
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChlorotrifluoroethylidenes: an efficient and convenient approach to their synthesis†
Abhay Atmaram Upareab,
Pradip K. Gadekarb,
Kiran Jadhavb,
H. Sivaramakrishnanb and
Selvaraj Mohana Roopan *a
*a
aChemistry of Heterocycles and Natural Product Research Lab, Department of Chemistry, School of Advanced Science, Vellore Institute of Technology, Vellore – 632014, Tamil Nadu, India. E-mail: mohanaroopan.s@vit.ac.in; Tel: +91-0416-220-2313
bDepartment of Process Development, Piramal Enterprises Ltd., Lighthall A Wing, Hiranandani Business Park, Sakivihar Road, Chandivali, Andheri (East), Mumbai 400072, India
First published on 5th May 2020
Abstract
A convenient one step synthesis of chlorotrifluoroalkyl olefins starting from aldehydes was developed. The stable reagent 2-((1-chloro-2,2,2-trifluoroethyl)sulfonyl)benzothiazole was prepared from readily available benzothiazole-2-thiol and halothane. This method comprises using stable 2-((1-chloro-2,2,2-trifluoroethyl)sulfonyl)benzothiazole according to the Julia procedure and presents new opportunities for the synthesis of trifluoroalkylidene derivatives.
1. Introduction
The use of fluorinated compounds in medicinal chemistry, agrochemical fields and material science applications has had a remarkable impact on industrial and academic research. The presence of a fluorine (–F) or a trifluoromethyl (–CF3) moiety often leads to improved lipophilicity, metabolic stability, bioavailability and binding selectivity.1 Hence, the incorporation of a –F or –CF3 group into organic compounds is a popular research area in organofluorine chemistry.2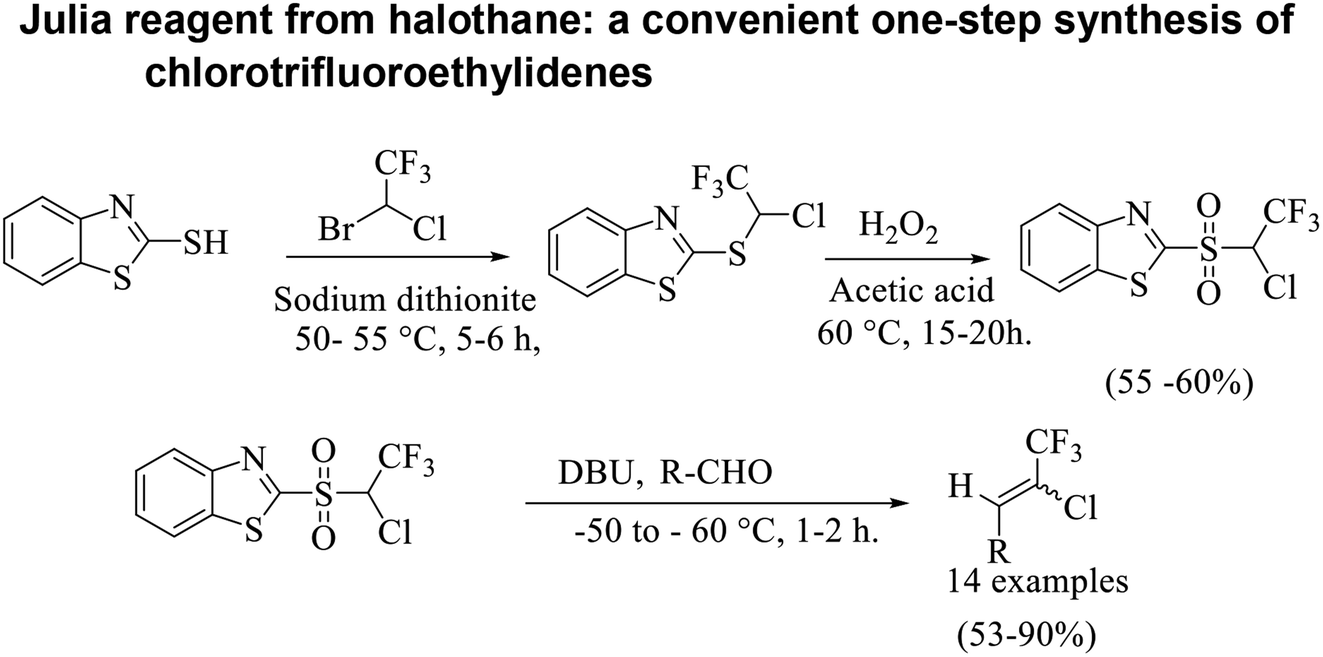
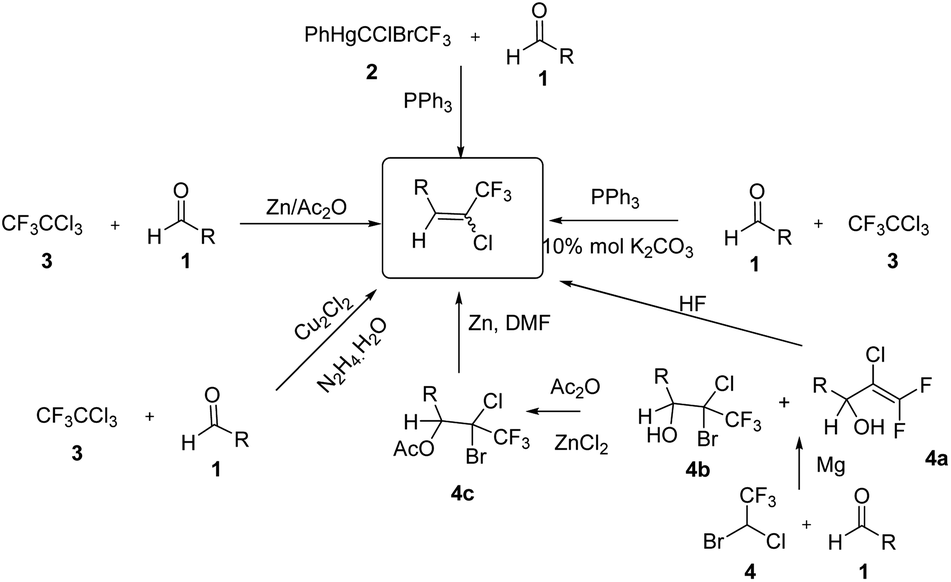
The photo stable chlorotrifluoroethylidene (R–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CClCF3) moiety, is an important structural unit which is present in pyrethroid insecticides.3 The use of chlorotrifluoroethylidene was reported in the synthesis of various heterocycles containing a –CF3 group such as substituted benzofurans.4 The copper-catalyzed double thiolation reaction of 1,4-dihalides with sulfides has been reported for the selective synthesis of 2-trifluoromethyl benzothiophenes and benzothiazoles via chlorotrifluoroethylidene.5 In addition, the synthesis of 1-aryl-3,3,3-trifluropropynes were reported using sodamide and the corresponding chlorotrifluoroethylidene.5 Moreover, from the subsequent alkynes prepared from this alkylidine various heterocyles can be synthesize such as aryl trifluoromethyl-1,2,3-triazoles.6–8 Various methods for the synthesis of chlorotrifluoroethylidenes have been reported in the literature (Scheme 1). The chlorotrifluoroethylidene group has been synthesized using aldehydes and CF3CCl2ZnCl, which was prepared from CF3CCl3 and zinc powder. However, the selective formation of the chlorotrifluoroethylidene moiety requires an excess of zinc powder and acetic anhydride.9 The most common method for the synthesis of chlorotrifluoroethylidene derivatives is the reaction of aldehydes or ketones with fluorinated phosphorous based reagents such as PPh3CClCF3 or (EtO)2(O)PCClCF3COOEt. A major drawback of this approach is the difficulty in accessing their phosphoranes that requires an ozone depleting CFC reagent and which is not readily available.10 The synthesis of chlorotrifluoroethylidene compounds was also reported using organomercury reagents. This procedure requires a high reaction temperature and a long reaction time (71 h).11 A one-pot procedure was also reported for the synthesis of said alkene without isolation of the hydrazone by reacting the aldehyde with hydrazine hydrate followed by reaction with CF3CCl3 in the presence of copper(I) chloride.8 Toshiyuki and co-workers reported the synthesis of chlorotrifluoroethylidene by reacting aldehydes with halothane under Grignard reaction condition (Scheme 1).12 Hu and co-workers report a one-pot synthesis of chlorotrifluoroethylidene compounds using CF3CCl3 and triphenylphosphine.13
CClCF3) moiety, is an important structural unit which is present in pyrethroid insecticides.3 The use of chlorotrifluoroethylidene was reported in the synthesis of various heterocycles containing a –CF3 group such as substituted benzofurans.4 The copper-catalyzed double thiolation reaction of 1,4-dihalides with sulfides has been reported for the selective synthesis of 2-trifluoromethyl benzothiophenes and benzothiazoles via chlorotrifluoroethylidene.5 In addition, the synthesis of 1-aryl-3,3,3-trifluropropynes were reported using sodamide and the corresponding chlorotrifluoroethylidene.5 Moreover, from the subsequent alkynes prepared from this alkylidine various heterocyles can be synthesize such as aryl trifluoromethyl-1,2,3-triazoles.6–8 Various methods for the synthesis of chlorotrifluoroethylidenes have been reported in the literature (Scheme 1). The chlorotrifluoroethylidene group has been synthesized using aldehydes and CF3CCl2ZnCl, which was prepared from CF3CCl3 and zinc powder. However, the selective formation of the chlorotrifluoroethylidene moiety requires an excess of zinc powder and acetic anhydride.9 The most common method for the synthesis of chlorotrifluoroethylidene derivatives is the reaction of aldehydes or ketones with fluorinated phosphorous based reagents such as PPh3CClCF3 or (EtO)2(O)PCClCF3COOEt. A major drawback of this approach is the difficulty in accessing their phosphoranes that requires an ozone depleting CFC reagent and which is not readily available.10 The synthesis of chlorotrifluoroethylidene compounds was also reported using organomercury reagents. This procedure requires a high reaction temperature and a long reaction time (71 h).11 A one-pot procedure was also reported for the synthesis of said alkene without isolation of the hydrazone by reacting the aldehyde with hydrazine hydrate followed by reaction with CF3CCl3 in the presence of copper(I) chloride.8 Toshiyuki and co-workers reported the synthesis of chlorotrifluoroethylidene by reacting aldehydes with halothane under Grignard reaction condition (Scheme 1).12 Hu and co-workers report a one-pot synthesis of chlorotrifluoroethylidene compounds using CF3CCl3 and triphenylphosphine.13
 | ||
| Scheme 1 Reported methods for synthesis of chlorotrifluoroethylidene derivatives. | ||
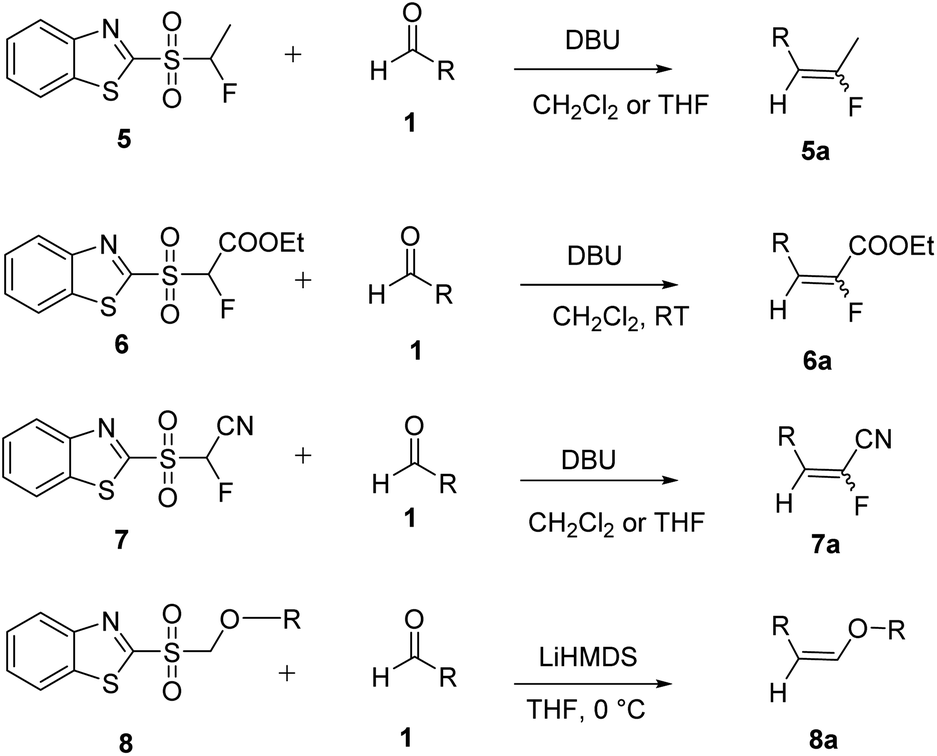
Julia and co-workers reported a one-step olefination reaction as an alternative to the HWE reaction.14 According to this methodology the sulfone reacts with aldehydes or ketones to afford alkenes (Scheme 2). A number of synthetic applications of this reaction have been reported in the literature.15–18 The selective preparation of fluoroalkenoates was reported using a modified Julia fluoroolefination.19 Similarly the use of alkyl substituted-(1,3-benzothiazol-2-ylsulfonyl)fluoroacetates as reagents for the synthesis of substituted fluoroacrylates was reported (Scheme 2).20 Various Julia olefination reactions have been reported in literature based on benzothiazole sulfones (Scheme 2).21
 | ||
| Scheme 2 Benzothiazole based reagents for olefination reaction. | ||
2. Results and discussion
Based on the Julia olefination using benzothiazole sulfones a retro-synthetic analysis of the compound of interest i.e. chlorotrifluoroethylidene leads to the readily available materials benzothiazole-2-thiol and halothane (Scheme 3). Herein, we report a synthetic procedure for addition of chlorotrifluoromethyl group at the aldehyde using 2-((1-chloro-2,2,2-trifluoroethyl)sulfonyl)1,3-benzothiazole a sulfone reagent 11. In an attempt to explore the scope and limitations of this methodology, a set of diverse aldehydes were reacted with sulfone reagent 11 in THF at −50 to −60 °C using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as base to give the corresponding olefins in good to moderate yields (Table 1). 2-[(1-Chloro-2,2,2-trifluoroethyl)sulfanyl]-1,3-benzothiazole 10 was synthesized using the reported procedure, wherein 2-mercapto benzothiazole 9 was reacted with an excess of halothane 4 in DMF, in the presence of sodium bicarbonate and sodium dithionite at 55 °C.22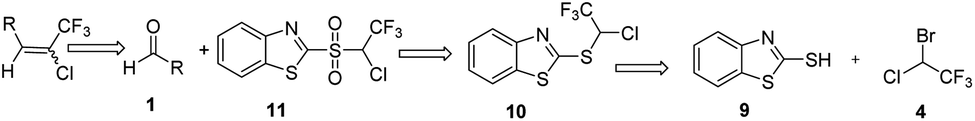 | ||
| Scheme 3 Retro-synthesis of chlorotrifluoroethylidene derivatives. | ||
| Entry no. | Substrate no. | Substrate | Product no. | Product | Yield (%) | E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z ratio* :Z ratio* |
|---|---|---|---|---|---|---|
| a Reagents and conditions: 11 (1.0 equiv.), DBU (2.1 equiv.), THF (15–20 w/v), −50 to −60 °C, RT, 1 h. *Relative ratio the of E/Z determined by 19F NMR for the isolated compounds. | ||||||
| 1 | 12 | 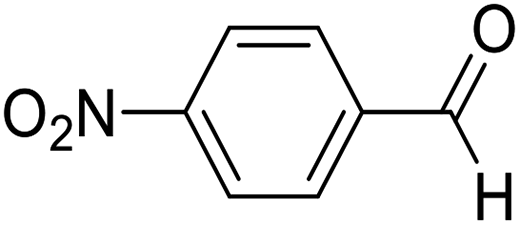 |
12a |  |
80 | 81:19 |
| 2 | 13 | 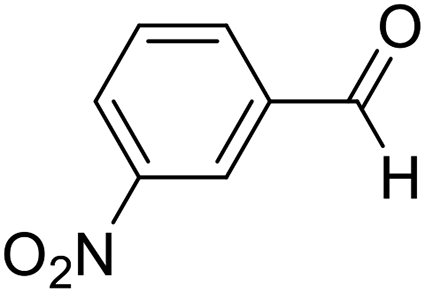 |
13a |  |
78 | 80:20 |
| 3 | 14 | 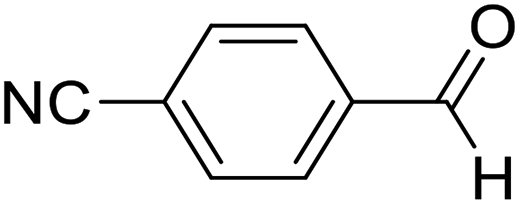 |
14a | 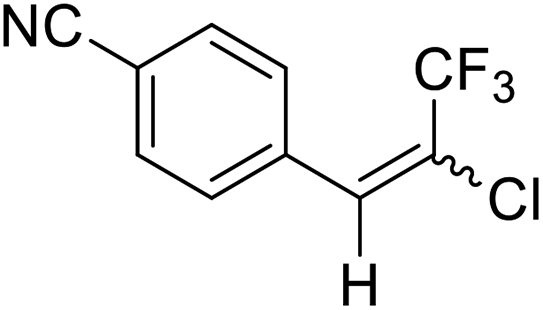 |
82 | 65:35 |
| 4 | 15 | 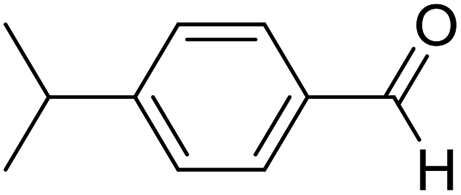 |
15a | 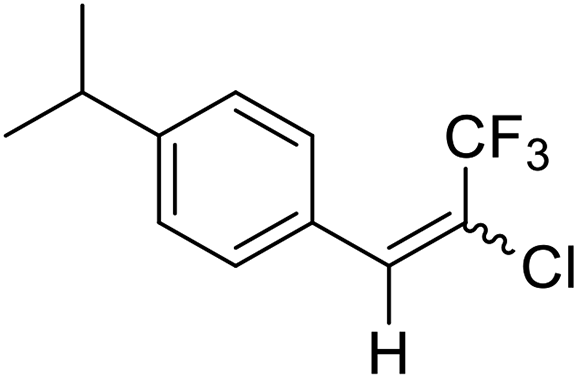 |
59 | 70:30 |
| 5 | 16 | 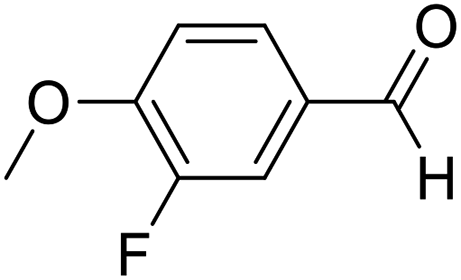 |
16a |  |
73 | 64:36 |
| 6 | 17 | 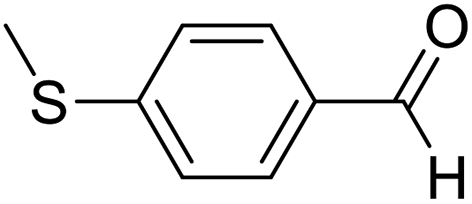 |
17a |  |
64 | 74:26 |
| 7 | 18 | 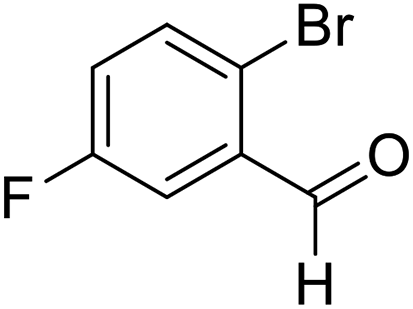 |
18a | 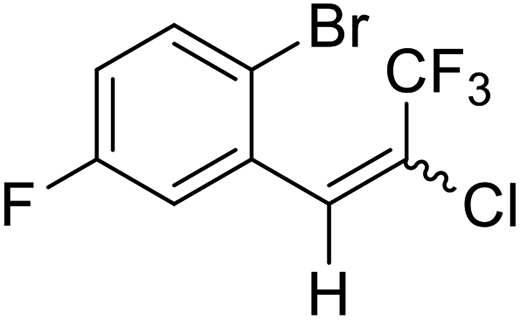 |
84 | 92:8 |
| 8 | 19 |  |
19a |  |
85 | 73:27 |
| 9 | 20 |  |
20a |  |
92 | 49:51 |
| 10 | 21 | 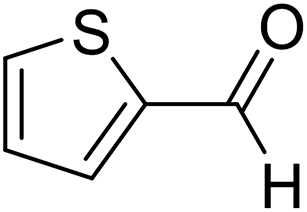 |
21a | 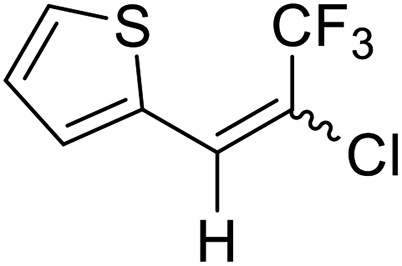 |
53 | Na* |
| 11 | 22 | 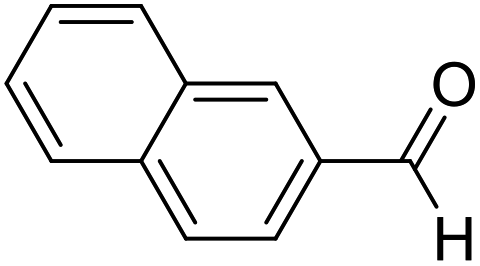 |
22a |  |
85 | 76:24 |
| 12 | 23 | 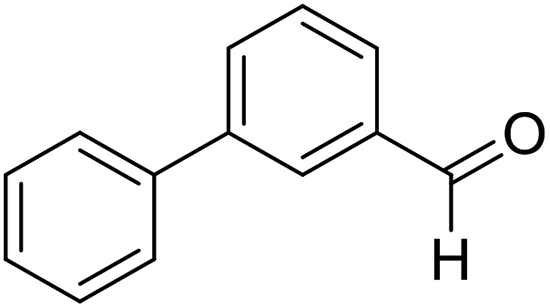 |
23a | 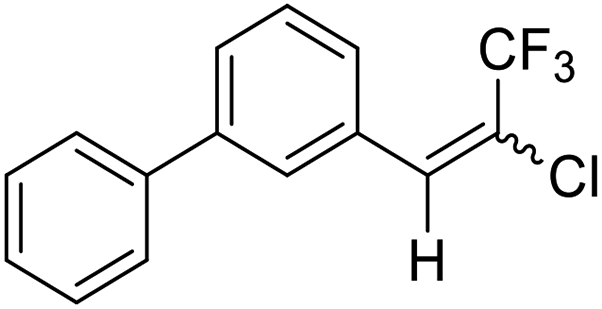 |
90 | 75:25 |
| 13 | 24 |  |
24a | 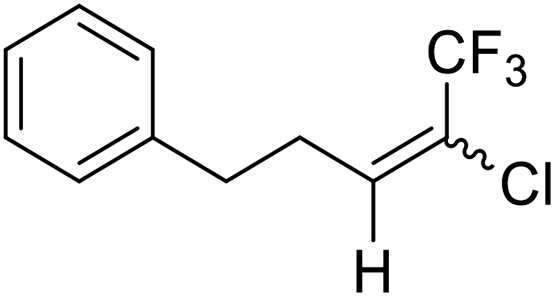 |
80 | 70:30 |
| 14 | 25 | 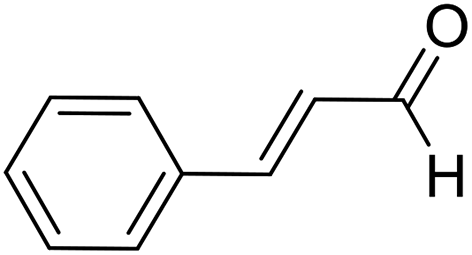 |
25a | 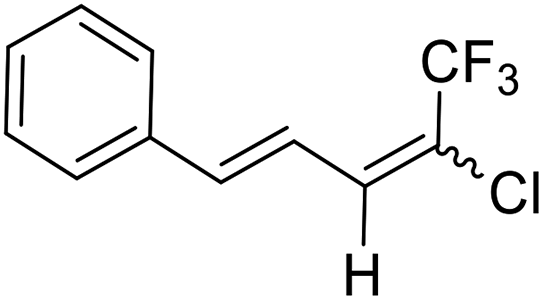 |
78 | 72:28 |
Various methods were investigated for the oxidation of the compound 10. Oxidation with mCPBA in CH2Cl2 did not proceed to completion under varied reaction conditions such as the molar ratio, time and temperature. Oxidation using OXONE® and sodium bicarbonate in a THF/water mixture did not proceed to completion even with the use of 10 equiv. of OXONE®, leaving an unconverted monooxidized intermediate. Oxidation with H2O2 and acetic acid proceeded well but in low yield. Hence, further optimization of the reaction conditions such as temperature and the molar ratio of H2O2 were studied. It was observed that when to a solution of compound 10 (1.0 equiv.) in acetic acid (3 volumes), 50 wt% H2O2 solution (15–20 equiv.) was added in drop-wise manner at 60 °C resulted in completion of reaction in a good yield to afford compound 11. The reaction mass was concentrated to 1 volume under vacuum and then ice water was added to precipitate the product sulfone 11, which was isolated by filtration, post neutralization. Recrystallization of the sulfone from 3 volumes of IPA resulted in the isolation of a white solid in 55–60% yield. Alternatively, it can be extracted in ethyl acetate post neutralization and column purified.
Lequeux and co-workers19 have extensively studied the olefination of carbonyl compounds and reported the effect of various bases, temperature and solvent on the E/Z selectivity. In the optimization of reaction conditions for olefination of aldehydes viz. compound 12 with sulfone reagent 11, among the different bases tried such as CsCO3, LDA, NaHMDS, DBU and potassium tert-butoxide, only DBU gave the product in good yield in solvent THF at −50 to −60 °C (Scheme 4). The structure and configuration of the isolated compounds were confirmed by 1H, 13C and 19F NMR spectroscopy. The assignment of E/Z isomers was done based on 19F NMR δ values (E isomer δ value is −61 and Z isomer δ value is −67). 19F NMR spectroscopy showed the “E” isomer as the major product. Compounds 12–14 (Table 1) containing electron-withdrawing groups on the aromatic ring gave good yields, as did halogen substituted compounds 18 and 19. However, it was observed that electron-donating substituents on the aromatic ring such as compounds 15, 16 and 17 resulted in low yields. This methodology also worked well for heterocyclic compound 20; however, a low yield was obtained in the case of compound 21. The naphathyl 22 and biphenyl 23 compounds also resulted in good yields of the corresponding olefins.
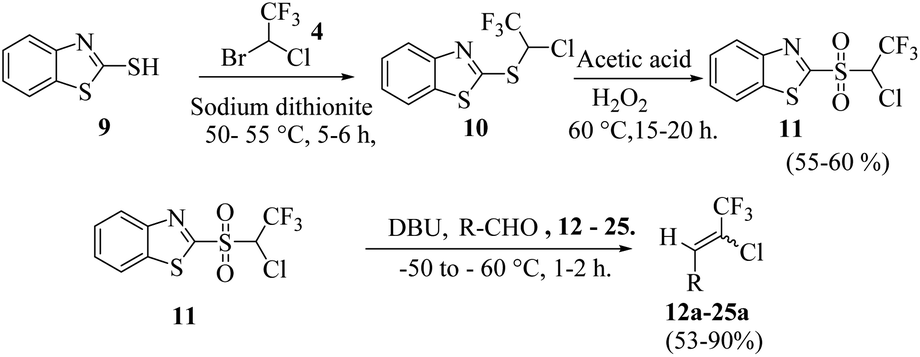 | ||
| Scheme 4 Synthesis of chlorotrifluoroethylidene derivatives. | ||
We then turned our attention towards the olefination of ketones. However, we found that this methodology under, the optimized conditions, did not work for ketones.19 The selected compounds were 4-nitroacetophenone, 4-fluoroacetophenone, 4-methoxyacetophenone and benzophenone examined and that the reaction was unsuccessful. The sulfone reagent 11, itself decomposed under the reaction conditions, as observed by TLC during reaction monitoring. The fact that the reaction failed for aromatic ketones is not clearly understood by us but may be due to a retroreaction as noted by Lequeux and co-workers.14,19
3. Conclusions
In conclusion, we have reported a method for the synthesis of R–CHCClCF3 compounds under mild conditions using a new benzothiazole sulfone halothane based reagent. This stable reagent is easy to synthesize from readily available starting materials. This method allows the preparation of a variety of chlorotrifluoroethylidene derivatives in moderate to good yields with the biggest advantage that it provides the “E” isomer as the major product compared to the Wittig methodology where the “Z” isomer is major product.
4. Experimental section
4.1. Procedure for preparation of 2-[(1-chloro-2,2,2-trifluoroethyl)sulfanyl]-1,3-benzothiazole (10)
2-Bromo-2-chloro-1,1,1-trifluoroethane (Halothane) (167.0 g, 847.7 mmol) was added dropwise to a stirred suspension of 1,3-benzothiazole-2-thiol 9 (50.0 g, 299 mmol), sodium bicarbonate (50.0 g, 595 mmol) and 85% sodium dithionite (104.0 g, 597 mmol) in 200.0 ml of DMF at 25 to 30 °C within 30 minutes. The reaction mixture was heated and stirred for 5–6 h at 55 °C. The reaction mass was poured into 200 ml water and extracted with 3 × 100 ml ethyl acetate. Combined organic layer was washed with water followed by brine and dried over anhydrous sodium sulphate. The solvent was removed under vacuum and further purified by column chromatography to yield 2-[(1-chloro-2,2,2-trifluoroethyl)sulfanyl]-1,3 benzothiazole 58.3 g.Yield: 69%; Pale yellow liquid; 1H NMR (300 MHz, CDCl3): 8.12–8.09 (d, J = 9 Hz, 1H), 8.04–8.01 (d, J = 9 Hz, 1H), 7.57–7.54 (t, 1H), 7.52–7.43 (t, 1H), 7.26–7.19 (q, J = 6 Hz, 15 Hz, 1H) meets with reported compound; 19F NMR(282.4 MHz, DMSO-d6): −71.42 (s, CF3, −3F) (reported −73.09 at 188 MHz in CDCl3);22 GC-MS (ESI 70 eV +ve) m/z 284 (M + 1), 283, 248, 228, 184, 166, 135, 122, 108, 69, 50; GC% purity 99.52%.
4.2. Procedure for preparation of 2-[(1-chloro-2,2,2-trifluoroethyl)sulfonyl]-1,3 benzothiazole (11)
H2O2 50 wt% solution (220 g. 3.23 mol) was added dropwise to a stirred solution of 2-[(1-chloro-2,2,2-trifluoroethyl)sulfanyl]-1,3 benzothiazole 10 (51.0 g 179.76 mmol) in acetic acid (160.0 2.67 mmol) over a period of 8 h at 60 °C. The completion of reaction is monitored by TLC. The reaction mass was concentrated to 1 volume under vacuum. The pH of the reaction mass adjusted to 8 to 7 using saturated sodium bicarbonate solution and product was extracted with ethyl acetate. The organic layer was washed with water followed by brine and dried over anhy-drous sodium sulphate. The solvent removed under vacuum and purified by column chromatography to yield 2-[(1-chloro-2,2,2-trifluoroethyl)sulfonyl]-1,3-benzothiazole 32 g.Yield: 56%; white solid; mp 100–102 °C; 1H NMR (300 MHz, DMSO-d6): 8.47–8.42 (m, 1H), 8.41–8.34 (m, 1H), 7.84–7.80 (m, 1H), 7.79–7.74 (m, 1H), 7.62–7.55 (q, J = 6 Hz, 15 Hz, 1H);
13C NMR (75 MHz, DMSO-d6): 161.5 (Ar C, benzthiazole C, NC–S, −1C), 152.4 (Ar C ring junction attached to nitrogen, −1C), 137.9 (Ar C ring junction attached to sulphur, −1C), 129.6, 129.0, 125.9, 124.2 (Ar C, −4C), 126.9, 123.2, 119.4, 115.7 (q, JCF = 280.5 Hz, CF3, −1C), 69.4, 69.0, 68.5, 68.1 (q, JCF = 33.0 Hz, aliphatic C, Cl–C–CF3, −1C); 19F NMR (282.4 MHz, DMSO-d6): −66.05 (s, CF3, −3F); GC-MS (ESI 70 eV +ve) m/z 284 (M + 1), 315, 198, 170, 134, 117, 90, 69, 50; HRMS (ESI +ve) m/z [M + H]+ calcd for C9H6ClF3NO2S2: 315.9471, found 315.9475, m/z [M + Na]+ C9H5ClF3NNaO2S2: 337.9292, found 337.9294; GC% purity 94.1%.
4.3. Representative procedure for preparation of olefin
:5) to yield (E)-1-(2-chloro-3,3,3-trifluoroprop-1-en-1-yl)-4-nitrobenzene 12a. 1.28 g.Mixture E/Z 12a: yield: 80%; yellow oil; 19F NMR (282.4 MHz, DMSO-d6): −61.05 (s, CF3, −3F, E−isomer), −67.70 (s, CF3, −3F, Z-isomer); GC-MS (ESI 70 eV +ve) m/z 252(M + 1), 251, 221, 185, 169, 143, 120, 101, 75, 50 (similar fragments for both isomers-E/Z = 83.40:16.60); GC purity: 84.65%, RT – 33.85 min, E-isomer + 11.46% RT – 33.92 min, Z-isomer.
Pure E isomer of 12a: yellow oil; 1H NMR (300 MHz, DMSO-d6): δ 8.29–8.28 (d, J = 3 Hz, 2H), 7.92 (s, 1H), 7.62–7.59 (d, J = 9 Hz, 2H); 13C NMR (75 MHz, DMSO-d6): 147.9 (Ar C attached to NO2, −1C), 139.3(Ar C attached to olefinic bond –CC, −1C), 137.5(3)–137.50 (Olefinc C attached to Ar ring –CC, −1C), 130.1(4), 130.1(1) (Ar C meta to NO2, −2C), 124.1, 123.9 (Ar C ortho to NO2, −2C), 125.9, 122.2, 118.6, 115.0 (q, JCF = 272.25 Hz, CF3, −1C) 122.0, 121.5, 121.0, 120.5(5) (q, JCF = 37.5 Hz, olefinic C, =C–CF3Cl, −1C); GC-MS (ESI 70 eV +ve) m/z 252(M + 1), 251, 221, 185, 169, 143, 120, 101, 75, 50; GC purity: 99.65% (RT – 33.85 min, E-isomer).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Madhusudan Sarwate and Sachin Kakale from Navin Fluorine International Limited, India for their support in recording spectroscopic data. The authors are also thankful to Dr Owen Chambers and Dr G. S. K Surya Prakash for their valuable guidance.Notes and references
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- S. B. Vallejo, B. Lantano and B. A. Postigo, Chem.–Eur. J., 2014, 20, 1–25 CrossRef.
- M. Fujita, K. Kondo and T. Hiyama, Bull. Chem. Soc. Jpn., 1987, 60, 4385–4394 CrossRef CAS.
- C. Wang, L. H. Chen, C. L. Deng and X. G. Zhang, Synthesis, 2013, 45, 000A–000G Search PubMed.
- C. L. Li, X. G. Zhang, R. Y. Tang, P. Zhong and P. Li, J. Org. Chem., 2010, 75, 7037–7040 CrossRef CAS PubMed.
- H. Tamejiro, S. K. Ichi and F. Makoto, Bull. Chem. Soc. Jpn., 1989, 62, 1352–1354 CrossRef.
- G. Meazza and G. Zanardi, J. Fluorine Chem., 1991, 55, 199–206 CrossRef CAS.
- V. N. Korotchenko, A. V. Shastin, V. G. Nenajdenko and E. S. Balenkova, Tetrahedron, 2001, 57, 7519–7527 CrossRef CAS.
- M. Fujita and T. Hiyama, Tetrahedron Lett., 1986, 27, 3655–3658 CrossRef CAS.
- D. J. Burton, Z. Yang and W. Qiu, Chem. Rev., 1996, 96, 1641–1715 CrossRef CAS PubMed.
- D. Seyferth and D. C. Mueller, J. Am. Chem. Soc., 1971, 93, 3714–3720 CrossRef CAS.
- (a) T. Takagi, A. Takesue, A. Isowaki, M. Koyama, A. Ando and I. Kumadaki, Chem. Pharm. Bull., 1995, 4, 1071–1075 CrossRef; (b) W. Dmowski, J. Fluorine Chem., 2011, 132, 504–511 CrossRef CAS.
- M. W. Chen, X. Zhang, P. Zhong and M. L. Hu, Synth. Commun., 2009, 39, 756–763 CrossRef CAS.
- D. Chevrie, T. Lequeux, J. P. Demoute and S. Pazenok, Tetrahedron Lett., 2003, 44, 8127–8130 CrossRef CAS.
- R. Bellingham, K. Jarowicki, P. Kocienski and V. Martin, Synthesis, 1996, 285–296 CrossRef CAS.
- H. Hilpert and B. Wirz, Tetrahedron, 2001, 57, 681–694 CrossRef CAS.
- N. D. Smith, P. Kocienski and S. D. Street, Synthesis, 1996, 652–666 CrossRef CAS.
- A. B. Charrette and H. Lebel, J. Am. Chem. Soc., 1996, 118, 10327–10328 CrossRef.
- E. Pfund, C. Lebargy, J. Rouden and T. Lequeux, J. Org. Chem., 2007, 72, 7871–7877 CrossRef CAS PubMed.
- B. Zajc and S. Kake, Org. Lett., 2006, 8, 4457–4460 CrossRef CAS PubMed.
- G. K. S. Prakash and Z. Zhang, Progress in Fluorine Science, 2017, 289–337 CAS.
- Y. Pustovit, A. Alexeenko, S. Trofymchuk, O. Lukin and A. A. Tolmachev, Synthesis, 2010, 7, 1159–1165 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02481c |
| This journal is © The Royal Society of Chemistry 2020 |