
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlobally stabilized bent carbon–carbon triple bond by hydrogen-free inorganic–metallic scaffolding Al4F6†
Ying-ying Xuea,
Ying Zhanga,
Zhong-hua Cui *b and
Yi-hong Ding*ac
*b and
Yi-hong Ding*ac
aLaboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, P. R. China. E-mail: yhdd@jlu.edu.cn
bInstitute of Atomic and Molecular Physics, Jilin University, Changchun 130023, P. R. China. E-mail: zcui@jlu.edu.cn
cKey Laboratory of Carbon Materials of Zhejiang Province, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, P. R. China
First published on 3rd July 2020
Abstract
For over 100 years, known bent C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C compounds have been limited to those with organic (I) and all-carbon (II) scaffoldings. Here, we computationally report a novel type (III) of bent CC compound, i.e., C2Al4F6-01, which is the energetically global minimum isomer and bears an inorganic–metallic scaffolding and unexpected click reactivity.
C compounds have been limited to those with organic (I) and all-carbon (II) scaffoldings. Here, we computationally report a novel type (III) of bent CC compound, i.e., C2Al4F6-01, which is the energetically global minimum isomer and bears an inorganic–metallic scaffolding and unexpected click reactivity.
1. Introduction
Carbon, the element with 2s22p2 electron configuration, has three different hybridization forms of the valence atomic orbitals, i.e., sp, sp2 and sp3 hybrid orbitals, with significantly more acute bond angles and decreased homo-atomic bond strength (see Schemes 1a and b).1 Understandably, bending a CC bond is in most cases geometrically and electronically unfavourable. In fact, in acyclic or less strained cyclic compounds, the ∠CCR angle of CC is strictly 180° (e.g., acetylene in Scheme 1c) or very close to 180° (e.g., acyclic alkynes1b,2 and other substituted compounds3). A significantly bent CC without additional coordination is only possible when it is embedded in highly strained or cyclic scaffoldings. As for most exotic species,4 the conceptual and synthetic challenges of developing compounds with a significantly bent CC bond have been long attempted.5 The first postulation of a bent CC constrained in an aromatic framework as an reactive intermediate was published more than one century ago (in 1902).6 Since then, related species with bent CC bonds in organic scaffolding (see type I in Scheme 1c) have witnessed fruitful achievements, including applications in the pharmaceutical chemistry, materials chemistry, natural products synthesis, and organometallic chemistry.5c,7 Cycloalkynes or angle-strained alkynes have received great attention due to their closeness to alkenes (obtained by simply losing two ligands).5b–5h,7 Besides, mono-cyclic all-carbon molecules, i.e., cyclocarbon[n], present the second class (see type II in Scheme 1c), each of which possess alternating CC and C–C bonds.5a,8 After lengthy pursuit, the first ring-shaped molecule of pure carbon, i.e., cyclocarbon (C18), was recently synthesized, representing a breakthrough and possibly advancing potential molecular-scale electronic applications (e.g., semiconductors).5a,9
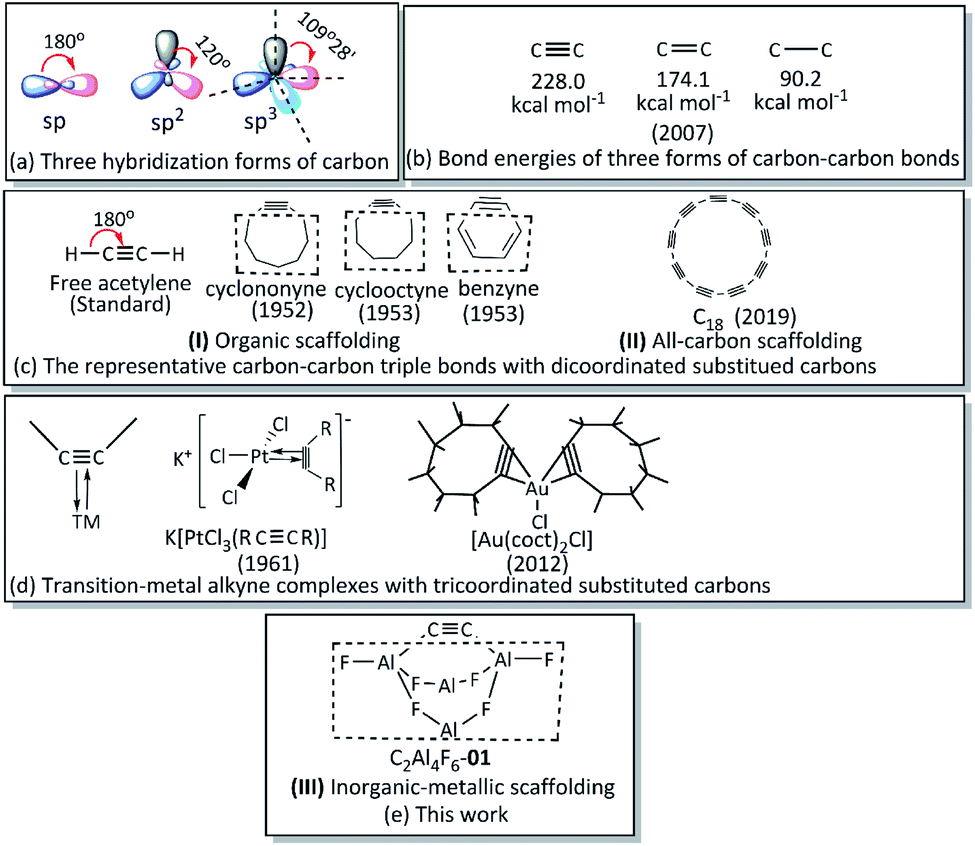 | ||
| Scheme 1 Hybridization forms and homo-nuclear bond energies of carbon, and representative compounds with carbon–carbon triple bonds. | ||
One should be aware that when an alkyne perpendicularly interacts with a transition metal (TM) complex, so-called “transition-metal alkyne complexes” form (see Scheme 1d).10 However, rather than the strain in type I and II, the synergistic bonding interactions between CC and TM in these complexes bend the CC, and the coordination number of the carbon atoms is increased to three rather than two in I and II. Thus, such compounds are not the topic of the present work. Moreover, one should note that in numerous cases, the TM-CC interactions may be so strong that the hybridization state of the ethynyl carbon atoms can change from sp to sp2 upon coordination, greatly reducing or even diminishing the CC feature.10a,10d,10g
Can a significantly bent CC bond be stabilized by a non-carbon-based scaffolding while maintaining the dicoordinate carbons? We speculated that fluorinated metal dicarbides might possess the desired bent CC bond if metal fluorides can form stable and closed structures. During our study on F-persubstituted dicarbalanes (C2Al4F6), by means of an extensive structural search and high-level energy calculations, we fortunately found a novel type (III) in which a bent CC bond can be globally stabilized by the novel scaffolding Al4F6 that is hydrogen-free and non-carbon-based. The global structure of C2Al4F6-01 has an interesting “flower-basket” shape (with an Al–F alternative 8-member ring tray and a C–C handle). The nature and reactivity of CC within the global C2Al4F6-01 was further studied via numerous analytic methods.
2. Theoretical methods
Due to the good balance between reliability and computational cost, the density function theory (DFT) method is now indispensable for studying molecules and materials, though debates still exist.11 First, to obtain the global structure, we reasonably assumed that the fluorine atoms act as ligands around the C2Al4-core. We then applied our locally developed “skeleton-ligand cluster-growth” method12 at the level of B3LYP13/6-31G(d), which has been shown to be quite cost-effective for initial large-scale isomeric searches. Further, those with energies lower than 20 kcal mol−1 were refined at the composite CBS-QB3 (ref. 14) level, which reliably gives accurate thermochemical properties in numerous fields. Second, similar to most DFT methods, the presently applied B3LYP is of single-reference nature and is constructed empirically via parameterization. The geometries, energetics and T1 diagnostics15 of the former two lowest-energy isomers were computed using wave function-based methods, i.e., CCSD(T)/aug-cc-pVTZ//CCSD/cc-pVTZ. The single-point CBS-QB3 energies were recomputed at CCSD/cc-pVTZ and CCSD/6-311G(2d,d,p) geometries. Their geometries were also optimized at the M062X/6-311G(2d,d,p) level.Natural bond orbital (NBO)16 analysis and adaptive natural density partitioning (AdNDP) analysis17 were conducted to understand their electronic structures at the B3LYP/6-311G(2d,d,p) level. The AdNDP analysis was analyzed by the Multiwfn program.17b All these calculations were performed using the commercial Gaussian 16 (ref. 18) and Gaussian 09 (ref. 19) packages.
3. Results and discussion
An amazingly large number, i.e., 28600, of C2Al4F6 isomers were obtained as local energy minima at the B3LYP/6-31G(d) level. For brevity and easy discussion, we only show the first two low-energy isomers with the respective bent and linear forms of CC, i.e., 01 and 02, at the CBS-QB3 level (see Fig. 1). Notably, the zero-point energy (ZPE)-corrected CBS-QB3 energy of 01 is lower than that of 02 by 2.4 kcal mol−1. However, the relative Gibbs free energy between 01 and 02 is very close, with the latter lower by 0.6 kcal mol−1. This suggests the profound influence of the Gibbs free energies on the stability of 01 and 02. Thus, we performed additional calculations at the CBS-QB3 level using the costly CCSD/cc-pVTZ and CCSD/6-311G(2d,d,p)-optimized geometries. At the two CBS-QB3//CCSD levels, the Gibbs free energy of 01 is slightly more stable than 02 by 3.5 and 5.4 kcal mol−1, respectively. Thus, 01 can be viewed as the global minimum.
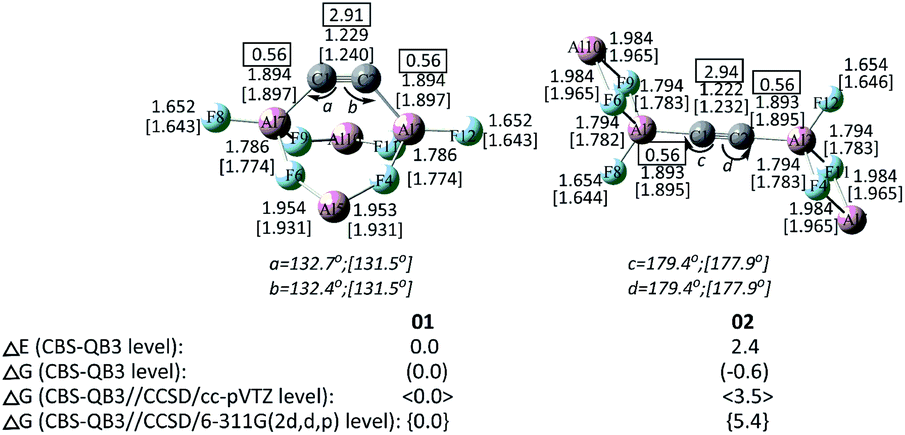 | ||
| Fig. 1 The key geometrical parameters (distances in Å and angles in (°)) at the CBS-QB3 and CCSD(T)/aug-cc-pVTZ//CCSD/cc-pVTZ (in []) levels. The Wiberg bond indexes are shown in rectangular boxes. The relative CBS-QB3 energies (in kcal mol−1) with zero-point correction (ΔE) and the relative Gibbs free energies (ΔG) of the former two isomers of C2Al4F6 at different levels are shown. | ||
Besides, since the T1 values of 01 and 02, i.e., 0.0137 and 0.0136, respectively, are considerably smaller than the recommended threshold value 0.02,15 there should be negligible multi-reference characters for both structures. Note that the main bond distances and angles of C2Al4F6-01 and 02 are consistent among the CBS-QB3, CCSD and M062X levels (see Table S1†). Thus, for consistency, the following discussions are based on the CBS-QB3 values unless otherwise specified.
Bonding features of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C bond and C–Al bonds in C2Al4F6-01 and 02
C bond and C–Al bonds in C2Al4F6-01 and 02
By comparing the corresponding typical triple and double carbon–carbon bond distances, i.e., 1.198 Å in HCCH and 1.327 Å in H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 at the B3LYP/6-311G(2d,d,p) level (see Fig. S7†), both 01 and 02 with very short CC distances (1.229 and 1.222 Å, respectively) can be viewed as containing a CC moiety, which is further supported by their large WBI values of 2.91 and 2.94, respectively. The CC bond of 01 is heavily bent, with two acute ∠CCAl angles of 132.7° and 132.4°. By contrast, 02 has an almost linear CC bond with a ∠CCAl angle of 179.4°.
CH2 at the B3LYP/6-311G(2d,d,p) level (see Fig. S7†), both 01 and 02 with very short CC distances (1.229 and 1.222 Å, respectively) can be viewed as containing a CC moiety, which is further supported by their large WBI values of 2.91 and 2.94, respectively. The CC bond of 01 is heavily bent, with two acute ∠CCAl angles of 132.7° and 132.4°. By contrast, 02 has an almost linear CC bond with a ∠CCAl angle of 179.4°.
The Al–F bonds in both 01/02 can be categorized into three classes, i.e., 1.652/1.654, 1.786/1.794, and 1.954/1.984 Å, respectively, with increasing bond distances. The first two are comparable to the corresponding terminal (1.638 Å) and bridge (1.821 Å) Al–F bonds of the model Al2F6 (see Fig. S7†). The third type with the longer Al–F distance can be viewed as the dative bonding, indicating the presence of two Al+-ions in 01/02. The low-valent Al+-subunits20 are effectively stabilized by the neighbouring F → Al donor–acceptor interactions rather than the electron-sharing one. The situation is quite similar to the reference molecule Al2F4,21 which has a ground ionic structure Al+[AlF4]− with the two types of Al–F bonds, i.e., 1.993 Å between Al+ and F and 1.648/1.783 Å between Al3+ and F (see Fig. S7†). Compared to the typical C–Al single bond of the model molecule Al(CH3)3 (1.967 Å), the C–Al bonds of 01 and 02, i.e., 1.894, 1.894, 1.893 and 1.893 Å, can be viewed as covalent single bonding. The somewhat shortened (smaller by 3.7%) bond of C–Al could result from the additional interaction between the Al-center and the similar CC π bond in 01 and 02.
Note that the isomer 02 has a rather small low frequency (2.2 cm−1). Its local minimum was confirmed at the MP2/6-311G(2d,d,p) level (5.4 cm−1) (see Fig. S10†). The small low frequency indicates that 02 is a very floppy structure due to the two Al+-ions stabilized by dative bonding. In fact, in bonds associated with easy rotation or wagging, very small low frequencies are evident. For example, in a small H-terminated cluster of graphene with 6 benzene units and 6 CC units, the first two imaginary frequencies are as small as 7.1 cm−1 and 7.2 cm−1 at the B3LYP/6-311G(2d,d,p) level, which correspond to the up-and-down wagging of the benzene unit.
The main molecular orbitals of isomers 01 and 02 are shown in Fig. 2. Both possess one σ bond, HOMO-4 (01) and HOMO-4 (02), and two π bonds (HOMO-2 and HOMO-3 for 01 and HOMO-2 and HOMO-3 for 02). Each has two σ C–Al bonds (HOMO-10 and HOMO-11 in 01 and HOM0-12 and HOMO-15 in 02). The widely used and efficient method, namely adaptive natural density partitioning (AdNDP) analysis,17 was adopted at the B3LYP/6-311G(2d,d,p) level. The one 2c–2e σ (two centers and two electrons) bond and two 2c–2e π bonds with high occupation numbers (close to 2.0) support the presence of a triple CC bond and the high occupation numbers (close to 2.0) of two 2c–2e C–Al bonds well support the presence of the two C–Al the single bonds in both 01 and 02. The detailed orbital and AdNDP analysis can be found in Fig. S2 and S3.†
 | ||
| Fig. 2 The selected molecular orbitals of C2Al4F6-01 and 02 at the level of B3LYP/6-311G(2d,d,p). “ON” denotes the occupation number on the localized orbital. | ||
Based on the above structural and electronic analysis, we can deduce that the connection between C and Al should be of the “electron-sharing” type for both 01 and 02 (see 1a and 2a in Scheme 2) rather than the “electron dative” type (see 1b′ and 2b′ in Scheme 2). According to the “electron-sharing” mode, if C2 is removed, the radical centers should be positioned at the neighbouring Al centers. This is well supported by the optimized Al2F3˙ and an Al–Al connected Al4F6 structure (see 1a′ and 2a′ in Scheme 2). However, the optimized fragments based on the “electron dative” mode, i.e., Al2F3+ and Al4F62+, differ rather dramatically from the structures of 01 and 02.
 | ||
| Scheme 2 The path of eliminating C2 and C22− in C2Al4F6-01 and 02 at the level of B3LYP/6-311G(2d,d,p). | ||
Reactivity of CC in C2Al4F6-01 and 02
Further, the presence of CC within the 01 and 02 isomers of C2Al4F6 is consistent with the computational observation that the CC bond can undergo the addition of two H2 molecules as well as [3 + 2] click reactions with HN3. For known cycloalkynes, there is a clear linear correlation (R2 = 0.995, see Fig. S11†) between the Gibbs free energy barriers and the bending angles of CC, i.e., the higher bending degree of CC, the more reactive it becomes. The HN3 click reactivity of 01 and 02 was a great surprise. Both have comparable barrier heights (20.3 and 21.2 kcal mol−1, respectively) despite the significantly different bending angles of CC. The unexpected click reactivity of CC can be ascribed to the involvement of the neighbouring acid Al centers of 02 (see Fig. S4†). One of the Al+ atoms of 02 could flip and attach to the nitrogen atom with the lone pair electrons of the HN3 unit. Such additional interaction lowers the barrier of 02 with linear CC, approaching that of 01 with the bent CC bond. Compared with known cycloalkynes (see Fig. 3), the HN3 click barrier (20.3 kcal mol−1) of 01 lies between cycloheptyne (C7H10, 17.3 kcal mol−1) and cyclooctyne (C8H12, 22.4 kcal mol−1), indicating the feasible existence of 01 at least via spectroscopic detection.
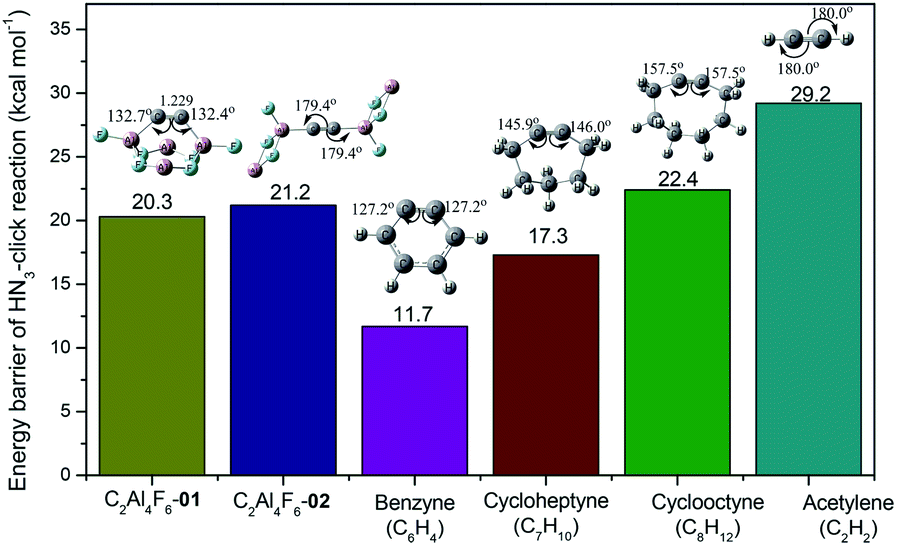 | ||
| Fig. 3 Gibbs free energy barriers (in kcal mol−1) of the HN3 click reaction with C2Al4F6-01, C2Al4F6-02, benzyne, cycloheptyne, cyclooctyne and acetylene at the CBS-QB3 level. | ||
Interconversion between C2Al4F6-01 and 02
For the intrinsic stability, we attempted to identify the isomerization of the global isomer of C2Al4F6-01. The lowest barrier is associated with an indirect conversion to C2Al4F6-02 by sequentially breaking the F → Al+ dative bond via an intermediate C2Al4F6-18 (see Fig. 4). The barrier is as high as 14.8 kcal mol−1. In addition, at the B3LYP/6-311G(2d,d,p) level, we obtained an optimized complex by adding two BH3NH3 to 01 (see Fig. S6†), which comprises four dative bonds, i.e., two C → BH3 and two NH3 → C. This indicates that the bent CC bond in 01 does have the “hidden carbene” feature as was proposed very recently.3a
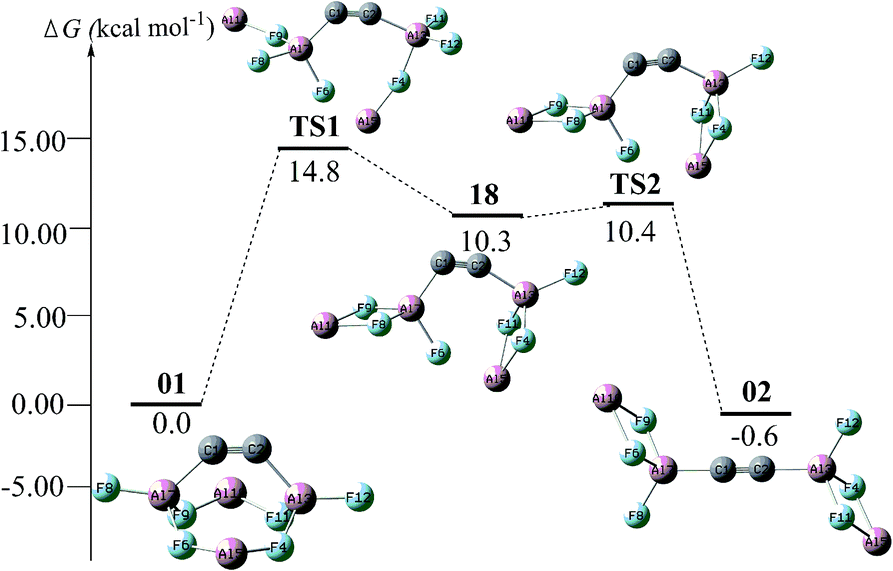 | ||
| Fig. 4 The conversion pathways between C2Al4F6-01 and C2Al4F6-02 with the Gibbs free energy barriers at the CBS-QB3 level. | ||
The unique stability of isomer 01 could be attributed to the balance of two opposite effects. On one hand, bending the CC bond leads to an energetic destabilization. On the other hand, the recombination of the two Al2F3 units causes energetic stabilization. Fig. 4 vividly shows this energetic change during the interconversion between 01 and 02.
Implications
Two computational facts from the present work deserve the interest of the chemical community. First, our global isomeric search unexpectedly identified a bent CC bond (both C-atoms are in dicoordination) supported by a scaffolding that is neither of the two known carbon-based systems, i.e., type-I and II. 01 presents the first example of a bent CC bond stabilized by an inorganic metal cluster composed of Al and F, despite the typical expectation that a polyatomic cluster such as C2Al4F6 could sufficiently undergo complex structural rearrangement to avoid a global bent CC. Second, we observed an inverse click reactivity for CC between the inorganic metal-supported structures 01 (bent CC) and 02 (linear CC) due to the active involvement of the attached Al-atoms. We postulated that this structure could be versatile in such metal–inorganic CC compounds.
A large number of organometallic fluorides have been synthesized,22 among which various C, Al, and F-based species are known.23 Numerous general synthetic methods for generating angle-strained cycloalkynes have been reported.5e In our study, the lowest-energy C2Al4F6-01 with the bent CC bond has a high likelihood to be synthesized in future. Here, we tentatively supposed a possible synthetic method via the photochemical reactions of compounds C2Al4F6HCl or C2Al4F6CO. We calculated the adsorption energy for removing HCl from C2Al4F6HCl at 273.15 K, which is 20.0 kcal mol−1 at the CBS-QB3 level. The process of removing CO from C2Al4F6CO was predicted to be exothermic by 9.3 kcal mol−1 at the CBS-QB3 level.
4. Conclusion
The unexpected finding of a bent CC bond stabilized globally within the chemical formula C2Al4F6 contrasts sharply with the current knowledge of 6-vertex dicarbalanes, i.e., C2Al4R6 with R = H and CH3 shows a distorted octahedral structure as the lowest energy isomer with the two carbons being well separated.24 Besides providing of the great possibility to regulate the ground shape of dicarbalanes by substituent engineering, the strong tendency to form the triply bonded CC moiety indicates that a variety of low-lying bent CC with inorganic–metallic scaffoldings could be found in similar dicarbon metal fluorides (i.e., C2MxFy, M = heavier than group 13).
In summary, in this study, through our locally developed “skeleton-ligand cluster-growth” method, we report the first example of a main-group metal–inorganic compound isomer (C2Al4F6-01) with a globally stabilized bent carbon–carbon triple bond. Via bonding analysis, we determined that C2Al4F6-01 exhibits a salt-like character with two [–AlF3]− and two Al+ units.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Key Research and Development Program of China (No. 2016YFB0701100) and the National Natural Science Foundation of China (No. 21473069, 21773082, 11922405). The authors are very grateful to the reviewers' invaluable comments and suggestions to improve our work.Notes and references
- (a) L. Pauling, J. Am. Chem. Soc., 1931, 53, 1367–1400 CrossRef CAS; (b) M. B. Smith and J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, 6th edn, 2007, pp. 3–136 Search PubMed; (c) Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC press, Boca Raton, 2007, pp. 147–154 CrossRef.
- S. Patai, The Chemistry of Carbon-Carbon Triple Bonds, Wiley, New York, 1978, pp. 1–199 Search PubMed.
- (a) P. V. Bijina and C. H. Suresh, ChemPhysChem, 2018, 19, 3266–3272 CrossRef CAS PubMed; (b) D. C. Georgiou, L. L. Zhao, D. J. D. Wilson, G. Frenking and J. L. Dutton, Chem.–Eur. J., 2017, 23, 2926–2934 CrossRef CAS PubMed; (c) D. C. Georgiou, B. D. Stringer, C. F. Hogan, P. J. Barnard, D. J. D. Wilson, N. Holzmann, G. Frenking and J. L. Dutton, Chem.–Eur. J., 2015, 21, 3377–3386 CrossRef CAS PubMed.
- (a) C. A. Dyker and G. Bertrand, Nat. Chem., 2009, 1, 265–266 CrossRef CAS; (b) R. Hoffmann and H. Hopf, Angew. Chem., Int. Ed., 2008, 47, 4474–4481 CrossRef CAS PubMed.
- (a) K. Kaiser, L. M. Scriven, F. Schulz, P. Gawel, L. Gross and H. L. Anderson, Science, 2019, 365, 1299–1301 CrossRef CAS PubMed; (b) A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940–3943 CrossRef CAS PubMed; (c) H. H. Wenk, M. Winkler and W. Sander, Angew. Chem., Int. Ed., 2003, 42, 502–528 CrossRef CAS PubMed; (d) T. Kawase, Y. Seirai, H. R. Darabi, M. Oda, Y. Sarakai and K. Tashiro, Angew. Chem., Int. Ed., 2003, 42, 1621–1624 CrossRef CAS PubMed; (e) A. Krebs and J. Wilke, Angle strained cycloalkynes, Wittig Chemistry, Springer, Berlin, Heidelberg, 1983, pp. 189–233 Search PubMed; (f) J. D. Roberts Jr, H. E. Simmons, L. A. Carlsmith and C. W. Vaughan, J. Am. Chem. Soc., 1953, 75, 3290–3291 CrossRef; (g) A. T. Blomquist and L. H. Liu, J. Am. Chem. Soc., 1953, 75, 2153–2154 CrossRef CAS; (h) A. T. Blomquist, L. H. Liu and J. C. Bohrer, J. Am. Chem. Soc., 1952, 74, 3643–3647 CrossRef CAS.
- R. Stoermer and B. Kahlert, Ber. Dtsch. Chem. Ges., 1902, 35, 1633–1640 CrossRef CAS.
- (a) K. Uchida, Y. Minami, S. Yoshida and T. Hosoya, Org. Lett., 2019, 21, 9019–9023 CrossRef CAS; (b) T. Harris and I. V. Alabugin, Mendeleev Commun., 2019, 29, 237–248 CrossRef CAS; (c) A. E. Goetz, T. K. Shah and N. K. Garg, Chem. Commun., 2015, 51, 34–45 RSC; (d) T. H. Poole, J. A. Reisz, W. Zhao, L. B. Poole, C. M. Furdui and S. B. King, J. Am. Chem. Soc., 2014, 136, 6167–6170 CrossRef CAS; (e) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140–3152 RSC; (f) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef CAS; (g) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed; (h) H. Pellissier and M. Santelli, Tetrahedron, 2003, 59, 701–730 CrossRef CAS.
- (a) C. Neiss, E. Trushin and A. Görling, ChemPhysChem, 2014, 15, 2497–2502 CrossRef CAS PubMed; (b) F. Diederich, Nature, 1994, 369, 199–207 CrossRef CAS; (c) V. Parasuk, J. Almölf and M. W. Feyereisen, J. Am. Chem. Soc., 1991, 113, 1049–1050 CrossRef CAS; (d) F. Diederich, Y. Rubin, C. B. Knobler, R. L. Whettern, K. E. Schriver, K. N. Houk and Y. Li, Science, 1989, 245, 1088–1090 CrossRef CAS PubMed.
- D. Castelvecchi, Nature, 2019, 572, 426 CrossRef CAS PubMed.
- (a) K. D. J. Parker and M. D. Fryzuk, Organometallics, 2015, 34, 2037–2047 CrossRef CAS; (b) F. D. Rochon and C. Tessier, Inorg. Chim. Acta, 2007, 360, 3533–3542 CrossRef CAS; (c) M. A. Bennett and H. P. Schwemlein, Angew. Chem., Int. Ed., 1989, 28, 1296–1320 CrossRef; (d) R. S. Dickson and P. J. Fraser, Compounds Derived from Alkynes and Carbonyl Complexes of Cobalt, Advances in Organometallic Chemistry, Academic Press, 1974, vol. 12, pp. 323–377 Search PubMed; (e) A. L. Beauchamp, F. D. Rochon and T. Theophanides, Can. J. Chem., 1973, 51, 126–131 CrossRef CAS; (f) M. A. Bennett, G. B. Robertson, P. O. Whimp and T. Yoshida, J. Am. Chem. Soc., 1971, 93, 3797–3798 CrossRef CAS; (g) G. R. Davies, W. Hewertson, R. H. B. Mais and P. G. Owston, Chem. Commun., 1967, 9, 423–424 RSC; (h) J. Chatt, R. G. Guy and L. A. Duncanson, J. Chem. Soc., 1961, 827–834 RSC.
- (a) S. Ghosh, P. Verma, C. J. Cramer, L. Gagliardi and D. G. Truhlar, Chem. Rev., 2018, 118, 7249–7292 CrossRef CAS PubMed; (b) P. R. C. Kent and G. Kotliar, Science, 2018, 361, 348–354 CrossRef CAS PubMed; (c) S. Hammes-Schiffer, Science, 2017, 355, 28–29 CrossRef CAS PubMed; (d) M. G. Medvedev, I. S. Bushmarinov, J. W. Sun, J. P. Perdew and K. A. Lyssenko, Science, 2017, 355, 49–52 CrossRef CAS PubMed; (e) K. P. Kepp, Science, 2017, 356, 496b CrossRef PubMed; (f) M. G. Medvedev, I. S. Bushmarinov, J. W. Sun, J. P. Perdew and K. A. Lyssenko, Science, 2017, 356, 496c CrossRef PubMed.
- (a) Y. H. Ding, Skeleton-ligand Cluster-growth Isomeric Search Algorithm, Jilin University, Changchun, China, 2014 Search PubMed; (b) Y. Y. Xue and Y. H. Ding, Chem. Commun., 2019, 55, 6373–6376 RSC; (c) X. X. Bo, H. F. Zheng, J. F. Xin and Y. H. Ding, Chem. Commun., 2019, 55, 2597–2600 RSC; (d) Y. W. Sun, H. Y. Wang and Y. H. Ding, RSC Adv., 2019, 9, 40772–40780 RSC; (e) H. F. Zheng, S. Yu, T. D. Hu, J. Xu and Y. H. Ding, Phys. Chem. Chem. Phys., 2018, 20, 26266–26272 RSC; (f) J. Xu, X. Zhang, S. Yu, Y. H. Ding and K. H. Bowen, J. Phys. Chem. Lett., 2017, 8, 2263 CrossRef CAS PubMed; (g) Y. Y. Xue, J. J. Sui, J. Xu and Y. H. Ding, ACS Omega, 2017, 2, 5407–5414 CrossRef CAS PubMed; (h) J. J. Sui, J. Xu and Y. H. Ding, Dalton Trans., 2016, 45, 56–60 RSC; (i) Z. H. Cui, Y. H. Ding, J. L. Cabellos, E. Osorio, R. Islas, A. Restrepo and G. Merino, Phys. Chem. Chem. Phys., 2015, 17, 8769–8875 RSC; (j) X. Y. Zhang and Y. H. Ding, RSC Adv., 2015, 5, 27134–27139 RSC.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- (a) J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef CAS; (b) J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS.
- T. J. Lee and P. R. Taylor, Int. J. Quantum Chem., 1989, 36, 199–207 CrossRef.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, version 3.1, University of Wisconsin, Madison, WI, 1995 Search PubMed.
- (a) D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC; (b) T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) H. W. Roesky and S. S. Kumar, Chem. Commun., 2005, 32, 4027–4038 RSC; (b) M. N. S. Rao, H. W. Roesky and G. Anantharaman, J. Organomet. Chem., 2002, 646, 4–14 CrossRef.
- T. Pankewitz, W. Klopper, P. Henke and H. Schnöckel, Eur. J. Inorg. Chem., 2008, 4879–4890 CrossRef CAS.
- (a) S. Singh and H. W. Roesky, J. Fluorine Chem., 2007, 128, 369–377 CrossRef CAS; (b) J. Pinkas and H. W. Roesky, J. Fluorine Chem., 2003, 122, 125–150 CrossRef CAS; (c) B. Neumülle, Coord. Chem. Rev., 1997, 158, 69–101 CrossRef.
- (a) H. Hatop, H. W. Roesky, T. Labahn, A. Fischer, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2000, 19, 937–940 CrossRef CAS; (b) C. Schnitter, K. Klimek, H. W. Roesky, T. Albers, H. G. Schmidt, C. Röpken and E. Parisini, Organometallics, 1998, 17, 2249–2257 CrossRef CAS; (c) Y. X. Chen, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1997, 119, 2582–2583 CrossRef CAS; (d) G. Natta, G. Allegra, G. Perego and A. Zambelli, J. Am. Chem. Soc., 1961, 83, 5033 CrossRef; (e) G. Gundersen, T. Haugen and A. Haaland, J. Organomet. Chem., 1973, 54, 77–86 CrossRef CAS.
- A. A. A. Attia, A. Lupan and R. B. King, Inorg. Chem., 2015, 54, 11377–11384 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Method details, Cartesian coordinates and total energies computed of key structures of C2Al4F6 at the CBS-QB3 level, and some important information of the reactivity. See DOI: 10.1039/d0ra02280b |
| This journal is © The Royal Society of Chemistry 2020 |