 5′-leader sequences
Given the ability of RsmG to bind to precursor rRNAs, we hypothesized that RsmG could bind to rRNA segments located within the 5′-leader. Filter binding assays were performed to determine the affinities of RsmG towards different 5′-leader fragments. Two RNA fragments containing sequences from RNaseIII cut-site to RNaseE (RIII-RE) and RNaseG (RIII-RG) cut-sites, respectively, were designed. These two RNAs may resemble two nascent rRNA fragments present at different stages of 16S rRNA transcription. In addition, an RNA that carried the sequence from RNaseE cut site to that of RNaseG (RE-RG) was used.RIII-RE showed a tighter affinity (Kd = 0.12 ± 0.03 μM) to RsmG in comparison to RIII-RG (Kd = 1.1 ± 0.1 μM) and RE-RG (Kd = 0.7 ± 0.3 μM) (Fig. 3a–c). This data demonstrates that RsmG can bind to unprocessed precursor 16S rRNA away from its methylation site. The affinities of RsmG to 5′-leader RNA fragments were lower than that to 5′-domain rRNA, suggesting that 5′-leader sequences are not the primary binding site of RsmG. Significantly higher binding affinity of RsmG to RIII-RE compared to the other two RNA fragments suggests that RsmG preferentially binds within 17S rRNA nts −115 and −66, before the rest of the 5′-leader is transcribed. The lower affinity for the full leader sequence can presumably be due to the formation of a secondary structure that sequesters the exposed binding site of RsmG in RIII-RE. During ribosome biogenesis, the binding of RsmG to 5′-leader between RNaseIII and RNaseE cut sites may prevent the formation of non-native secondary structures as the transcription proceeds to form the entire 5′-domain structure, including the methylation site of RsmG. These filter binding assays were performed in the absence of magnesium ions. We were unable to observe a significant binding of RsmG to these leader RNAs in the presence of 4–20 mM [Mg2+], perhaps due to the existence of non-native higher-order secondary structures formed in the presence of Mg2+.
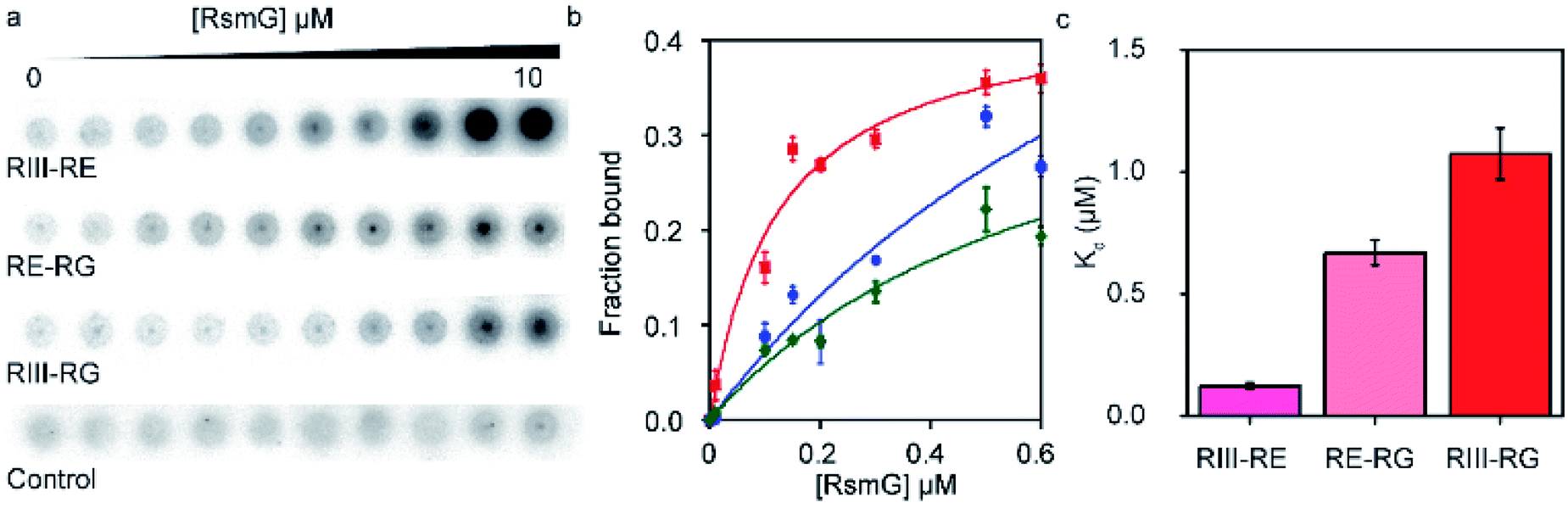 | ||
| Fig. 3 RsmG forms additional contacts with different 5′-leader sequences. (a) Nitrocellulose membrane of the filter binding assay performed to determine Kds for complexes of RsmG and different 5′-leader fragments (RIII-RE, RIII-RG and RE-RG) in HK buffer (80 mM HEPES pH 7.6, 330 mM KCl). (b) Fraction bound vs. [RsmG] curves for RIII-RE (red), RE-RG (green) and RIII-RG (blue). The curves represent the least square fit to quadratic equation. (c) Kd values obtained for different 5′-leader fragments (RIII-RE, RIII-RG, and RE-RG). | ||
3.3 RsmG forms equally productive complexes with both mature and premature 30S 5′-domain rRNAs
Mature 16S rRNA may not be the optimal substrate for the RsmG methyltransferase activity. We tested whether RsmG activity differs for the three 16S rRNA constructs including a mature and two unprocessed precursor rRNAs. Mature (5D21) and unprocessed precursor 16S rRNAs (RIII5D and RE5D) were used in a reverse transcriptase-based assay to determine the activity of RsmG in each case. In the presence of methylation, m7G would be reduced by sodium borohydride followed by β-elimination with aniline that results in RNA strand scission at its 5′-phosphate. The reverse transcriptase stop due to methylation-dependent strand scission was used to detect the activity of RsmG. Only a slight difference in RsmG activity between premature and mature 16S rRNAs was observed (Fig. 4a and b). Regardless of the differential affinities of RsmG based on the maturation status of 5′-domain rRNA, RsmG forms productive complexes with each RNA construct used. This result indicates that the active site of RsmG is correctly positioned at the methylation site located in helix 18 regardless of the maturation status of small subunit 16S rRNA. It should be noted that the RsmG concentration required for activity assays are well above the binding affinities detected for respective RNAs. Therefore, the influence of differential affinities of RsmG to various 5′-domain rRNAs may not be reflected in detecting its activity due to the addition of saturating concentrations of RsmG in these activity assays.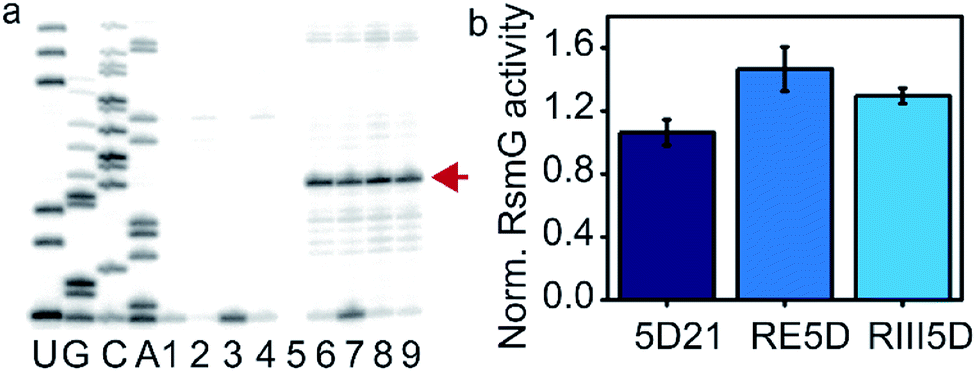 | ||
| Fig. 4 RsmG is active on both mature and mature 5′-domain rRNAs. (a) Reverse transcriptase-based activity assay for RsmG. Sequencing reactions are labeled as U, G, C, A. Lanes 1 and 2 are pause control lanes for 5D21 and RIII5D, respectively. Reaction controls (without SAM) for 5D21 (Lane 4) and RIII5D (Lane 5), respectively. RsmG methylation reactions of 5D21 (Lanes 6 and 7) and RIII5D (Lanes 8 and 9) are shown (with SAM). The strong pause indicated by the arrow (red) is the reverse transcriptase stop observed at G527. (b) The enzymatic activity of RsmG on different 30S 5′-domain rRNA constructs is shown. | ||
3.4 Ribosomal assembly proteins differentially stabilize RsmG complexes formed by mature and precursor 30S 5′-domain rRNA
The binding of modification enzymes can impact the hierarchical binding of r-proteins. Many rRNA modification enzymes are sub-optimally active on both the 16S rRNA and native 30S ribosomes, suggesting that they bind preferentially to 30S assembly intermediates with unknown combinations of r-proteins. In Thermus thermophilus, 16S rRNA serves as a better substrate for RsmG compared to the fully assembled 30S ribosome, indicating that RsmG can bind to rRNA before all or several 5′-domain-binding proteins are bound and possibly before processing of the 5′-leader sequence.25 Surprisingly, in E. coli, RsmG is capable of methylating fully assembled 30S ribosomes, but not the 16S rRNA.9 The lack of G527 methylation in 16S rRNA substrates may be due to the impaired folding of 16S helix 18 at 2 mM Mg2+ concentration. Ribosomal proteins uS4 and uS12 form contacts with helix 18 during the early stages of the 30S assembly. In addition, pseudouridine synthase, RsuA that is responsible for Ψ516 binds to the same helix during 30S biogenesis.26 Although ribosomal proteins bS16, uS17, and bS20 bind to 30S 5′-domain away from helix 18 (Fig. 1b), RsmG binding can be influenced by their presence.27 We hypothesized that early binding ribosomal proteins can influence the binding preference of RsmG to either premature or mature rRNA.FRET titration experiments were carried out for 5D21, RE5D, and RIII5D at both 4 mM and 20 mM Mg2+ concentrations in the presence of different primary assembly proteins (uS4, uS17) and secondary assembly protein bS16 in combination with primary assembly proteins uS4 and bS20. Binding cooperativity (−ΔGlink) of RsmG with r-proteins was calculated and compared for various 5′-domain RNAs (Fig. 5a and b). At 20 mM Mg2+, binding of RsmG to 5D21 was slightly anti-cooperative (−0.6 ± 1.0 kJ mol−1) with uS4, while significant cooperativity was observed with proteins uS4, bS16, and bS20 in combination (2.8 ± 1.3 kJ mol−1). However, binding of RsmG to either RIII5D or RE5D was found to be anti-cooperative with all protein combinations that were tested. Even though assembly proteins generally increase the compactness, their influence can be less pronounced at higher Mg2+ concentrations due to the ability of Mg2+ to stabilize RNA structures. Therefore, we carried out the same set of experiments at 4 mM Mg2+ concentration to reveal the bona fide effect of assembly proteins on RsmG binding to various mature and premature 5′-domain RNAs. At 4 mM Mg2+, binding of RsmG and uS4 to 5D21 was cooperative (3.6 ± 1.4 kJ mol−1), while significant anti-cooperativity was observed between the binding of RsmG and uS17 (−8.5 ± 0.8 kJ mol−1). Proteins that stabilize native-like 5′-domain structures strengthen RsmG–rRNA complexes, whereas protein uS17 that stabilizes a non-native 5′-domain assembly intermediate is antagonist for RsmG binding. These results indicate that RsmG preferably bind to native-like 5′-domain complexes. However, a different trend was observed for RsmG binding to RIII5D at 4 mM Mg2+. Interestingly, ribosomal proteins do not significantly influence RsmG activity at both 4 and 20 mM [Mg2+] suggesting that regardless of the presence of ribosomal assembly proteins, RsmG binds to its active site in the correct orientation that is necessary for its methylation activity (Fig. S3†). The levels of methylation in both mature and premature 5′-domain rRNAs are not influenced by the binding cooperativity between RsmG and r-proteins. This lack of preference is likely due to the ability of RsmG–rRNA complexes to dynamically interchange between active and inactive forms.
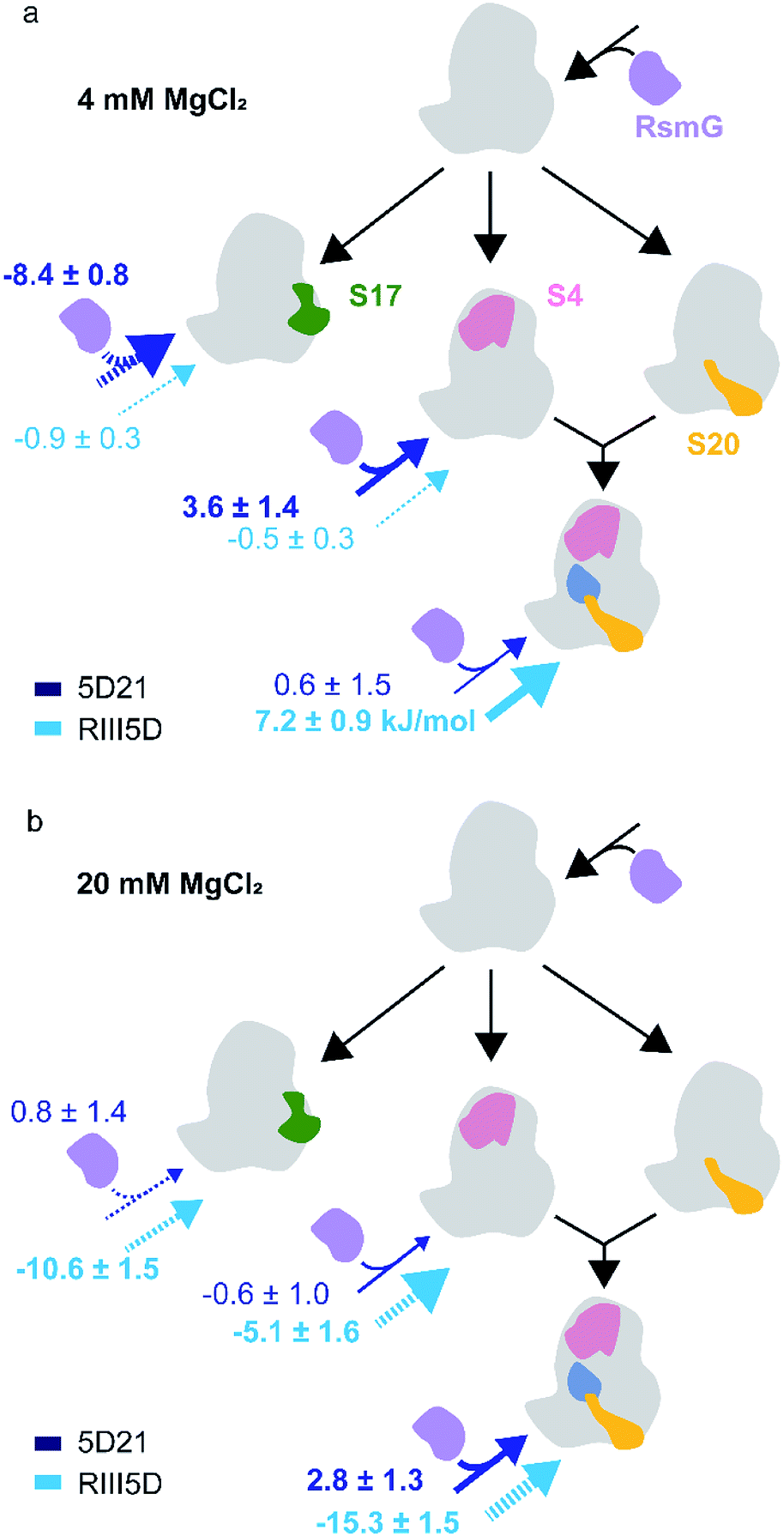 | ||
| Fig. 5 Ribosomal assembly proteins differentially stabilize RsmG binding to mature and premature 5′-domain 16S rRNA. The ΔGlink values (kJ mol−1) calculated for the binding of RsmG and r-proteins to various mature (5D21; dark blue) and premature (RIII5D; light blue) 5′-domain rRNA constructs, at (a) 4 mM (HKM4 buffer) and (b) 20 mM [Mg2+] (HKM20 buffer) traced to Nomura assembly map are shown. The solid and dashed arrows represent positive and negative cooperativity, respectively. Errors for ΔGlink was obtained by propagating errors from least square fittings. | ||
3.5 RsmG preferably binds to a native-like helix 18 structure
Divalent cations help RNAs fold to their native structures by stabilizing native tertiary contacts between various regions of RNA strands.28,29 The 30S 5′-domain rRNA forms all its native contacts in the presence of Mg2+, even in the absence of 5′-domain-binding r-proteins. Although in general the presence of Mg2+ ions increases the compactness of the 5′-domain rRNA, different regions of the 5′-domain require dissimilar Mg2+ concentrations to form native RNA–RNA contacts.30 RsmG binding site (G527) is located adjacent to the three nucleotides on the tip of the 530 loop that forms helix 18 pseudoknot (Fig. 6b). We performed the same FRET titrations with 5D21 and RIII5D at 4 mM Mg2+ and compared the binding affinities of RsmG with those at 20 mM Mg2+ (Fig. 6a). We did not observe a significant difference in binding affinities of RsmG for the mature 5D21 construct, while the premature RIII5D demonstrated more than a 100-fold increase in Kd at 4 mM Mg2+ implying a drastic loss of its affinity towards RsmG.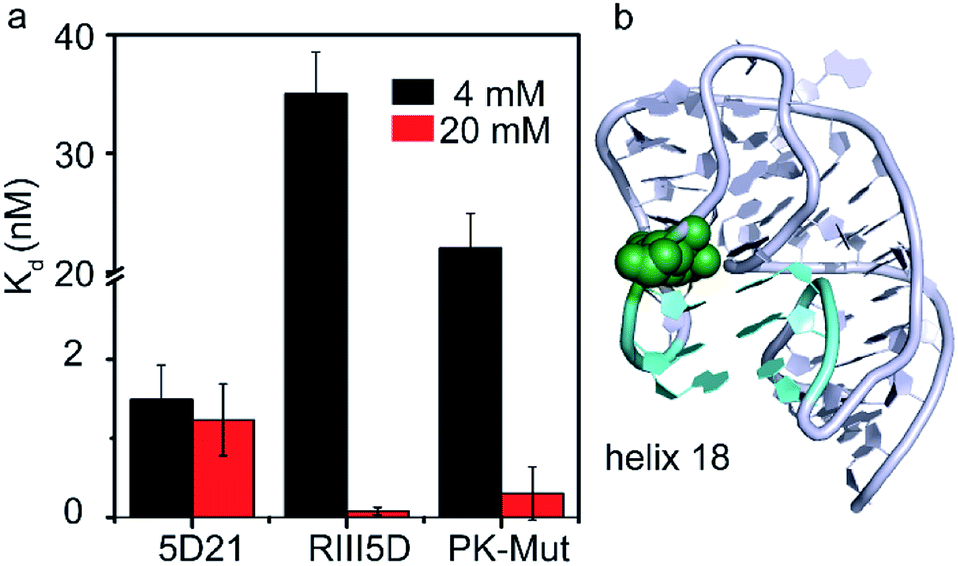 | ||
| Fig. 6 RsmG preferably binds to a compact pseudoknot structure at helix 18. (a) The comparison of Kd values of wild-type (5D21 and RIII5D) and pseudoknot mutant (PK-Mut) at 4 (HKM4 buffer) and 20 mM [Mg2+] (HKM20 buffer). PK-Mut is the helix 18 pseudoknot mutant at positions G505 and C507 (G505A and C507U respectively). (b) The crystal structure (ribbon) of helix 18 region of 16S rRNA 5′-domain, with the pseudoknot formed. Base pairs in the helix 18 pseudoknot (cyan sticks) and G527 (green spheres) are shown (PDB ID 2I2P). | ||
A clear distinction of the binding affinity of RsmG to the mature 5D21 was not observed at both 4 mM and 20 mM Mg2+ concentration. In mature 5′-domain rRNA, the ability of the tip of helix 18 to form native tertiary contacts with the lower parts of the helix even at 4 mM Mg2+ (Adilakshmi et al. 2005)30 may have caused similar affinity of RsmG to mature 5′-domain rRNA at both Mg2+ concentrations. Furthermore, this result also illustrated the preference of RsmG to 5′-domain native-like structure. To further test the nature of preferred RsmG–rRNA complex, we constructed a 5′-domain mutant that cannot form a stable pseudoknot structure. In this mutant (PK-Mut), two nucleotides out of the three (at the base of helix 18) that are base-paired in pseudoknot are mutated. Disruption of the pseudoknot decreased helix 3 motions in S4–rRNA complexes and stabilized an intermediate S4–rRNA complex.31 Binding affinities of RsmG to PK-Mut was compared to 5D21 at varying Mg2+ concentrations to investigate the preference of RsmG to native-like 5′-domain structure over an intermediate state in which 16S helix 3 is flipped away from helix 18. At 4 mM Mg2+, PK-Mut showed a 10-fold increase in Kd compared to the wild-type 5D21. The preference of flipped intermediate when pseudoknot formation is impaired especially at low Mg2+ may have caused lower stability of the RsmG–rRNA complex. All these data suggest that RsmG prefers to bind to a more compact native-like 5′-domain, in which helix 3 is stacked at the base of bent helix 18. In vitro, all native contacts of 16S 5′-domain are formed at Mg2+ concentration higher than the physiological concentration, perhaps due to the lack of crowded environment in vitro.30 Surprisingly, at 20 mM Mg2+, the Kd values are not significantly different between PK-Mut and 5D21. It is possible that pseudoknot mutations can sample native-like structure.31 However, the magnesium ion concentration that is required to fold helix 18 to a native-like structure can be relatively higher. Although the pseudoknot is not formed in the mutant rRNA, a more compact bent structure may still be sampled by helix 18 structure at high Mg2+ as also observed in twister ribozyme.32 In addition to extra contacts with the 5′-leader RNA, the inability to form helix 3 may have also contributed to the formation of stable complexes of RsmG with premature rRNAs at high Mg2+. However, non-native like helix 18 conformations at low Mg2+ may cause significantly lower binding affinities of RsmG to premature rRNA despite its additional contacts with 5′-leader RNAs.
Various ribosome assembly factors are known to bind to premature rRNA both in bacteria and in eukaryotes. Binding of ribosome assembly factors prevent misfolding of rRNA or premature binding of r-proteins. These trans-acting factors will also prevent mis-assembled or partially assembled ribosomes from participating in translation by preventing premature subunits association or blocking the decoding site and the peptide exit tunnel.33 Deficiencies in ribosome assembly and translation machinery can cause bacterial growth defects. Although RsmG deletion strains do not exhibit any growth defects at 37 °C, RsmG deletion caused mild resistance to streptomycin and resulted in mildly hyper-accurate ribosomes in B. subtilis.8,9 These observations suggest a role for RsmG to maintain the structural integrity near the 30S decoding site. The binding preference of RsmG to 5′-domain rRNA complexed with uS4 or bS16 in combination with uS4 and bS20 is also indicative of its preference to bind to native 5′-domain complex, in which the tip of the pseudoknotted helix 18 interacts with helix 3 that is stacked at the base of helix 18.23,31,34 The strong correlation between RsmG and uS12 that bind to helix 18 pseudoknot in terms of streptomycin resistance may have arisen due to such binding preference of RsmG. RsmG binding and its methyltransferase activity is potentially important to distinguish rRNA–protein complexes that have already formed helix 18 pseudoknot accurately. Unavailability of RsmG may generate a pool of sub optimally active ribosomes in which the decoding site is slightly deformed.
The tighter RsmG binding to unprocessed precursor small subunit rRNA compared to the mature 16S rRNA suggest the ability of RsmG to distinguish premature RNA from its mature counterpart. Binding anti-cooperativity between the RsmG and r-protein to premature rRNA suggests that RsmG binding to its active site may be unfavorable and thus delayed until the 5′-leader is processed. On the other hand, binding cooperativities between RsmG and r-proteins implies the ability of RsmG to influence the loading of r-proteins to rRNA by generating low free energy intermediates for r-proteins to bind to. Changes in the binding cooperativity between RsmG and r-proteins, depending on the maturation status of small subunit rRNA, illustrate the capability of RsmG to direct assembly flux through various parallel pathways suggested in Mulder et al. 2010 (ref. 35) and Talkington et al. 2005,36 depending on the processing status of the 5′-leader. Furthermore, RsmG may be involved in regulating co-transcriptional assembly of 30S ribosomes that is found to be deviating from the thermodynamically preferred pathways in the presence of 5′-leader (Rodgers and Woodson 2019).37
4 Conclusions
In conclusion, our experiments illustrate that RsmG prefers to bind to a native-like 5′-domain rRNA (Fig. 7). In addition, the thermodynamic preference of RsmG binding is changed as the 30S assembly progresses, depending on the rRNA maturation status. We propose that RsmG binding can uniquely change the energy landscape of 30S assembly based on the level of 16S rRNA maturation.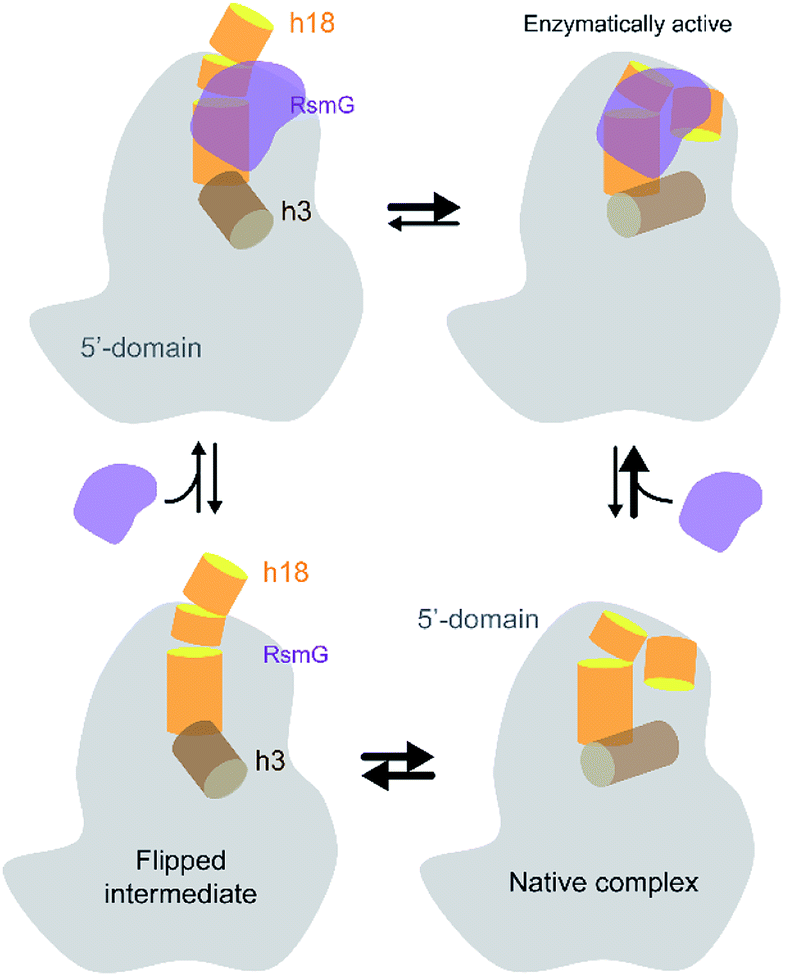 | ||
| Fig. 7 The current working model for RsmG binding is shown. RsmG-bound to flipped intermediate and native-like complexes are in equilibrium with each other. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank NSF-REU (CHE-1659571) for Caitlin Hawkin's summer research support and Prof. Gloria Culver for providing over-expression vectors for ribosomal proteins.Notes and references
- W. A. Held, B. Ballou, S. Mizushima and M. Nomura, J. Biol. Chem., 1974, 249, 3103–3111 CAS.
- S. Mizushima and M. Nomura, Nature, 1970, 226, 1214–2018 CrossRef CAS.
- K. Inoue, S. Basu and M. Inouye, J. Bacteriol., 2007, 189, 8510–8518 CrossRef CAS PubMed.
- Z. Xu, H. C. O'Farrell, J. P. Rife and G. M. Culver, Nat. Struct. Mol. Biol., 2008, 15, 534–536 CrossRef CAS PubMed.
- D. L. Lafontaine, T. Preiss and D. Tollervey, Mol. Cell. Biol., 1998, 18, 2360–2370 CrossRef CAS PubMed.
- B. Roy-Chaudhuri, N. Kirthi, T. Kelley and G. M. Culver, Mol. Microbiol., 2008, 68, 1547–1559 CrossRef CAS PubMed.
- Q. Guo, Y. Yuan, Y. Xu, B. Feng, L. Liu, K. Chen, M. Sun, Z. Yang, J. Lei and N. Gao, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13100–13105 CrossRef CAS PubMed.
- K. Nishimura, S. K. Johansen, T. Inaoka, T. Hosaka, S. Tokuyama, Y. Tahara, S. Okamoto, F. Kawamura, S. Douthwaite and K. J. Ochi, Bacteriol, 2007, 189, 6068–6073 CrossRef CAS PubMed.
- S. Okamoto, A. Tamaru, C. Nakajima, K. Nishimura, Y. Tanaka, S. Tokuyama, Y. Suzuki and K. Ochi, Mol. Microbiol., 2007, 63, 1096–1106 CrossRef CAS PubMed.
- B. Springer, Y. G. Kidan, T. Prammananan, K. Ellrott, E. C. Böttger and P. Sander, Antimicrob. Agents Chemother., 2001, 45, 2877–2884 CrossRef CAS PubMed.
- A. Benítez-Páez, S. Cárdenas-Brito, M. Corredor, M. Villarroya and M. E. Armengod, Biomedica, 2014, 34, 41–49 CrossRef.
- H. Demirci, F. V. Murphy, E. L. Murphy, J. L. Connetti, A. E. Dahlberg, G. Jogl and S. T. Gregory, Antimicrob. Agents Chemother., 2014, 58, 4308–4317 CrossRef PubMed.
- K. Nishimura, T. Hosaka, S. Tokuyama and K. Ochi, J. Bacteriol., 2006, 189, 3876–3883 CrossRef PubMed.
- M. Finken, P. Kirschner, A. Meier and E. C. Böttger, Mol. Microbiol., 1993, 9, 1239–1246 CrossRef CAS PubMed.
- C. Cazenave and O. C. Uhlenbeck, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6972–6976 CrossRef CAS PubMed.
- T. Powers and H. F. Noller, RNA, 1995, 1, 194–209 CAS.
- A. M. Popova and J. R. Williamson, J. Am. Chem. Soc., 2014, 136, 2058–2069 CrossRef CAS PubMed.
- T. Siibak and J. Remme, RNA, 2010, 16, 2023–2032 CrossRef CAS PubMed.
- J. Brosius, T. J. Dull, D. D. Sleeter and H. F. Noller, J. Mol. Biol., 1981, 148, 107–127 CrossRef CAS PubMed.
- R. A. Young and J. A. Steitz, Cell, 1979, 17, 225–234 CrossRef CAS.
- D. Ginsburg and J. A. Steitz, J. Biol. Chem., 1975, 250, 5647–5654 CAS.
- M. P. Deutscher, Prog. Mol. Biol. Transl. Sci., 2009, 85, 369–391 CAS.
- S. C. Abeysirigunawardena and S. A. Woodson, RNA, 2016, 21, 1859–1865 CrossRef PubMed.
- M. Romanowski, J. Bonanno and S. Burley, Proteins: Struct., Funct., Genet., 2002, 47, 563–567 CrossRef CAS PubMed.
- S. T. Gregory, H. Demirci, R. Belardinelli, T. Monshupanee, C. Gualerzi, A. E. Dahlberg and G. Jogl, RNA, 2009, 15, 1693–1704 CrossRef CAS PubMed.
- J. Wrzesinski, A. Bakin, K. Nurse, B. G. Lane and J. Ofengand, Biochemistry, 1995, 34, 8904–8913 CrossRef CAS PubMed.
- V. Ramakrishnan, Science, 1986, 231, 1562–1564 CrossRef CAS PubMed.
- D. Leipply and D. E. Draper, Biochemistry, 2010, 49, 1843–1853 CrossRef CAS PubMed.
- A. M. Soto, V. Misra and D. E. Draper, Biochemistry, 2007, 46, 2973–2983 CrossRef CAS PubMed.
- T. Adilakshmi, P. Ramaswamy and S. A. Woodson, J. Mol. Biol., 2005, 351, 508–519 CrossRef CAS PubMed.
- H. Kim, S. C. Abeysirigunawarden, K. Chen, M. Mayerle, K. Ragunathan, Z. Luthey-Schulten, T. Ha and S. A. Woodson, Nature, 2014, 506, 334–338 CrossRef CAS PubMed.
- S. Panja, B. Hua, D. Zegarra, T. Ha and S. A. Woodson, Nat. Chem. Biol., 2017, 13, 1109–1114 CrossRef CAS PubMed.
- K. Karbstein, Trends Cell Biol., 2013, 23, 242–250 CrossRef CAS PubMed.
- P. Ramaswamy and S. A. Woodson, J. Mol. Biol., 2009, 392, 666–677 CrossRef CAS PubMed.
- A. M. Mulder, C. Yoshioka, A. H. Beck, A. E. Bunner, R. A. Milligan, C. S. Potter, B. Carragher and J. R. Williamson, Science, 2010, 330, 673–677 CrossRef CAS PubMed.
- M. W. Talkington, G. Siuzdak and J. R. Williamson, Nature, 2005, 438, 628–632 CrossRef CAS PubMed.
- M. L. Rodgers and S. A. Woodson, Cell, 2019, 79, 1370–1381 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02732d |
| ‡ Current address: James L. Winkle College of Pharmacy, University of Cincinnati, Cincinnati, Ohio 45267, USA. |
| This journal is © The Royal Society of Chemistry 2020 |