DOI:
10.1039/D0RA02728F
(Paper)
RSC Adv., 2020,
10, 17026-17036
A manganese(III) Schiff base complex immobilized on silica-coated magnetic nanoparticles showing enhanced electrochemical catalytic performance toward sulfide and alkene oxidation†
Received
24th March 2020
, Accepted 16th April 2020
First published on 30th April 2020
Abstract
In this study, a novel Mn(III)–Schiff base complex was synthesized and characterized. The structure of this complex was determined to be a deformed octahedral coordination sphere by single-crystal X-ray diffraction analysis. The Mn(III)–Schiff base complex was supported on silica-coated iron magnetic nanoparticles via axial coordination by one-step complex anchoring to produce a heterogenized nanocatalyst. After this, the complex was characterized by Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), transmission electron microscopy (TEM), energy-dispersive X-ray spectroscopy (EDX), thermogravimetric analysis (TGA), vibrating sample magnetometry (VSM), and powder X-ray diffraction (XRD). Moreover, atomic absorption spectroscopy was used to determine the amount of the loaded metal. The heterogenized nanocatalyst effectively catalyzed the oxidation of a broad range of sulfides and alkenes with H2O2 in the presence of a glassy carbon electrode, applying voltage to the reaction mixture. The results showed that the application of a potential to the reaction mixture could significantly decrease the reaction time when compared with the case of similar chemical oxidation reactions. In addition, an excellent value of turnover frequency (17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750 h−1) was achieved for the electrochemical oxidation of styrene. Moreover, the nanocatalyst showed good recoverability without significant loss of its activity within six successive runs in the electrochemical oxidation of methyl phenyl sulfide and cyclooctene. The electrochemical properties and stability of Fe3O4@SiO2-[MnL(OAc)] were investigated by cyclic voltammetry measurements and chronoamperometry technique.
750 h−1) was achieved for the electrochemical oxidation of styrene. Moreover, the nanocatalyst showed good recoverability without significant loss of its activity within six successive runs in the electrochemical oxidation of methyl phenyl sulfide and cyclooctene. The electrochemical properties and stability of Fe3O4@SiO2-[MnL(OAc)] were investigated by cyclic voltammetry measurements and chronoamperometry technique.
Introduction
Over the past few years, several studies have been reported on the applications of Schiff-base ligands and their transition metal complexes in homogeneous and heterogeneous catalysis.1–5 Schiff base complexes show excellent catalytic activity in various reactions. Since manganese complexes are highly active in oxidation processes, they have been widely studied as homogeneous catalysts for olefin epoxidation in the presence of various oxygen donors.6–9 However, they suffer from some disadvantages such as the problem of separation and recovery from the reaction mixture at the end of the process.10 Hence, designing catalytic systems of Schiff base complexes with easy recyclability is still a challenging task.
One of the widely used models of catalytic systems is the oxidation of organic substrates using Schiff base complexes immobilized on the surface of a solid support. In addition, solid supports allow easy separation and recovery of the catalyst from the reaction mixture such that the catalyst can be reused several times, which is an approach to the green chemistry.11–14
In recent decades, iron oxide magnetic nanoparticles (Fe3O4) have attracted interest due to their chemical resistance, superparamagnetic features, high level contact, cost-effectiveness, and environmental friendliness.15–18 Because of the special features of these magnetic nanoparticles, they can be used in different fields such as magnetic resonance imaging (MRI),19 sensing,20 pharmacology,21 and catalysis.22–24 Due to these particular characteristics of the iron oxide magnetic nanoparticles, they can be developed as a practical solid support having stability under reaction conditions and easy separation by an external magnet.25–29
Comprehensively, the electrocatalytic performances of transition metal complexes in the oxidation processes and electrochemical oxidation methods have become popular research topics. To date, many reports of electrocatalytis in industrially important reactions are available.30–32
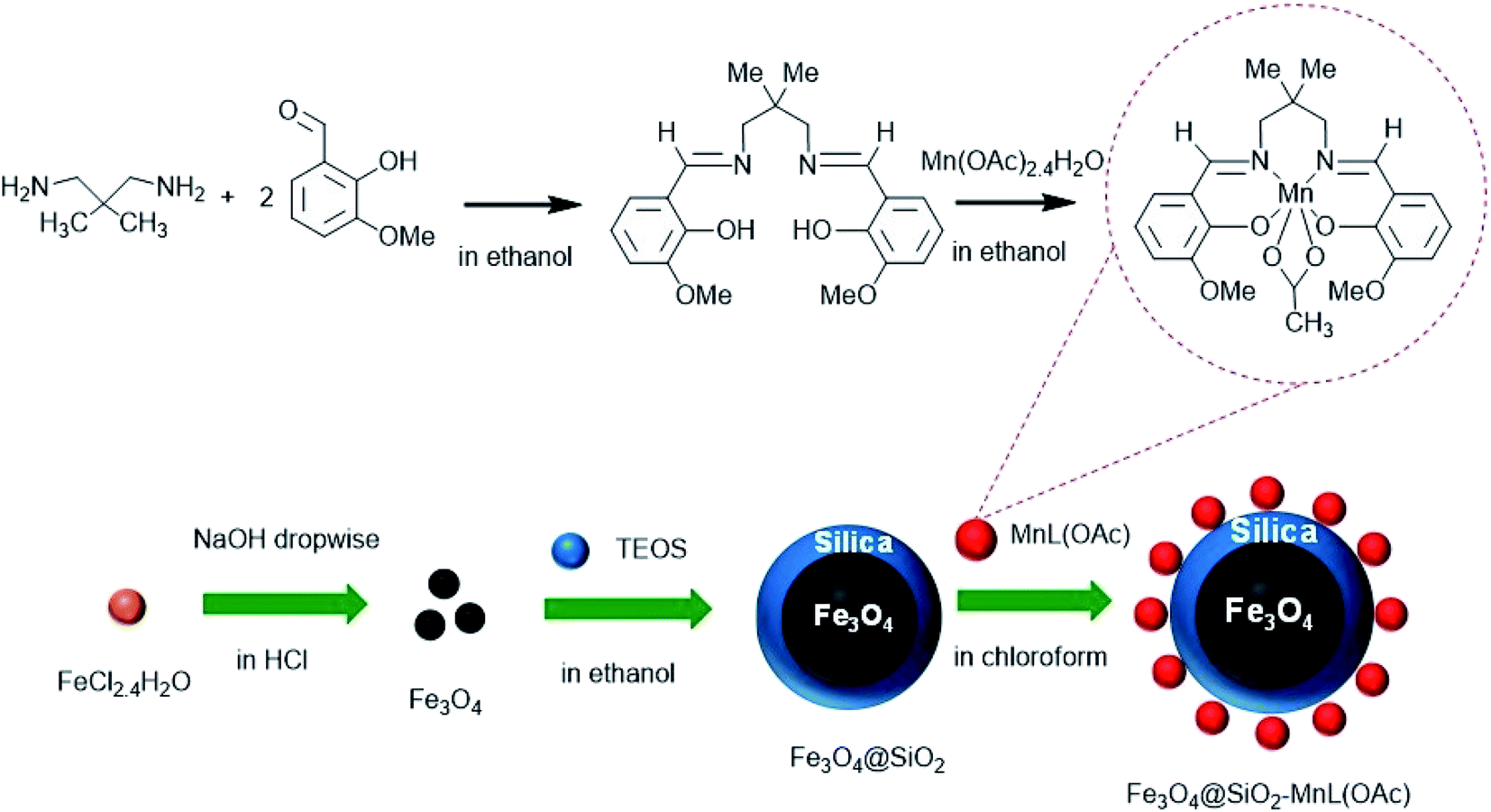
The study reported herein focused on the synthesis and immobilization of a Mn–Schiff base complex on the surface of silica-coated Fe3O4 magnetic nanoparticles (MNPs) to obtain a heterogenized nanocatalyst. Furthermore, in our pervious investigation, the catalytic activity of another Mn–Schiff base complex supported on silica-coated magnetic nanoparticles has been investigated in the chemical oxidation of sulfides and alkenes with hydrogen peroxide.32 In this study, the catalytic activity of the prepared heterogenized nanocatalyst was examined in the electrochemical oxidation of sulfides and alkenes in the presence of a glassy carbon electrode as a new modified procedure. Moreover, the reusability and the electrochemical properties of the heterogenized nanocatalyst were tested.
Experimental
Chemicals
The chemicals, reagents, and solvents were purchased from Aldrich, Merck or Fluka chemical companies. 2,2-Dimethylpropylenediamine, 2-hydroxy-3-methoxybenzaldehyde, manganese(II) acetate tetrahydrate, iron(II) chloride tetrahydrate, hydrogen peroxide (30% solution in water), imidazole, and all the organic substrates, including sulfides and alkenes, were used as received from commercial suppliers without further purification.
Instruments
The UV-Vis absorption spectra were acquired using the Unico 4802 double beam spectrometer. 1H NMR and 13C NMR analyses were carried out by the 300 MHz BRUKER spectrometer in DMSO. Infrared spectra were obtained using KBr pellets by Unicam Matson 1000 FTIR in the 400–4000 cm−1 range. Scanning electron microscopy (SEM) images and energy dispersive X-ray (EDX) spectra were obtained by the VEGA3 TESCAN field emission scanning electron microscope using an HV voltage of 20 kV. Transmission electron microscopy (TEM) images were obtained via the Zeiss EM-1030 transmission electron microscopy device using a voltage of 100 kV. Thermogravimetric analysis (TGA/DTGA) was performed using the Mettler Toledo instrument. Magnetic properties were examined by the BHV-55 vibrating sample magnetometer (VSM). X-ray powder diffraction (XRD) was carried out using the Philips PW 3710 diffractometer with Cu Kα radiation. A Varian (AA240) atomic absorption spectrometer was used for metal determination. The Agilent 7890B instrument with an SAB-5 capillary column (phenyl methyl siloxane; 30 m × 0.32 mm × 0.25 μm) and a flame ionization detector was used to perform gas chromatography experiments (GC). The electrochemical studies of catalysts were carried out in a conventional three-electrode cell powered by an electrochemical system consisting of an EG&G model 273 potentiostat/galvanostat and a Solartron model 1255 frequency response analyzer. The system was run by a PC using the M270 and M398 commercial software packages via a GPIB interface for impedance measurements. A glassy carbon (GC) disc electrode and graphite electrode were used as the working and counter electrodes, respectively. The potentials are relative to the aqueous Ag/AgCl placed in a separate compartment containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAH) in anhydrous acetonitrile.
Preparation of bis(2-hydroxy-3-methoxybenzaldehyde)-2,2-dimethylpropanediimine (H2L)
H2L was prepared according to a previously described procedure.33 An ethanolic solution (20 mL) containing 2,2-dimethylpropylenediamine (1 mmol, 0.102 g) and 2-hydroxy-3-methoxybenzaldehyde (2 mmol, 0.304 g) was refluxed for 1 h. The yellow H2L crystals were filtered from the reaction mixture, washed, and dried at 80 °C. Anal. calc. for C21H26N2O4 (MW = 370.44 g mol−1). Yield: 92%. Mp: 110 °C. Required: C, 68.21; H, 7.16; N, 7.49%. Found: C, 68.28; H, 7.07; N, 7.56%. 1H NMR (DMSO-d6, 300 MHz), δ(ppm): 14.14 (s, 2H, C–OH); 6.7–6.95 (m, 6H, aromatic C–H); 8.32 (s, 2H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH); 3.91 (s, 6H, OCH3); 3.48 (s, 4H, NCH2); 1.07 (s, 6H, CCH3) ppm. 13C NMR (DMSO-d6, 300 MHz), δ(ppm): 165.78 (CN); 113.76–152.02 (aromatic C); 67.29 (OCH3); 56.01 (CH2); 37.48 (CH2CCH3); 24.23 (CH3) ppm. IR, νmax, cm−1: ν(O–H)arom 3510; νsym(C–H)arom 2960; νsym(CN) 1628; νsym(CC) 1528; νsym(C–O) 1054 cm−1 (KBr disk). UV-Vis, λmax, nm (ε, mol−1 dm3 cm−1): 221 (2900); 270 (1600) (in a 5 × 10−4 M CHCl3 solution).
CH); 3.91 (s, 6H, OCH3); 3.48 (s, 4H, NCH2); 1.07 (s, 6H, CCH3) ppm. 13C NMR (DMSO-d6, 300 MHz), δ(ppm): 165.78 (CN); 113.76–152.02 (aromatic C); 67.29 (OCH3); 56.01 (CH2); 37.48 (CH2CCH3); 24.23 (CH3) ppm. IR, νmax, cm−1: ν(O–H)arom 3510; νsym(C–H)arom 2960; νsym(CN) 1628; νsym(CC) 1528; νsym(C–O) 1054 cm−1 (KBr disk). UV-Vis, λmax, nm (ε, mol−1 dm3 cm−1): 221 (2900); 270 (1600) (in a 5 × 10−4 M CHCl3 solution).
Preparation of the Mn(III)–Schiff base complex [MnL(OAc)]
[MnL(OAc)] was synthesized by adding Mn(II) acetate (Mn(OAc)2·4H2O, 1.2 mmol, 0.2 g) to an ethanolic solution (20 mL) of the prepared Schiff base ligand (H2L, 1 mmol, 0.37 g), and the resulting mixture was refluxed for 2 h. The dark green precipitate of [MnL(OAc)] was filtered, washed with cold ethanol, and dried at 80 °C. C23H27MnN2O6 (MW = 482.41 g mol−1). Yield: 92%. Mp: 222 °C. IR, νmax, cm−1: νsym(CN) 1617; νsym(CC) 1549; νsym(C–O) 1070 cm−1 (KBr disk). UV-Vis, λmax, nm (ε, mol−1 dm3 cm−1): 240 (3800); 280 (1400); 390 (780) (in a 5 × 10−4 M CHCl3 solution).
Crystal structure determination
Crystals were obtained from the CH2Cl2:MeOH (1:1) solution. X-ray diffraction data were acquired via the Oxford Sapphire CCD diffractometer using Mo Kα radiation, λ = 0.71073 Å, at 293(2) K by the ω–2θ method. The structure was solved by direct methods and refined by the full-matrix least-squares method on F2 using the SHELX2017 (ref. 34) program package. The analytical absorption corrections were applied (Table 1) (CrysAlisPro 1.171.38.43 software),35 and the maximum and minimum transmissions were 0.921 and 0.747, respectively. The hydrogen atoms were located from the electron density maps, and their positions were constrained in the refinement with appropriate AFIX restraints as implemented in SHELX. The [MnL(OAc)] structure has been deposited at the Cambridge Crystallographic Data Centre, and the deposition number is CCDC 1977428.†
Table 1 Crystal data and structure refinement of [MnL(OAc)]
| Empirical formula |
C23H27MnN2O6 |
| Formula weight |
482.40 |
| Temperature; K |
293(2) |
| Wavelength; Å |
0.71073 |
| Crystal system |
Monoclinic |
| Space group |
P21/c |
| Unit cell dimensions; Å, ° |
a = 9.1400(9) |
| b = 12.8784(12) |
| c = 19.5628(19) |
| α = 90 |
| β = 95.859(11) |
| γ = 90 |
| Volume; Å3 |
2290.7(4) |
| Z |
4 |
| Density (calculated); mg m−3 |
1.399 |
| Absorption coefficient; mm−1 |
0.617 |
| F(000) |
1008 |
| Crystal size; mm |
0.611 × 0.411 × 0.146 |
| Theta range for data collection |
2.240–28.656° |
| Index ranges |
−12 ≤ h ≤ 11, −16 ≤ k ≤ 16, 25 ≤ l ≤ 25 |
| Reflections collected |
15348 |
| Independent reflections |
5222 [R(int) = 0.0377] |
| Completeness to theta |
25.242°, 100.0% |
| Absorption correction |
Analytical |
| Max. and min. transmission |
0.921 and 0.747 |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
5222/0/289 |
| Goodness-of-fit on F2 |
1.055 |
| Final R indices [I > 2sigma(I)] |
R1 = 0.0418, wR2 = 0.0949 |
| R indices (all data) |
R1 = 0.0591, wR2 = 0.1020 |
| Largest diff. peak and hole; e Å−3 |
0.379 and −0.416 |
Preparation of Fe3O4@SiO2 MNPs
The Fe3O4 MNPs were synthesized according to the following procedure. In a typical procedure, FeCl2·4H2O (4 g, 20 mmol) was dissolved in HCl (60 mL, 1 M), and to this mixture, an aqueous solution of NaOH (60 mL, 3 M) was added dropwise until black precipitates appeared. The resulting precipitates were filtered, washed several times with distilled water until neutral pH was achieved, and then dried at 100 °C.
In the next step, the prepared Fe3O4 MNPs were dispersed in ethanol (50 mL) by 30 min sonication. To this suspension, 8 mL of TEOS and 24 mL of aqueous ammonia (25%) were slowly added, and the mixture was sonicated for 10 min followed by stirring for 24 h at room temperature. The dark brown precipitates of Fe3O4@SiO2 were separated by an external magnet, washed with distilled water, and dried at 100 °C.36
Preparation of Fe3O4@SiO2-[MnL(OAc)]
To prepare the heterogenized nanocatalyst, the as-prepared silica-coated Fe3O4 MNPs (Fe3O4@SiO2, 1 g) were dispersed in 50 mL chloroform by sonication. Subsequently, to this suspension, 0.6 g of [MnL(OAc)] was added, and the mixture was stirred for 12 h at room temperature. The Fe3O4@SiO2-[MnL(OAc)] precipitates were then filtered, washed with chloroform, and finally dried at 80 °C.32 A schematic of the synthesis routes of Fe3O4@SiO2-[MnL(OAc)] is shown in Scheme 1.
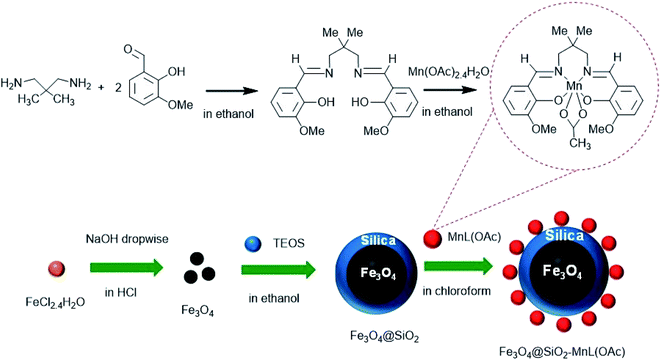 |
| | Scheme 1 Schematic of the preparation of Fe3O4@SiO2-[MnL(OAc)]. | |
The quantity of [MnL(OAc)] immobilized via axial coordination was determined by measuring the manganese content of the prepared catalyst Fe3O4@SiO2-[MnL(OAc)] by AAS. The average particle size was calculated by the Debye–Scherrer equation.
Catalytic reactions
The oxidation reactions of sulfides and alkenes were carried out in a 25 mL glass beaker containing the prepared heterogenized catalyst (0.0017 mmol), substrate (0.51 mmol of sulfide or 0.255 mmol of alkene), imidazole (0.17 mmol), and H2O2 (2.55 mmol for sulfide oxidation and 1.275 mmol for alkene oxidation) in ethanol (1 mL) in the presence of a glassy carbon electrode, applying a voltage of 60 V to the reaction mixture for 90 second. The progress of the reaction was monitored by GC.
Recycling procedure
After completion of the reaction, the catalyst was separated from the reaction mixture by an external magnet, washed 3 times with ethanol, and dried at 80 °C.
Results and discussion
Characterization of the Mn(III)–Schiff base complex [MnL(OAc)]
The results of the diffraction experiment and the structure refinement of [MnL(OAc)] are summarized in Table 1. The selected bond lengths and angles are presented in Table S1.†
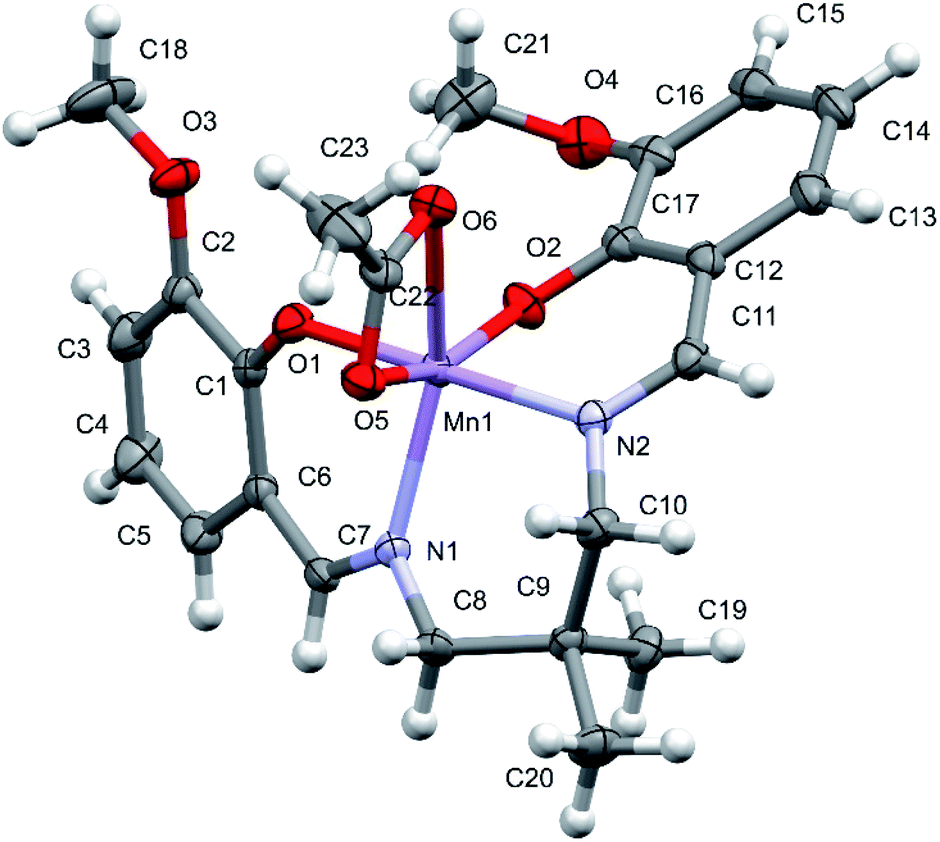
Note that one molecule of the [MnL(OAc)] complex constitutes the asymmetric unit of the reported structure (Fig. 1). The deformed octahedral coordination sphere of Mn(III) is formed by the four-dentate L ligand and the bidentate acetate ligand (OAc). The shortest bonds within the coordination sphere involve both phenolate O atoms, with the Mn1–O1 and Mn1–O2 distances of 1.8630(14) and 1.9251(15) Å, respectively, and the significantly longer Mn1–O5 [2.1188(16) Å] is formed by the acetate ligand. The Mn1–N2 and Mn1–N1 bond distances are 1.9925(16) and 2.1289(17) Å, respectively, reflecting a smaller electrostatic component when compared with the case of the Mn–O (phenolate) bonds. Moreover, the Mn1–N1 bond is significantly longer than the Mn1–N2 bond, and this seems to reflect the internal rigidity of the L ligand with two imine moieties. This was further supported by the values of −15.2(3)° and −16.6(3)° for the torsion angles C1–C6–C7–N1 and C17–C12–C11–N2, respectively, reflecting the internal strain caused by the four-dentate coordination of L. The Mn1–O6 bond involves the second O atom of acetate and is the longest bond (2.2870(16) Å) in the coordination sphere. This is due to the positioning of the O5 atom in the corner of the octahedron and rigid valence geometry of the carboxylate group of acetate. Since the four-dentate coordination of L makes the pairs of the O and N atoms of L occupy the cis positions in the coordination sphere, the significant angular deformations of the octahedral geometry are related to the position of the acetate ligand. The largest angular deformation from the ideal octahedral geometry arises from the bidentate coordination of acetate ion, resulting in the O5–Mn1–O6 angle of 59.37(6)°. In addition, the trans angles involving the acetate O atoms are significantly deviated from the expected angle of 180°, with the O2–Mn1–O5 angle of 159.56(6)° and N1–Mn1–O6 angle of 150.84(6)°. Contrarily, the O1–Mn1–N2 angle of 178.81(7)° is close to the theoretical value. The other cis angles within the coordination sphere range from 87.67(7)° (O2–Mn1–N2) to 108.61(7)° (O2–Mn1–N1).
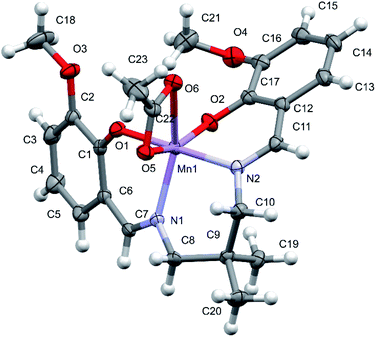 |
| | Fig. 1 Crystal structure of the manganese(III) Schiff base complex [MnL(OAc)]. | |
The described deformation of the Mn coordination sphere strongly affects the conformation of the four-dentate L ligand and the formed chelate rings. The dihedral angle between the two phenyl rings of L is 56.36(11)°. The consecutive torsion angles describing the conformation of the central bridge moiety C6–C7–N1–C8–C9–C10–N2–C11–C12 are 172.5, 136.9, 61.9, −72.0, and −115.9°. The two O–C–C–C–N–Mn1 chelate rings have an envelope conformation, whereas the Mn1–N1–C8–C9–C10–N2 ring has a chair conformation. In the detected architecture of the reported complex, the dihedral angles between the acetate plane and the O1 and O2 phenolate rings are 62.5 and 69.0°, respectively. The hydrogen bond geometry (Å and °) for [MnL(OAc)] is presented in Table S2.†
Characterization of the heterogeneous nanocatalyst (Fe3O4@SiO2-[MnL(OAc)])
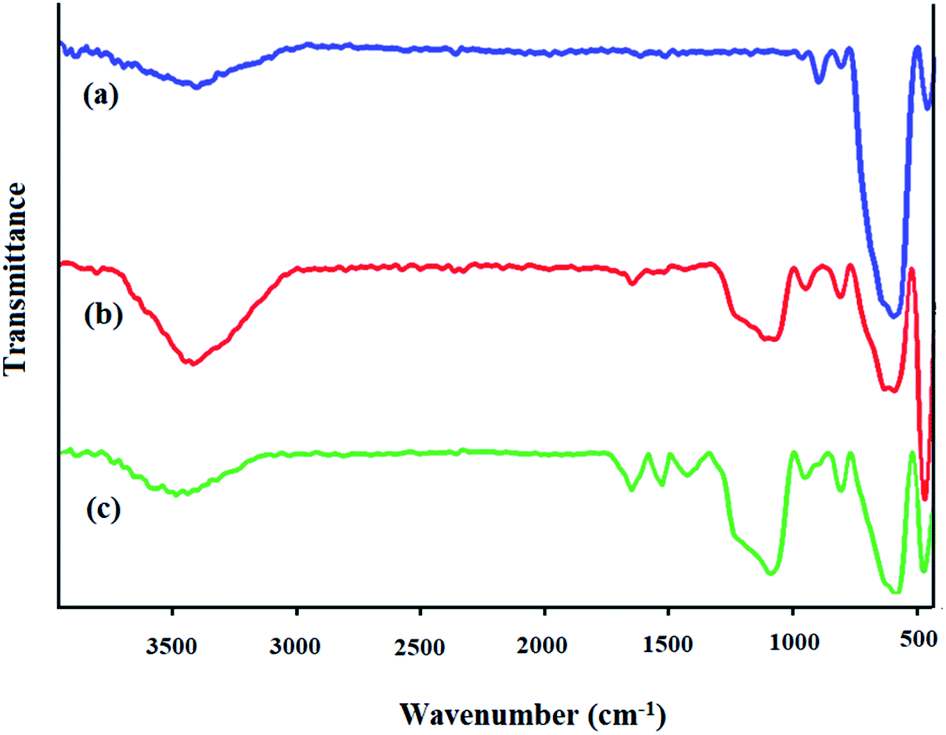
FTIR spectra. The FTIR spectra of Fe3O4, Fe3O4@SiO2, and the prepared Fe3O4@SiO2-[MnL(OAc)] heterogenized nanocatalyst are shown in Fig. 2. In the FTIR spectra of Fe3O4 (Fig. 2a), the characteristic peak at around 3400 cm−1 is related to the stretching vibration of OH. In addition, the absorption band at 580 cm−1 is attributed to the Fe–O bond in the Fe3O4 magnetic nanoparticles. In the FTIR spectrum of Fe3O4@SiO2 (Fig. 2b), the characteristic band at 575 cm−1 is attributed to the Fe–O stretching vibration. The strong absorption band at around 1000–1100 cm−1 can be assigned to the vibration modes of Si–O–Si and prove that the Fe3O4 nanoparticles were successfully coated with silica layers. The band at 3400 cm−1 is associated with the vibrations of hydrogen bonding related to the water molecules and the hydroxyl group.37,38 In the FTIR spectrum of Fe3O4@SiO2-[MnL(OAc)] (Fig. 2c), new bands were observed at 1600 and 1650 cm−1 for CN, which could be due to the coordination of the N atom of the azomethine group to the Mn metal; on the other hand, the band at 1500 cm−1 can be assigned to CC stretching in the L ligand in Fe3O4@SiO2-[MnL(OAc)]. The band at 480 cm−1 could be assigned to the Mn–O bond formed by the axial ligation of the hydroxyl group with the Mn–salen complex.39
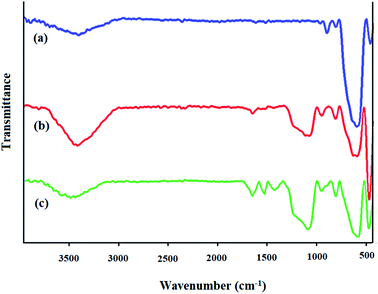 |
| | Fig. 2 FTIR spectra of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Fe3O4@SiO2-[MnL(OAc)]. | |
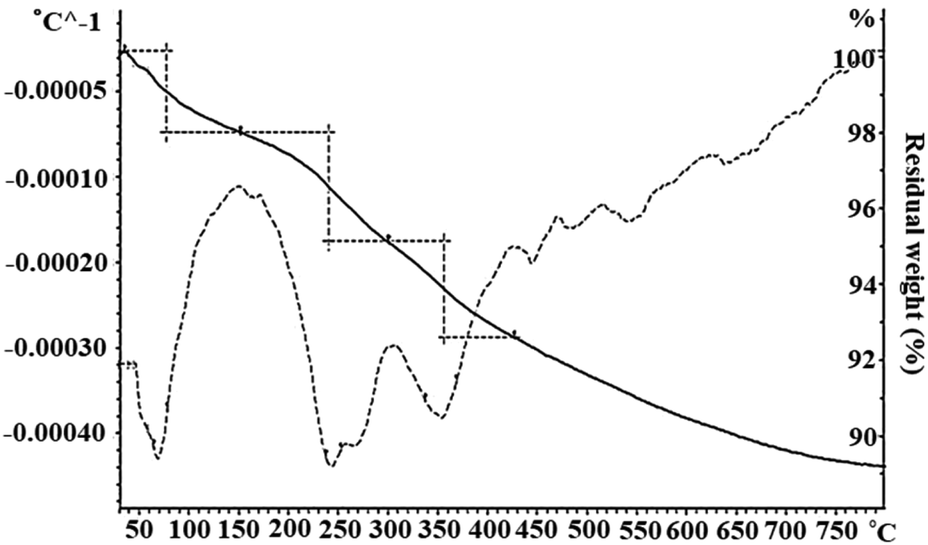
TGA/DTGA analysis. The thermal stability of the Schiff base complex immobilized on Fe3O4@SiO2 via axial coordination was analyzed by TGA/DTGA thermograms. Fig. 3 shows the thermogravimetric analysis curves, depicting the variations in the residual mass of the sample with the increasing temperature. The first stage of weight loss below 100 °C corresponds to the removal of physically and chemically adsorbed water. The absolute mass loss in the range of 240–360 °C can be associated with the release of the Mn(III)–Schiff base complex from the surface of Fe3O4@SiO2 and the decomposition of the Mn(III)–Schiff base complex. Based on these results, the grafting of the Schiff base complex on the surface of Fe3O4@SiO2 was verified.26
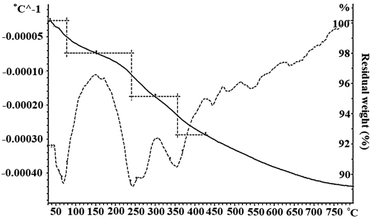 |
| | Fig. 3 The TGA/DTGA thermogram of Fe3O4@SiO2-[MnL(OAc)]. | |
SEM and TEM images. The SEM image of Fe3O4 illustrated that the nanoparticles have an approximately smooth spherical morphology (Fig. 4a). After coating the Fe3O4 MNPs with the silica shell (Fig. 4b) and immobilizing [MnL(OAc)] via axial coordination (Fig. 4c), the morphology of the obtained powder was preserved.
 |
| | Fig. 4 SEM images of (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Fe3O4@SiO2-[MnL(OAc)]. | |
Moreover, Fig. 5 displays the TEM image of Fe3O4@SiO2-[MnL(OAc)] with clear evidence of the spherical shape and thus confirms the well-defined core–shell structure with the shell thickness of ∼30.4 nm.
 |
| | Fig. 5 TEM image of Fe3O4@SiO2-[MnL(OAc)]. | |
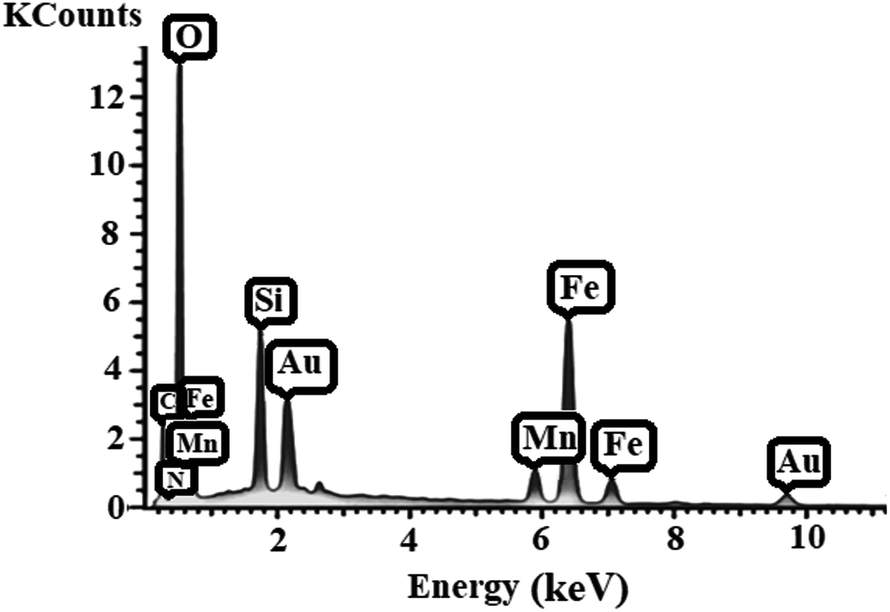
EDX analysis. Energy-dispersive X-ray spectroscopy (EDX) was conducted to obtain the local information of the different elements of [MnL(OAc)] on the surface of Fe3O4@SiO2 (Fig. 6), and the results confirmed the tertiary character of Fe3O4@SiO2-[MnL(OAc)].
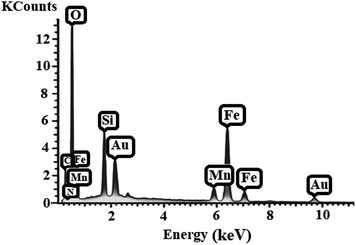 |
| | Fig. 6 The energy-dispersive X-ray spectroscopy (EDX) spectrum of Fe3O4@SiO2-[MnL(OAc)]. | |
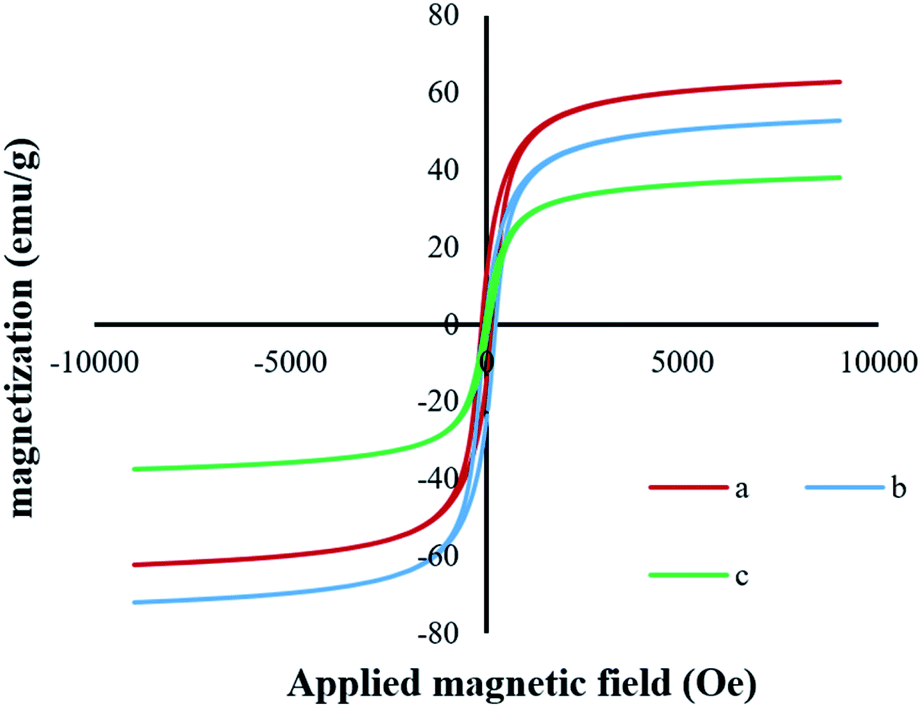
VSM analysis. The magnetic hysteresis loops (Fig. 7a–c) were measured using a vibrating sample magnetometer at room temperature, and the S-Shaped hysteresis loop, typical of magnetite, indicated ferromagnetic behavior.40 The plot of magnetization versus magnetic field (M–H loop) provided the magnetization saturation values of 62.21, 49.67, and 30.63 emu g−1 for Fe3O4, Fe3O4@SiO2, and Fe3O4@SiO2-[MnL(OAc)], respectively (Fig. 7c). The observed decrease in the saturation magnetization of Fe3O4@SiO2 as compared to that of the pure Fe3O4 MNPs could be due to the interaction between the iron core and the silica shell, which decreased the magnetic moment.41 Moreover, the saturation magnetization of silica-coated Fe3O4 significantly decreased with the attachment of the Mn–Schiff base complex via axial coordination. However, in the case of both the core–shell magnetic nanoparticles and the heterogenized nanocatalyst, these values were still sufficiently large, facilitating the easy separation of nanoparticles by an external magnet.
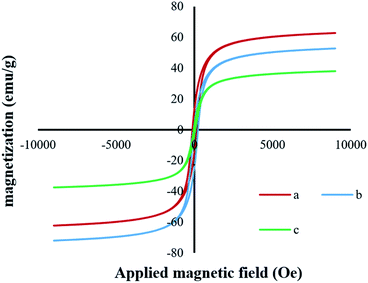 |
| | Fig. 7 Magnetization curves at 300 K for (a) Fe3O4, (b) Fe3O4@SiO2, and (c) Fe3O4@SiO2-[MnL(OAc)]. | |
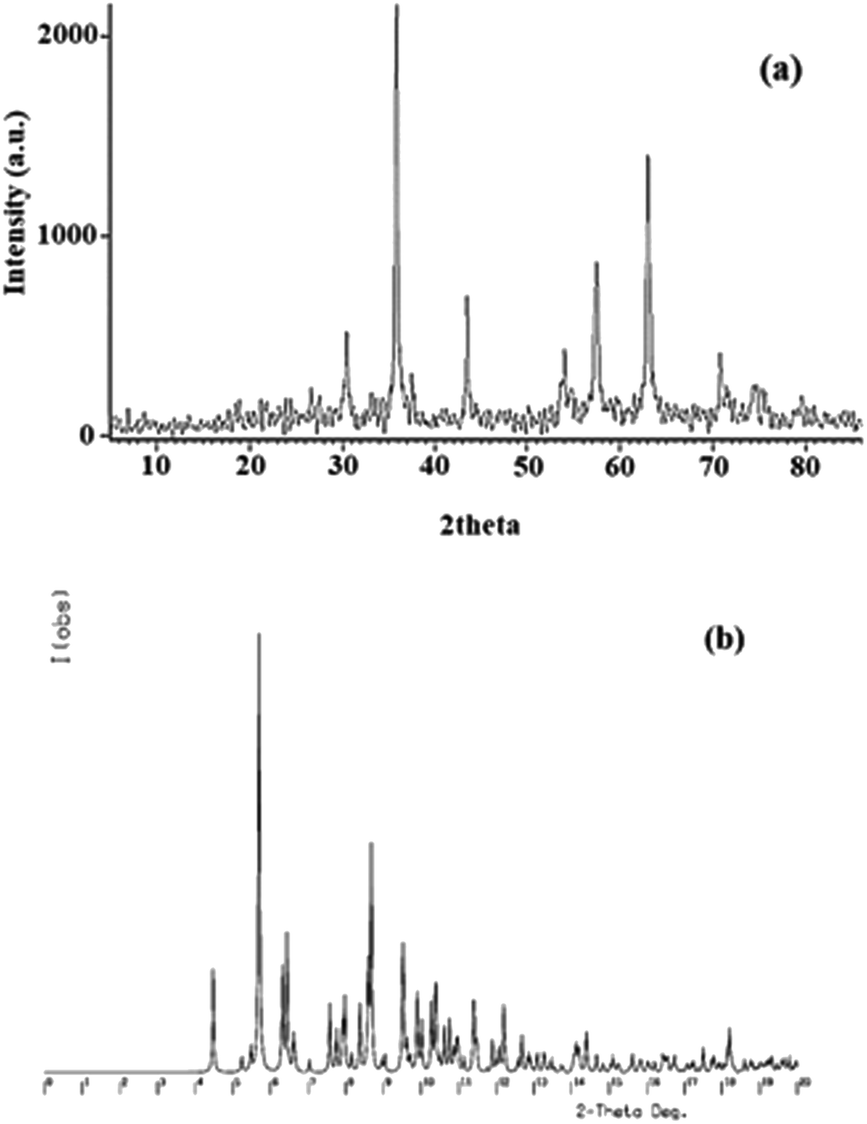
X-ray diffraction. The X-ray diffraction patterns of Fe3O4@SiO2-[MnL(OAc)] in the 2theta range of 5–90° are displayed in Fig. 8a. The diffraction peaks at 2theta = 31°, 36°, 44°, 54°, 58°, 63°, 71°, and 75° corresponded to (2 2 0), (3 1 1), (4 0 0), (4 2 2), (5 1 1), (4 4 0), (6 2 0), and (5 3 3) planes. The pattern is in accordance with the standard database of cubic inverse spinel Fe3O4 in the JCPDS file (JCPDS card no. 19-629); this reveals that during the preparation of the heterogenized magnetic nanocatalyst, the structure of Fe3O4 MNPs remains unchanged.42 Based on the Scherrer equation, the average particle size of Fe3O4@SiO2-[MnL(OAc)] was 54.5 nm. The powder diffraction pattern of [MnL(OAc)] (Fig. 8b) was simulated using Cu Kα radiation to ensure the compatibility of the simulated pattern with the experimental XRD pattern obtained for the nanocatalyst. A comparison of these patterns confirms the dispersion of the catalyst on the Fe3O4@SiO2 support and lack of the [MnL(OAc)] substructure corresponding to the reported monocrystalline state.
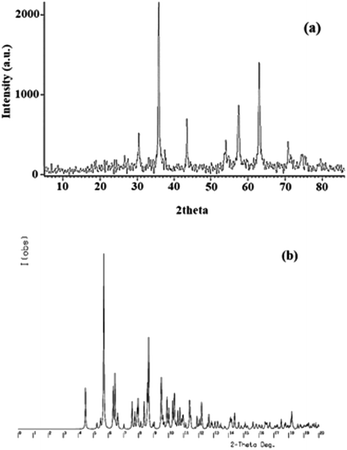 |
| | Fig. 8 (a) The powder XRD patterns of Fe3O4@SiO2-[MnL(OAc)]. (b) The simulated powder diffraction pattern based on the crystal structure of the [MnL(OAc)] complex. | |
Atomic absorption spectroscopy (AAS). The metal content of the heterogenized catalyst was determined by the flame atomic absorption spectroscopy (FAAS), which revealed that each gram of the nanohybrid catalyst contained 540 μmol of Mn–Schiff base.
Electrochemical catalytic studies
Electrochemical oxidation of methyl phenyl sulfide and cyclooctene by the synthesized nanocatalyst. In the presence of an external voltage, the electrochemical oxidations of methyl phenyl sulfide and cyclooctene were investigated. Imidazole was used as a co-catalyst of [MnL(OAc)]. In the absence of the catalyst or in the presence of Fe3O4@SiO2, no significant products (methyl phenyl sulfoxide or cyclooctene oxide) were obtained after 90 s (Table 2, entries 1 and 2). In the presence of unsupported [MnL(OAc)], methyl phenyl sulfoxide and cyclooctene oxide were obtained in a significant yield (Table 2, entry 3). Generally, in the case of methyl phenyl sulfide oxidation, two products, i.e. methyl phenyl sulfoxide and sulfone, are obtained.43 However, in the present reaction, methyl phenyl sulfoxide was obtained as the sole product. Moreover, the Schiff base complex still had good activity after immobilization on the Fe3O4@SiO2 MNPs (Table 2, entry 4); thus, the supported catalyst was preferred over the unsupported catalyst due to its recyclability. Note that in the absence of voltage (potential = 0 V), methyl phenyl sulfide and cyclooctene were not electrochemically oxidized to the desired products by Fe3O4@SiO2-[MnL(OAc)] even after 100 s.
Table 2 The electrochemical catalytic oxidation of methyl phenyl sulfide and cyclooctene
| Entry |
Catalyst |
Oxidation of methyl phenyl sulfide |
Oxidation of cyclooctene |
| Conversiona (%) |
Selectivityb (%) |
Conversionc (%) |
Selectivityd (%) |
| Reaction conditions: cat. (0.0017 mmol), molar ratio of cat.:imidazole:MePhS:H2O2: is 1:100:300:1500, solvent: EtOH (1 mL), stirred at r.t., potential: 60 V, and time: 90 s. GC yield based on the starting reactant methyl phenyl sulfide. Methyl phenyl sulfoxide is the sole product. Reaction conditions: cat. (0.0017 mmol), molar ratio of cat.:imidazole:cyclooctene:H2O2 is 1:100:150:750, solvent: EtOH (1 mL), stirred at r.t., potential: 60 V, and time: 90 s. GC yield based on the starting reactant cyclooctene. Cyclooctene oxide is the sole product. |
| 1 |
No catalyst |
0 |
0 |
0 |
0 |
| 2 |
Fe3O4@SiO2 |
0 |
0 |
0 |
0 |
| 3 |
[MnL(OAc)] |
95 |
100 |
95 |
100 |
| 4 |
Fe3O4@SiO2-[MnL(OAc)] |
96 |
100 |
97 |
100 |
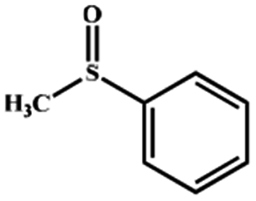
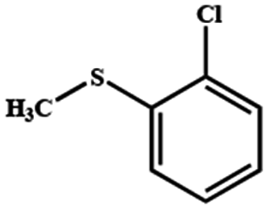
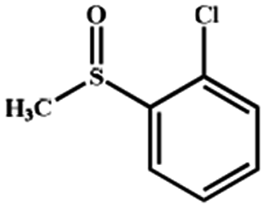
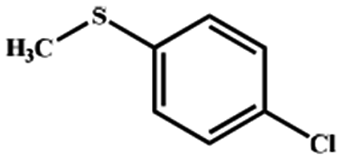
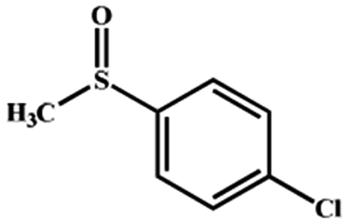
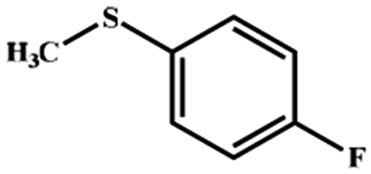
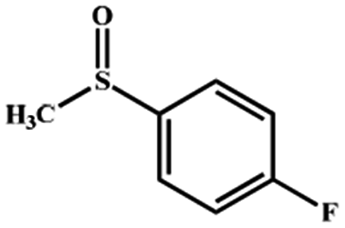
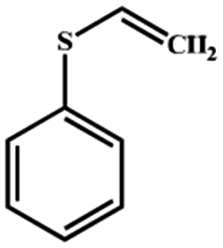
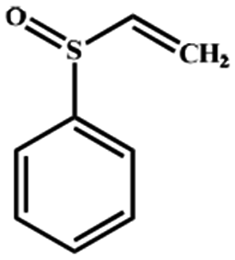
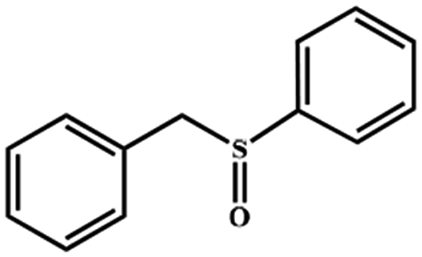
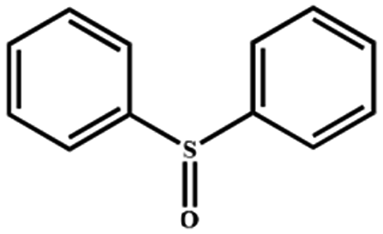
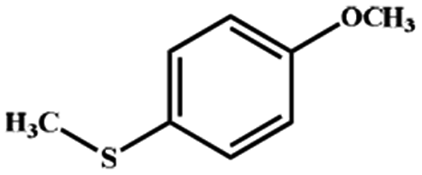
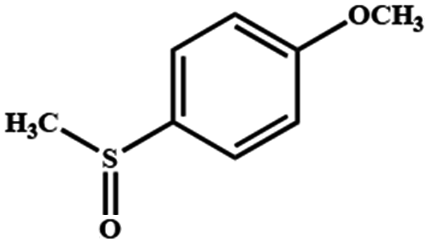
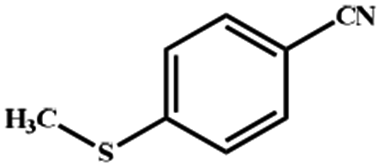
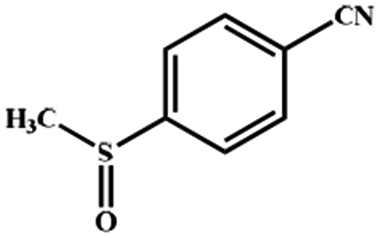
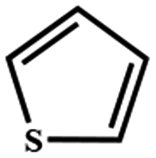
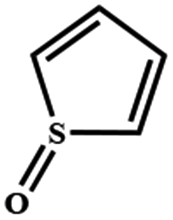
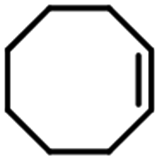
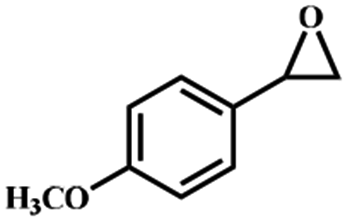
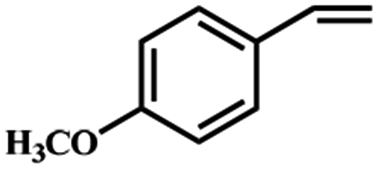
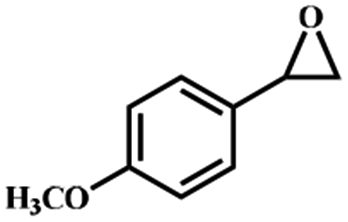
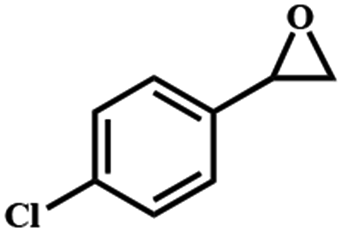
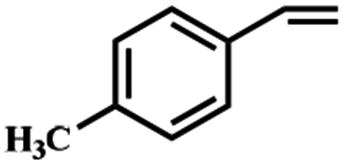
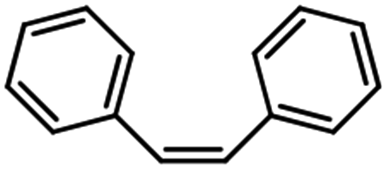
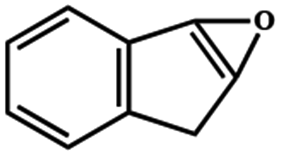
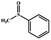
Electrochemical oxidation of different sulfides by the heterogenized nanocatalyst. In order to further explore the oxidation potential of the heterogenized nanocatalyst, a wide variety of sulfides (Table 3) were oxidized to their corresponding oxidation products in the presence of Fe3O4@SiO2-[MnL(OAc)]. Excellent conversions of sulfides and selectivities of sulfoxides were obtained in the electrochemical oxidation of sulfides (Table 3, entries 1–11). According to the results, the presence of electron-withdrawing groups, such as a vinyl group in phenyl vinyl sulfide, directly attached to the S atom in the sulfide structure can dramatically decrease the sulfoxide yield of the electrochemical oxidation reaction (a 54% conversion) (Table 3, entry 5) as compared to the case of methyl phenyl sulfide (Table 3, entry 1). In addition, thiophene had a lower oxidation yield than the linear sulfides; this may be due to the presence of the S atom in the ring that can make the oxidation of thiophene difficult (Table 3, entry 11).
Table 3 Electrochemical oxidation of various sulfides catalyzed by Fe3O4@SiO2-[MnL(OAc)]
| Entry |
Substrate |
Major product |
Conversiona (%) |
Selectivityb (%) |
| Reaction conditions: cat. (0.0017 mmol), molar ratio of cat.:imidazole:MePhS:H2O2: is 1:100:300:1500, solvent: EtOH (1 mL), room temperature, potential: 60 V, and time: 90 s. GC yield based on the starting sulfide. |
| 1 |
 |
 |
96 |
100 |
| 2 |
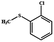 |
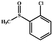 |
93 |
81 |
| 3 |
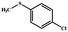 |
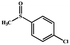 |
96 |
100 |
| 4 |
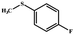 |
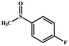 |
91 |
90 |
| 5 |
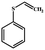 |
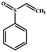 |
54 |
100 |
| 6 |
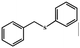 |
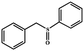 |
95 |
90 |
| 7 |
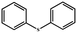 |
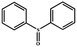 |
100 |
100 |
| 8 |
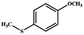 |
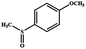 |
89 |
85 |
| 9 |
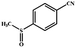
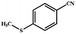 |
 |
100 |
100 |
| 10 |
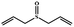
 |
 |
100 |
85 |
| 11 |
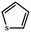 |
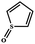 |
70 |
100 |
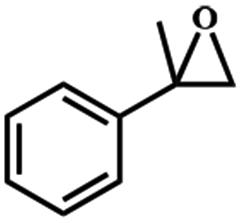
Turn over frequency. In order to estimate the longevity of the proposed heterogeneous catalytic system, the turnover frequency (TOF) was studied. The catalyst exhibited excellent activity for the oxidation of styrene in an ethanol solution under mild conditions (the molar ratio of cat.:imidazole:styrene:H2O2 was 1:13300:20000:100000) with the TOF of 17750 h−1 for Fe3O4@SiO2-[MnL(OAc)], which is one of the highest values reported to date for heterogenized Schiff base complexes.44
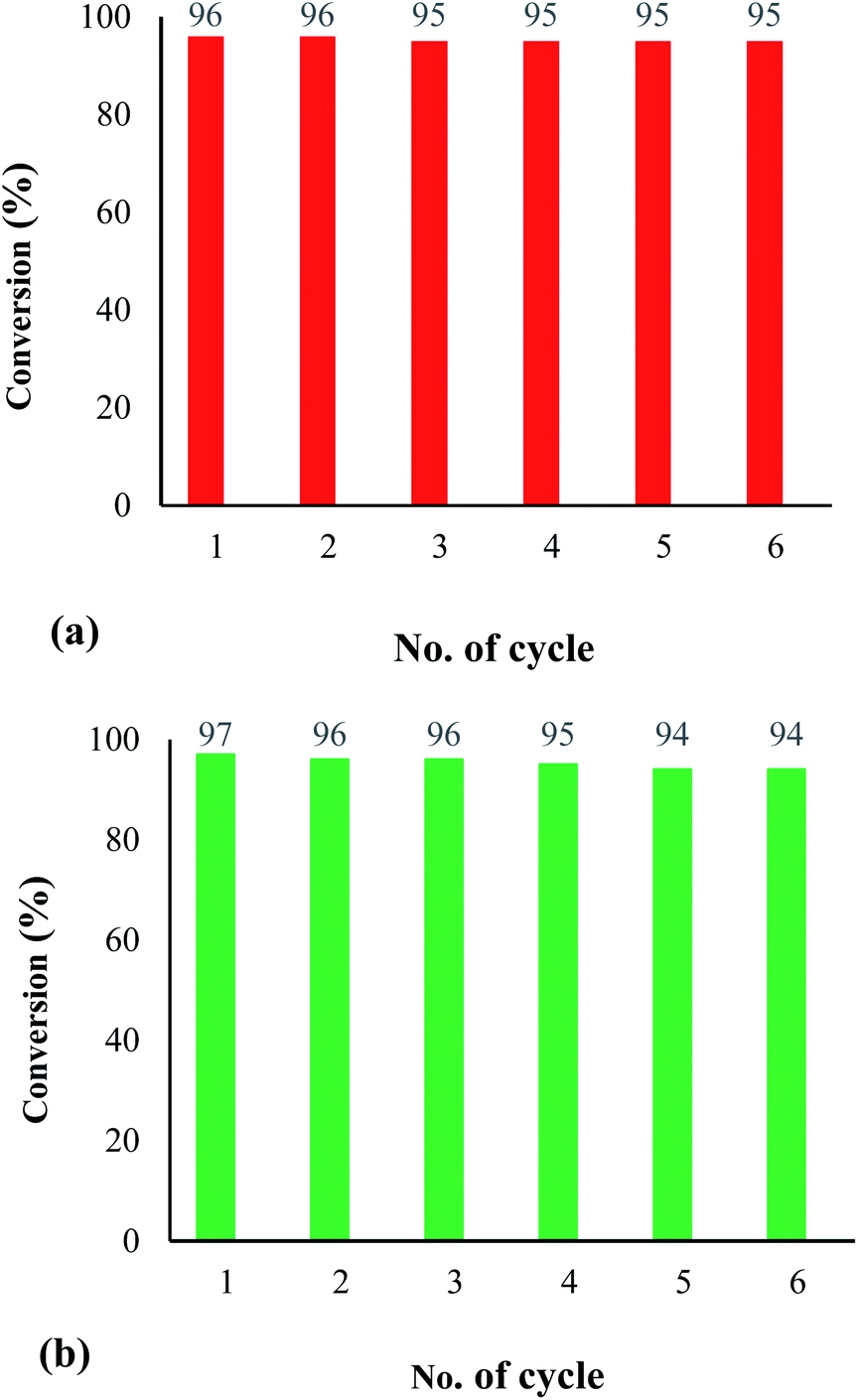
Reusability of the heterogenized nanocatalyst. The recovery and reusability of the Fe3O4@SiO2-[MnL(OAc)] heterogenized magnetic nanocatalyst were examined. For this, the catalyst was used in the electrochemical oxidation of methyl phenyl sulfide (Fig. 9a) and cyclooctene (Fig. 9b). In both catalytic reactions, the catalyst showed remarkable results with no significant loss of activity after 6 runs. These observations illustrate the advantage of the proposed heterogeneous system over a homogeneous system.
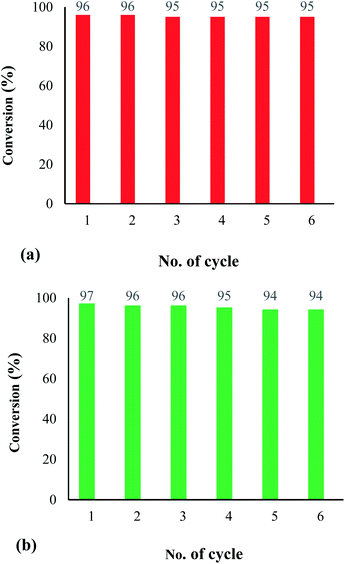 |
| | Fig. 9 Recycling of the catalytic system for the electrochemical oxidation of methyl phenyl sulfide (a) (reaction conditions: cat. (0.0017 mmol), molar ratio of cat.:imidazole:sulfide:H2O2: is 1:100:300:1500, solvent: EtOH (1 mL), room temperature, potential: 60 V, and time: 90 s) and cyclooctene (b) (reaction conditions: cat. (0.0017 mmol), molar ratio of cat.:imidazole:alkene:H2O2 is 1:100:150:750, solvent: EtOH (1 mL), room temperature, potential: 60 V, and time: 90 s). | |
Plausible mechanism. A mechanism for the electrochemical oxidation of sulfides and alkenes catalyzed by Fe3O4@SiO2-[MnL(OAc)] in the presence of a potential was proposed (see ESI, Scheme S1†). However, the precise mechanism of the abovementioned electrochemical oxidations remains unclear.
Comparison with previous studies. A comparison of the obtained results with those reported in the literature and our previous study shows a higher catalytic activity of the prepared heterogenized nanocatalyst and a significantly lower reaction time (Table 5).
Table 5 Comparison of the activities of different complexes in the oxidation reaction
| Entry |
Catalyst |
Substrate |
Potential |
Oxidant |
Time |
Conversion (%) |
Ref. |
| 1 |
MnP2-S1-Mag |
Cyclooctene |
None |
PhIO |
1 h |
78 |
45 |
| 2 |
MnP3-S2-Mag |
Cyclooctene |
None |
PhIO |
1 h |
51 |
45 |
| 3 |
Fe3O4@SiO2-Im@[MnT(4-OMeP)P] |
Cyclooctene |
None |
n-Bu4NHSO5 |
3 h |
88.5 |
46 |
| 4 |
Fe3O4@SiO2–NH2@[MnTPFPP] |
Cyclooctene |
None |
n-Bu4NHSO5 |
20 h |
97 |
47 |
| 5 |
[Fe3O4@SiO2@NH2@MnTCPP(OAc)] |
Cyclooctene |
None |
O2 |
60 min |
83 |
26 |
| 6 |
Fe3O4@SiO2-[MnL(OAc)] |
Methyl phenyl sulfide |
None |
UHP |
5 min |
100 |
32 |
| 7 |
Fe3O4@SiO2-[MnL(OAc)] |
Cyclooctene |
None |
UHP |
4 h |
100 |
32 |
| 8 |
Fe3O4@SiO2-[MnL(OAc)] |
Methyl phenyl sulfide |
60 V |
H2O2 |
90 s |
96 |
This work |
| 9 |
Fe3O4@SiO2-[MnL(OAc)] |
Cyclooctene |
60 V |
H2O2 |
90 s |
97 |
This work |
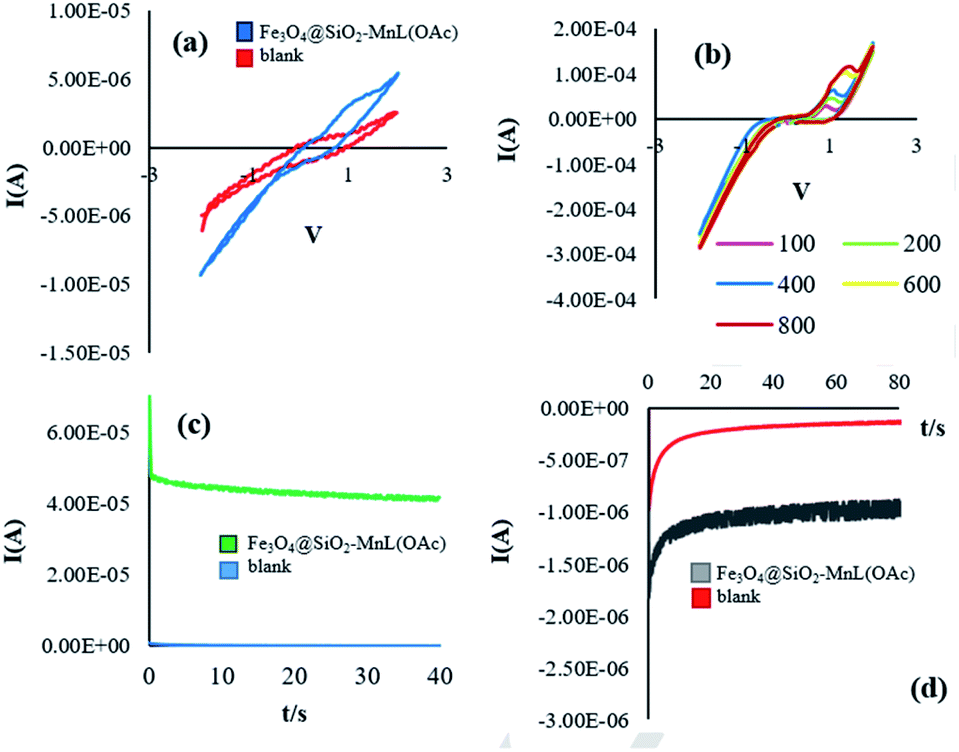
Electrochemical studies. Cyclic voltammetry measurements and chronoamperometry technique were used to investigate the electrochemical properties of the prepared Fe3O4@SiO2-[MnL(OAc)] heterogenized nanocatalyst in the oxidation of methyl phenyl sulfide (Fig. 10). CV experiments were performed for a blank reaction (with no catalyst) and a catalytic reaction (in the presence of the prepared heterogenized nanocatalyst) by cycling the potential of the glassy carbon (GC) electrode in the potential range of −3 to 3 V in ethanol at the scan rate of 100 mV s−1 (Fig. 10a). A comparison of the electrochemical oxidation reaction in the presence of Fe3O4@SiO2-[MnL(OAc)] with the blank reaction confirms that the presence of the nanocatalyst is necessary for the electrochemical oxidation of methyl phenyl sulfide. With an increase in the scan rate above 100 mV s−1, the reversible CV curve shifted towards positive potentials (Fig. 10b). These effects showed that with an increase in the scan rate, the rate of the electrochemical oxidation of methyl phenyl sulfide decreased. The chronoamperometry experiments were performed to determine the electrochemical activity and stability of Fe3O4@SiO2-[MnL(OAc)] in the electrochemical oxidation of methyl phenyl sulfide (Fig. 10c and d). The comparison of the current intensities at the oxidation or reduction potentials for blank and catalytic reactions shows a higher rate of the oxidation reaction in the presence of the heterogenized nanocatalyst.
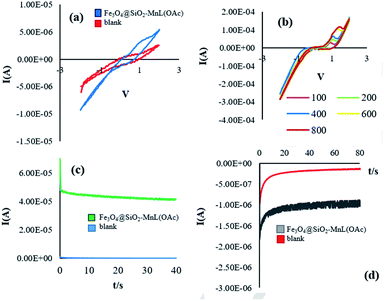 |
| | Fig. 10 The voltammetric behavior of Fe3O4@SiO2-[MnL(OAc)] and blank at 100 mV s−1 (a). Cyclic voltammograms of Fe3O4@SiO2-[MnL(OAc)] at different scan rates (100, 200, 400, 600, and 800 (mV s−1)) (b), chronoamperograms of Fe3O4@SiO2-[MnL(OAc)] and blank at the oxidation potential (c), and chronoamperograms of Fe3O4@SiO2-[MnL(OAc)] and blank at the reduction potential in EtOH for the oxidation of methyl phenyl sulfide (d). Reaction conditions: cat. (0.0017 mmol), molar ratio of cat.:imidazole:sulfide:H2O2: is 1:100:300:1500, solvent: EtOH (1 mL), room temperature, potential: 60 V, and time: 90 s. | |
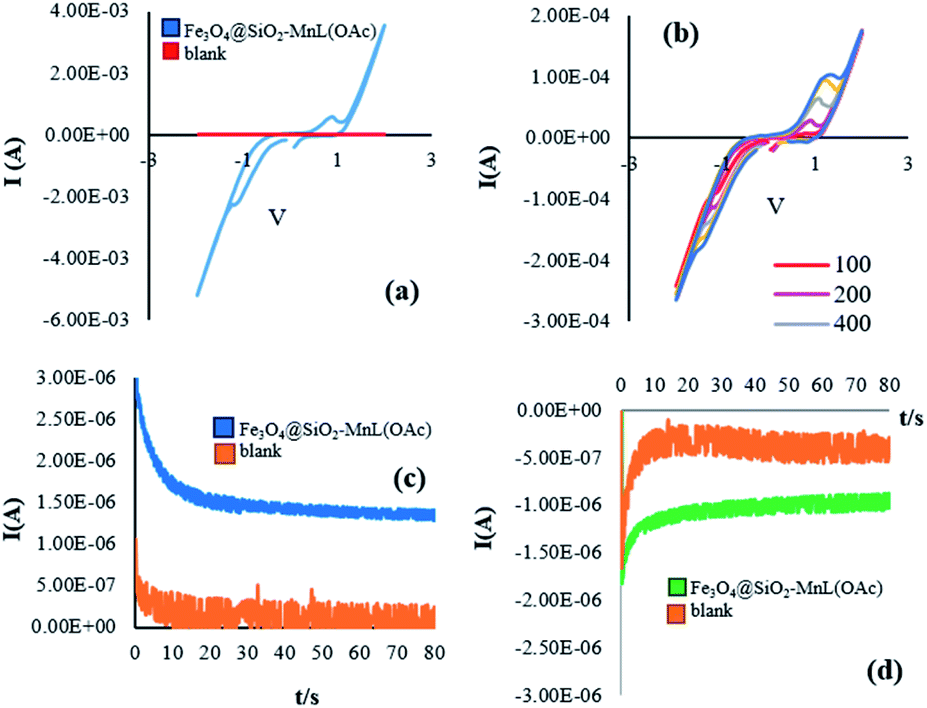
Similar results were obtained for the electrochemical oxidation of cyclooctene in the presence of Fe3O4@SiO2-[MnL(OAc)] (Fig. 11a–d).
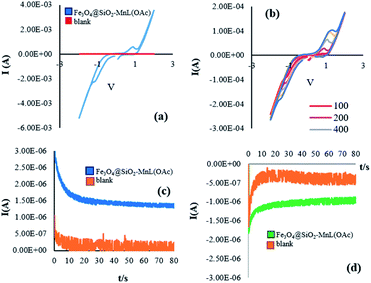 |
| | Fig. 11 The voltammetric behavior of Fe3O4@SiO2-[MnL(OAc)] and blank at 100 mV s−1 (a). Cyclic voltammograms of Fe3O4@SiO2-[MnL(OAc)] at different scan rates (100, 200, 400, 600, and 800 (mV s−1)) (b). Chronoamperograms of Fe3O4@SiO2-[MnL(OAc)] SiO2 and blank at the oxidation potential (c) and chronoamperograms of Fe3O4@SiO2-[MnL(OAc)] and blank at the reduction potential in EtOH for the oxidation of cyclooctene (d). Reaction conditions: cat. (0.0017 mmol), molar ratio of cat.:imidazole:alkene:H2O2 is 1:100:150:750, solvent: EtOH (1 mL), room temperature, potential: 60 V, and time: 90 s. | |
Conclusions
In this study, a manganese complex of a tetradentate Schiff base ligand [MnL(OAc)] was prepared and characterized by single crystal X-ray analysis. The synthesized complex was anchored on the surface of silica-coated Fe3O4 MNPs via an axial coordination, as proved by standard techniques. The dispersion of [MnL(OAc)] on the silica-coated Fe3O4 MNPs was confirmed by the simulation of the powder diffraction pattern of the complex. The prepared heterogenized nanocatalyst showed a notable activity in the electrochemical oxidation of sulfides and alkenes with H2O2 in the presence of a glassy carbon electrode, applying 60 V voltage, and the excellent turnover frequency of 17750 h−1 was obtained for the oxidation of styrene. The ternary Fe3O4@SiO2-[MnL(OAc)] catalytic system has at least the same catalytic efficacy as [MnL(OAc)] itself, having the great advantage of easy recovery from the reaction mixture. The heterogenized nanocatalysts were successfully reused for six consecutive times without loss of activity in the electrochemical oxidation of methyl phenyl sulfide and cyclooctene. Eventually, the significant electrochemical properties and stability of Fe3O4@SiO2-[MnL(OAc)] were confirmed by cyclic voltammetry measurements and chronoamperometry techniques.
Conflicts of interest
There are no conflicts of interest to declare”.
Acknowledgements
This work was supported by the Center for International Scientific Studies & Collaboration (CISSC), Ministry of Science Research and Technology. AW acknowledges the Nicolaus Copernicus University for partly supporting the research under the Center for Excellence in Science and the interdisciplinary BRAIN research team.
References
- Y. Wu, W. Wang, L. Liu, S. Zhu, X. Wang, E. Hu and K. Hu, ChemistryOpen, 2019, 8, 333–338 CrossRef CAS PubMed.
- H. Tavallaei, M. Jafarpour, F. Feizpour, A. Rezaeifard and A. Farrokhi, ACS Omega, 2019, 4, 3601–3610 CrossRef CAS PubMed.
- S. S. Kumbar, K. M. Hosamani, G. C. Gouripur and S. D. Joshi, R. Soc. Open Sci., 2018, 5, 172416–172435 CrossRef PubMed.
- H. Sua, S. Wua, Z. Lia, Q. Huob, J. Guana and Q. Kana, Appl. Organomet. Chem., 2015, 29, 462–467 CrossRef.
- D. S. Nesterov, O. V. Nesterova, M. F. C. G. da Silva and A. J. L. Pombeiro, Catal. Sci. Technol., 2015, 5, 1801–1812 RSC.
- Y. Tohru, I. Kiyomi, N. Takushi and M. Teruaki, Bull. Chem. Soc. Jpn., 1994, 67, 2248–2256 CrossRef.
- C. Choe, L. Yang, Z. A. Lv, W. L. Mo, Z. Q. Chen, G. X. Li and G. C. Yin, Dalton Trans., 2015, 44, 9182–9192 RSC.
- K. Jin, J. H. Maalouf, N. Lazouski, N. Corbin, D. Yang and K. Manthiram, J. Am. Chem. Soc., 2019, 141, 6413–6418 CrossRef CAS PubMed.
- J. Rai, B. Basumatary, S. Bhandary, M. Murugavel and J. Sankar, Dalton Trans., 2019, 48, 7394–7402 RSC.
- S. Xu, J. Du, H. Li and J. Tang, Ind. Eng. Chem. Res., 2017, 56, 15030–15037 CrossRef CAS.
- M. Kazemnejadi, S. A. Alavi G., Z. Rezazadeh, M. A. Nasseri, A. Allahresani and M. Esmaeilpour, Green Chem., 2019, 21, 1718–1734 RSC.
- J. Rakhtshah and F. Yaghoobi, Int. J. Biol. Macromol., 2019, 139, 904–916 CrossRef CAS PubMed.
- Y. Leng, D. Lu, C. Zhang, P. Jiang, W. Zhang and J. Wang, Chem.–Eur. J., 2016, 22, 8368–8375 CrossRef CAS PubMed.
- A. Nodzewska, A. Wadolowska and M. Watkinson, Coord. Chem. Rev., 2019, 382, 181–216 CrossRef CAS.
- L. Wu, A. M. Garcia, Q. Li and S. Sun, Chem. Rev., 2016, 116, 10473–10512 CrossRef CAS PubMed.
- H. R. E. Seedi, R. M. E. Shabasy, S. A. M. Khalifa, A. Saeed, A. Shah, R. Shah, F. J. Iftikhar, M. M. A. Daim, A. Omri, N. H. Hajrahand, J. S. M. Sabir, X. Zou, M. F. Halabi, W. Sarhann and W. Guo, RSC Adv., 2019, 9, 24539–24559 RSC.
- S. Palanisamy and Y.-M. Wang, Dalton Trans., 2019, 48, 9490–9515 RSC.
- M. Chandrappa, G. V. Shiva Reddy, R. Fazlur, B. Narasimha Murthy, P. Kumar Pullela and S. G. Kumar, Chem. Pap., 2017, 71, 2445–2453 CrossRef CAS.
- R. A. Revia and M. Zhang, Mater. Today, 2016, 19, 157–168 CrossRef CAS PubMed.
- N. Z. Knežević, I. Gadjanskiab and J.-O. Durand, J. Mater. Chem. B, 2019, 7, 9–23 RSC.
- T. Gong, Z. Dong, Y. Fu, T. Gong, L. Deng and Z. Zhang, J. Mater. Chem. B, 2019, 7, 5861–5872 RSC.
- Q. Zhang, X. Yang and J. Guan, ACS Appl. Nano Mater., 2019, 2, 4681–4697 CrossRef CAS.
- M. B. Gawande, P. S. Brancoa and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- M. Bhardwaj, H. Sharma, S. Paul and J. H. Clark, New J. Chem., 2016, 40, 4952–4961 RSC.
- M. Bagherzadeh and A. M. Manesh, RSC Adv., 2016, 6, 41551–41560 RSC.
- S. Rayati, F. Nejabat and F. Panjiali, Catal. Commun., 2019, 122, 52–57 CrossRef CAS.
- M. Sarkheil, M. Lashanizadegan and M. Ghiasi, J. Mol. Struct., 2019, 1179, 278–288 CrossRef CAS.
- M. Farahi, B. Karami, R. Keshavarz and F. Khosravian, RSC Adv., 2017, 7, 46644–46650 RSC.
- M. Esmaeilpour and J. Javidi, J. Chin. Chem. Soc., 2015, 62, 614–626 CrossRef CAS.
- X. Chia and M. Pumera, Nat. Catal., 2018, 1, 909–921 CrossRef CAS.
- S. Poudyala and S. Laursen, Catal. Sci. Technol., 2019, 9, 1048–1059 RSC.
-
(a) S. Rayati, E. Khodaei, M. Jafarian and A. Wojtczak, Polyhedron, 2017, 133, 327–335 CrossRef CAS;
(b) S. Rayati, E. Khodaei, S. Shokoohi, M. Jafarian, B. Elmi and A. Wojtczak, Inorg. Chim. Acta, 2017, 466, 520–528 CrossRef CAS.
-
(a) F. Heshmatpour, S. Rayati, M. Afghan Hajiabbas and B. Neumüller, Z. Anorg. Allg. Chem., 2011, 637, 1224–1228 CrossRef CAS;
(b) F. Heshmatpour, S. Rayati, M. Afghan Hajiabbas, P. Abdolalian and B. Neumuller, Polyhedron, 2012, 31, 443–450 CrossRef CAS.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- CrysAlisPro 1.171.38.43 software, Rigaku OD, 2015 Search PubMed.
- P. B. Shete, R. M. Patil, R. S. Ningthoujam and S. H. Pawar, New J. Chem., 2013, 37, 3784–3792 RSC.
- C. Yuan, Z. Huang and J. Chen, Catal. Lett., 2011, 141, 1484–1490 CrossRef CAS.
- L. F. V. Ferreira, I. F. Machado, A. Gama, F. Lochte, R. P. Socoteanu and R. Boscencu, J. Photochem. Photobiol., A, 2020, 15, 112152–112177 CrossRef.
-
(a) H.-A. Chu, W. Hillier, N. A. Law and G. T. Babcock, Biochim. Biophys. Acta, 2001, 1503(5), 69–82 CrossRef CAS PubMed;
(b) A. Rezaeifard, M. Jafarpour, A. Farrokhi, S. Parvin and F. Feizpour, RSC Adv., 2016, 6, 64640–64650 RSC.
- C. Dey, A. Das and M. M. Goswami, ChemistrySelect, 2019, 4, 12190–12196 CrossRef CAS.
- I. Dayana, T. Sembiring, A. P. Tetuko, K. Sembiring, N. Maulida, Z. Cahyarani, E. A. Setiadi, N. S. Asri, M. Ginting and P. Sebayang, J. Mol. Liq., 2019, 294, 111557–111563 CrossRef CAS.
- L. Jia, T. Zhou, J. Xu, F. Li, Z. Xu, B. Zhang, S. Guo, X. Shen and W. Zhang, Nanomaterials, 2017, 7, 333–345 CrossRef PubMed.
- S. Rayati, F. Nejabat and S. Zakavi, Inorg. Chem. Commun., 2014, 40, 82–86 CrossRef CAS.
- T. A. G. Duarte, A. P. Carvalho and L. M. D. R. S. Martins, Catal. Sci. Technol., 2018, 8, 2285–2288 RSC.
- G. M. Ucoski, F. S. Nunesa, G. D- Silva, Y. M. Idemori and S. Nakagaki, Appl. Catal., A, 2013, 459, 121–130 CrossRef CAS.
- M. Bagherzadeh and A. Mortazavi-Manesh, RSC Adv., 2016, 6, 41551–41560 RSC.
- M. Bagherzadeh and A. Mortazavi-Manesh, J. Coord. Chem., 2015, 68, 2347–2360 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1977428. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02728f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Elham Khodaeia,
Parinaz Nafarieha,
Majid Jafariana,
Bahareh Elmia and
Andrzej Wojtczak
*a,
Elham Khodaeia,
Parinaz Nafarieha,
Majid Jafariana,
Bahareh Elmia and
Andrzej Wojtczak