
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-dimensional graphene oxide cross-linked by benzidine as an efficient metal-free photocatalyst for hydrogen evolution†
Xin Zhou,
Shi-Cong Cui* and
Jin-Gang Liu *
*
Key Lab for Advanced Materials, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Shanghai, 200237, P. R. China. E-mail: shicongcui@ecust.edu.cn; liujingang@ecust.edu.cn
First published on 14th April 2020
Abstract
The use of low-cost photocatalysts to split water into H2 fuel via solar energy is highly desirable for the production of clean energy and a sustainable society. Here three-dimensional graphene oxide (3DG) porous materials were prepared by cross-linking graphene oxide (GO) sheets using aromatic diamines (benzidine, 2,2′-dimethyl-4,4′-biphenyldiamine, 4,4′-diaminodiphenylmethane) that reacted with the carboxyl groups of the GO sheets at room temperature. The prepared 3DG porous materials were used as efficient metal-free photocatalysts for the production of H2 via water splitting under full-spectrum light, where the photocatalytic activity was highly dependent on the cross-linker and the 3DG reduction level. It was also found that the 3DG prepared with benzidine as the linker demonstrated a significantly higher H2 evolution rate than the 3DGs prepared using 2,2′-dimethyl-4,4′-biphenyldiamine and 4,4′-diaminodiphenylmethane as the cross-linkers. The photoactivity was further tuned by varying the mass ratio of GO to benzidine. Among the prepared 3DG materials, 3DG-3, with an intermediate C/O ratio of 1.84, exhibited the highest H2 production rate (690 μmol g−1 h−1), which was significantly higher than the two-dimensional GO (45 μmol g−1 h−1) and the noncovalent 3DG synthesized by a hydrothermal method (128 μmol g−1 h−1). Moreover, this study revealed that the 3DG photocatalytic performance was favored by effective charge separation, while it could be further tuned by changing the reduction level. In addition, these results could prompt the preparation of other 3D materials and the application of new types of photocatalysts for H2 evolution.
Introduction
The utilization of solar energy and photocatalysts to split water into H2 provides a low-cost and environmentally benign way of H2 fuel production and a promising solution to solar energy storage as well.1 In the past few decades, various metal-based semiconductor photocatalysts have been extensively studied for photocatalytic water splitting with the aim of producing H2.2–7 Recently, researchers have been working to replace the expensive and rare-metal semiconductors by developing low-cost and sustainable, environment-friendly materials for efficient H2 production. Carbon-based materials, such as graphite carbon nitride8–10 and carbon nanotubes,11,12 have been studied as metal-free semiconductor photocatalysts for water splitting. These carbon-based metal-free catalysts are expected to replace the metal-based photocatalysts owing to their wide distribution in nature.Graphene, a two-dimensional (2D) carbon nanomaterial, has attracted much attention for the H2 evolution via water splitting. Yeh et al.13 reported that graphene oxide (GO) with moderate oxidation level can be used as a photocatalyst for the production of H2 from water. When GO is used as a semiconductor photocatalyst, the electronic properties of graphene are adjusted and the state density is altered by introducing vacancies14 or different oxidation levels,15 thereby forming a semiconductor band gap. It was found that the band gap of GO increased with increasing oxygen content, resulting in a decrease in photocatalytic activity. Putri et al.16 tailored GO with different oxidation degrees for the photocatalytic H2 evolution and demonstrated that GO with an intermediate C/O ratio exhibited the highest photocatalytic activity by loading the optimum oxygen content on GO. At present, there are limited studies on the effect of the GO reduction degree on its photocatalytic performance for H2 production.
In graphene-supported metal-containing semiconductor photocatalyst composites, graphene has been frequently used as a carrier or matrix material to promote the transfer and separation of photogenerated electron–hole pairs.17–20 However, conventional graphene-based photocatalysts tend to agglomerate when they are reduced.21–23 Three-dimensional graphene aerogel (3DGA) effectively solved this problem and has been widely used in the form of 3DGA-based photocatalysts in recent years. 3D graphene not only retains the large specific surface area and the good electrical conductivity of the 2D graphene nanosheets24 but provides an effective multidimensional electron transport pathway, which is favorable for the separation of photogenerated electron–hole pairs from the composite photocatalysts.25 Ho et al. used vitamin C as a reducing agent and cross-linker to synthesize a photocatalytic composite (NGH–Au), which consisted of 3D graphene hydrogel (NGH), while TiO2 nanorods and Au nanoparticles served as the photoactive materials.26 The generated composite exhibited a higher H2 production efficiency than the corresponding 2D graphene composite (RGO–Au) under ultraviolet-visible (UV-vis) light irradiation.
Furthermore, Chen et al. discovered that a carbon-only 3D graphene oxide (3DG), apart from its role as an electronic reservoir accepting and transferring photogenerated electrons produced by the semiconductor,27 can be directly used as a photocatalyst to generate H2 via water splitting under UV-vis light.28 It was also found that the calcination treatment can enhance the photoactivity of the 3DG for H2 evolution. This photocatalyst was synthesized by a hydrothermal reduction method, which has been widely used to prepare 3D graphene materials. However, those synthetic processes usually require high temperature and high pressure conditions, which could be uncontrollable and energy consuming.
In this study, we presented a simple and effective method to obtain a covalent 3DG network using an aromatic diamine as the covalent linker to functionalize GO at room temperature. The method relied on the condensation reaction of the carboxyl groups on GO with aromatic diamines (benzidine, 2,2′-dimethyl-4,4′-biphenyldiamine (DMPDA), 4,4′-diaminodiphenylmethane (MDA)), in the presence of a carbodiimide. The photocatalytic performance of the prepared 3DG as the metal-free photocatalyst for H2 evolution was investigated, and it was proven that 3DG-3—with benzidine as the linker—remarkably enhanced the H2 evolution rate (690 μmol g−1 h−1), which was about 5.4 times higher than that of the noncovalent 3DG network synthesized by a conventional hydrothermal method (128 μmol g−1 h−1). Tuning the H2 production performance of the catalyst by varying the reduction level on the prepared covalent 3DG network was also discussed.
Experimental
Materials
Natural flake graphite (99 wt% purity) with average particle diameter of 40 μm was purchased from Sinopharm Chemical Reagent Co., Ltd. Shanghai, China. All other reagents and solvents were purchased from Adamas Reagent Co., Ltd. and were used without further purification.Characterization
The Fourier-transform infrared (FTIR) spectra were collected using a Shimadzu FTIR (IR Prestige-21) spectrometer. X-ray powder diffraction (XRD) was performed with a Rigaku Dmax-3C diffractometer using Cu Kα radiation (λ = 0.15408 nm) at 40 kV and 20 mA. The UV-vis absorption spectra of the samples were recorded using a Shimadzu UV-2600 spectrophotometer. The photoluminescence (PL) spectra were recorded on a fluorospectrophotometer (Horiba Fluoromax-4) at room temperature. The chemical states of the surface elements were analyzed by X-ray photoelectron spectroscopy (XPS, Thermo Fisher ESCALAB 250Xi). The specific surface area was measured using a Micromeritics ASAP 2460 surface area and porosity analyzer. The Raman spectra were obtained on a confocal microscopy Raman spectrometer (excitation at 532 nm). The morphology of the 3DG materials was examined using scanning electron microscopy (SEM) (S-3400N).Preparation of 3DG
GO was synthesized from natural graphite by modifying the method developed by Marcano et al.29 To increase the oxygen-containing groups on GO, it was subjected to secondary oxidation. Double oxidized GO was synthesized using a modification of the Hummers' method.30 In particular, the synthesized GO was mixed with deionized water and ultrasonically dispersed to form a 1.5 mg mL−1 GO suspension. Subsequently, appropriate amounts of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 0.3 mmol) and N-hydroxysuccinimide (NHS, 0.3 mmol) were added to 100 mL of the GO suspension. The organic diamine (0.22 mmol) was dissolved in 2.0 mL DMF and then added to the GO suspension. The mixed solution was stirred at room temperature for 24 h to promote the cross-linking of GO with the diamine monomer. The cross-linked GO was washed three times, each with N,N-dimethylformamide (DMF) and deionized water, to remove the unreacted diamine monomer, and separated by a high speed centrifuge. The cross-linked GO powder was then freeze-dried for 2 days. The products prepared using powdery GO and benzidine, DMPDA, and MDA were labelled as GO-benzidine, GO-DMPDA, and GO-MDA, respectively. In addition, a series of five 3DG GO-benzidine composites were prepared with varying benzidine amounts, i.e., 40, 60, 80, 100, and 120 mg, in DMF and mixed with the above GO suspension. The produced 3DG composites were designated as 3DG-1, 3DG-2, 3DG-3, 3DG-4, and 3DG-5, respectively.The control sample of noncovalent 3DG (3DGA) was obtained by a hydrothermal method.31 Typically, 50 mL of a GO solution (2 mg mL−1) was sealed in a 100 mL Teflon-lined autoclave, heated to 180 °C, and maintained at this temperature for 12 h. After cooling, the produced 3DGA was freeze-dried.
Photocatalytic measurements for H2 evolution
The photocatalytic experiments were conducted in a gas-sealed system using a heat-resistant glass cell with a quartz window on top. Typically, a 10 mg sample was dispersed in a triethanolamine (TEOA) aqueous solution (50 mL, volume ratio of water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TEOA = 4:1) using a magnetic stirrer, and the resulting mixture was irradiated with a 300 W Xe lamp (CEL HXF300) under inert atmosphere. A water-cooling circulator was used to control the reaction at 25 °C. The reaction was carried out for 10 h and the generated H2 gas was analyzed by a gas chromatograph (SHIMADZU GC-2014) equipped with a thermal conductivity detector.
:TEOA = 4:1) using a magnetic stirrer, and the resulting mixture was irradiated with a 300 W Xe lamp (CEL HXF300) under inert atmosphere. A water-cooling circulator was used to control the reaction at 25 °C. The reaction was carried out for 10 h and the generated H2 gas was analyzed by a gas chromatograph (SHIMADZU GC-2014) equipped with a thermal conductivity detector.
Results and discussion
Preparation and characterization of 3DGs
The 3DG samples were obtained by the chemical modification of GO with different amounts of benzidine (3DG-1, 3DG-2, 3DG-3, 3DG-4, and 3DG-5), where the GO sheets were linked by the reaction of their carboxyl groups on both sides with the aromatic diamine groups of the linker (Fig. 1). The first indication of the GO reduction was the conversion of the aqueous GO solution from yellow to brown after the addition of benzidine (Fig. 1). The functional groups on GO and 3DG were determined by FTIR spectroscopy (Fig. 2a). Specifically, for GO, the absorption peaks at 3435, 1735, 1630, 1396, 1220, and 1074 cm−1 corresponded to the hydroxyl group of the water molecule (O–H), carboxyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), aromatic skeleton carbon rings (CC), deformation vibrations of the O–H bond (CO–H), C–OH stretching, and epoxy (C–O–C) moiety, respectively. After the cross-linking of GO with benzidine, the characteristic bending vibration peak of the amide (–NH–) group appeared at 1550 cm−1 (amide II band, 1460–1554 cm−1),32 while the relative intensity of the CO peak was significantly reduced compared to the starting material (GO). Thus, it could be inferred that the condensation reaction between the carboxyl group and the amino group formed the amide bond, indicating that benzidine was covalently attached to GO. Moreover, the observed peaks at 1497 and 808 cm−1 could be ascribed to the C–N and N–H stretching vibrations of benzidine, respectively.
O), aromatic skeleton carbon rings (CC), deformation vibrations of the O–H bond (CO–H), C–OH stretching, and epoxy (C–O–C) moiety, respectively. After the cross-linking of GO with benzidine, the characteristic bending vibration peak of the amide (–NH–) group appeared at 1550 cm−1 (amide II band, 1460–1554 cm−1),32 while the relative intensity of the CO peak was significantly reduced compared to the starting material (GO). Thus, it could be inferred that the condensation reaction between the carboxyl group and the amino group formed the amide bond, indicating that benzidine was covalently attached to GO. Moreover, the observed peaks at 1497 and 808 cm−1 could be ascribed to the C–N and N–H stretching vibrations of benzidine, respectively.
 | ||
| Fig. 1 Preparation scheme of the 3DG materials with benzidine as the cross-linker. | ||
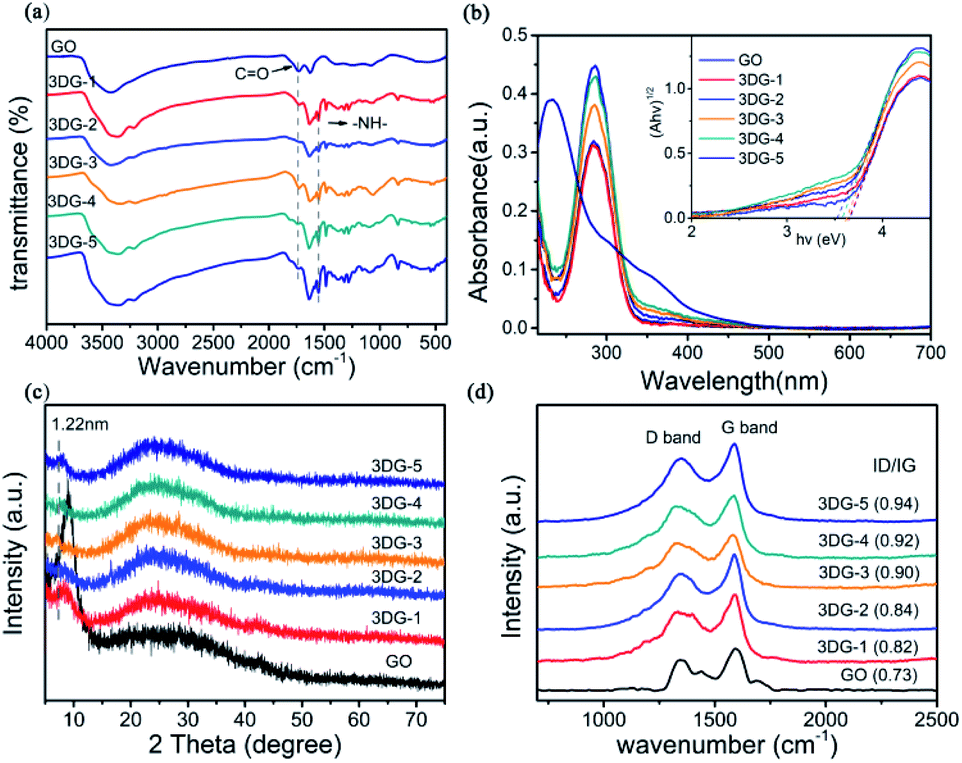 | ||
| Fig. 2 Spectroscopic characterization of GO and 3DG (3DG-1, 3DG-2, 3DG-3, 3DG-4, and 3DG-5) with successively increasing reducing properties. (a) FTIR spectra (b) UV-vis absorption spectra recorded in ethanol. The inset shows the (Ahv)1/2 vs. hv plots for GO and the five 3DGs. (c) XRD patterns. (d) Raman spectra. The numbers in brackets represent the D/G intensity ratio of the samples. | ||
The UV-vis absorption spectra of 3DG in ethanol are depicted in Fig. 2b. Two main characteristic peaks were observed for the GO solution: one at 230 nm, caused by the π–π* transition of the aromatic structure, and a shoulder around 300 nm due to the n–π* transitions of the carbonyl groups.33 For the 3DG-1 solution, the absorption peak shifted from 230 to 282 nm. By increasing the mass of benzidine to 120 mg, the same peak further shifted to 287 nm, indicating the reduction of GO and the appearance of electronic conjugation in the graphene sheets due to the covalent interactions.34,35 In addition, the inset of Fig. 2b displays the square of the absorption energy (Ahv, where A is the absorbance) against the photon energy (hv). Moreover, the Kubelka–Munk plots36,37 implied that the band gap energy value of GO was 2.4–4.3 eV for the semiconductor photocatalyst.13 As the reduction degree increased, the 3DG-1, 3DG-2, 3DG-3, 3DG-4, and 3DG-5 samples displayed decreasing band gap energies of 3.65, 3.65, 3.60, 3.57, and 3.50 eV, respectively. This result was consistent with the relationship between the oxygen density and the energy band of the reduced GO.38,39
Furthermore, the covalent functionalization of GO with benzidine could also be observed by the corresponding XRD patterns (Fig. 2c). The characteristic sharp peak of GO at 9° corresponded to an interlayer spacing of 0.98 nm, while the prepared 3D functionalized graphene-based networks exhibited diffraction peaks at smaller diffraction angles (8.4°, 8.2°, 7.2°, 8°, and 7.8° for 3DG-1, 3DG-2, 3DG-3, 3DG-4, and 3DG-5, respectively). According to the Bragg equation, the d001 spacing of 3DG-1, 3DG-2, 3DG-3, 3DG-4, and 3DG-5 was calculated to be 1.07, 1.05, 1.22, 1.10, and 1.13 nm, respectively, suggesting an increasing enlargement in the interlayer distance compared to GO, which could be attributed to the cross-linking of benzidine in the GO sheets by the amide formation reaction.40,41 In addition, a broad peak (002) appeared at 24.6°, indicating the efficient reduction of GO by benzidine.31
The reduction degrees of the five 3DG materials were further compared by Raman spectroscopy (Fig. 2d), which is a well-known, powerful tool for characterizing graphite materials. The common characteristic Raman bands of GO are the D band (1350 cm−1), resulting from the sp3 carbon atom in the defect structure, and the G band (1591 cm−1) corresponding to the in-plane vibration mode of the sp2 hybridized carbon. Based on Fig. 2d, all five 3DG samples revealed the D- and G-bands of the GO skeleton structure. Interestingly, the Raman spectra of GO cross-linked with various benzidine amounts exhibited higher D/G intensity ratios than GO (0.73), while the D/G intensity ratio increased by increasing the benzidine mass (40, 60, 80, 100, and 120 mg) from 0.82 to 0.94 for 3DG-1 to 3DG-5, respectively. The increase in the D/G intensity ratio in the Raman spectrum of the prepared 3GDs also suggested that the number of the sp3 domains increased.42
The composition and chemical status of GO and 3DGs were also analysed by XPS. The C 1s XP spectrum of GO (Fig. 3a) could be decomposed into four peaks that corresponded to the unoxidized graphite carbon skeleton (C–C, 284.6 eV), epoxy/hydroxyl carbon (C–O, 286.2 eV), carbonyl (CO, 288.1 eV), and carboxylate functional group (O–CO, 289.5 eV).43 After cross-linking with benzidine, a new signal corresponding to the C–N group of 3DG-3 appeared at 285.6 eV, while the peak intensity of the CO and O–CO groups decreased substantially (Fig. 3b). The cross-coupling reaction between the carboxyl group of GO and the benzidine amine to form amide bonds was also confirmed by the C 1s spectra of the other 3DG samples (ESI, Fig. S5†).44 Different N-containing species in the 3DGs could be revealed by the N 1s XPS spectra (Fig. 3c), where the broad peak could be divided into two components with binding energies at 399.9 and 402.1 eV that were assignable to the N-atom of the amide (OC–N) and imine groups (CN), respectively. The presence of the (OC–N) peak confirmed the chemical reaction between GO and benzidine, which has been suggested by the C 1s XP spectrum as well. Furthermore, the results of the N 1s XPS analysis of the other 3DG samples were consistent to the above results (ESI, Fig. S6†). Moreover, the C/O atomic ratio increased from 1.42 for GO to 1.80 for 3DG-1, indicating the reduction of GO by benzidine (Fig. 3d), while the C/O ratio increased gradually from 3DG-1 to 3DG-5, highlighting that the reduction degree of the 3DGs increased by increasing the benzidine amount during the reaction.
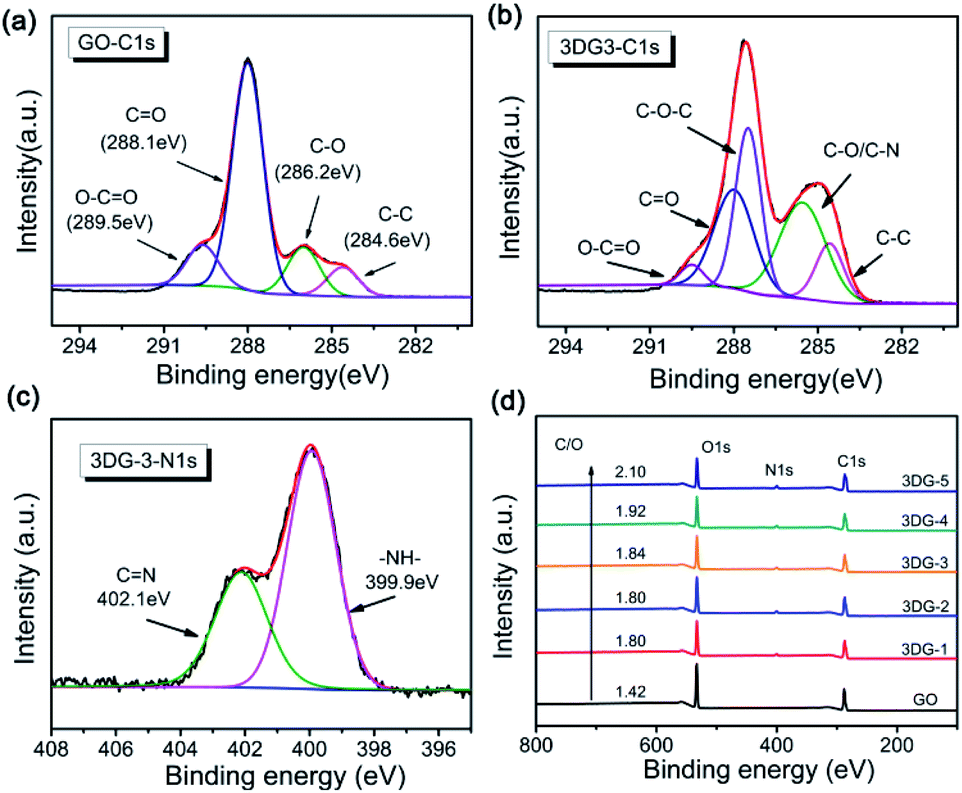 | ||
| Fig. 3 C 1s XPS spectra of (a) GO and (b) 3DG-3. (c) N 1s XPS spectra of 3DG-3. (d) XPS survey spectrum of GO and the 3DG samples. | ||
Morphology characterizations
The morphology of GO and the 3DGs (3DG-1 to 3DG-5) were revealed by SEM images (Fig. 4). 2D GO displayed a very large anisometric shape with face-to-face stacking of the sheets, resulting in the interlayer aggregation with narrow interlayer spacing. As shown in Fig. 4a, the surface of the sheet of GO was quite smooth. In contrast, the surface of the benzidine cross-linked GO samples was rougher and possessed highly porous surfaces (Fig. 4b–f), indicating that treating 2D GO flakes with rigid benzidine molecules hinders the face-to-face stacking. However, no significant differences in the morphology of the five samples were observed. | ||
| Fig. 4 SEM images of (a) GO, (b) 3DG-1, (c) 3DG-2, (d) 3DG-3, (e) 3DG-4, and (f) 3DG-5. | ||
To improve our understanding in the morphology of the five prepared materials, N2 adsorption–desorption isotherms were recorded (Fig. 5). The specific surface area of the synthesized GO was 4.14 m2 g−1, which was significantly lower than the theoretical specific surface area of graphene (2600 m2 g−1). This common problem was caused by the face-to-face aggregation of the GO sheets during the wet processing. Instead, the specific surface area of the 3DGs increased from 3DG-1 (92.67 m2 g−1) to 3DG-3 (154.24 m2 g−1) and then declined to 3DG-5 (135.08 m2 g−1). The obvious increase in the surface area of the 3DGs indicated that the face-to-face stacking was effectively limited by the rigid cross-linkers derived from benzidine. Thus, with the appropriate amount of benzidine in the 3DGs, the benzidine molecules could serve as pillaring agents, thus separating the graphene sheets to form a porous network between them. On the contrary, benzidine excess leads to the graphene sheets stacking through π–π interactions. Consequently, as the amount of benzidine increased, the specific surface area of the 3DGs gradually increased and then decreased, while 3DG-3 exhibited the highest specific surface area.
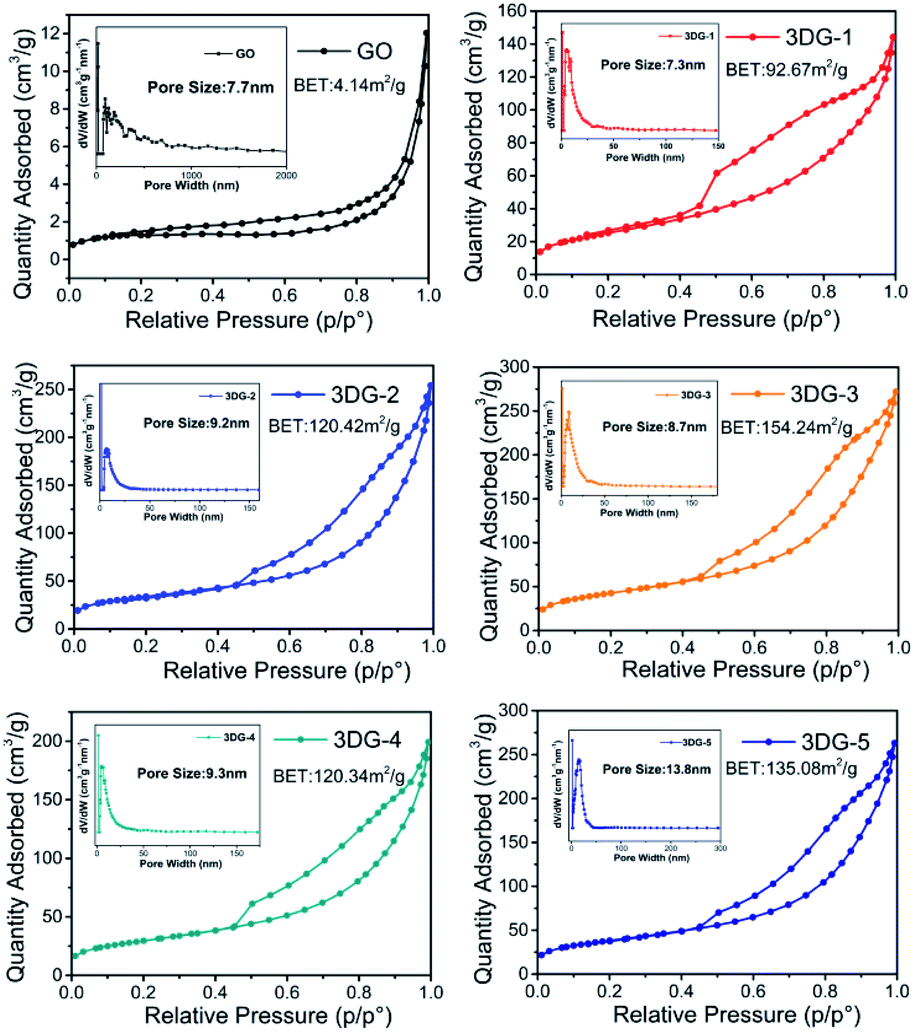 | ||
| Fig. 5 N2 adsorption–desorption isotherm pore size distribution of the prepared 3DG samples. | ||
Photocatalytic H2 evolution
The photocatalytic H2 evolution properties of the 3DGs prepared with different diamines (GO-benzidine, GO-DMPDA, GO-MDA) were evaluated under full-spectrum irradiation in the presence of a TEOA solution as the sacrificial agent. The H2 evolution rate of the three 3DG samples bearing a unique 3D network structure were significantly enhanced relative to the control GO, especially for GO-benzidine (Fig. S3†). The enhanced specific surface area of these three samples (Fig. S4†) apparently increased their adsorption capacity and the number of active sites, which in turn improved the photocatalytic performance. Surprisingly, GO-MDA that had the largest specific surface area among the three samples did not yield the highest H2 evolution rate. For the synthesized GO-DMPDA and GO-MDA, they all showed larger specific surface area than GO-benzidine (Fig. S4†), but their photocatalytic activity was lower than GO-benzidine. This suggests that the specific surface area of 3DGs does not play decisive role on the photocatalytic H2 production.While, the reduction degree of GO was found to be an important factor for the H2 evolution in the photoinduced water splitting process with the GO-based catalyst.15 The aromatic diamine molecules of MDA, DMBPDA and benzidine were employed both as a pillar to separate the GO sheets and as a reducing agent to reduce GO to prepare the 3DGs. Thus, the reduction degree of the prepared 3DGs varied with the diamine linker, among which benzidine turned out to be the most suitable cross-linker with appropriate reducibility, rendering the GO-benzidine 3DG with the highest H2 evolution rate. Tuning the reducibility by variation of the structure of p-phenylene diamine was previously noticed as well.47
Next, the reduction level of GO in the 3DGs was then further regulated by altering the mass ratio of GO to benzidine, which afforded a series of five 3DGs (3DG-1 to 3DG-5) with increasing benzidine amounts. The results from the above C/O ratio and the band gap analysis revealed that the reduction degree of the 3DGs was positively correlated with the increase in the benzidine mass. To exclude the effect of the specific surface area on the H2 production rate, the evolution rate (μmol g−1 h−1) was further divided by each sample's specific surface area value. 3DG-5 with relative large specific surface area was found to show the lowest H2 production rate (3.25 μmol g−1 m−2 h−1) among the five 3DGs. And the photocatalytic activity of the H2 evolution followed the order of 3DG-3 > 3DG-2 > 3DG-1 > 3DG-4 > 3DG-5 (Table 1).
| Sample | BET (m2 g−1) | ID/IG | Band gap (eV) | C/O (%) | H2 evolved (μmol g−1 h−1) | H2 evolved (μmol m−2 h−1) |
|---|---|---|---|---|---|---|
| 3DG-1 | 92.67 | 0.82 | 3.65 | 1.80 | 360 | 3.88 |
| 3DG-2 | 120.42 | 0.84 | 3.65 | 1.80 | 480 | 3.98 |
| 3DG-3 | 154.24 | 0.90 | 3.60 | 1.84 | 690 | 4.49 |
| 3DG-4 | 120.34 | 0.92 | 3.57 | 1.92 | 400 | 3.32 |
| 3DG-5 | 135.08 | 0.94 | 3.50 | 2.10 | 440 | 3.25 |
For the prepared GO-benzidine samples, 3DG-3 exhibited the best photocatalytic activity, suggesting the best oxygen content loaded with C/O ratio of 1.84 that is consistent with the previous observation.16 3DG-3 exhibited an H2 evolution rate of 690 μmol g−1 h−1, which was remarkably higher than that of GO (45 μmol g−1 h−1) and 3DGA (125 μmol g−1 h−1) that was hydrothermally synthesized under the same experimental conditions (Fig. 6a). Instead, the most reduced sample (3DG-5), which had the lowest energy band (3.50 eV) and absorbed the most light, exhibited the poorest photocatalytic performance. The excessive reduction of the oxygen-containing groups on the GO surface led to a small exposed area on 3DG-5, thus inhibiting the access of the hydrophilic water molecules, which did not favor the evolution of the photocatalytic H2 production. Nevertheless, the least reduced sample (3DG-1) also did not show the best photoactivity. Although 3DG-1 had the largest energy band gap (3.65 eV) among the prepared 3DGs, it only absorbed high-energy light in the short wavelength range. In addition, because of the high electronegativity of the oxygen groups, it would have a negative effect when used as recombination center.48 Therefore, higher oxygen coverage on the GO sheets also inhibited H2 production. It was demonstrated that the covalent cross-linking method could effectively regulate the structure of the 3DG catalyst, thus achieving the optimum photocatalytic activity for 3DG-3. The high efficient H2 evolution rate as observed with 3DG-3 is comparable to those excellent metal-free photocatalysts reported (Table S1†).
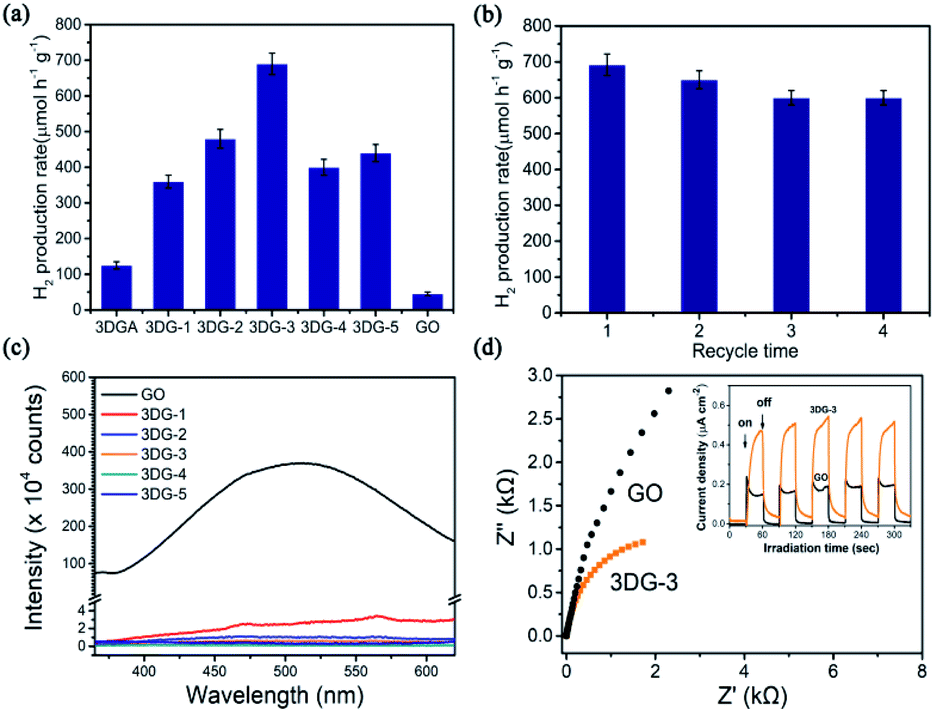 | ||
| Fig. 6 (a) Photocatalytic H2 evolution by water splitting over a series of 3DG samples synthesized at different conditions, GO, and 3DGA. (b) Cycling measurement of H2 evolution over 3DG-3. (c) Fluorescence spectra (λex = 325 nm) of GO and 3DG-1, 3DG-2, 3DG-3, 3DG-4 and 3DG-5. (d) Electrochemical research-impedance (EIS) plot and photocurrent-time dependence of GO and 3DG-3. | ||
It has been reported that the specific surface area is an important factor affecting the catalytic activity.45 But there are also some other factors, such as the oxidation degree of graphene,16 the distribution of active sites46 and so on. For the photocatalytic H2 evolution catalysed by 3DGs, we found that the reduction degree of 3DGs, the proper C/O ratio, rather than its surface area plays a major role in H2 production.
The catalytic stability of 3DG-3 was additionally evaluated by measuring repeated cycles of photocatalytic H2 production under the same reaction conditions. After four cycles of continuous exposure to full-spectrum light, no significant decline in photoactivity was observed over time (Fig. 6b).
Plausible mechanism of the photocatalytic H2 production
The significant enhancement of the photocatalytic H2 production by 3DG-3 compared to the 2D GO was due to the greater absorption of UV-vis light by the 3DGs,49 while 3DG-3 had a more efficient separation of the photoinduced charge carriers under UV light excitation based on PL spectroscopic and photochemical measurements. The PL emission spectrum of 3DG-3 revealed a very weak fluorescence, while GO showed a strong intrinsic fluorescence emission peak at 500 nm (Fig. 6c), indicating that the barrier of charge recombination in 3DG-3 was larger than that in GO.50 Furthermore, 3DG-3 showed a much smaller arch radius than GO in the EIS spectrum (Fig. 6d), suggesting that its interface charge transfer rate was faster.51 Moreover, compared to GO, the photocurrent density of the 3DG-3 electrode was significantly increased, indicating its more efficient photogenerated charge carrier separation,52 which may be due to the unique porous structure. The 3D interconnected network of the porous nanosheets promoted the mass transfer and shortened the diffusion length of the charge transfer, which in turn promoted the electron repositioning at the edge of the surface, thereby hindering the charge recombination.45Although it has been reported that the GO band gap changes with the oxygen concentration, studies have shown that GO with different degrees of oxidation have similar conduction band edge levels of −0.75 eV (vs. Ag/AgCl).15 As shown in Fig. 2b, the energy gap (Eg) of 3DG-3 was 3.60 eV for an indirect transition. Based on the above results and literature reports, we could infer the valence band position of 3DG-3 (3.60 eV) and propose a plausible mechanistic diagram of the photocatalytic reaction (Fig. 7). In particular, under irradiation, the GO vacancies are excited. Then, water molecules are reduced to H2 by the photoinduced electrons (e−), while the holes (h+) are captured by the sacrificial agent (TEOA) on the 3DG surface.
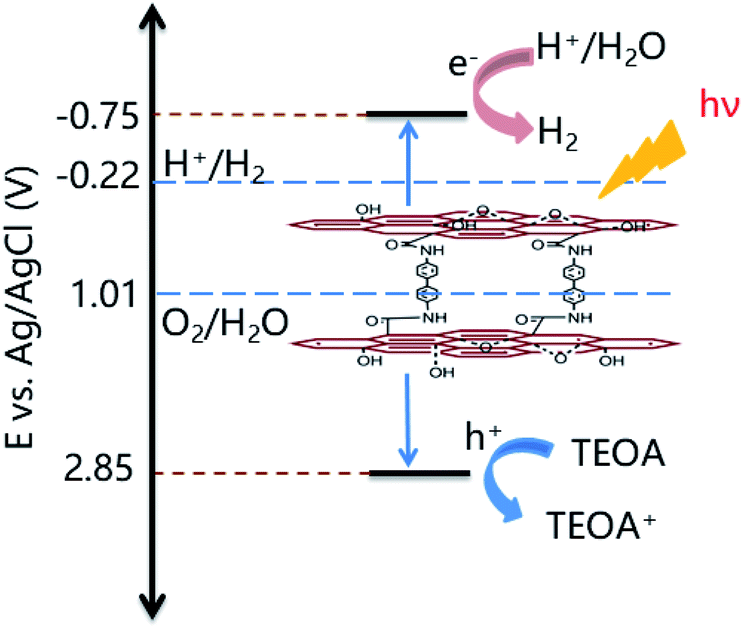 | ||
| Fig. 7 Plausible mechanism for the photocatalytic H2 production catalyzed by the 3DG-3 porous material. | ||
Conclusions
In summary, we presented a facile method to prepare covalently linked 3DG network composites using aromatic diamines as the cross-linker, while the prepared 3DG were applied as metal-free photocatalyst for the efficient evolution of H2. It was proven that the photocatalytic performance of the 3DGs was highly dependent on the linker and the reduction degree of GO. The benzidine-linked 3DGs exhibited much higher H2 evolution rate than the DMPDA and MDA-linked 3DG catalysts. Furthermore, tuning the reduction degrees of GO in the 3DG catalyst was achieved by varying the amount of the introduced benzidine. Thus, the optimized 3DG-3 catalyst with an intermediate C/O ratio of 1.84 exhibited the highest H2 evolution rate (690 μmol g−1 h−1) among the prepared 3DGs, which was about 15.3 higher than that of the 2D GO (ca. 45 μmol g−1 h−1) and 5.4 times higher than that of the noncovalent 3DG (128 μmol g−1 h−1) synthesized by a conventional hydrothermal method under similar experimental conditions. This work thus provided a new strategy for improving the photocatalytic activity of 3DG using the benzidine linker both as a pillar and a reducing agent to covalently assemble GO layers in the corresponding 3DG networks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the NSF of China (No. 21571063 to SCC, 21571062 to JGL), the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning to JGL.Notes and references
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- Q. Lu, Y. Yu, Q. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca and H. Idriss, Nat. Chem., 2011, 3, 489–492 CrossRef CAS PubMed.
- X. Zhou, Z. Luo, P. Tao, B. Jin, Z. Wu and Y. Huang, Mater. Chem. Phys., 2014, 143, 1462–1468 CrossRef CAS.
- J. Zhang, J. Yu, Y. Zhang, Q. Li and J. R. Gong, Nano Lett., 2011, 11, 4774–4779 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- W. Cui, Q. Liu, N. Cheng, A. M. Asiri and X. Sun, Chem. Commun., 2014, 50, 9340–9342 RSC.
- K. Xie, H. Wu, Y. Meng, K. Lu, Z. Wei and Z. Zhang, J. Mater. Chem. A, 2015, 3, 78–82 RSC.
- T. F. Yeh, J. M. Syu, C. Cheng, T. H. Chang and H. Teng, Adv. Funct. Mater., 2010, 20, 2255–2262 CrossRef CAS.
- Y. Wu, N. Yi, L. Huang, T. Zhang, S. Fang, H. Chang, N. Li, J. Oh, J. A. Lee, M. Kozlov, A. C. Chipara, H. Terrones, P. Xiao, G. Long, Y. Huang, F. Zhang, L. Zhang, X. Lepro, C. Haines, M. D. Lima, N. P. Lopez, L. P. Rajukumar, A. L. Elias, S. Feng, S. J. Kim, N. T. Narayanan, P. M. Ajayan, M. Terrones, A. Aliev, P. Chu, Z. Zhang, R. H. Baughman and Y. Chen, Nat. Commun., 2015, 6, 6141–6150 CrossRef CAS PubMed.
- T. F. Yeh, F. F. Chan, C. T. Hsieh and H. Teng, J. Phys. Chem. C, 2011, 115, 22587–22597 CrossRef CAS.
- L. K. Putri, B. J. Ng, K. H. Tan, F. S. Lim, W. J. Ong, W. S. Chang and S. P. Chai, Catal. Today, 2018, 315, 93–102 CrossRef CAS.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577–583 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604–607 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z. R. Tang and Y. J. Xu, J. Phys. Chem. C, 2014, 118, 5299–5308 CrossRef CAS.
- W. Wan, Y. Lin, A. Prakash and Y. Zhou, J. Mater. Chem. A, 2016, 4, 18687–18705 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- B. Wang, L. Si, J. Geng, Y. Su, Y. Li, X. Yan and L. Chen, Appl. Catal., B, 2017, 204, 316–323 CrossRef CAS.
- R. Zhang, W. Wan, D. Li, F. Dong and Y. Zhou, Chin. J. Catal., 2017, 38, 313–320 CrossRef CAS.
- M. Gao, C. K. N. Peh, W. L. Ong and G. W. Ho, RSC Adv., 2013, 3, 13169–13177 RSC.
- R. Wang, K. Q. Lu, F. Zhang, Z. R. Tang and Y. J. Xu, Appl. Catal., B, 2018, 233, 11–18 CrossRef CAS.
- Y. Lu, B. Ma, Y. Yang, E. Huang, Z. Ge, T. Zhang, S. Zhang, L. Li, N. Guan, Y. Ma and Y. Chen, Nano Res., 2017, 10, 1662–1672 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- B. J. Hong, O. C. Compton, Z. An, I. Eryazici and S. T. Nguyen, ACS Nano, 2012, 6, 63–73 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- Q. Y. Cheng, D. Zhou, Y. Gao, Q. Chen, Z. Zhang and B. H. Han, Langmuir, 2012, 28, 3005–3010 CrossRef CAS PubMed.
- L. Li, B. Song, L. Maurer, Z. Lin, G. Lian, C. C. Tuan, K. S. Moon and C. P. Wong, Nano Energy, 2016, 21, 276–294 CrossRef CAS.
- J. I. Paredes, S. Villar Rodil, A. Martinez Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- X. Zhao, Q. Zhang, D. Chen and P. Lu, Macromolecules, 2010, 43, 2357–2363 CrossRef CAS.
- M. A. Khan, S. Mutahir, F. Wang, W. Lei, M. Xia and S. Zhu, J. Hazard. Mater., 2019, 367, 293–303 CrossRef CAS PubMed.
- M. A. Khan, S. Mutahir, F. Wang, J. W. Zhou, W. Lei and M. Xia, Sol. Energy, 2019, 186, 204–214 CrossRef CAS.
- J. Ito, J. Nakamura and A. Natori, J. Appl. Phys., 2008, 103, 113712 CrossRef.
- H. Huang, Z. Li, J. She and W. Wang, J. Appl. Phys., 2012, 111, 054317 CrossRef.
- H. Yang, C. Shan, F. Li, D. Han, Q. Zhang and L. Niu, Chem. Commun., 2009, 3880–3882 RSC.
- W. Li, X. Z. Tang, H. B. Zhang, Z. G. Jiang, Z. Z. Yu, X. S. Du and Y. W. Mai, Carbon, 2011, 49, 4724–4730 CrossRef CAS.
- Z. Wang, X. Zhou, J. Zhang, F. Boey and H. Zhang, J. Phys. Chem. C, 2009, 113, 14071–14075 CrossRef CAS.
- W. S. Hung, C. H. Tsou, M. De Guzman, Q. F. An, Y. L. Liu, Y. M. Zhang, C. C. Hu, K. R. Lee and J. Y. Lai, Chem. Mater., 2014, 26, 2983–2990 CrossRef CAS.
- B. Song, J. I. Choi, Y. Zhu, Z. Geng, L. Zhang, Z. Lin, C. c. Tuan, K. s. Moon and C. p. Wong, Chem. Mater., 2016, 28, 9110–9121 CrossRef CAS.
- Q. Liang, Z. Li, X. Yu, Z. H. Huang, F. Kang and Q. H. Yang, Adv. Mater., 2015, 27, 4634–4639 CrossRef CAS PubMed.
- W. D. Wei, X. Y. Liu, S. C. Cui and J. G. Liu, RSC Adv., 2017, 7, 25650–25656 RSC.
- J. R. Quinn, F. W. Foss Jr, L. Venkataraman and R. Breslow, J. Am. Chem. Soc., 2007, 129, 12376–12377 CrossRef CAS PubMed.
- K. Bothe and J. Schmidt, J. Appl. Phys., 2006, 99, 013701–013711 CrossRef.
- Y. S. Jun, E. Z. Lee, X. Wang, W. H. Hong, G. D. Stucky and A. Thomas, Adv. Funct. Mater., 2013, 23, 3661–3667 CrossRef CAS.
- A. Du, S. Sanvito, Z. Li, D. Wang, Y. Jiao, T. Liao, Q. Sun, Y. H. Ng, Z. Zhu, R. Amal and S. C. Smith, J. Am. Chem. Soc., 2012, 134, 4393–4397 CrossRef CAS PubMed.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed.
- Y. Hou, F. Zuo, A. P. Dagg, J. Liu and P. Feng, Adv. Mater., 2014, 26, 5043–5049 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional figures. See DOI: 10.1039/d0ra00383b |
| This journal is © The Royal Society of Chemistry 2020 |