
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal decomposition and diffusion of methane in clathrate hydrates from quantum mechanics simulations
Dezhou Guo†
 ,
Hongwei Wang†,
Yidi Shen and
Qi An*
,
Hongwei Wang†,
Yidi Shen and
Qi An*
Department of Chemical and Materials Engineering, University of Nevada-Reno, Reno, Nevada 89557, USA. E-mail: qia@unr.edu
First published on 14th April 2020
Abstract
Clathrate hydrates are ice-like crystalline substances in which small gas molecules are trapped inside the polyhedral cavities of water molecules. They are of great importance in both scientific research and the petroleum industry because of their applications in modern energy and environmental technologies. To achieve an atomistic-level understanding of the diffusion and decomposition of trapped molecules in clathrate hydrate, we used methane hydrates (MHs) as the prototype system and examined the methane diffusion and decomposition mechanism by employing quantum mechanics (QM) and quantum mechanics molecular dynamics (QMD) simulations. Our QMD simulations illustrated that the initial decomposition reaction in MHs initiates from hydrogen transfer among water molecules and attacks by fragments of O and OH on CH4 molecules are responsible for the destruction of the methane molecules. Next, our QM simulations revealed that the methane molecule prefers to escape from the ice cage through the hexagonal face at low temperature. To suppress the methane diffusion, we demonstrated that the diffusion barrier is significantly enhanced by adding electron or hole carriers. This is because the extra electrons and holes enhance the electrostatic interaction between methane and water molecules, leading to an increased diffusion barrier. Thus, the clathrate hydrates could be stabilized by adding extra free electron or hole carriers.
1. Introduction
Clathrate hydrates are ice-like nonstoichiometric crystalline compounds in which small non-polar molecules or polar molecules with large hydrophobic moieties are encapsulated in hydrogen-bonded three-dimensional network consisting of polyhedral cages of water molecules.1,2 Clathrate hydrates are of great interest in both scientific research and industry because of their important energy applications.3 Huge deposits of natural gas in the form of clathrate hydrates are present in petrochemical production lines and hostile environments such as marine sediments and permafrost regions. Clathrate hydrates exhibit high stability under these complex nonequilibrium environments, which is a self-preservation effect. This self-preservation effect arises from the coating ice layer that prevents the further dissociation of the hydrate.4 Clathrate hydrates provide new energy sources of natural gas in the future as the current accessible fossil fuel resources have become depleted. Another important application is the storage and transportation of natural gas in the form of frozen hydrates, which is considered as an alternative to conventional storage approaches such as liquefied or compressed natural gas.1 Moreover, the CO2 hydrates have potential applications in the CO2 sequestration to stabilize the atmospheric concentrations of greenhouse gases.5Among the gas hydrates, methane hydrates (MHs) are especially attractive for many countries with limited fossil energy resources as they are abundantly detected in seafloor and permafrost zones and the estimated total amounts of carbon are twice than those in the current proved fossil fuels on earth.6 Therefore, MHs are one of the most promising energy alternatives to the fossil energies in the future for a long period.7,8 However, the exploitation techniques of MHs still confront many problems. Particularly, the rapid extract of MHs from the sea floor inevitably increases the local temperature, leading to fast chemical reactions and causing serious safety issues. Moreover, the increasing temperature may destabilize the structures of MHs so that large-scale methane molecules would enter atmosphere, aggravating global warming and resulting in dramatic geology problems.6 The safety issues are also of serious concerns for the engineering applications of MHs in energy storage and transportation, and petrochemical production. In particular, the accidental fire should be prevented as the natural gas hydrates are stored and transported using huge tankers. Thus, it is essential to understand the diffusion and decomposition mechanism of methane in MHs under external stimuli (e.g. heat, shock, or impact), which is crucial to prevent the spontaneous ignition and explosion of natural gas under extraction, storage and transportation.
For exploiting and utilizing the enormous energy resources contained in clathrate hydrates, it is of great importance to control the stability and combustion of clathrate hydrates, which are closely related to the diffusion and decomposition processes of trapped molecules. Many experiments have been conducted to investigate the decomposition and combustion behavior of the clathrate hydrates.9–11 For example, the combustion experiments on tetrahydrofuran (THF) hydrate indicated that the flame propagates in the direction against the air flow at a constant velocity of 0.2 mm s−1.12 Another combustion experiment was performed to investigate the propagation of a flame above a methane hydrate cake in an air laminar boundary layer, and the flame propagation velocity was determined to be ∼2 mm s−1.13 Recently, the combustion of methane hydrates has attracted intensive attention because complete combustion of methane hydrate is difficult to achieve by direct combustion.14 The reason is that the methane hydrate flame contains a high percentage of water vapor, leading to a low flame temperature. For example, the maximum flame temperature achieved experimentally is 1615 K with the water to methane molar ratio (H2O/CH4 = 1.4).15 In addition, Hwang et al.16 found that methane diffusion flame diluted with an appropriate amount of H2O may cause the enhancement of overall reaction rate. This might be because the radicals produced by water molecules, such as H, O, and OH, participate in the methane reactions and modify the methane reaction pathways from the reactants to the products. The involvement of radicals decelerates the methane oxidation reaction rate and leads to the unstable flame and the flame extinction. However, the fundamental mechanisms associated with decomposition processes of CH4 remain not fully understood. This is due to the complex environment and coupling of thermal, chemical, and mechanical degree of freedom, which is extremely difficult to unravel experimentally.
In theoretical studies, the atomistic mechanisms of nucleation, structure stabilities, and chemical and physical properties of MHs are extensively examined by employing molecular dynamics (MD) and quantum mechanics (QM) simulations.17–24 Previous MD simulations revealed that both water and methane molecules play an important role in the nucleation process of MHs. In particular, Guo et al. demonstrated that the 512 cage is able to absorb the methane molecules nearby and these methane molecules would in turn significantly prolong the lifetime of the labile clusters.17,18 Jacobson et al. claimed that clathrate cages only form and aggregate on the guest methane clusters, which causes the initiation of the nucleation.19,20 Khan showed that the CH4 molecule, stabilized in the 512 cages, has a stabilization energy of around 7 kcal mol−1.21,22 Ida et al. showed that the proton of methane in the cavities could form the weaken hydrogen bond with four water molecules based on the electronic states of enclathrated 512 and 51262 cages.23 Besides MHs, Willow et al. showed that the presence of methane could block the released hydrogen gases in hydrogen hydrates.24 However, many aspects remain unclear of the diffusion mechanism and decomposition reaction mechanism of methane in MHs, which is important for their energy applications.
In this work, we employed the quantum mechanics (QM) simulations to illustrate the diffusion and decomposition mechanism of methane in clathrate hydrates. Firstly, in order to examine the plausible decomposition mechanisms between CH4 and H2O molecules, we carried out the quantum molecular dynamic (QMD) simulations by heating up the methane clathrate system. Then we examined the diffusion mechanism of methane in clathrate using the QM simulations. The CH4 diffusion is expected to occur at much lower temperature than its decomposition, even before the melting of clathrate. Finally, to improve the stability of the MH, we added extra electrons or holes to the MH and examined how these free carriers influence the diffusion mechanism and barriers. Our study provides important information to understand the thermal decomposition and diffusion mechanisms of clathrates.
2. Simulation methods
2.1 Quantum mechanics simulations
All the simulations were performed using the periodic DFT code Vienna ab initio simulation package (VASP).25–27 The Perdew–Burke–Ernzerhof functional and the projector augmented-wave (PAW) method were applied for the exchange–correlation interaction and the core-valence interaction, respectively.28,29 The 2s22p2 electrons, 1s1 electrons and 2s22p4 electrons were constructed as the valence states for the C, H, and O elements, respectively in the pseudopotentials. The electron partial occupancies were determined using the tetrahedron method with Blöchl corrections.30 The kinetic energy cutoff was set to 500 eV for the plane-wave expansions. The convergence criteria were set to 1 × 10−5 eV and 1 × 10−2 eV·Å−1 for the electronic self-consistent field (SCF) procedure and ionic relaxation loop, respectively. The Brillouin zone integration was performed on Γ-centered symmetry-reduced 1 × 1 × 1 Monkhorst–Pack k-point grid meshes because of the large unit cell of the MH. We considered the van der Waals interactions using the DFT-D3 method with the Becke–Johnson damping approach.31We applied the Born–Oppenheimer molecular dynamics (BO-MD) approach implemented in the VASP package to examine the decomposition reactions during the cook-off simulations. Firstly, the equilibrium crystal structure was obtained at ambient conditions by heating the optimized system from 20 K to 300 K within 2 ps. Then, the system is equilibrated at 300 K for 2 ps using the NVT ensemble (constant volume, constant temperature, and constant number of atoms) with the Nose–Hoover thermostat. Finally, to accelerate the dissociation process of methane molecules, the system was heated from 300 K to 4350 K over a period of 30 ps with a constant heating rate of 135 K ps−1. A timestep of 0.5 fs was applied for integrating the equations of motion in the QMD simulations.
To analyze the fragments in the QMD simulations, we applied the molecular fragment recognition analysis algorithm using the connectivity matrix and bond orders at 0.1 ps intervals.32 Independent molecules are identified if their bond orders are smaller than 0.3, and then were assigned with specific ID numbers to track the reaction paths. The cutoff values of 0.3 are tested to make sure that the fragments during the reactions are appropriately described. The breaking or formation of bonds arising from thermal fluctuations was avoided by a time window of 1.0 ps in which every bond must exist for at least 1.0 ps. The bond orders were computed using the reactive force field (ReaxFF)33 for C–H–O systems by converting the QMD trajectory to the ReaxFF trajectory.
2.2 Methane diffusion
There are three common crystal structures of clathrate hydrates, such as type sI, type sII and type sH. Among these three types of structures, the MH crystal prefers to adopt the type I structure with two small 512 cages and six large 51262 cages per unit cell because of the size of guest molecules and specific temperature-pressure formation conditions.2 Thus, we selected the sI cubic structure MH, in which one methane molecule is trapped in each cage. The unit cell of methane hydrate consists of 46 water molecules, forming two types of cages: two small (512) cages which have the shape of a pentagonal dodecahedron, and six large (51262) ones which have a hexagonal truncated trapezohedron. In order to examine the methane diffusion mechanism, as well as the effects of free carriers (electrons and holes), we first constructed a single 51262 cage with one methane molecule inside as the initial state. Then we constructed the final state with the methane molecule outside the cage. Finally, we carried out the climbing image nudged elastic band (CI-NEB) method to determine the transition state by finding the saddle points and minimum energy paths between the initial and final states.34,35 Five intermediate images were optimized along the reaction path, with the constraints implemented by adding spring forces along the band between images and projecting out the component of the force because of the potential perpendicular to the band. The spring force constant was set up to −5.0 eV Å−2. The transition state was confirmed by the frequency calculations using the density functional perturbation theory approach.36 The migration mechanism was examined at zero temperature.To examine the effects of electrons (or holes), we repeated the CI-NEB calculations by adding (or removing) one addition electron in the system. The extra electrons (or holes) are simulated by adding a homogeneous background-charge offset the unbalanced charge. It is possible to add or subtract a certain number of electrons even fractional charge. To illustrate the mechanism of electron (or hole) effects, we performed the analyses of charge density difference between charged and neutral systems. The VESTA software37 is used to visualize crystal structures and plot charge densities. In our simulations, we added electrons or holes into the neutral system, which may lead to the interaction of periodic charged images. Thus, we create a large supercell with the lattice parameter of 25 Å along three perpendicular axes. Then we add one clathrate molecule into this supercell. To check the energy convergence of the simulation box, we expanded the supercell twice along one direction and added two electrons or holes to the system. The calculated electronic energies for electron and hole states in the large supercell are almost identical to the small supercell with the difference by less than 0.1%. Thus, our current model is accurate to predict the charged systems.
3. Results and discussion
3.1 MH crystal structure
The unit cell of MHs was optimized using a conjugate gradients method in the VASP package with a plane-wave basis set, as shown in Fig. 1.25–27 The optimized cubic cell parameters are a = b = 11.44 Å, and c = 11.43 Å (at 0 K), which lead to a density of 0.92 g cm−3, agreeing well with the X-ray experimental values of a = b = c = 12.00 Å, leading to a density of 0.81 g cm−3 at 273 K and 0.1 MPa.2 Thus, the DFT-D3 methods can accurately describe the crystal structure of MHs. | ||
| Fig. 1 The DFT-optimized crystal structure of methane hydrate (type sI) from two view directions to show the structure clearly. The C, H and O atoms are represented by brown, white and red balls, respectively. The structure is plotted using the VESTA visualization software.37 | ||
3.2 Reaction mechanisms at high temperatures from QMD simulations
To uncover the chemical reactions of the methane hydrate, we first examined the important fragments including HO, O and CO2 as representatives during the QMD cook-off simulation, as shown in Fig. 2. During the initial heating-up process, from 300 to 2000 K, no chemical reactions are observed. At ∼2000 K, we observed the first reaction in which the hydrogen atom is transferred between two water molecules, leading to the formation of OH and (H3O)+. This hydrogen transfer phenomenon was commonly observed in the hydrated materials in QMD simulations.38 As the temperature continuously increases to ∼3400 K, hydrogen transfer between water molecules occurs more frequently and rapidly, producing a large number of isolated H, HO, H3O and O fragments. However, all CH4 molecules remain inert during this period. This is because of the strong C–H bond with the bond energy of ∼98.7 kcal mol−1.39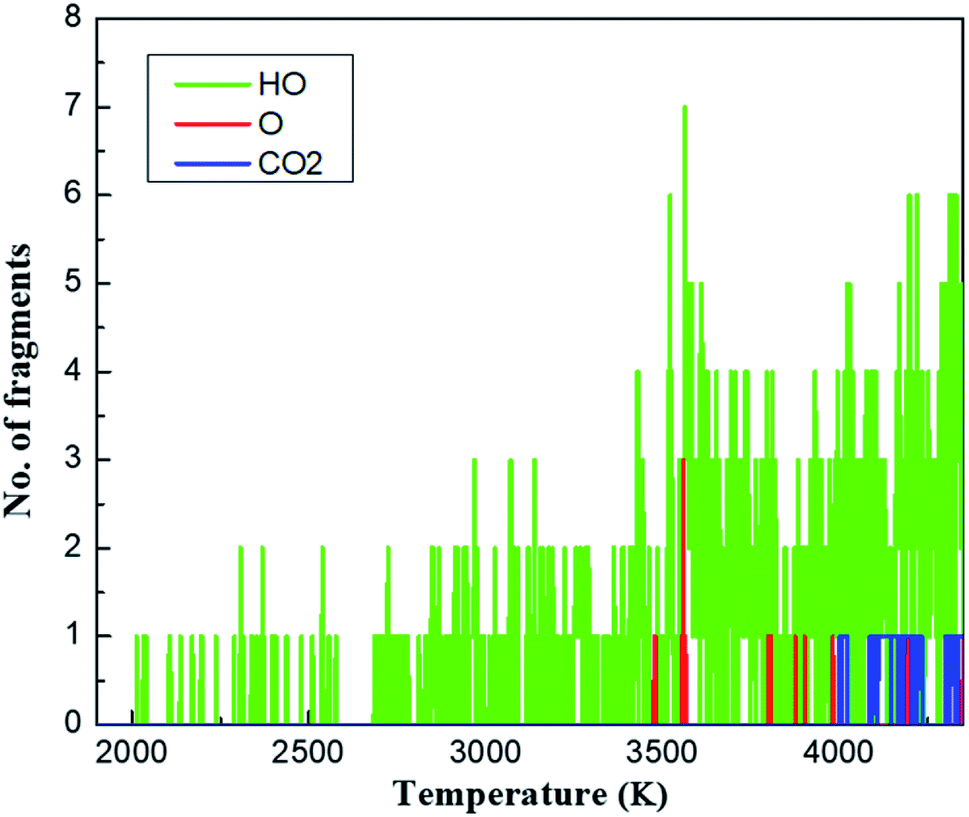 | ||
| Fig. 2 Analysis of species of HO, O and CO2 for the decomposition process heated from 300 K to 4350 K over 30 ps. | ||
To illustrate the details of how the methane molecules are decomposed, we plot key snapshots at several temperatures during the cook-off simulation, as shown in Fig. 3. The initial reaction of CH4 molecule, occurring at 3480 K, is a bimolecular reaction in which one O2− ion attacks CH4 molecule, leading to the formation of OH− and CH3 (Fig. 3a). This mutilated CH3 becomes more chemically active than CH4: it first combines with one OH fragment and forms methanol (CH3OH) at 3520 K (Fig. 3b); then it is attacked by another OH− ion and loses its second H atom (Fig. 3c); after the third attacking by OH−, the carbon atom loses all of its hydrogen atoms at 3570 K and forms the second C–O bond (Fig. 3f); the intermediate CH2O2 product finally turns into CO2 at 4000 K (Fig. 3g). We only observed one CH4 molecule converting into the final product CO2. While other seven CH4 molecules remain intact because of the strong C–H bond in CH4. Our QMD simulation indicated that the interactions between CH4 and fragments of O and OH are responsible for the thermal decomposition pf methane molecules through the continuous assaults of O or OH to the carbon atom and its affiliated hydrogen atoms. In addition, in order to obtain detailed chemical reactions information, we heat up the system with a gradually linearly increased temperature from 300 K to above 4000 K. In fact, the chemical reactions would happen at a lower temperature in real experiment than our simulations here due to the long timescale, as discussed by Ye et al.40 The initial decomposition temperature of CH4 in my QMD simulations is 3480 K which is higher than the experimental measured flame temperature (∼1500 K).14,15 This is due to the extremely high heating rate in our QMD simulations.
 | ||
| Fig. 3 The decomposition reaction mechanisms of CH4 in methane hydrate during cook-off QMD simulations from 300 K to 4350 K: (a) methane molecule loses its first H due to O2− attack; (b) the first C–O formation, leading to the formation of CH3OH; (c and d) methane molecule loses its second H; (e) the second C–O bond formation; and (f and g) CO2 formation. The dotted green oval represents the key chemical reaction sites of the reacted methane fragments. | ||
3.3 Methane migration mechanism and electron excitation impaction
Methane diffusion plays an important role in the stability of MHs, as well as CH4 exploitation from MHs. To uncover how the methane molecule escapes from the hydrate cage, we calculated the energy barrier of the escaping process from a 51262 cage through QM simulations. Two plausible diffusion paths were considered here, as shown in Fig. 4: (I) the migration via the hexagonal face (Fig. 4b), and (II) the diffusion through pentagonal ring (Fig. 4c) by breaking and reforming a hydrogen bond of that pentagonal ring.24 The relationship between energy and reaction coordinate (the summation of all atom movements) of these two possible pathways are calculated from CI-NEB and shown in Fig. 4a. For the hexagonal path I, the diffusion barrier is only 0.25 eV, which is much lower than that of the pentagonal path II (0.58 eV). Thus, the lowest barriers for methane diffusion is through hexagonal face, which is consistent with previous periodic calculations.41,42 The transition state structures at both paths, shown in Fig. 4, indicate that two hydrogen bonds are broken for path I while in path II three hydrogen bonds are broken to have enough space for CH4 diffusion. This explains the lower diffusion barrier for hexagonal path I. | ||
| Fig. 4 The diffusion mechanism of methane: (a) minimum energy path of hexagonal (blue) and pentagonal (black) migration path as a function of the summation of displacements of all atoms including four hydrogen ones and one carbon one. The total displacements were obtained from CI-NEB calculations; (b1–b3) the initial, transition state, and final configurations of hexagonal escape path; and (c1–c3) the initial, transition state, and final configurations of pentagonal escape path. | ||
The drilling in offshore regions to access the natural gas may lead to charge and discharge of clathrate hydrates. Thus, it is important to examine how electrons and holes affect the diffusion process. To illustrate how electron excitation affects the CH4 migration behavior of MHs, we first computed the diffusion path I at extra-electron and electron-deficient (hole) conditions, respectively. Fig. 5a displays the energies for charged (and discharged) systems and the comparison with the neutral state. The e− represents the state with one extra electron, while h+ stands for the state with one extra hole (or one electron deficient). The diffusion barrier for one extra electron is increased from 0.25 to 0.35 eV, suggesting that adding electron will make the cage more stable. More promising, in the hole state the diffusion barrier is significantly increased to 0.61 eV, much higher than the neutral state and e− state. Our results suggest that the storage of the CH4 in the clathrate can be stabilized by adding electron (charging) or hole (discharging) to the MHs system. The discharging is more effective than charging in stabilizing MHs.
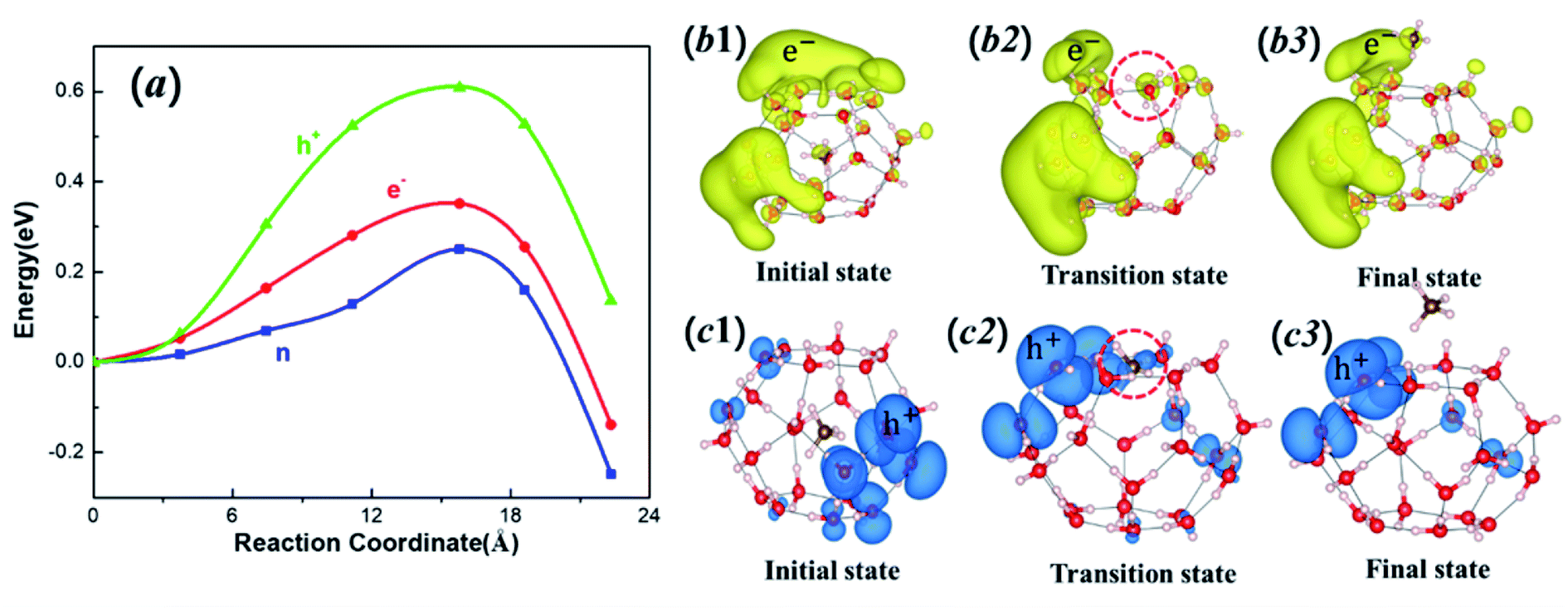 | ||
| Fig. 5 (a) Energy of electron excited (red), hole excited (green) and the ground state (blue) through hexagonal path as a function of the summation of displacements of all atoms. Here n, e− and h+ represent the results for the ground state, one-electron excited state and one-hole excited state, respectively. (b1–b3) The partial charge densities for electron excited (yellow isosurfaces) migration state; (c1–c3) the partial charge densities for hole excited (blue isosurfaces) migration state. The isosurface value for plotted hole and electron densities is set to 0.004 e Å−3. | ||
In order to explain the increased energy barrier by electrons and holes, we analyzed the charge density difference for the initial, transition and final states by subtracting the charge density of the neutral system to the charged (and discharged) systems. As shown in Fig. 5b1–b3, the extra electron is distributed on both water molecule and CH4 molecule. As the CH4 migrates to the transition state (Fig. 5b2), the repulsion is increased by the electrostatic interaction between CH4 and nearby H2O molecular, leading to an increased energy barrier. Under the hole condition, the positive charge is also distributed on both CH4 and water molecules (Fig. 5c1–c3), leading to an increased barrier due to the electrostatic repulsion. For the e− case, the negative charges are more delocalized than the h+ case, leading to a weaker electrostatic repulsion while for the h+ case the positive charges are mainly concentrated in the long pair nonbonding orbitals of oxygen atoms, making a stronger electrostatic repulsion force between water and methane molecules, leading to a higher energy barrier.
4. Conclusions
In summary, we have performed QM and QMD simulations to provide a detailed atomic mechanism revealing the decomposition process, methane migration mechanism, as well as the influence of the electron and hole carriers. This mechanism will be useful for understand the diffusion during the rapid extract and exploit processes and decomposition during combustion of MHs. Our major findings are:(1) Chemical reactions in MHs initiate from the hydrogen transfer among water molecules and the reaction rates are increased dramatically with the accelerated temperature. The fragments from the water dissociation attack the methane molecules, leading to the initial decomposition reaction of CH4 + O2− → CH3+ + HO− and the fast destruction of methane molecule leads to the final product of CO2 in our QMD simulations.
(2) The diffusion mechanism of methane molecule prefers to move through hexagonal face rather than through pentagonal side. Charge characteristics are helpful to increase the stability of clathrate for the storage of methane molecules because of the enhanced electrostatic repulsion force between water and methane molecules. The energy barrier of extra electron (or hole) is higher than that of neutral system due to the localized partial charge distribution. The hole has a more significant effect than the extra electron.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is supported by the American Chemical Society Petroleum Research Fund (PRF# 58754-DNI6).References
- X. Li, C. Xu, Y. Zhang, X. Ruan and Y. Wang, Investigation into gas production from natural gas hydrate: a review, Appl. Energy, 2016, 172, 286–322 CrossRef CAS.
- A. Lenz and L. Ojamae, Structures of the I-, II- and H-methane clathrates and the ice-methane clathrate phase transition from quantum-chemical modeling with force-field thermal corrections, J. Phys. Chem. A, 2011, 115, 6169–6176 CrossRef CAS PubMed.
- E. D. Sloan Jr, Fundamental principles and applications of natural gas hydrates, Nature, 2003, 426, 353–359 CrossRef PubMed.
- S. Takeya, W. Shimada, Y. Kamata, T. Ebinuma, T. Uchida, J. Nagao and H. Narita, In situ X-ray diffraction measurements of the self-preservation effect of CH4 hydrate, J. Phys. Chem. A, 2001, 105, 9756–9759 CrossRef CAS.
- N. Goel, In situ methane hydrate dissociation with carbon dioxide sequestration: current knowledge and issues, J. Pet. Sci. Eng., 2006, 51, 169–184 CrossRef CAS.
- H. Huo, Y. Liu, Z. Zheng, J. Zhao, C. Jin and T. Lv, Mechanical and thermal properties of methane clathrate hydrates as an alternative energy resource, J. Renewable Sustainable Energy, 2011, 3, 063110 CrossRef.
- C. A. Koh, A. K. Sum and E. D. Sloan, Gas hydrates: unlocking the energy from icy cages, J. Appl. Phys., 2009, 106, 061101 CrossRef.
- S. Patchkovskii and J. S. Tse, Thermodynamic stability of hydrogen clathrates, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14645–14650 CrossRef CAS PubMed.
- S. Y. Misyura and I. G. Donskoy, Dissociation of natural and artificial gas hydrate, Chem. Eng. Sci., 2016, 148, 65–77 CrossRef CAS.
- M. Iwata and T. Ueda, Combustion behavior of a combustible clathrate hydrate in a laminar boundary layer over a hydrate, Proceedings of the 4th International Conference on Gas Hydrates, 2002, pp. 1059–1062 Search PubMed.
- Y. Nakamura, R. Katsuki, T. Yokomori, R. Ohmura, M. Takahashi, T. Iwasaki, K. Uchida and T. Ueda, Combustion characteristics of methane hydrate in a laminar boundary layer, Energy Fuels, 2009, 23, 1445–1449 CrossRef CAS.
- M. Iwata and T. Ueda, Combustion behavior of a combustible clathrate hydrate in a laminar boundary layer over a hydrate, Proceedings of the 4th International Conference on Gas Hydrates, 2002, pp. 1059–1062 Search PubMed.
- Y. Nakamura, R. Katsuki, T. Yokomori, R. Ohmura, M. Takahashi, T. Iwasaki, K. Uchida and T. Ueda, Combustion characteristics of methane hydrate in a laminar boundary layer, Energy Fuels, 2009, 23, 1445–1449 CrossRef CAS.
- F. Wu and Y. Chao, A study of methane hydrate combustion phenomenon using a cylindrical porous burner, Combust. Sci. Technol., 2016, 188, 1983–2002 CrossRef CAS.
- O. C. Kwon, Research at new energy system lab, presented at the Pacific Rim Workshop on Deep Ocean Power Science, Honolulu, Hawaii, 2014 Search PubMed.
- D. J. Hwang, J. W. Choi, J. Park, S. I. Keel, C. B. Ch and D. S. Noh, Numerical study on flame structure and NO formation in CH4-O2-N2 counterflow diffusion flame diluted with H2O, Int. J. Eng. Res., 2004, 28, 1255–1267 CAS.
- G. Guo, M. Li, Y. Zhang and C. Wu, Why can water cages adsorb aqueous methane? A potential of mean force calculation on hydrate nucleation mechanisms, Phys. Chem. Chem. Phys., 2009, 11, 10427–10437 RSC.
- G. Guo, Y. Zhang and H. Liu, Effect of methane adsorption on the lifetime of a dodecahedral water cluster immersed in liquid water: a molecular dynamics study on the hydrate nucleation mechanisms, J. Phys. Chem. C, 2007, 111, 2595–2606 CrossRef CAS.
- L. C. Jacobson, W. Hujo and V. Molinero, Amorphous precursors in the nucleation of clathrate hydrates, J. Am. Chem. Soc., 2010, 132, 11806–11811 CrossRef CAS PubMed.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Phys. Chem. B, 2010, 114, 13796–13807 CrossRef CAS PubMed.
- A. Khan, Theoretical studies of CH4(H2O)20, (H2O)21, (H2O)20 and fused dodecahedral and tetrakaidecahedral structures: how do natural gas hydrates form?, J. Chem. Phys., 1999, 110, 11884 CrossRef CAS.
- A. Khan, Stabilization of hydrate structure H by N2 and CH4 molecules in 435663 and 512 cavities, and fused structure formation with 51268 cage: a theoretical study, J. Phys. Chem. A, 2001, 105, 7429–7434 CrossRef CAS.
- T. Ida, M. Mizuno and K. Endo, Electronic state of small and large cavities for methane hydrate, J. Comput. Chem., 2002, 23, 1071–1075 CrossRef CAS PubMed.
- S. Y. Willow and S. S. Xantheas, Enhancement of hydrogen storage capacity in hydrate lattices, Phys. Lett., 2011, 525–526, 13–18 Search PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. Paier, R. Hirschl, M. Marsman and G. Kresse, The Perdew-Burke-Ernzerhof exchange-correlation functional applied to the G2-1 test set using a plane-wave basis set, J. Chem. Phys., 2005, 122, 234102 CrossRef PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Improved tetrahedron method for brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- D. Guo and Q. An, Thermal stability and detonation properties of potassium 4,4′-bis(dinitromethyl)-3,3′-azofurazanate, an environmentally friendly energetic three-dimensional metal-organic framework, ACS Appl. Mater. Interfaces, 2019, 11, 1512–1519 CrossRef CAS PubMed.
- L. Liu, Y. Liu, S. V. Zybin, H. Sun and W. A. Goddard III, ReaxFF-lg: correction of the ReaxFF reactive force field for London dispersion, with applications to the equations of state for energetic materials, J. Phys. Chem. A, 2011, 115, 11016–11022 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jonsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- S. Baroni, P. Giannozzi and A. Testa, Green's-function approach to linear response in solids, Phys. Rev. Lett., 1987, 58, 1861–1864 CrossRef CAS PubMed.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- D. Guo, Q. An, S. V. Zybin, W. A. Goddard III, F. Huang and B. Tang, The co-crystal of TNT/CL-20 leads to decreased sensitivity toward thermal decomposition from first principles based reactive molecular dynamics, J. Mater. Chem. A, 2015, 3, 5409–5419 RSC.
- R. T. Morrison and R. N. Boyd, Química orgánica, Pearson Educación, 5th edn, 1998, p. 395 Search PubMed.
- C. Ye, Q. An, W. A. Goddard III, T. Cheng, W. Liu, S. V. Zybin and X. Ju, Initial decomposition reaction of di-tetrazinetetroxide (DTTO) from quantum molecular dynamics: implications for a promising energetic material, J. Mater. Chem. A, 2015, 3, 1972–1978 RSC.
- Q. Li, B. Kolb, G. Román-Pérez, J. M. Soler, F. Yndurain, L. Kong, D. C. Langreth and T. Thonhauser, Ab initio energetics and kinetics study of H2 and CH4 in the SI clathrate hydrate, Phys. Rev. B, 2011, 84, 153103 CrossRef.
- G. Román-Pérez, M. Moaied, J. M. Soler and F. Yndurain, Stability, adsorption, and diffusion of CH4, CO2, and H2 in clathrate hydrates, Phys. Rev. Lett., 2010, 105, 145901 CrossRef PubMed.
Footnote |
| † D. Z. G. and H. W. W. contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |