
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine/water-mediated deprotective oxidation of allylic ethers to access α,β-unsaturated ketones and aldehydes†
Yuntian Xuea,
Yaolong Yana,
Kezhi Jianga,
Weifeng Chen*a and
Lei Yang *ab
*ab
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 311121, China. E-mail: lyang@hznu.edu.cn; chenwf@hznu.edu.cn
bEngineering Research Center of High Performance Polymer and Molding Technology, Ministry of Education, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, China
First published on 14th April 2020
Abstract
The first iodine/water-mediated deprotective oxidation of allylic ethers to access α,β-unsaturated ketones and aldehydes was achieved. The reaction tolerates a wide range of functionalities. Furthermore, this protocol was found to be applicable to the oxidative transformation of allylic acetates. The proposed mechanism involves an oxygen transfer from solvent water to the carbonyl products.
Introduction
The organic functional group transformation of allylic ethers into α,β-unsaturated ketones or aldehydes is a very useful tool in natural product synthesis and pharmaceutical synthesis and has attracted increasing interest from synthetic chemists and biochemists in recent years.1–6 As a result, many useful methods, which can be summarized into two categories, have been developed to achieve this transformation. Traditionally, such transformation was achieved via a two-step method, i.e. deprotection of allylic ethers into allylic alcohols followed by oxidation in the presence of additional oxidants (Scheme 1a).1,2 However, the decreased step economy of the process reduced the attractiveness of this approach. Another attractive method, which has been less explored to date, is the direct oxidative transformation of allylic ethers to their corresponding α,β-unsaturated ketones or aldehydes (Scheme 1b).3–6 In this regard, 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ),3 oxo-ammonium salt,4 etc.5,6 could be used as the oxidants. Although significant improvements have been made in this research area, the use of toxic oxidants, the narrow substrate scope and the generation of stoichiometric amounts of organic waste restrict their further application. Moreover, most of these reported methods are not suitable for the simple allyl alkyl ethers. Thus, the development of new methods or nontoxic reagents for enabling such a functional group transformation is highly desirable. On the other hand, molecular iodine has been used extensively in various reactions as a green and environmentally benign reagent or catalyst,7 because iodine is inexpensive, readily available, and nontoxic. Inspired by our ongoing research on the synthesis of (±)-cassumunin C and (±)-latifolin using iodine-mediated aromatic propargylation as the key step,8 we herein report the first iodine-mediated direct transformation of allylic ethers to α,β-unsaturated ketones or aldehydes in a biphasic solvent system.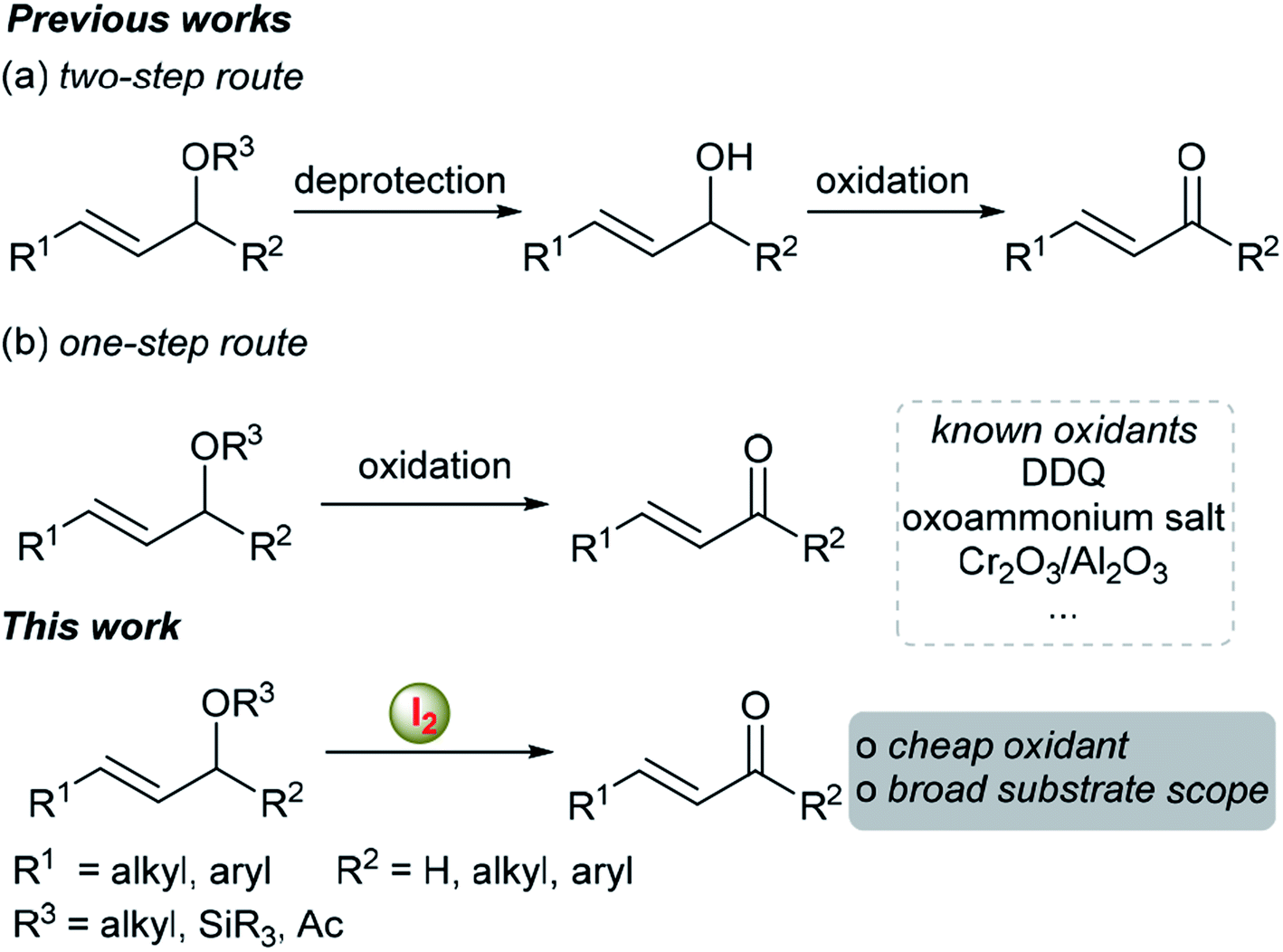 | ||
| Scheme 1 Transformation of allylic ethers into α,β-unsaturated ketones or aldehydes. | ||
Results and discussion
The studies were initiated with (E)-(3-(benzyloxy)but-1-en-1-yl)benzene 1a as a model substrate. As shown in Table 1, it was found that 1a when heated with molecular iodine (1.6 eq.) in 1,4-dioxane/water (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)9 at the refluxing temperature produced benzylideneacetone 2a in 78% yield after 2 h (entry 1). Inspired by this result, we began to tune the reaction conditions to further improve the yield of 2a. Knowing that the presence of water would be critical to the oxidative process, a variety of biphasic solvent systems was first screened (entries 2–7). A lower efficiency was observed when using other different biphasic solvents investigated, thus indicating the important role of the solvents in the reaction.
:1)9 at the refluxing temperature produced benzylideneacetone 2a in 78% yield after 2 h (entry 1). Inspired by this result, we began to tune the reaction conditions to further improve the yield of 2a. Knowing that the presence of water would be critical to the oxidative process, a variety of biphasic solvent systems was first screened (entries 2–7). A lower efficiency was observed when using other different biphasic solvents investigated, thus indicating the important role of the solvents in the reaction.
|
||||
|---|---|---|---|---|
| Entry | I2 (eq.) | Solvent | Time (h) | Yieldb (%) |
| a General conditions: 1a (0.1 mmol), I2 (1.6 eq.), solvent (3.6 mL), at refluxing temperature, under air.b Isolated yields.c Under Ar.d At room temperature. | ||||
| 1 | 1.6 | 1,4-Dioxane/H2O (5:1) |
2 | 78 |
| 2 | 1.6 | Toluene/H2O (5:1) |
2 | 18 |
| 3 | 1.6 | THF/H2O (5:1) |
2 | 40 |
| 4 | 1.6 | MeOH/H2O (5:1) |
2 | 15 |
| 5 | 1.6 | DMF/H2O (5:1) |
2 | 26 |
| 6 | 1.6 | DMSO/H2O (5:1) |
2 | 51 |
| 7 | 1.6 | DCM/H2O (5:1) |
2 | 12 |
| 8c | 1.6 | 1,4-Dioxane/H2O (5:1) |
2 | 63 |
| 9d | 1.6 | 1,4-Dioxane/H2O (5:1) |
2 | 0 |
| 10 | 1.6 | 1,4-Dioxane | 2 | 0 |
| 11 | 1.6 | H2O | 2 | 35 |
| 12 | 0 | 1,4-Dioxane/H2O (5:1) |
2 | 0 |
| 13 | 1.6 | 1,4-Dioxane/H2O (5:1) |
24 | 91 |
| 14 | 0.5 | 1,4-Dioxane/H2O (5:1) |
24 | 72 |
| 15 | 2.0 | 1,4-Dioxane/H2O (5:1) |
24 | 79 |
| 16 | 1.6 | 1,4-Dioxane/H2O (1:1) |
24 | 63 |
| 17 | 1.6 | 1,4-Dioxane/H2O (2:1) |
24 | 72 |
| 18 | 1.6 | 1,4-Dioxane/H2O (10:1) |
24 | 90 |
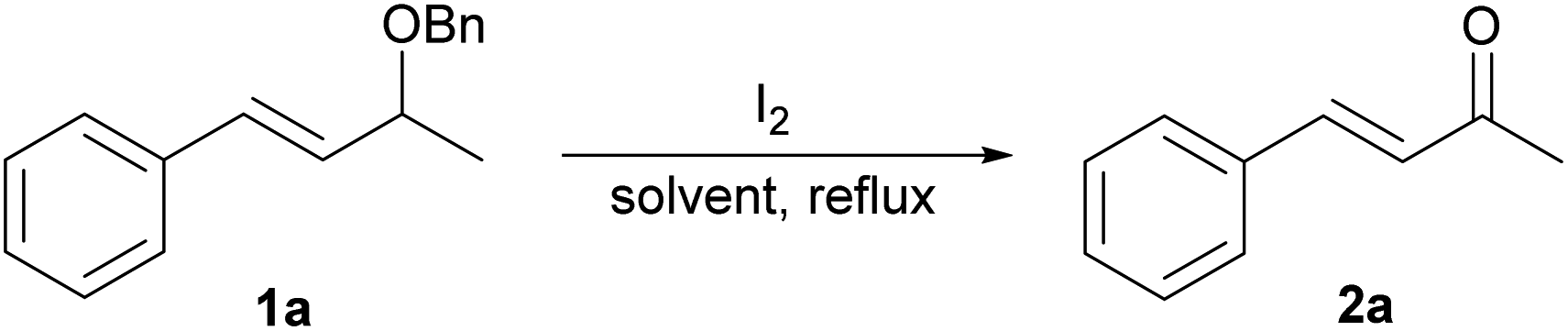
When the reaction was carried out under an argon instead of air atmosphere, the desired product was provided in 63% yield (entry 8). Temperature was also crucial to this transformation because the reaction could not occur at all when it was carried out at room temperature (entry 9). Control experiments demonstrated that the use of single 1,4-dioxane solvent did not give the desired product and the reaction could be performed in pure water, although this process becomes sluggish (entries 10 and 11). Furthermore, no product was observed in the absence of molecular iodine (entry 12). To our delight, we observed complete conversion to the desired ketone 2a by extending the reaction time to 24 h (entry 13). In order to study the potential activity of iodine as a catalyst, the reaction was performed by using a lower loading of iodine. However, an obvious decreased yield of 2a was obtained using 0.5 eq. of iodine (entry 14). In addition, an increased amount of 2.0 eq. iodine also gave negative improvement on the yield of 2a (entry 15). It is noteworthy that increasing the amount of 1,4-dioxane in the biphasic solvent system gave a positive effect on the product yield and the best ratio of 1,4-dioxane to water in current transformation is 5:1 (entries 16–18).
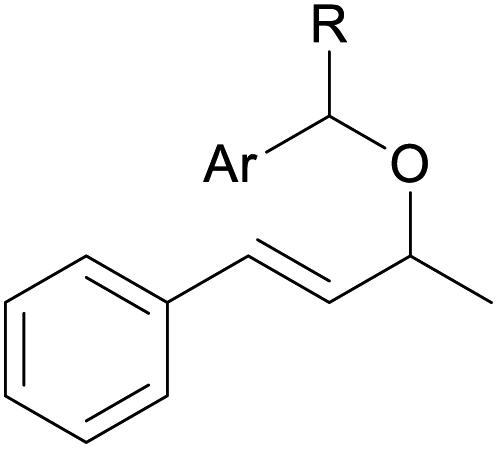
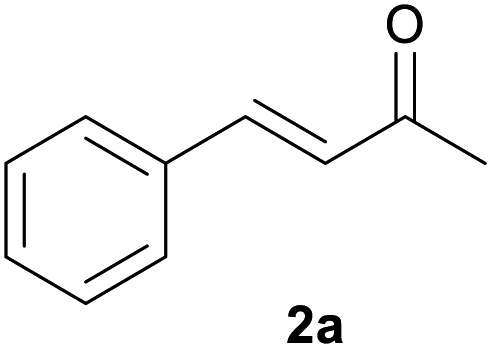
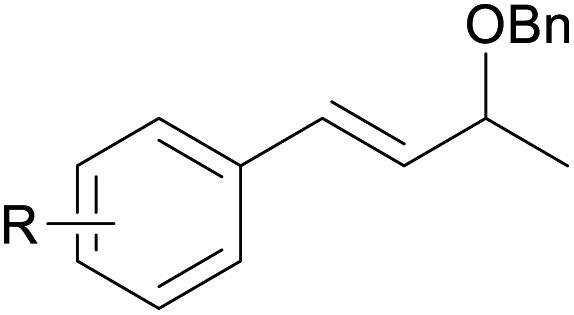
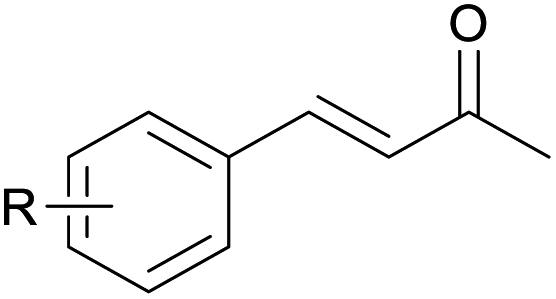
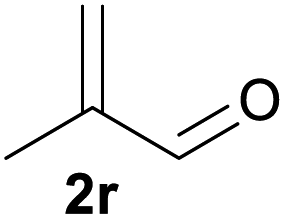
With the optimized reaction conditions in hand, we next explored the generality of this transformation on a range of allylic ethers (Tables 2 and 3). We initially scrutinized the effect of benzyl ether groups on the reaction. As shown in Table 2, allylic ethers bearing primary or secondary benzyl ether groups with either electron-donating or electron-withdrawing groups on phenyl ring of the benzyl ether groups were found to be suitable substrates for this transformation, affording ketone 2a in 85–92% yields (entries 1–5). In addition, electronic and steric changes to the allyl ethers by introducing a substituent on the aromatic ring adjacent to olefin had little to no effect on the yields of ketone products (entries 6–10). Moreover, a heterocycle-containing allyl benzyl ether also proved amenable to deprotective oxidation under the standard reaction conditions (entry 11). Interestingly, allyl ethers bearing two benzyl ether groups proved to be a suitable substrate for this protocol and afforded the desired 1,3-diketone product 2l in high yield (entry 12). In order to further demonstrate the generality of this methodology, other allylic ethers 1m–1q, e.g. cinnamyl benzyl ethers 1m–1o, which would be sterically different to 1a–1k, were then investigated. To our delight, steric or electronic changes arisen from the allylic moiety had no dramatic effect on the success of this transformation. The desired products cinnamaldehyde 2m (entries 13–15) and chalcone 2p (entries 16 and 17) were produced in good yields. Finally, we found that our methodology was not limited to the aromatic allyl system. For instance, aliphatic allyl benzyl ethers 1r and 1s were also compatible with this protocol (entries 18 and 19), allowing access to 2-methylacrolein 2r and ketone 2s in 91% and 87% isolated yields, respectively.
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb |
| a General conditions: 1 (0.1 mmol), I2 (1.6 eq.), 1,4-dioxane/H2O (5:1, 3.6 mL), at refluxing temperature, 24 h, under air.b Isolated yields.c I2 (3.2 eq.).d I2 (6.4 eq.). |
|||
 |
 |
||
| 1 | 1a R = H, Ar = Ph | 91 | |
| 2 | 1b R = H, Ar = 4-MeC6H4 | 91 | |
| 3 | 1c R = H, Ar = 4-BrC6H4 | 92 | |
| 4 | 1d R = H, Ar = 4-OMeC6H4 | 90 | |
| 5 | 1e R = Me, Ar = Ph | 85 | |
 |
 |
||
| 6 | 1f R = 4-Me | 2f R = 4-Me | 92 |
| 7 | 1g R = 4-Br | 2g R = 4-Br | 91 |
| 8 | 1h R = 2-OMe | 2h R = 2-OMe | 88 |
| 9 | 1i R = 3-OMe | 2i R = 3-OMe | 89 |
| 10 | 1j R = 4-OMe | 2j R = 4-OMe | 90 |
| 11 | 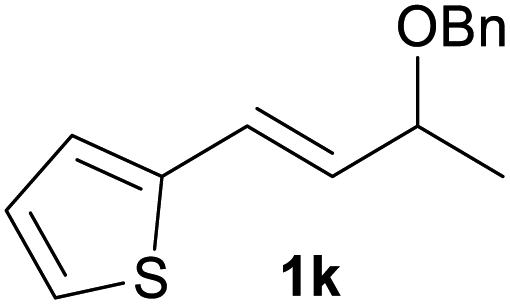 |
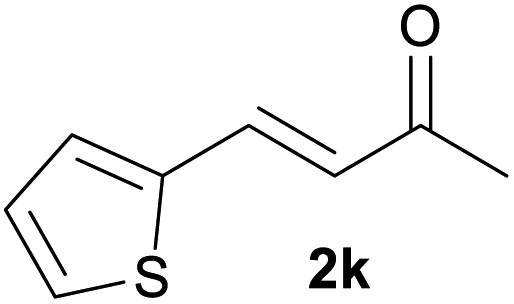 |
51 |
| 12c | 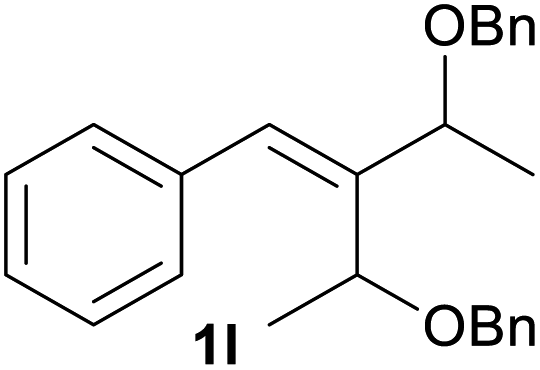 |
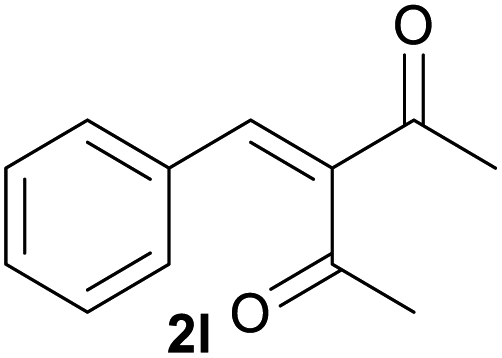 |
89 |
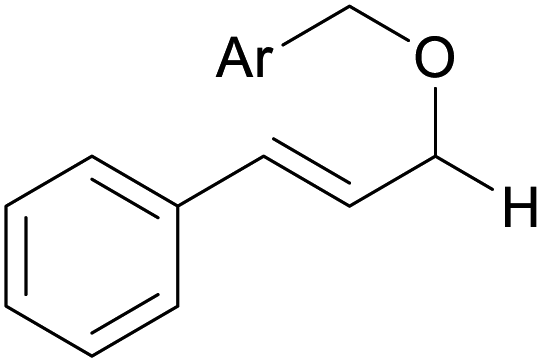 |
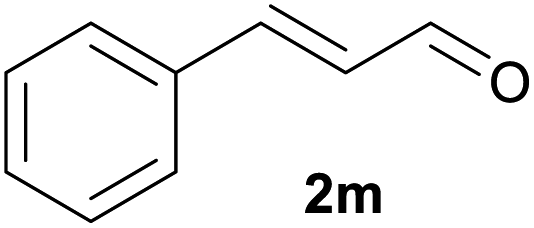 |
||
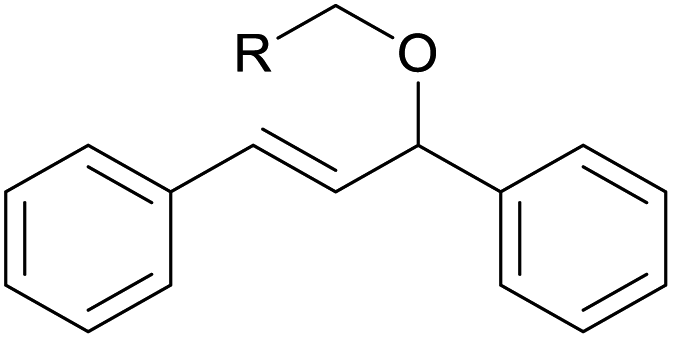
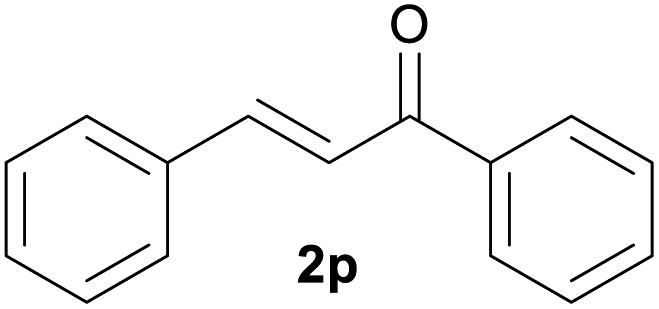
| 13d | 1m Ar = Ph | 92 | |
| 14d | 1n Ar = 4-BrC6H4 | 91 | |
| 15d | 1o Ar = 4-OMeC6H4 | 90 | |
 |
 |
||
| 16 | 1p R = Ph | 88 | |
| 17 | 1q R = 2-naphthyl | 89 | |
| 18 | 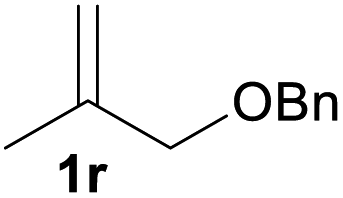 |
 |
91 |
| 19 | 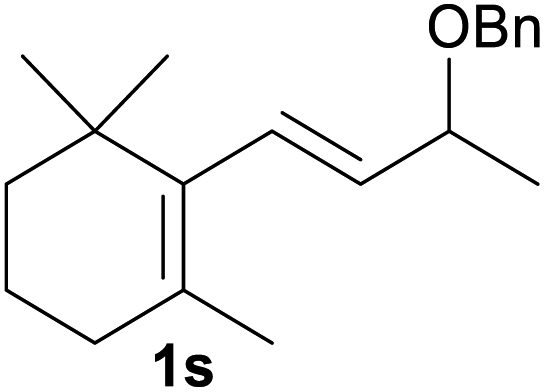 |
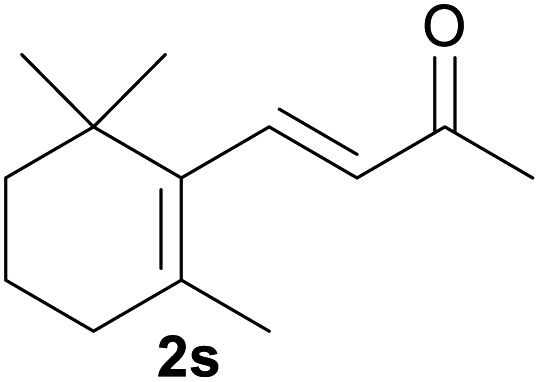 |
87 |
|
|||||
|---|---|---|---|---|---|
| Entry | Ether | R1 | R2 | Product | Yieldb |
| a General conditions: 1 (0.1 mmol), I2 (1.6 eq.), 1,4-dioxane/H2O (5:1, 3.6 mL), at refluxing temperature, 24 h, under air.b Isolated yields. |
|||||
| 1 | 1t | Me | SiEt3 | 2a | 95 |
| 2 | 1u | Me | SiiPr3 | 2a | 85 |
| 3 | 1v | Me | SiMe2tBu | 2a | 0 |
| 4 | 1w | Me | Ac | 2a | 47 |
| 5 | 1x | H | Ac | 2m | 56 |
| 6 | 1y | Me | Me | 2a | 61 |
| 7 | 1z | Me | Allyl | 2a | 52 |
| 8 | 1aa′ | Ph | Me | 2p | 68 |
| 9 | 1ab′ | Ph | Allyl | 2p | 64 |
Encouraged by the above results, we were interested in the possibility that other protective groups could serve as suitable replacements for benzylic groups of the allyl benzyl ethers. The results are summarized in Table 3. Considering its widely usage as a protective group and the required common conversion of allyl silyl ethers to enones10 to organic and medicinal chemists, we first investigated the transformation of several allyl silyl ether compounds. The allyl silyl ethers with small trialkylsilyl groups (entries 1–2), i.e. SiEt3 and Si(iPr)3, resulted in the desired product 2a in high to excellent yields, whereas the allyl silyl ether bearing a SiMe2tBu group resulted in no reaction (entry 3). This interesting effect of silyl protective groups in iodine/water-mediated oxidation of allyl silyl ethers would be useful in multistep organic synthesis. Notably, allyl esters, which are unreactive substrates under oxoammonium salt catalysis,4a were also valid substrates and delivered the corresponding ketone 2a and cinnamaldehyde 2m in 47% and 56% yields, respectively (entries 4 and 5). We next explored whether allyl ethers with other alkoxyl groups could be underwent oxidation under the reaction conditions (entries 6–9). Indeed, two methyl ethers (1y and 1aa′) and two allyl ethers (1z and 1ab′) were all proceeded smoothly and could be converted into 2a and 2p, albeit with moderate yields.
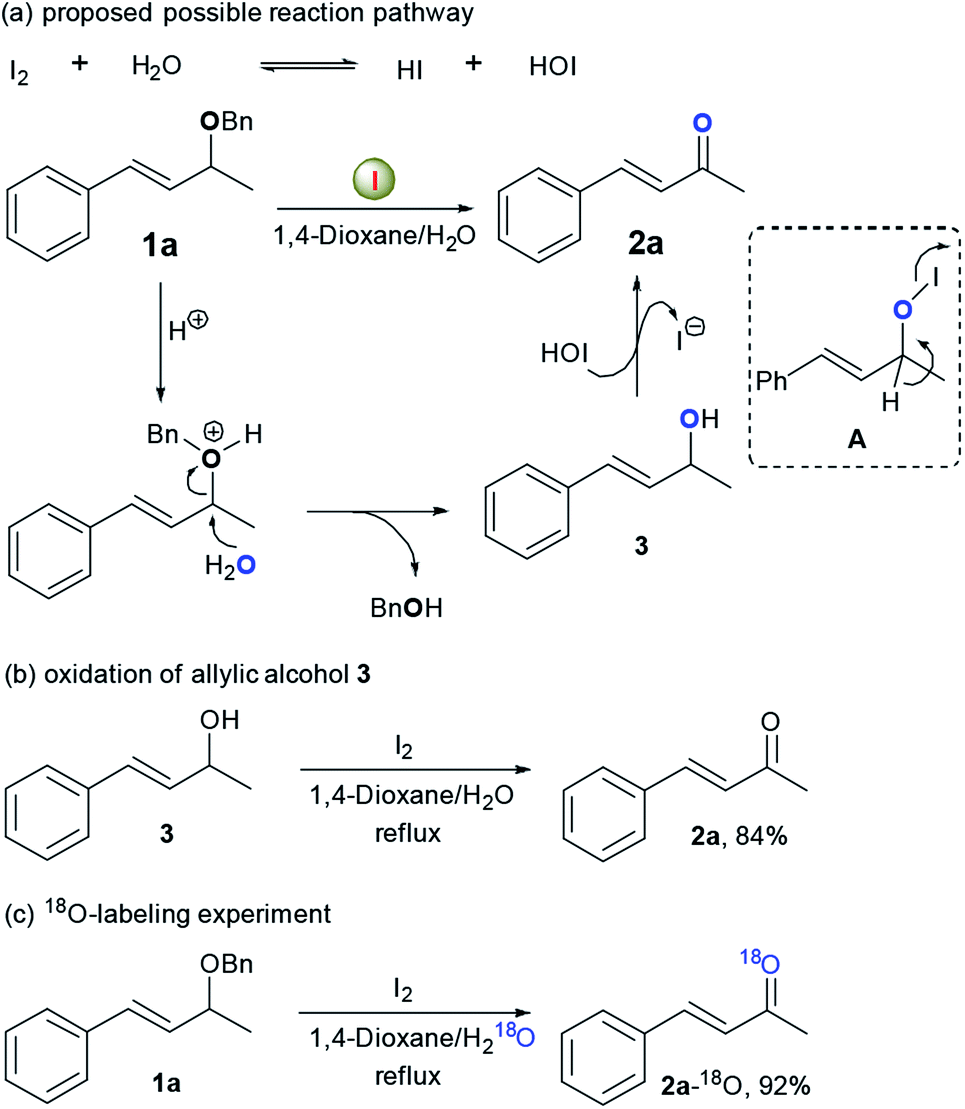
On the basis of our above results and the mechanistic understanding from the I2-catalysed reaction,7b,11 a possible reaction mechanism is proposed using 1a as the substrate (Scheme 2). Initially, disproportionation of I2 with water may generate an equilibrium mixture with hydroiodic acid (HI) and hydroiodous acid (HOI).11,12 The protonation of benzylic ether group in 1a with the generated HI provides an oxonium ion intermediate, which is subsequently attacked by water as a nucleophilic reagent, leading to the formation of allylic alcohol 3 and benzyl alcohol.13,14 Finally, 3 is oxidized to afford α,β-unsaturated ketone 2a in the presence of electrophilic iodine species via intermediates such as A.15 The proposed reaction mechanism was further convinced by following experiments.14 First, to gain a better understanding of the possible reaction intermediates, we monitored the oxidation of 1a under optimal conditions by GC-MS. Surprisingly, full conversion of 1a was observed only after 30 min under the standard reaction conditions, yielding allylic alcohol 3 (65% GC yield), desired ketone 2a (30% GC yield) and a large amount of benzyl alcohol. After 24 h, the allylic alcohol 3 was completely disappeared. These results clearly proved the possibility of above proposed deprotection/oxidation sequence, and also indicated that the deprotection step is faster than the following alcohol oxidation step under current reaction conditions. Moreover, the oxidation of 3 to 2a (84% yield) could be effectively achieved under standard conditions. Finally, 18O-labeling experiment revealed that the oxygen atom of ketone group in the formed α,β-unsaturated ketone originated from the 18O-labeled water instead of the starting ether 1a, thus giving a very facile method for the preparation of 18O-labeled α,β-unsaturated ketones and aldehydes.
 | ||
| Scheme 2 Mechanistic considerations. | ||
Conclusions
In summary, we have disclosed a protocol for the direct protective oxidation of a variety of allylic ethers to their corresponding aldehydes or ketones promoted by molecular iodine in the presence of water. Our protocol is practically simple and proceeds under ligand- and metal-free conditions. In addition, allylic esters can also undergo this oxidative process. The suggested mechanism involves an oxygen transfer from the solvent water to the carbonyl products. Further applications of this protocol are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21202031 and 21372231) and the Hangzhou Normal University. We acknowledge the excellent support from the analysis and test center of the key laboratory in HZNU. We thank Prof. Z. Li in our key laboratory for GC-MS analysis.Notes and references
-
(a) R. E. Ireland and M. G. Smith, J. Am. Chem. Soc., 1988, 110, 854–860 CrossRef CAS
; (b) J. Aleu, B. Bergamo, E. Brenna, C. Fuganti and S. Serra, Eur. J. Org. Chem., 2000, 3031–3038 CrossRef CAS
-
(a) E. Brenna, C. Fuganti, S. Ronzani and S. Serra, Helv. Chim. Acta, 2001, 84, 3650–3666 CrossRef CAS
-
(a) E. Lee-Ruff and F. J. Ablena, Can. J. Chem., 1989, 67, 699–702 CrossRef CAS
-
(a) C. B. Kelly, J. M. Ovian, R. M. Cywar, T. R. Gosselin, R. J. Wiles and N. E. Leadbeater, Org. Biomol. Chem., 2015, 13, 4255–4259 RSC
- M. M. Heravi, D. Ajami and M. Ghassemzadeh, Synthesis, 1999, 3, 393–394 CrossRef
-
(a) T. Nishiguchi and M. Bougauchi, J. Org. Chem., 1989, 54, 3001–3002 CrossRef CAS
- For a seminal report, see:
(a) H. Hibbert, J. Am. Chem. Soc., 1915, 37, 1748–1763 CrossRef CAS
-
(a) Y. Dai, Y. He, Y. Wang, H. Jiang, Z. Li and W. Chen, Synthesis, 2014, 46, 3041–3046 CrossRef CAS
- H. Kikuchi, K. Kogure and M. Toyoda, Chem. Lett., 1984, 341–344 CrossRef CAS
-
(a) H. Tanaka, T. Hasegawa, M. Iwashima, K. Iguchi and T. Takahashi, Org. Lett., 2004, 6, 1103–1106 CrossRef CAS PubMed
- For reviews, see: M. Breugst and D. von der Heiden, Chem. Eur. J., 2018, 24, 9187–9199 CrossRef CAS PubMed
- In fact, the actual iodine-containing species in aqueous obtained are pH-dependent, see:
(a) I. Lengyel, R. Epstein and K. Kustin, Inorg. Chem., 1993, 32, 5880–5882 CrossRef CAS
- The formation of benzyl alcohol was observed by GC-MS and TLC analysis at the early reaction stage.
- See the ESI† for more details.
-
(a) J. Barluenga, M. Marco-Arias, F. González-Bobes, A. Ballesteros and J. M. González, Chem. Commun., 2004, 2616–2617 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02625e |
| This journal is © The Royal Society of Chemistry 2020 |