
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA compatibility study on the glycosylation of 4,4′-dihydroxyazobenzene†
Jonathan Berry,
Guillaume Despras * and
Thisbe K. Lindhorst*
* and
Thisbe K. Lindhorst*
Otto Diels Institute of Organic Chemistry, Christiana Albertina University of Kiel, Otto-Hahn-Platz 3/4, 24118 Kiel, Germany. E-mail: tklind@oc.uni-kiel.de
First published on 5th May 2020
Abstract
Photoresponsive glycoconjugates based on the azobenzene photoswitch are valuable molecules which can be used as tools for the investigation of carbohydrate–protein interactions or as precursors of shape-switchable molecular architectures, for example. To access such compounds, glycosylation of 4,4′-dihydroxyazobenzene (DHAB) is a critical step, frequently giving heterogeneous results because DHAB is a challenging glycosyl acceptor. Therefore, DHAB glucosylation was studied using nine different glycosyl donors, and reaction conditions were systematically varied in order to find a reliable procedure, especially towards the preparation of azobenzene bis-glucosides. Particular emphasis was put on glucosyl donors which were differentiated at the primary 6-position (N3, OAc) for further functionalisation. The present study allowed us to identify suitable glycosyl donors and reaction conditions matching with DHAB, affording the bis-glycosylated products in fair yields and good stereocontrol.
Introduction
Since their historical use as dyes,1 azobenzene derivatives have received great interest as photosensitive molecular switches.2 This is due to the fact that the azobenzene N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond gives rise to two isomeric forms, a trans (E) and a cis (Z) isomer, which can be reversibly interchanged by light of different wavelengths.2 Moreover, substitution of the phenyl rings allows modification and fine-tuning of the photochromic properties of azobenzene photoswitches.
N double bond gives rise to two isomeric forms, a trans (E) and a cis (Z) isomer, which can be reversibly interchanged by light of different wavelengths.2 Moreover, substitution of the phenyl rings allows modification and fine-tuning of the photochromic properties of azobenzene photoswitches.
The phenyl rings of azobenzene derivatives can be further functionalised in order to include the photoswitch into various molecular architectures and probes. Hence, the azobenzene hinge has found applications in liquid crystals,3 fast information-transmitting materials,4 photoswitchable catalysts,5 or actuators.6 Azobenzene photoswitches were also extensively used in supramolecular chemistry, in particular in light-driven molecular machines,7 and importantly, they were incorporated into biomolecules,2b,8 such as peptides9 and proteins,10 nucleic acids,11 lipids,12 and also carbohydrates3b–d,13,14,15,16 to enable photocontrol of molecular features and biological activity.
As the distance between the two para-positions of azobenzene varies between roughly 9 Å in the trans state and 5.5 Å in the cis form (Fig. 1a), isomerisation of the NN double bond offers an opportunity to switch the relative orientation of molecular moieties ligated to both ends of the azobenzene. In glycobiology, this has been employed to study carbohydrate–lectin interactions in relation to the spatial presentation of the connected glyco ligands. In 2002, Jayaraman et al. first described the synthesis of azobenzene glycoconjugates and their investigation in lectin binding studies.17 Since then, azobenzene glycoconjugates have gained increasing interest in the glycosciences. Our group has recently mounted azobenzene glycoconjugates onto gold surfaces in the form of glyco-SAMs (self-assembled monolayers).18 Here, the azo group was employed as a molecular hinge in order to switch the orientation of α-D-mannoside ligands terminating the SAMs (Fig. 1b). By photoisomerization, the orientation of the sugar ligands on the surface can be altered, allowing for the control of mannose-specific adhesion of bacterial cells.19 More recently, we have introduced photoresponsive glycoazobenzene macrocycles (Fig. 1c), which are shape-switchable and capable of reversible modification of chiroptical or solubility properties.20 Along the same lines, J. Xie and colleagues reported the synthesis of azobenzene glycomacrolactones.21
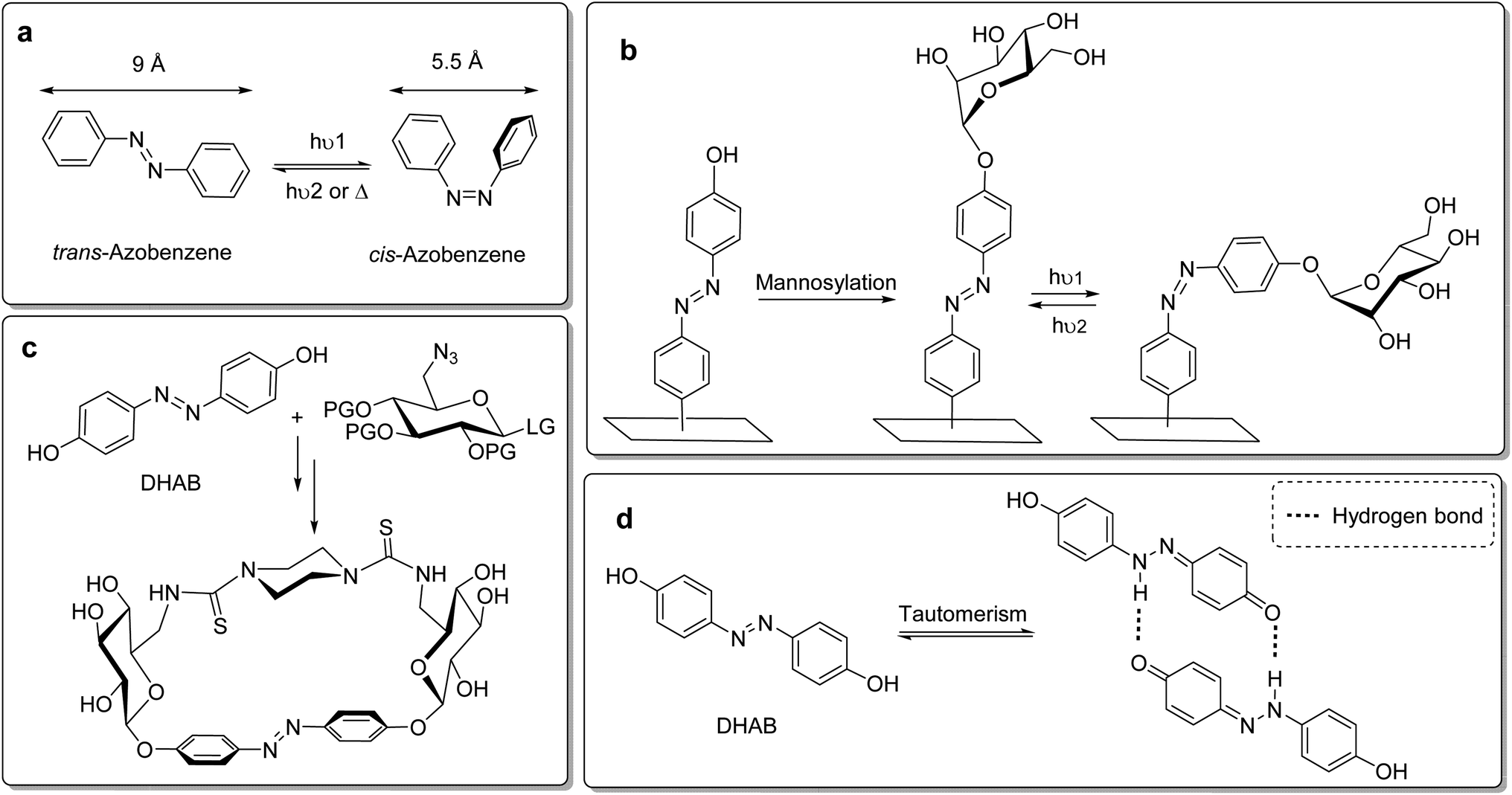 | ||
| Fig. 1 Photoisomerisation of azobenzene and examples of photoresponsive glycoconjugates. (a) Light or heat-induced isomerisation of azobenzene; (b) mannosylation of hydroxyazobenzene mounted onto a surface leads to glyco-SAMs where glyco ligand orientation can be photoswitched; (c) glycosylation of 4,4′-dihydroxyazobenzene (DHAB) leads to shape-switchable glycoazobenzene macrocycles; (d) tautomeric equilibrium of DHAB with its hydrazoquinone form and possible intermolecular hydrogen bonding promoting the hydrazoquinone tautomer. | ||
In such work, the glycosylation of hydroxy-functionalised azobenzene served as a key step in the direct conjugation of the carbohydrate moiety and the photoswitchable azobenzene unit. We have typically employed O-glycosyl trichloroacetimidates as glycosyl donors to achieve azobenzene glycosides in good yields.18,22 However, the bis-glycosylation of 4,4′-dihydroxyazobenzene (DHAB), as it was used for the preparation of azobenzene glycomacrocycles for example (Fig. 1c), is frequently impaired by poor to moderate yields.10d,20a This is due to the decreased nucleophilicity of the hydroxy groups in para-positions of the azobenzene, which is based on a tautomerism with the corresponding hydrazoquinone form, affecting both para-OH groups in DHAB (Fig. 1d). This tautomerism is promoted by intermolecular hydrogen bonds.23
We figured that, in order to improve the yields of DHAB bis-glycosides and to broaden the scope of azobenzene glycosides, we would have to study alternative glycosylation methods rather than to modify the azobenzene acceptor diol. Consequently, we commenced a study on the bis-glycosylation of DHAB, employing different glucosyl donors with variation of the anomeric leaving group as well as protecting groups. We were especially interested to improve the yields of DHAB bis-glycosylation with 6-azido-6-deoxy-functionalised glucosyl donors, as 6-functionalisation enables incorporation of the glycoazobenzene switch into larger molecular constructs.20a
Results and discussion
Preparation of glucosyl donors
Four common types of glucosyl donors were investigated in the glycosylation of DHAB, thioglucosides, glucosyl bromides, glucosyl sulfoxides, and O-glucosyl acetimidates (Chart 1). In most cases, the sugar hydroxy groups were protected as acetyl or benzoyl esters, respectively, to guarantee the stereoselective formation of 1,2-trans-glucosides based on the participating effect of the 2-O-acyl group. Two of the employed donors were silylated to achieve so-called super-armed glycosyl donors (vide infra).24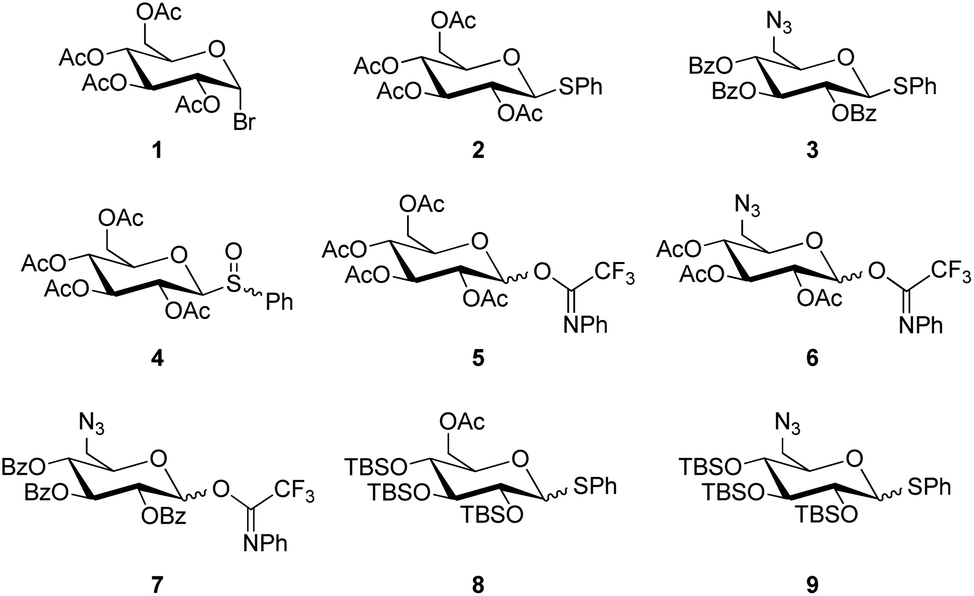 | ||
| Chart 1 Structures of glycosyl donors used in the present study. | ||
The glucosyl donors 1, 2, 4, 5 and 6 were prepared starting from D-glucose according to standard procedures (cf. ESI†).
The preparation of the 6-azido-6-deoxy-modified glucosyl donors 3 and 7 started from levoglucosan (Scheme 1), which was first perbenzoylated with benzoyl chloride in pyridine to give 11 in 92% yield. Subsequent opening of the protected 1,6-anhydroglucose with (phenylthio)trimethylsilane (TMSSPh) in the presence of zinc iodide at 120 °C under microwave heating gave thioglucoside 12 in 98% yield with complete β-selectivity. Then, the free primary hydroxy group was substituted by an azide under Bose-Mitsunobu25 conditions to afford 3 in 71% yield. Trichlorocyanuric acid (TCCA)-promoted hydrolysis of 3 in an acetone/water mixture yielded the reducing intermediate, which in turn reacted with (N-phenyl)trifluoroacetimidoyl chloride under basic conditions to provide the glucosyl donor 7 in 76% yield over two steps.
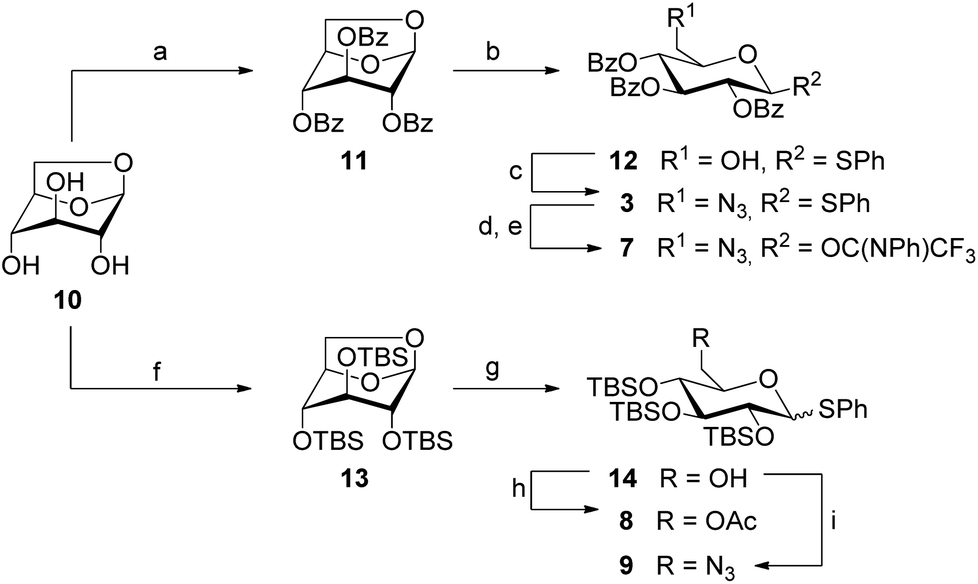 | ||
Scheme 1 Preparation of glucosyl donors 3 and 7–9. Reagents and conditions: (a) BzCl (6 eq.), pyridine, rt, 92%; (b) ZnI2 (4 eq.), TMSSPh (2.5 eq.), CH2Cl2, μW (150 W), 120 °C, 25 min, 98%; (c) PPh3 (1.5 eq.), DIAD (2.5 eq.), DPPA (1.5 eq.), THF, −15 °C to rt, 71%; (d) TCCA (1 eq.), acetone/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), rt; (e) ClC(NPh)CF3 (1.5 eq.), Cs2CO3 (1.5 eq.), CH2Cl2, rt, 76% over 2 steps; (f) TBSCl (6 eq.), NMI (7 eq.), I2 (2.5 eq.), pyridine, rt, 80%; (g) ZnI2 (4 eq.), TMSSPh (2.5 eq.), CH2Cl2, rt, 96%; (h) Ac2O (1.5 eq.), DMAP (0.2 eq.), pyridine, rt, 95%; (i) PPh3 (1.5 eq.), DIAD (2.5 eq.), DPPA (1.5 eq.), THF, −15 °C to rt, 85%; DIAD = diisopropyl azodicarboxylate; DPPA = diphenylphosphorylazide; TCCA = trichlorocyanuric acid; TBSCl = t-butyldimethylchlorosilane; NMI = N-methylimidazole; TMSSPh = (phenylthio)trimethylsilane. :1), rt; (e) ClC(NPh)CF3 (1.5 eq.), Cs2CO3 (1.5 eq.), CH2Cl2, rt, 76% over 2 steps; (f) TBSCl (6 eq.), NMI (7 eq.), I2 (2.5 eq.), pyridine, rt, 80%; (g) ZnI2 (4 eq.), TMSSPh (2.5 eq.), CH2Cl2, rt, 96%; (h) Ac2O (1.5 eq.), DMAP (0.2 eq.), pyridine, rt, 95%; (i) PPh3 (1.5 eq.), DIAD (2.5 eq.), DPPA (1.5 eq.), THF, −15 °C to rt, 85%; DIAD = diisopropyl azodicarboxylate; DPPA = diphenylphosphorylazide; TCCA = trichlorocyanuric acid; TBSCl = t-butyldimethylchlorosilane; NMI = N-methylimidazole; TMSSPh = (phenylthio)trimethylsilane. | ||
In analogy to the preparation of 11, persilylation of levoglucosan was envisaged for the synthesis of the silylated glucosyl donors 8 and 9. However, this reaction step required an optimization (cf. ESI†) to avoid the formation of the undesired 2,4-disilyated levoglucosan derivative. Finally, the persilylated levoglucosan derivative 13 was obtained in 80% yield by the addition of iodine to a mixture of 10, TBSCl and N-methylimidazole in pyridine.26
When the following ring opening step with 13 was performed under microwave heating as employed for the acyl-protected analogue 11, degradation was observed. Lowering the reaction temperature to 80 °C only slightly improved the reaction, but when the reaction was carried out at room temperature, it afforded the desired 6-OH-free thioglucoside 14 in 96% yield as an anomeric mixture (α:β = 1:4). This result complies with the higher reactivity of the armed silylated sugar derivatives in comparison to disarmed acylated analogues. The primary alcohol function in 14 was then acetylated to afford the glucosyl donor 8 in 95% yield, and the targeted 6-azido-6-deoxy glucosyl donor 9 was obtained in 85% under Bose-Mitsunobu conditions.
Glycosylations
Then, with nine different glucosyl donors in hand (cf. Chart 1), the bis-glycosylation of DHAB27 was investigated (Scheme 2). The results are summarized in Table 1. In previous work, O-glycosyl trichloroacetimidates were used for DHAB bis-glycosylation giving rise to yields between 30 and maximal 50%.10d,20a In order to improve the bis-glycosylation of DHAB, we first assessed glucosyl bromide 1, as silver salts, which are employed to activate Koenigs–Knorr-type glycosyl donors, should be compatible with an azobenzene acceptor. However, regardless of whether silver carbonate or silver oxide was used as promoter,28 only hydrolysis of the glucosyl donor occurred while the acceptor was nearly fully recovered (Table 1, entry 1). Next, we employed thioglucosides, as these donors present several great advantages; indeed thioglycosides are bench-stable, can be easily prepared in high yields and are compatible with a wide range of reaction conditions. When the acetyl-protected donor 2 was reacted under standard N-iodosuccinimide (NIS)/triflic acid (TfOH) activation,29 complete donor hydrolysis was observed (entry 2). When NIS was replaced by NBS (N-bromosuccinimide) a similar result was observed (entry 3). In both cases various DHAB-derived side products were formed, including the N-dehydro-N-iodohydrazoquinone tautomer (cf. ESI†). These results indicate that, the azophenol/hydrazoquinone tautomerism of DHAB (cf. Fig. 1d) not only effects a decreased oxygen nucleophilicity but also increases the nucleophilicity of the nitrogen atoms of the azo group. Hence, especially with soft electrophiles such as iodonium ions, N-halogenation of the azo group competes with the activation of the glycosyl donor and inhibits the reactivity of the acceptor.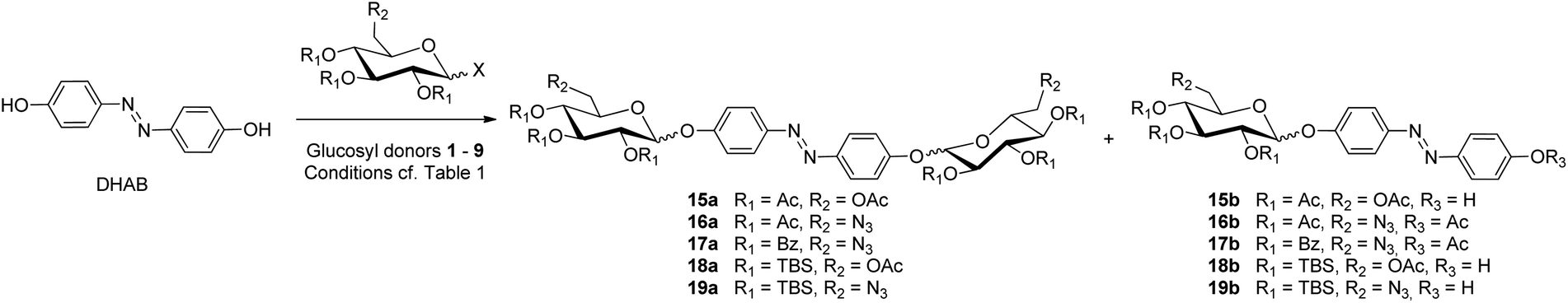 | ||
| Scheme 2 Glycosylation of DHAB with different glycosyl donors. The desired bis-glucosylated products are numbered with “a”, the monoglycosylated by-products with “b”. | ||
| Entry | Donorb | Promoter (eq./donor) | Base (eq./DHAB) | Solvent | Temp. | Bis-glycoside, yield, αβ:ββ, monoglycoside, yield, α:β |
|---|---|---|---|---|---|---|
| a All reactions were performed under dry conditions with addition of 3 Å molecular sieves (MS) and with [DHAB] = 0.05 mol × L−1 unless otherwise stated.b 2.2 eq. of donor were used unless otherwise stated.c 4 eq. of donor were used.d A solution of the acceptor was added to a mixture of donor and promoter; DTBMP = 2,6-di-tert-butyl-4-methylpyridine; DMTST = dimethyl(methylthio)sulfonium trifluoromethanesulfonate; [bmim][OTf] = 1-butyl-3-methylimidazolium trifluoromethanesulfonate; BSP = 1-benzenesulfinyl piperidine; NIS = N-iodosuccinimide; NBS = N-bromosuccinimide. | ||||||
| 1 | 1 | Ag2CO3 (1.1) or Ag2O (2) | — | MeCN | rt | Donor hydrolysis |
| 2 | 2 | NIS/TfOH (1.5/0.05) | — | MeCN | 0 °C | Donor hydrolysis |
| 3 | 2 | NBS/TfOH (1.5/0.1) | — | MeCN | rt | Donor hydrolysis |
| 4 | 3 | MeOTf (3.0) | DTBMP (2.2) | CH2Cl2 | rt | Donor hydrolysis |
| 5 | 3 | DMTST (1.5) | DTBMP (2.2) | CH2Cl2 | −30 °C | No activation |
| 6 | 4c | Tf2O (1.0) | DTBMP (3) | CH2Cl2 | −78 °C | Donor hydrolysis |
| 7 | 5 | BF3·OEt2 (1.1) | — | MeCN | rt | 15a, 68%, 0:1, 15b, not observed |
| 8 | 6 | BF3·OEt2 (1.1) | — | MeCN | rt | 16a, 48%, 0:1, 16b, 14%, 0:1 |
| 9 | 7 | BF3·OEt2 (1.1) | — | MeCN | rt | 17a, 38%, 0:1, 17b, 25%, 0:1 |
| 10 | 8 | MeOTf (3.0) | DTBMP (2.2) | CH2Cl2 | rt | 18a, 50%, 1:5, 18b, 20%, 1:10 |
| 11 | 8 | MeOTf (3) | DTBMP (2.2) | MeCN | rt | 18b, 17%, 1:5 |
| 12 | 8 | MeOTf (3) | DTBMP (2.2) | CH2Cl2/[bmim][OTf] (10:1) |
rt | 18a, 7%, 1:6, 18b, 31%, 1:12 |
| 13d | 8 | BSP/Tf2O (2.2/2.2) | DTBMP (2.2) | CH2Cl2/[bmim][OTf] (9:1) |
−90 °C | 18a, 18% 1:10, 18b, 12% 1:5 |
| 14 | 9 | MeOTf (3.0) | DTBMP (2.2) | CH2Cl2 | rt | 19a, 10%, 1:4, 19b, 14%, 1:8 |
As it was not possible to activate thioglycosides with halogenium-donating promoters, the benzoylated thioglucoside 3 was activated with methyl triflate (MeOTf)30 or dimethyl(methylthio)sulfonium triflate (DMTST).31 Bearing the azophenol/hydrazoquinone tautomerism in mind, we figured that acidic media may favor the formation of the hydrazoquinone tautomer. Therefore, we buffered the following glycosylation reactions with 2,6-di-tert-butyl-4-methylpyridine (DTBMP), which is known as an effective proton scavenger. Unfortunately, glycosylation of DHAB with 3 under these conditions (entries 4 and 5) failed. While the glucosyl donor was hydrolyzed or remained unreacted, again a number of DHAB-derived by-products were formed, presumably including O-methylated derivatives. The disappointing results with the thioglycosides led us to consider the glucosyl sulfoxide 4 as glycosyl donor, as it should be effective for the glycosylation of unreactive alcohols according to Kahne.32 When DHAB was treated with 4 in the presence of triflic anhydride (Tf2O) and DTBMP at −78 °C, the donor was completely consumed after 1 h, presumably transformed into the corresponding triflate, while DHAB remained unreacted (entry 6). Prolonging the reaction time and increasing the temperature led to the hydrolyzed donor and the unreacted acceptor. Even though Kahne et al. have described the successful glycosylation of sterically hindered and electronically deactivated phenols with sulfoxides, this method was not successful with DHAB, again underlining the limited reactivity of this azobenzene diol.
Next, we turned our attention to the O-(glucosyl)-N-phenyltrifluoroacetimidate donors. Indeed, the acetylated donor 5 under BF3-etherate activation afforded satisfying 68% of the desired bis-glycosylated compound 15a as a single ββ-anomer (entry 7). The monoglycosylated product 15b was not formed.
After this encouraging result, the same method was employed to introduce a 6-azido function into the azobenzene glycoside using the 6-azido-6-deoxy-functionalised glucosyl donor 6 (entry 8). This reaction was again successful, however with lower yields, 48% of 16a and 14% of the monoglycosylated 16b, again under complete β-selectivity. The benzoyl-protected analogue of 6, the acetimidate 7, under the same conditions led to slightly lower yields, affording the pure ββ-anomer 17a in 38% along with 25% of pure β-monoglycosylated 17b (entry 9).
At this stage of the study, the armed thioglycosides 8 and 9 were assessed for the bis-glycosylation of DHAB. As a conclusion from the results obtained with the disarmed thioglucosides (entries 2–5), MeOTf was chosen for the activation. In addition, in order to avoid acid-mediated intermolecular silyl transfer, the reaction medium was buffered with DTBMP. Satisfyingly, when DHAB was reacted with the armed donor 8 under these conditions (entry 10), 50% of the bis-glycosylated product 18a (αβ:ββ = 1:5) and 20% of the monoglycosylated compound 18b (α:β = 1:10) were obtained, however as anomeric mixtures. As DHAB is only partially soluble in CH2Cl2, 8 was employed under identical conditions but in acetonitrile (entry 11), and this reaction furnished the monoglycosylated product 19b in 17% yield as the sole glycoside along with methylated DHAB derivatives.33 This result indicates that the low solubility of DHAB in CH2Cl2 favors glycosylation over methylation of the phenol OH group as well as bis-glycosylation over monoglycosylation. Compared to the glycosylation in CH2Cl2, the α:β ratio of 19b which was obtained in acetonitrile is somewhat unexpected. As acetonitrile is known as a participating solvent, an α-nitrilium intermediate should occur, favoring the formation of the β-anomer.
Next several buffering bases were assessed (cf. ESI†). While none of the investigated bases led to a convincing improvement of the yields or anomeric selectivities, DTBMP was the base of choice to avoid the undesired O-methylation of DHAB. We also examined the use of the ionic liquid 1-butyl-3-methylimidazolium trifluoromethanesulfonate ([bmim][OTf]) as a co-solvent in CH2Cl2.34 In this solvent mixture, DHAB is completely dissolved and the activation of the thioglycoside 8 with MeOTf in the presence of DTBMP (entry 12) again favored the monoglycosylated product 19b (31%) with a good anomeric selectivity (α:β = 1:12), while 19a was isolated in only 7% yield. As observed for the reaction in acetonitrile, O-methylated derivatives of DHAB were formed. Interestingly, the promoter system 1-benzenesulfinyl piperidine (BSP)/Tf2O35 could efficiently activate donor 8 without reacting with the DHAB acceptor (entry 13). This reaction provided 19a in 18% yield (αβ:ββ = 1:10) and 19b in 12% (α:β = 1:5).
Then, the 6-azido-functionalised donor 9 was assessed under the glycosylation conditions which were found optimal for the armed donor 8 (entry 10). This reaction afforded the bis-glycoside 20a in 10% yield (αβ:ββ = 1:4) and the monoglycosylated compound 20b in 14% yield (α:β = 1:8) (entry 14). When the BSP/Tf2O system was applied with donor 9 only hydrolysis was observed. As observed with the acylated donors 6 and 7, the 6-azido group seems to have a detrimental effect on the glycosylation of DHAB. This is consistent with previous reports of glycosylations involving 6-azido-6-deoxy-glycosyl donors and different kinds of phenolic acceptors, yielding the products in low to moderate yields.10c,d,20a,36,37
Conclusions
We carefully investigated the glycosylation of 4,4′-dihydroxyazobenzene with a range of glycosyl donors displaying different reactivities. Our study provided useful data to delineate the scope and limitations of this glycosylation reaction and allows to foresee mismatch cases for DHAB. Four main features are underpinned by our results. First, DHAB is a poorly reactive acceptor, mainly because the nucleophilicity of the phenolic hydroxy groups is lowered by a tautomeric equilibrium between the azophenol and the hydrazoquinone form. Second, this tautomerism may increase the nucleophilicity of the nitrogen atoms in the azo bond, which leads to N-halogenation when halogenium-providing promoters are used. Third, when glycosylations are carried out in a buffered medium with electrophilic activators such as MeOTf or DMTST, the choice of the base is crucial to prevent competing O-methylation of DHAB. Fourth, the solubility of DHAB is an important parameter to control the degree of DHAB glycosylation. The lower the solubility of DHAB, the higher the yield of bis-glycosylated product.In conclusion, we believe our work reveals a new facet of the complexity of glycosylation reactions. Here, the azobenzene derivative DHAB was in the focus of our work and although glycosylation of DHAB remains challenging, we have identified glycosyl donors and activation methods compatible for the bis-glycosylation of this important photoswitch. In particular, the use of silyl-protected thioglycosides which are differentiated at the primary 6-position of the sugar ring is promising for the preparation of photoswitchable glycoconjugates and macrocycles. Efforts will be made for improving the glycosylation efficiency, regarding both stereoselectivity and degree of substitution, by varying the silyl protecting groups, the nature of the functional group at position 6 and further finely tune the reaction conditions.
Conflicts of interest
There are no conflicts of interest to declare about the authors.Acknowledgements
Financial support by the DFG (collaborative network SFB677) and FCI (Fonds der Chemischen Industrie) is gratefully acknowledged.Notes and references
- H. Zollinger, in Color Chemistry: Syntheses, Properties and Applications of Organic Dyes and Pigments, ed. P. M. Wallimann, Wiley-VCH, Weinheim, 2003, vol. 7, pp. 165–254 Search PubMed.
- (a) H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC; (b) A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- (a) L. Johnson, B. Ringstrand and P. Kaszynski, Liq. Cryst., 2009, 36, 176–185 CrossRef; (b) N. Laurent, D. Lafont, F. Dumoulin, P. Boullanger, G. Mackenzie, P. H. Kouwer and J. W. Goodby, J. Am. Chem. Soc., 2003, 125, 15499–15506 CrossRef CAS PubMed; (c) A. G. Cook, J. L. Wardell, N. J. Brooks, J. M. Seddon, A. Martínez-Felipe and C. T. Imrie, Carbohydr. Res., 2012, 360, 78–83 CrossRef CAS PubMed; (d) S. Abraham, S. Paul, G. Narayan, S. K. Prasad, D. S. Rao, N. Jayaraman and S. Das, Adv. Funct. Mater., 2005, 15, 1579–1584 CrossRef CAS.
- J. Gavía-Amorós and D. Velasco, Beilstein J. Org. Chem., 2012, 8, 1003–1017 CrossRef PubMed.
- P. Viehmann and S. Hecht, Beilstein J. Org. Chem., 2012, 8, 1825–1830 CrossRef CAS PubMed.
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071–1090 CrossRef CAS PubMed.
- (a) E. Wagner-Wysiecka, N. Łukasik and J. F. Biernat, J. Inclusion Phenom. Macrocyclic Chem., 2018, 90, 189–257 CrossRef CAS PubMed; (b) C. Gao, X. Ma, Q. Zhang, Q. Wang, D. Qu and H. Tian, Org. Biomol. Chem., 2011, 9, 1126–1132 RSC; (c) Y. Inoue, P. Kuad, Y. Okumura, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2007, 129, 6396–6397 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- (a) C. Renner, U. Kusebauch, M. Löweneck, A. G. Milbradt and L. Moroder, J. Pept. Res., 2005, 65, 4–14 CrossRef CAS PubMed; (b) G. A. Woolley, Acc. Chem. Res., 2005, 38, 486–493 CrossRef CAS PubMed.
- (a) H. Kaufman, S. M. Vratsanos and B. F. Erlanger, Science, 1968, 162, 1487–1489 CrossRef CAS PubMed; (b) I. Willner, S. Rubin and A. Riklin, J. Am. Chem. Soc., 1991, 113, 3321–3325 CrossRef CAS; (c) A. Müller and T. K. Lindhorst, Eur. J. Org. Chem., 2016, 1669–1672 CrossRef; (d) A. Müller, H. Kobarg, V. Chandrasekaran, J. Gronow, F. D. Sönnichsen and T. K. Lindhorst, Chem.–Eur. J., 2015, 21, 13723–13731 CrossRef PubMed.
- A. Heckel and G. Mayer, in The Chemical Biology of Nucleic Acids, ed. G. Mayer, Wiley-VCH, Chichester, 2010, vol. 13, pp. 279–306 Search PubMed.
- (a) J. Y. Wang, Q. F. Wu, J. P. Li, Q. S. Ren, Y. L. Wang and X. M. Liu, Mini-Rev. Med. Chem., 2010, 10, 172–181 CrossRef CAS PubMed; (b) A. Yavlovich, B. Smith, K. Gupta, R. Blumenthal and A. Puri, Mol. Membr. Biol., 2010, 27, 364–381 CrossRef CAS PubMed.
- (a) F. Hamon, F. Djedaini-Pilard, F. Barbot and C. Len, Tetrahedron, 2009, 65, 10105–10123 CrossRef CAS; (b) Y. Hu, R. F. Tabor and B. L. Wilkinson, Org. Biomol. Chem., 2015, 13, 2216–2225 RSC.
- H. Kobayashi, A. Friggeri, K. Koumoto, M. Amaike, S. Shinkai and D. N. Reinhoudt, Org. Lett., 2002, 4, 1423–1426 CrossRef CAS PubMed.
- (a) N. Drillaud, E. Banaszak-Léonard, I. Pezron and C. Len, J. Org. Chem., 2012, 77, 9553–9561 CrossRef CAS PubMed; (b) R. F. Tabor, M. J. Pottage, C. J. Garvey and B. L. Wilkinson, Chem. Commun., 2015, 51, 5509–5512 RSC.
- P. V. Jog and M. S. Gin, Org. Lett., 2008, 10, 3693–3696 CrossRef CAS PubMed.
- O. Srinivas, N. Mitra, A. Surolia and N. Jayaraman, J. Am. Chem. Soc., 2002, 124, 2124–2125 CrossRef CAS PubMed.
- (a) V. Chandrasekaran, H. Jacob, F. Petersen, K. Kathirvel, F. Tuczek and T. K. Lindhorst, Chem.–Eur. J., 2014, 20, 8744–8752 CrossRef CAS PubMed; (b) E. Fast, A. Schlimm, I. Lautenschläger, K. U. Clausen, T. Strunskus, C. Spormann, T. K. Lindhorst and F. Tuczek, Chem.–Eur. J., 2020, 26, 485–501 CrossRef CAS PubMed.
- T. Weber, V. Chandrasekaran, I. Stamer, M. B. Thygesen, A. Terfort and T. K. Lindhorst, Angew. Chem., Int. Ed., 2014, 53, 14583–14586 CrossRef CAS PubMed.
- (a) G. Despras, J. Hain and S. O. Jaeschke, Chem.–Eur. J., 2017, 23, 10838–10847 CrossRef CAS PubMed; (b) J. Hain and G. Despras, Chem. Commun., 2018, 54, 8563–8566 RSC.
- (a) C. Lin, S. Maisonneuve, R. Métivier and J. Xie, Chem.–Eur. J., 2017, 23, 14996–15001 CrossRef CAS PubMed; (b) C. Lin, S. Maisonneuve, C. Theulier and J. Xie, Eur. J. Org. Chem., 2019, 1770–1777 CrossRef CAS.
- V. Chandrasekaran, E. Johannes, H. Kobarg, F. D. Sönnichsen and T. K. Lindhorst, ChemistryOpen, 2014, 3, 99–108 CrossRef CAS PubMed.
- (a) N. Kurita, S. Nebashi and M. Kojima, Chem. Phys. Lett., 2005, 408, 197–204 CrossRef CAS; (b) G. Gabor, Y. F. Freí and E. Fischer, J. Phys. Chem. A, 1968, 72, 3266–3272 CrossRef CAS.
- M. Bols and C. M. Pedersen, Beilstein J. Org. Chem., 2017, 13, 93–105 CrossRef CAS PubMed.
- B. Lal, B. N. Pramanik, M. S. Manhas and A. K. Bose, Tetrahedron Lett., 1977, 23, 1977–1980 CrossRef.
- A. Bartoszewicz, M. Kalek, J. Nilsson, R. Hiresova and J. Stawinski, Synlett, 2008, 1, 37–40 Search PubMed.
- DHAB was prepared according to H. Tokuhisa, M. Yokoyama and K. Kimura, Bull. Chem. Soc. Jpn., 1996, 69, 2123–2130 CrossRef CAS.
- W. Koenigs and E. Knorr, Ber. Dtsch. Chem. Ges., 1901, 34, 957–981 CrossRef.
- (a) G. H. Vwneman, S. H. van Leeuwen and J. H. van Boom, Tetrahedron Lett., 1990, 31, 1331–1334 CrossRef; (b) P. Konradsson, U. E. Udodong and B. Fraser-Reid, Tetrahedron Lett., 1990, 31, 4313–4316 CrossRef CAS.
- H. Lönn, Carbohydr. Res., 1985, 139, 105–113 Search PubMed.
- P. Fügedi and P. J. Garegg, Carbohydr. Res., 1986, 149, 9–12 CrossRef.
- D. Kahne, S. Walker, Y. Cheng and D. Van Engen, J. Am. Chem. Soc., 1989, 111, 6881–6882 CrossRef CAS.
- O-Methylated DHAB derivatives could not be isolated after glycosylation as they were always obtained as mixtures with the corresponding glycosylated products. To confirm DHAB O-methylation, both the mono- and di-O-methylated derivatives were independently prepared and their NMR and TLC characteristics analysed (cf. ESI†). These data allowed us to confirm their structure as byproducts formed during the glycosylations.
- M. C. Galan, C. Brunet and M. Fuensanta, Tetrahedron Lett., 2009, 50, 442–445 CrossRef CAS.
- D. Crich and M. Smith, J. Am. Chem. Soc., 2001, 123, 9015–9020 CrossRef CAS PubMed.
- S. Sattin, M. Panza, F. Vasile, F. Berni, G. Goti, J. Tao, E. Moroni, D. Agard, G. Colombo and A. Bernardi, Eur. J. Org. Chem., 2016, 3349–3364 CrossRef CAS.
- H.-M. Chen and S. G. Withers, Carbohydr. Res., 2018, 467, 33–44 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02435j |
| This journal is © The Royal Society of Chemistry 2020 |