
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-assisted solvent- and catalyst-free production of well-defined poly(1-vinyl-2-pyrrolidone) for biomedical applications†
Paulina Maksym *ab,
Magdalena Tarnackaab,
Dawid Heczkoc,
Justyna Knapik-Kowalczukab,
Anna Mielańczykd,
Roksana Bernatbe,
Grzegorz Garbaczf,
Kamil Kaminski*ab and
Marian Paluchab
*ab,
Magdalena Tarnackaab,
Dawid Heczkoc,
Justyna Knapik-Kowalczukab,
Anna Mielańczykd,
Roksana Bernatbe,
Grzegorz Garbaczf,
Kamil Kaminski*ab and
Marian Paluchab
aInstitute of Physics, University of Silesia, ul. 75 Pułku Piechoty 1, 41-500 Chorzów, Poland. E-mail: paulina.maksym@smcebi.edu.pl; kamil.kaminski@smcebi.edu.pl; Tel: +48323497610
bSilesian Center of Education and Interdisciplinary Research, University of Silesia, ul. 75 Pulku Piechoty 1A, 41-500 Chorzow, Poland
cDepartment of Pharmacognosy and Phytochemistry, Medical University of Silesia in Katowice, School of Pharmacy with the Division of Laboratory Medicine in Sosnowiec, Jagiellonska 4, 41-200 Sosnowiec, Poland
dDepartment of Physical Chemistry and Technology of Polymers, Faculty of Chemistry, Silesian University of Technology, ul. M. Strzody 9, 44-100 Gliwice, Poland
eInstitute of Chemistry, University of Silesia, ul. Szkolna 9, 40-007 Katowice, Poland
fPhysiolution GmbH, Walther-Rathenau-Str. 49a, 17489 Greifswald, Germany
First published on 5th June 2020
Abstract
In this work, we developed a fast, highly efficient, and environmentally friendly catalytic system for classical free-radical polymerization (FRP) utilizing a high-pressure (HP) approach. The application of HP for thermally-induced, bulk FRP of 1-vinyl-2-pyrrolidone (VP) allowed to eliminate the current limitation of ambient-pressure polymerization of ‘less-activated’ monomer (LAM), characterized by the lack of temporal control yielding polymers of unacceptably large disperisites and poor result reproducibility. By a simple manipulation of thermodynamic conditions (p = 125–500 MPa, T = 323–333 K) and reaction composition (two-component system: monomer and low content of thermoinitiator) well-defined poly(1-vinyl-2-pyrrolidone)s (PVP) in a wide range of molecular weights and low/moderate dispersities (Mn = 16.2–280.5 kg mol−1, Đ = 1.27–1.45) have been produced. We have found that HP can act as an ‘external’ controlling factor that warrants the first-order polymerization kinetics for classical FRP, something that was possible so far only for reversible deactivation radical polymerization (RDRP) systems. Importantly, our synthetic strategy adopted for VP FRP enabled us to obtain polymers of very high Mn in a very short time-frame (0.5 h). It has also been confirmed that VP bulk polymerization yields polymers with significantly lower glass transition temperatures (Tg) and different solubility properties in comparison to macromolecules obtained during the solvent-assisted reaction.
Introduction
Sustainability-related studies in the polymer chemistry field have gained increasing attention and directed the main research focus to the production of well-defined materials using non-toxic solvents and catalytic systems. In fact, reversible deactivation radical polymerization (RDRP) of ‘pseudo-living’/controlled features allowed significant progress in the development of novel ‘greenest’ polymerization strategies induced by ‘external’ stimuli (e.g., light, ultrasound).1,2 Of these methods, the light-mediated processes have become an interesting alternative to thermally-induced ones, where the contribution of metal-based compounds has been significantly reduced and replaced by organocatalysts.3,4 It needs to be stressed that both photo-induced RDRPs namely Atom Transfer Radical Polymerization (photo-ATRP)5 and Reversible Addition-Fragmentation Chain-transfer Polymerization (photo-RAFT)6 gained a special interest and became universal and robust methods to polymerize a broad range of functional monomers. However, the main drawbacks of photo-RDRP strategies are related to the long reaction time, the high risk of degradation of reagents (during long-time irradiation), and, importantly, the difficult to scale polymerization process. In addition, the utilization of RDRP methods to polymerize several monomers, especially the ‘less-activated’ monomers (LAMs) (e.g., 1-vinyl-2-pyrrolidone (VP), vinyl chloride) result in lack of temporal control, poor system ‘livingness’ and production of polymers of broad molecular weight distributions (dispersity, Đ).7 Finally, due to some thermodynamic and kinetic limitations of RDRP approaches, the production of polymers of higher molecular weight (Mn) and predetermined structural parameters (narrow Đ) within a reasonable time-frame is still hardly possible. In this context, novel synthetic strategies providing fast polymerization in environmentally friendly media (without the use of toxic reagents) should be sought.One of the ways to address these issues is moving towards fast, highly efficient, and non-toxic strategy that uses a high-pressure (HP) as an ‘external’ stimulus. This interesting approach allows achieving control over polymerization due to changes in the density, intermolecular interactions, or viscosity. Since diffusivities are inversely proportional to the medium viscosity, the rate of termination can be reduced in the compressed systems. Importantly, the overall progress of polymerization also depends on the value of activation volume (ΔV), which in the majority cases is negative. Thus, at some thermodynamic conditions, polymerization is favored and proceeds much faster with respect to the reactions carried out at ambient conditions.8 Herein one can stress that such results were obtained for the HP controlled ATRP,9,10 RAFT11–15 and also FRP.16,17 Moreover, we have also applied high-pressure to control, ‘pseudo-living’ RAFT polymerization of sterically hindered imidazolium-based ionic liquids (IL) LAMs.18,19 The polymers produced in this way were characterized by tailored properties and excellent group fidelity with a wide range of Mn (up 530 kg mol−1) and narrow Đ ∼ 1.10 that was not achievable under ambient-pressure20 RAFT. Interestingly, we also proved that by appropriate selection of pressure (p = 500–1200 MPa) and system composition (a type of initiator, presence or absence of solvent) control over classical thermally-induced FRP could be achieved. Consequently, for FRP systems, the first-order polymerization kinetics and linear evolutions of the Mn with conversion were observed. Therefore, it seems to be crucial to see whether the application of high-pressure in the case of VP (LAM precursor of materials of high industrial importance), where conventional polymerization by RDRP fails, will be successful. It is worthwhile to note that VP possesses several structural limitations, including very high reactivity and high chain-transfer constant to the monomer that prevents the production of the well-defined poly(1-vinyl-2-pyrrolidone) (PVP) in a wide range of Mn by RDRP. Besides, among them, RAFT is the most effective VP polymerizing system.21 However, it can be stressed that despite the proper selection of chain transfer agents (CTA, i.e., xanthates and dithiocarbamates), reactions were poorly controlled, especially at higher monomer consumptions. As a consequence, PVP characterized by both moderate Mn and Đ (Mn = 6.4–53.0 kg mol; Đ = 1.13–2.30) have been obtained.22
In this work, we proposed a fast and versatile methodology for HP thermally-initiated FRP of VP (see Fig. 1S in the ESI,† Scheme 1). Our motivation was to develop the most straightforward and ‘greenest’ FRP strategy involving the use of only the monomer and thermoinitiator (of very low concentration, 0.01–0.15 wt%), allowing to maintain high control and efficiency of the process. The rate of polymerization was modulated by using a different initial concentration of thermoinitiator or varying thermodynamical parameters (p = 125–500 MPa, T = 323–333 K). We have found that well-defined PVP homopolymers in a wide range of molecular weight (Mn = 16.2–280.5 kg mol−1, Đ = 1.27–1.45) could be prepared even within 0.5 h. Additionally, also comprehensive thermodynamical and rheological measurements were performed on the selected synthesized system, and obtained results were compared to the ones measured for the commercially available samples. The characteristic of commercially supplied PVPs is presented in the ESI.† It can be stressed that although, at first sight, the application of HP seems to be a highly energy consuming process, recently published reports indicated that is not true, and the energy is consumed mostly during the compression stage.23 One can also notice that in industry pressure up to p = 1000 MPa is routinely used to preserve food.
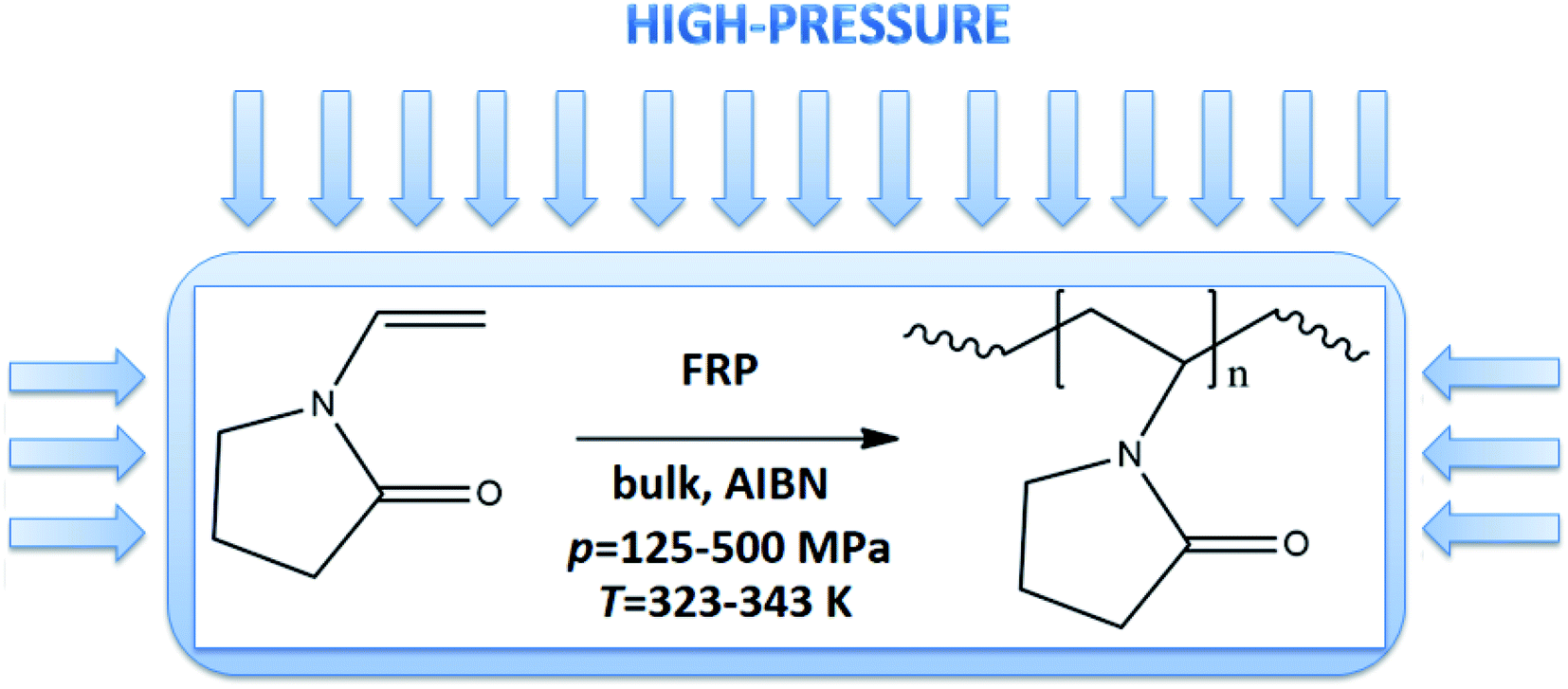 | ||
| Scheme 1 Synthetic route to produce well-defined PVPs under elevated-pressure. | ||
Results and discussion
VP polymerization at ambient- and elevated-pressures
A successful synthesis of well-defined PVP via HP FRP is strongly related to the appropriate selection of polymerization conditions. Lack of solvent, one type of initiator, low initiator concentration (0.15 wt%, 0.10 wt% and 0.01 wt% in respect to VP), variation in temperature (T = 323–343 K) and pressure (p = 125–500 MPa) have been proposed to minimize/eliminate the occurrence of the side reactions (e.g., chain transfer) and bimolecular termination characteristic for ambient-pressure systems.In preliminary experiments, ambient-pressure FRP of VP at T = 333 K with the presence of 0.10 and 0.15 wt% of AIBN were carried out. A kinetic study was performed by taking an aliquot of the reaction mixture at the required time point and conducting the 1H NMR (see Fig. 2S, ESI†) and SEC analysis. Note that the 1H and 13C NMR spectra of PVP, as well as SEC traces for polymers produced by both studied systems (ambient- and high-pressure), are collected in the ESI.† The data for the homopolymerization of VP performed at ambient-pressure are collected in Table 1. Fig. 1a and b show plots of ln([M]t/[M]0) versus time and molar masses Mn as well as plots of dispersity indices Đ against monomer conversion, respectively. As shown in Fig. 1a, polymerizations showed a similar course irrespective of the initial AIBN concentration. For the first 2 hours, the ln([M]t/[M]0) slop increased linearly, and then, the negative curvature of the kinetic plot was noted, indicating the domination of the chain transfer side reactions and chain termination process. Consequently, polymerizations had stopped at around 30% and 40% monomer consumption for 0.10 wt% and 0.15 wt% initial AIBN content, respectively.
| Ambient-pressure polymerization of VP | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.15 wt% of AIBN | 0.10 wt% of AIBN | ||||||||
| No. | Time [h] | Conv.a [%] | Mnb [kg mol−1] | Đb | No. | Time [h] | Conv.a [%] | Mnb [kg mol−1] | Đb |
| a Estimated by 1H NMR (600 MHz, CDCl3).b Estimated by SEC (DMF + 10 mM LiBr). | |||||||||
| Ia | 0.5 | 0.09 | 72.5 | 1.87 | IIa | 0.5 | 0.06 | 88.9 | 1.96 |
| Ib | 1 | 0.17 | 97.9 | 1.70 | IIb | 1 | 0.15 | 130.1 | 1.61 |
| Ic | 2 | 0.41 | 111.2 | 1.72 | IIc | 2 | 0.31 | 144.3 | 1.61 |
| Id | 3 | 0.43 | 109.4 | 1.78 | IId | 3 | 0.35 | 136.8 | 1.70 |
| Ie | 4 | 0.44 | 108.9 | 2.21 | IIe | 4 | 0.36 | 144.4 | 1.64 |
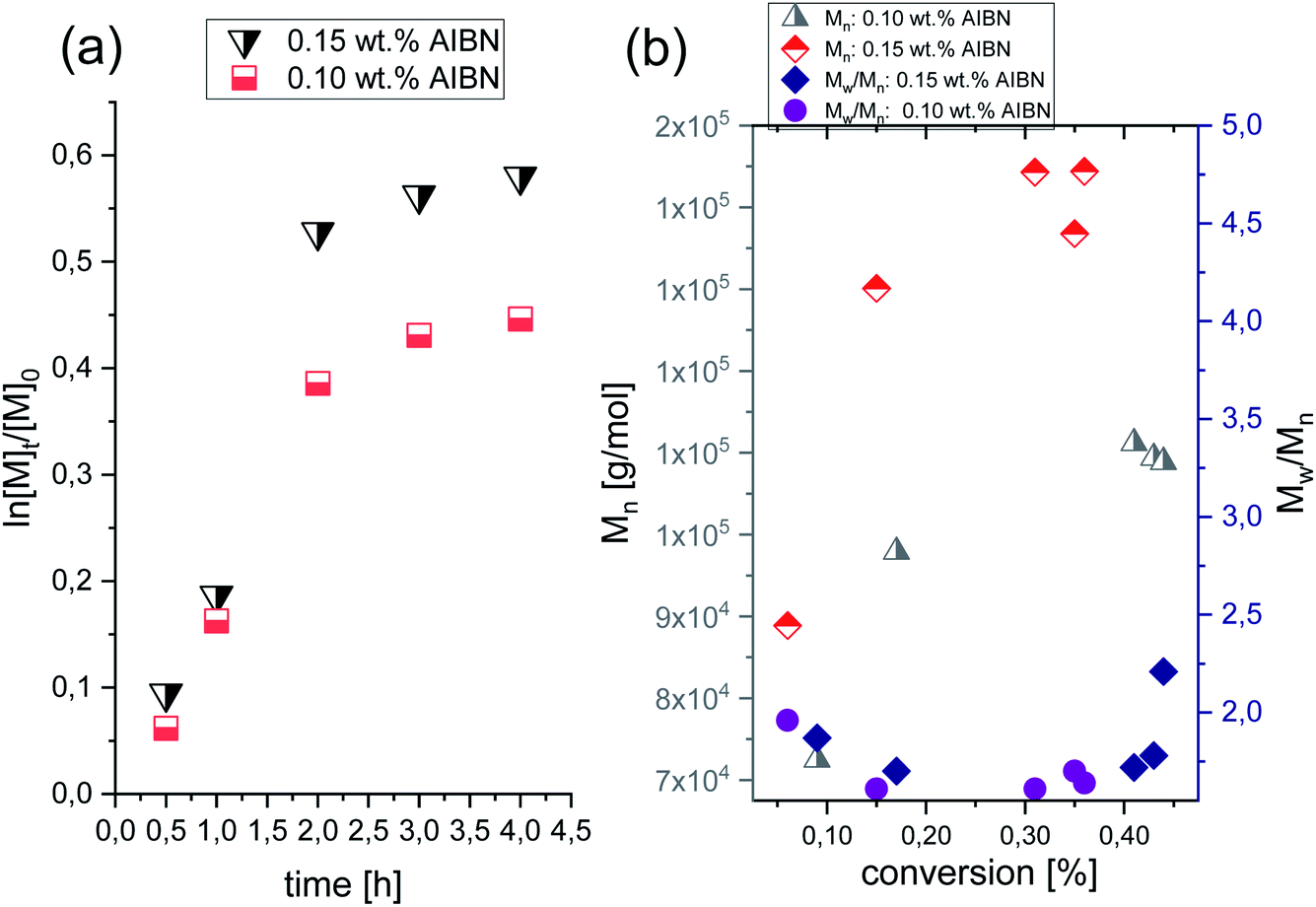 | ||
| Fig. 1 (a) Pseudo-first-order kinetic plot versus conversion for FRP of VP performed at varied AIBN initial content, at ambient-pressure; (b) Dependence of Mn vs. conversion and Đ vs. conversion of PVP produced at ambient-pressure. | ||
Fig. 1b demonstrates the typical dependencies of increasing Mn with the VP conversion for the uncontrolled FRP. The obtained polymers were characterized by Mn in the range of 72.2–144.4 kg mol−1 and as expected by high dispersities (Đ = 1.72–2.21). Such results indicated the lack of control over the chain-ends. One can note that our results are in good agreement with other data regarding conventional FRP of VP performed in bulk, an aqueous solution, or in organic solvents at atmospheric pressure conditions. A few examples of VP FRP, yielded PVP with relatively high molecular weight (Mn = 2.5–164.0 kg mol−1) and the high dispersity (Đ = 1.80–4.79).24–27
In this context, the application of elevated pressure as ‘external’ reaction force and controlling factor seems to be an excellent alternative to the current ambient pressure strategies. In order to find the optimal conditions for HP VP polymerization including acceleration of the process and gaining the control over the reaction, we have studied systems of the same content of AIBN (0.15 wt% and 0.10 wt% of AIBN) as in the case of synthesis carried out at p = 0.1 MPa also at different pressures (p = 125, 250, 500 MPa). Our preliminary measurements indicated that p = 250 MPa and T = 333 K are the most optimal conditions for VP polymerization. Besides, we also carried out reactions with very low AIBN concentration (0.01 wt%) to study the catalytic effect of the system's compression. The high-pressure polymerizations were conducted with the reaction time from 0.5 h up to 120 h depending on the initiator concentration (see Table 2). Note that the limiting value of conversion of VP for bulk processes vary between 60–70%. These values were reached after 5 h and 8 h for reactions with 0.15 wt% and 0.10 wt% AIBN respectively. However, the polymerization performed with the lowest AIBN content has stopped at ∼28% VP consumption.
| High-pressure polymerization of VP (250 MPa) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.15 wt% AIBN | 0.10 wt% AIBN | 0.01 wt% AIBN | ||||||||||||
| No. | Time [h] | Conv.a [%] | Mnb [kg mol−1] | Đb | No. | Time [h] | Conv.a [%] | Mnb [kg mol−1] | Đb | No. | Time [h] | Conv.a [%] | Mnb [kg mol−1] | Đb |
| a Estimated by 1H NMR (600 MHz, CDCl3).b Estimated by SEC (DMF + 10 mM LiBr). | ||||||||||||||
| 1a | 0.5 | 0.23 | 37.2 | 1.27 | 2a | 0.5 | 0.16 | 66.2 | 1.48 | 3a | 22 | 0.17 | 91.1 | 1.47 |
| 1b | 2 | 0.42 | 209.5 | 1.40 | 2b | 2 | 0.39 | 199.9 | 1.54 | 3b | 48 | 0.28 | 253.7 | 1.54 |
| 1c | 5 | 0.64 | 231.6 | 1.54 | 2c | 8 | 0.63 | 246.6 | 1.45 | 3c | 120 | 0.29 | 280.5 | 1.45 |
As can be seen from the data presented in Fig. 2a and b, HP FRP of VP shows first-order kinetic plots as ln([M]t/[M]0) increased linearly with a conversion for 0.15 wt% and 0.10 wt% AIBN concentration. Consequently, PVPs of Mn = 37.2–231.6 kg mol−1 (Đ = 1.27–1.54) and Mn = 66.2–246.6 kg mol−1 (Đ = 1.48–1.54) for systems with 0.15 wt% AIBN and 0.10 wt% have been obtained, respectively. Interestingly, the system with the lowest AIBN content showed negative curvature in the kinetic plot indicating the occurrence of the termination process (Fig. 2c). Taking into account very high Mn = 220–280.5 kg mol−1 and low/moderate dispersities (Đ ∼ 1.4) of produced in that way PVPs, we assume that too high system viscosity contributed to the end of this process rather than bimolecular termination. Fig. 3 presents the Mn and Đ of PVPs as a function of conversion. From the systems studied so far, the FRP with 0.10 wt% AIBN showed a linear relationship of Mn vs. conversion. It is worthwhile to mention that presented herein pressure-controlled strategy enabled us to produce PVP of very high Mn ∼ 200 kg mol−1, relatively narrow dispersity (up to Đ ∼ 1.40) within a very short reaction time (2 h, see example 1b in Table 2). On the other hand, PVPs obtained at ambient-pressure were characterized by much lower Mn (∼100 kg mol−1) and significantly broader dispersity indices (Đ = 1.70–2.21).
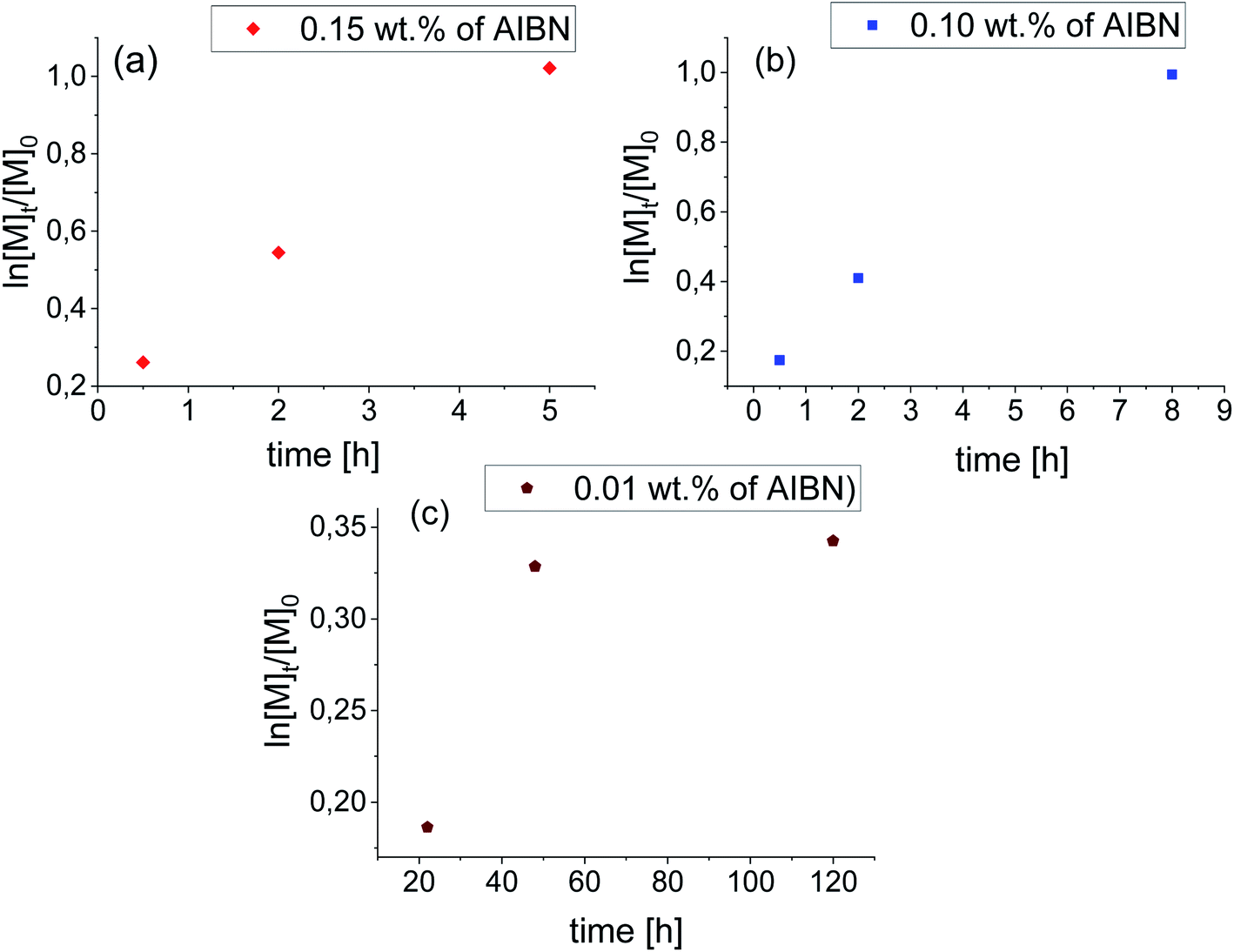 | ||
| Fig. 2 Pseudo-first-order kinetic plots versus conversion for FRP of VP at 250 MPa. | ||
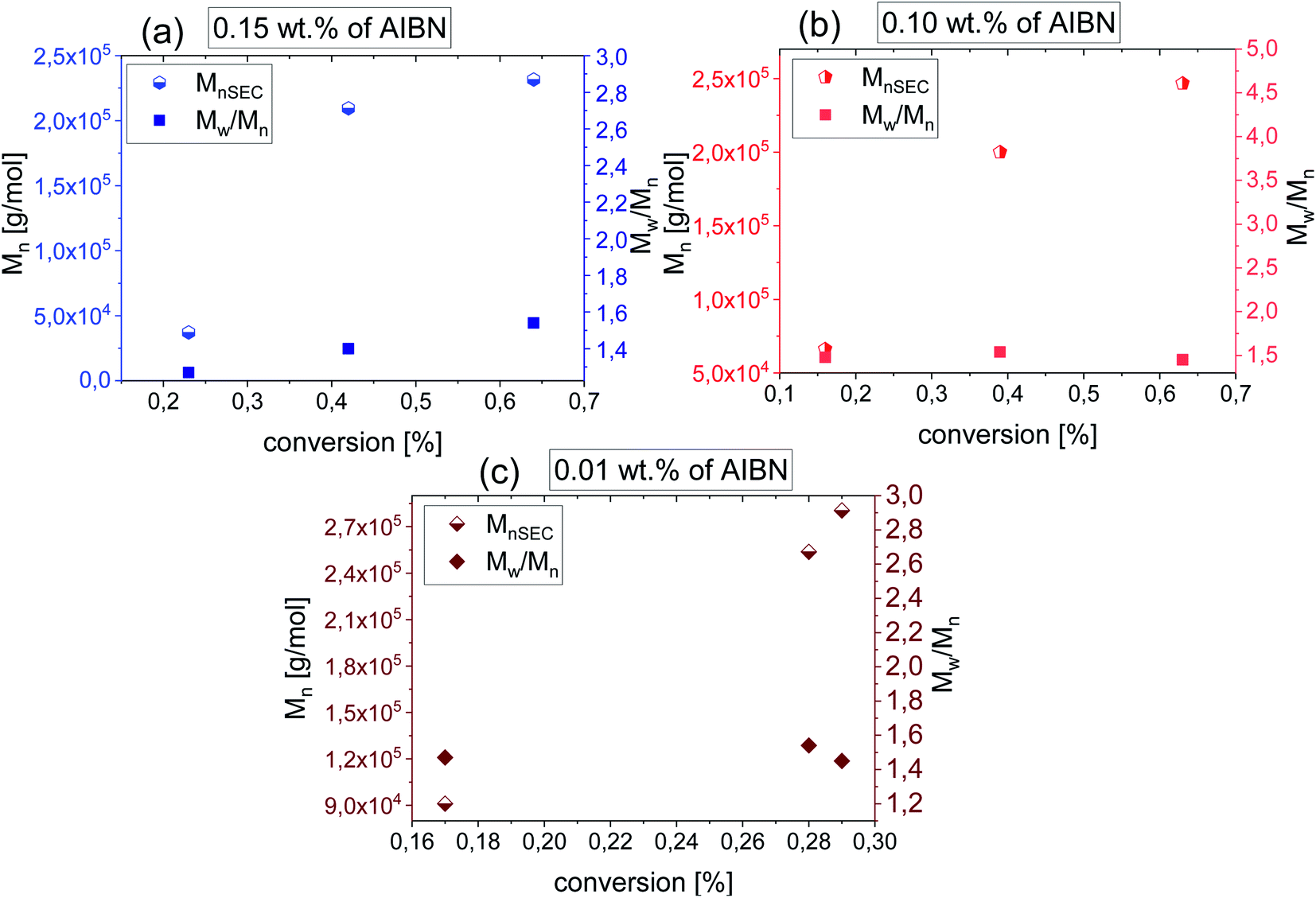 | ||
| Fig. 3 Dependence of Mn vs. conversion and Đ vs. conversion of PVP produced at varied AIBN concentration at 250 MPa. | ||
Also, from the SEC chromatograms (see Fig. 4), it can be seen that there is a huge difference in Mn and Đ values of the polymer produced via HP FRP and commercially available ones. The SEC trace of PVP obtained via high-pressure FRP revealed monomodal and symmetric shape indicating good control over the polymer characteristics. On the other hand, samples prepared via ambient-pressure FRP or commercial possessed unsymmetrical/bimodal shapes of SEC traces (see Fig. 5S in the ESI†).
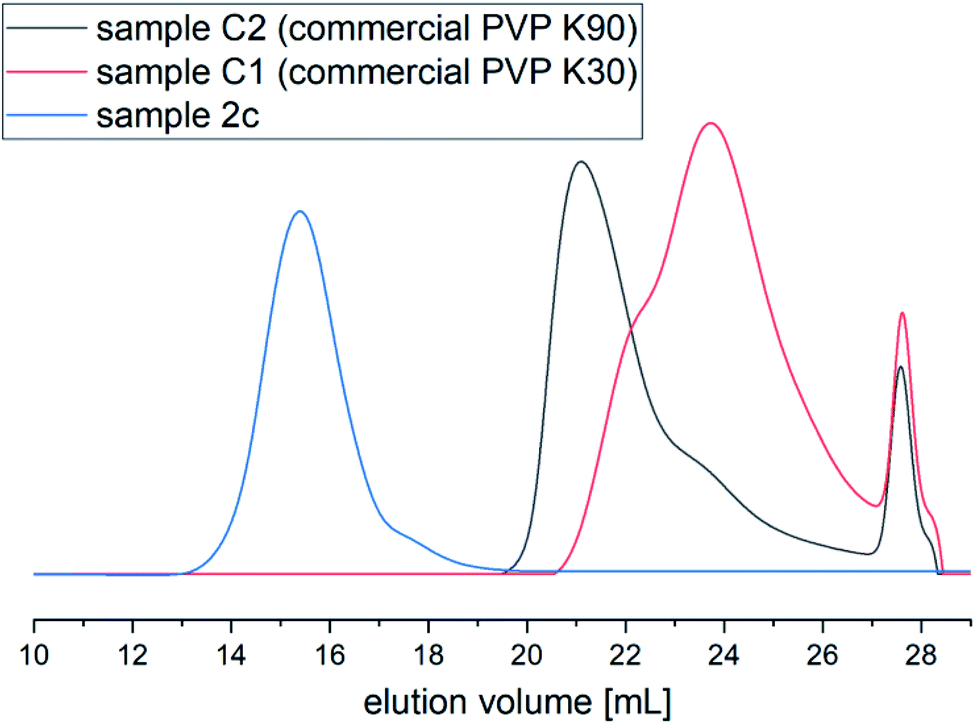 | ||
| Fig. 4 (a) SEC chromatograms of PVP produced under HP FRP (sample 2c) and commercially available samples C2 (PVP K90) and C1 (PVP K30); DMF+10 mM LiBr. | ||
To further investigate the impact of different temperature and pressure on the polymerization rate and properties of produced polymers, additional short-term reactions (0.5 h) for a selected system with 0.10 wt% AIBN were carried out (see Table 3). Reactions performed at T = +10 higher (T = 343 K), and p = 125 MPa allowed to obtain PVP with a very high Mn = 230.4 kg mol−1 and moderate dispersity Đ = 1.55 (65% VP conversion). On the other hand, lowering the temperature to T = 323 K and compression to p = 500 MPa resulted in a low VP conversion after 0.5 hours (4%) and production of well-defined polymer with a low Mn = 16.2 kg mol−1 (Đ = 1.38). Thus, collected data showed that we could modulate the reaction rate and molecular weight of polymers by changing both the thermodynamic parameters (p, T) or the concentration of the thermoinitiator.
Concluding this stage of our investigations, we found that the FRP process performed at ambient pressure proceeded with the uncontrolled path at the beginning of polymerization (fast termination, bimodal SEC traces of PVP samples). On the other hand, the high-pressure VP polymerization revealed the controlled nature of the reaction up to the end of the process. For HP systems, we observed a decrease in the termination rate (diffusion-controlled process) that resulted from the increase in system viscosity. Importantly, transfer reactions have a negative value of ΔV. Therefore, the pressure should have an increasing effect on the rate of these reactions. However, the proper selection of reaction conditions (lack of solvent) allowed to gain control over the FRP path and significantly reduce the transfer reactions and the biomolecular termination.
Thermal, rheological and solubility properties of synthesized PVPs
As a final point of our investigations, we characterized the dynamical and rheological properties of both produced and commercial PVPs with the use of DSC and rheological measurements. To compare properties between PVP samples one of the synthesized polymer (i.e., sample 2c presented in Table 2, Mn = 246.4 kg mol−1, Đ = 1.46) and two commercially available PVPs (Sigma-Aldrich; PVP K30 MnSEC ∼ 38.4 kg mol−1, Đ = 1.89 (sample C1) and PVP K90 MW 360 kg mol−1, MnSEC = 108.6 kg mol−1, Đ = 1.78 (sample C2), see ESI†) were examined.Fig. 5a, c and e present the mechanical storage (G′) and loss (G′′) modulus spectra of the measured polymers, which were obtained from the oscillation frequency sweep tests performed at different temperatures (426–452 K, 437–449 and 450–458 K for 2c, C1, C2, respectively). To better visualize the obtained data, the master curve was constructed for each sample using the time–temperature superposition (TTS) (see Fig. 5b, c and d). As a reference temperature (Tref) we have chosen T at which G′ and G′′ intersect at ω = 14 rad s−1. As can be seen, the mechanical response of the synthesized PVP polymer is more like commercial C2 sample. Both these samples exhibit (i) the glassy plateau (high frequency) (ii) three decades long transition regime (intermediate frequency) and (iii) the entanglement regime, also called rubbery plateau (low frequency). In the glassy and rubbery regime, G′ (storage or elastic moduli) surpasses G′′ (loss or viscous moduli) exhibiting a typical solid-like rheological behaviour. On the other hand, in the transition regime, the samples show a fluid-like behaviour since G′′ > G′.28 Intersections of the master curves of the G′ and G′′ delineate the boundaries between these regimes and correspond to the two important relaxation times.29 First, marked as τseg, is the relaxation time of one segment of the equivalent freely jointed chain, while the second is Rouse relaxation time of chain segments between entanglements (τe). The segmental relaxation times of the investigated polymers were estimated from the frequency of the crossover point of G′ and G′′ by employing the following relation τseg = 1/fcross. The temperature dependence of τseg obtained in this way is displayed in Fig. 6a. Since the τseg(T) usually follow the Vogel–Fulcher–Tammann (VFT) equation,30,31 we employed it to parametrized the obtained temperature dependences. The empirical VFT is expressed as follows:32–34
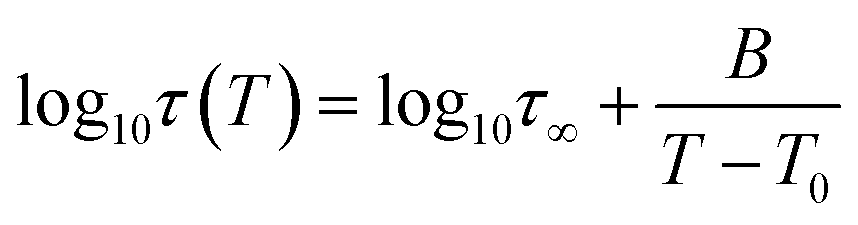 | (1) |
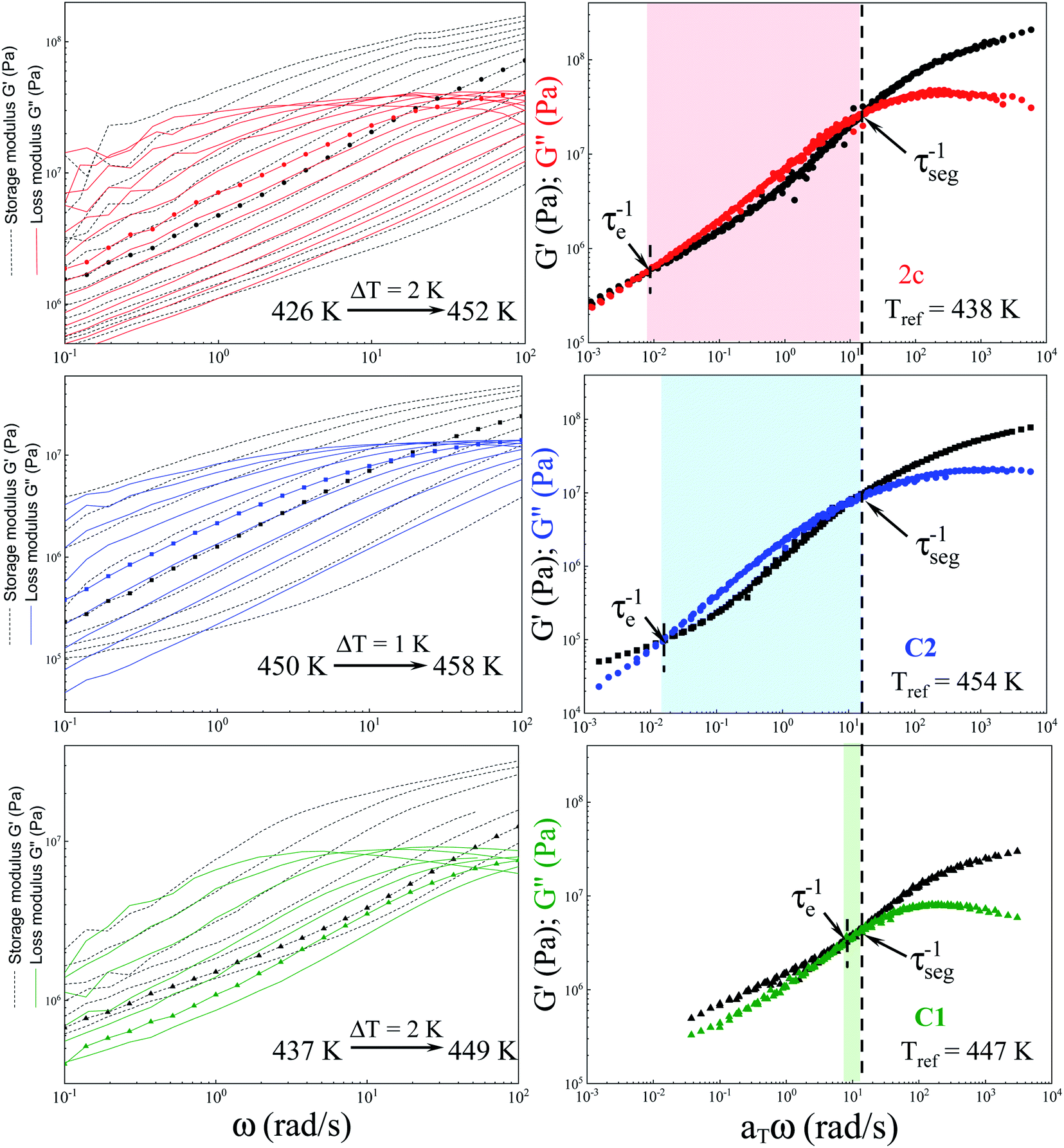 | ||
| Fig. 5 Mechanical loss G′′ (solid lines) and storage G′ (dashed lines) spectra of: (a) sample ‘2c’, (c) C2 and (e) C1 from the temperature region above Tg, Master curves of G′ (black symbols) and G′′ (coloured symbols) of: (b) sample ‘2c’, (d) C2 and (f) C1 obtained at a reference temperatures of Tref = T(τcross = 1.4 rad s−1). | ||
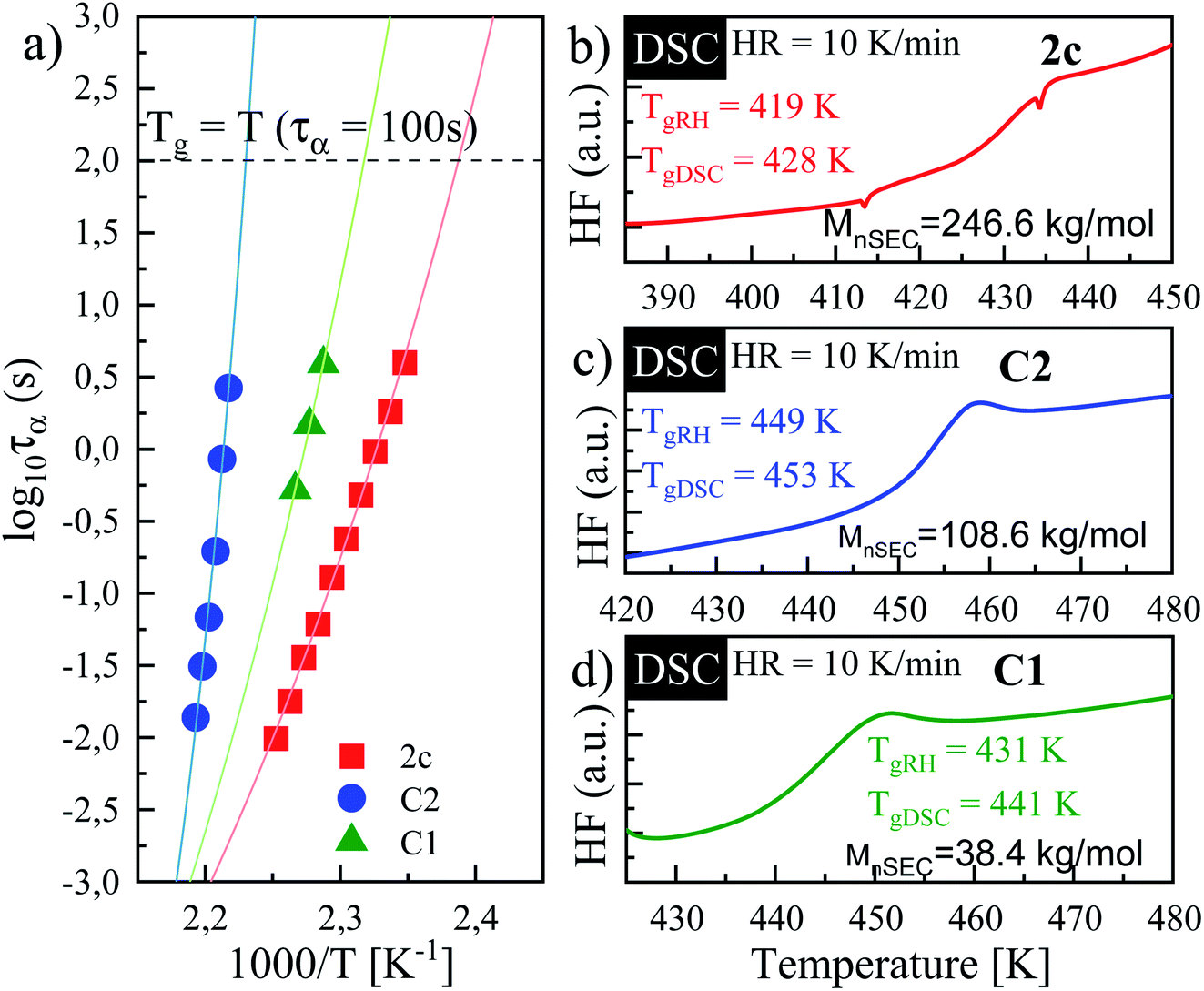 | ||
| Fig. 6 (a) Segmental relaxation times plotted as a function of temperature for PVP sample ‘2c’ (blue circles), C1 (green triangles) and C2 (red squares). Solid lines correspond to VFT fits of experimental data. DSC thermograms of: (b) 2c, (c) C2 and (d) C1. | ||
The solid lines in Fig. 6a correspond to VFT fits. From the extrapolation of the fits to temperatures at which τseg = 100 s, the glass transition temperatures of the examined polymers were estimated to be 419, 431 and 449 K for sample ‘2c’, C1 and C2, respectively. It is worth mentioning that these values are in good agreement with the Tgs obtained from the DSC measurement – see the DSC thermograms presented in Fig. 6b–d. Literature data reported that Tg of PVP increases with the molecular weight towards a limiting value (of 453 K) according to the Fox–Flory relation.35 Besides, Tg values decrease with a decrease in viscosity average molecular weight.20 Surprisingly, comparing examined Tg values, we found that synthesized herein PVP sample of Mn ∼ 246 kg mol−1 (2c) presents much lower Tg with respect to the commercial ones (C1, Mn ∼ 30 kg mol−1, and C2, Mn ∼ 108.6 kg mol−1). Such a scenario might be a result of reaction conditions in which the polymer was synthesized. Note that commercial samples were produced using solvent polymerization, whereas our samples from the monomer alone. As reported, PVPs prepared by γ-irradiation and thermal-decomposition of the initiator with the presence of water or aqueous solution have a much higher Tg than the ones produced in bulk (449 K vs. 405 K for γ-irradiation; 440 K vs. 386 K for thermal-decomposition of AIBN). Explanations of this phenomena should be sought in strong interactions between monomer and water molecule or other polar solvent forming both dipole and hydrogen bonds during polymerization. Another possible elucidation is related to the residual monomer that fails to polymerize in the glassy state and decreases the Tg value. However, in our case, each reaction mixture was purified by the highly-efficient ultrafiltration method following by precipitation, which allowed for the separation of the unreacted monomer from the reaction, mixture which was confirmed by 1H NMR measurements. Therefore, we assume that the main factor affecting the Tg value of produced PVP is connected to the condition applied for the polymerization (bulk polymerization). Interestingly, synthesized herein PVPs were characterized by different solubility properties than commercially supplied. It is well-known that PVP is soluble in water and other organic solvents, including alcohols (i.e., methanol, ethanol), and some chlorinated compounds such as chloroform or methylene chloride. On the other hand, it is essentially insoluble in hydrocarbons, ethers, ketones (i.e., acetone), and esters. Note that PVP produced with the lack of solvent (water) can also be dissolved in acetone, toluene or xylene. In our case, the main difference was in the solubility of synthesized polymers in acetone (see Table 1S in the ESI†). Nevertheless, our results are consistent with literature data showing the significant impact of the polymer production method on its thermomechanical and solubility properties.36
It should also be stressed that strategy proposed in this paper allowed us to obtain PVP homopolymers with a strictly defined Mn within the range of 16.2–280.5 kg mol−1, which makes it very promising considering great flexibility of these macromolecules across multiple applications. Note that PVP homopolymers, due to their unique combination of properties, i.e. good solubility in water and many organic solvents, chemical stability, biocompatibility, non-toxicity, and affinity to complex both hydrophobic and hydrophilic substances, are widely used for designing materials for different applications. One should mention the pharmaceutical industry and medicine, optical and electrical applications, adhesives, coatings and inks, agriculture, membranes, fibres and textiles.37 Among so many, the biomedical and pharmaceutical applications are particularly the most important. In fact, PVP is one of the best additives and pore-former agents in ultra-,38 macro-,39 micro- and nanofiltration40 membrane fabrication for biomedical applications, e.g. hemodialysis41 or drug release-controlling membranes.42 As polybase it can form interpolymer complexes (IPC) with polyacids. Keeping in mind that this complexation process is strongly dependent on the pH, these pH-sensitive materials were designed for biomedical applications, e.g. pH-controlled drug delivery.43 For the latter use, the PVP with low molecular weight is desirable since it forms complexes with both low molecular weight compounds and polymers, making insoluble substances soluble simultaneously improving their biocompatibility.36 On the other hand, PVP of high molecular weight (higher than Mn ∼ 300 kg mol−1) are less suitable for biomedical application, because they have a high viscosity in water and therefore dissolve too slowly, delaying dissolution of the active substance.44 Nevertheless, these materials are often used commercially for the clarification of beverages such as beer, vinegar, and grape wine.37 High molecular weight PVPs are also used as protective colloids and particle size regulators for suspension polymerization of styrene, vinyl acetate, and vinyl chloride.
Conclusions
In conclusion, the FRP of LAM monomer, VP, has been performed under high-pressure conditions (p = 125–500 MPa), extending the pool of ‘green’, solvent- and metal-free synthetic strategy of highly pure PVP. Our results demonstrated that a careful selection of conditions (a type of initiator, low initiator concentration, lack of solvent, pressure, temperature) is crucial for achieving PVP of well-defined structural parameters in a short reaction time-frame (0.5–2 h). Depending on the reaction conditions, well-defined polymers in a wide range of Mn = 16.2–280.5 kg mol−1 and the relatively low dispersities (Đ = 1.27–1.55) have been obtained. Note that produced PVP has the lowest dispersity values reported to date for FRP. We founded that HP acts as an ‘external’ controlling factor that provides the first-order polymerization kinetics for classical FRP, something that was possible so far only for RDRP systems. It was also confirmed that the method of polymer preparation strongly affects its physicochemical properties, including Tg and solubility. We also found that similar mechanical properties characterize PVP of low dispersity indices (produced herein) to that available commercially that are being sold as products of much higher dispersity. Note that the proposed PVP synthetic strategy is fully compatible with the current industrial polymerization possibilities. In this context, one can remind about high-pressure cells of large capacity that are used for the food pasteurization process (p = 1 GPa). We believe that presented herein the novel synthetic methodology opens an alternative tool of preparation of well-defined PVP of high purity using the ‘greenest’ synthetic strategy to date.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. M. and R. B. are thankful for financial support from the Polish National Science Centre within SONATA project (DEC-2018/31/D/ST5/03464). D. H. is thankful for financial support from Medical University of Silesia within Research for Young Scientists (Contract No. KNW-2-O-08/D/9/N).References
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS.
- Z. Wang, Z. Wang, X. Pan, L. Fu, S. Lathwal, M. Olszewski, J. Yan, E. A. Enciso, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 275–280 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- X. Pan, C. Fang, M. Fantin, N. Y. Malhotra, W. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- N. L. Zutty and R. D. Burkhart, in Polymerization and Polycondensation Processes, American Chemical Society, ed. N. A. J. Platzker, Van Nostrand Reinhold, New York, 1962 Search PubMed.
- L. Mueller, W. Jakubowski, K. Matyjaszewski, J. Pietrasik, P. Kwiatkowski, W. Chaladaj and J. Jurczak, Eur. Polym. J., 2011, 47, 730–734 CrossRef CAS.
- P. Kwiatkowski, J. Jurczak, J. Pietrasik, W. Jakubowski, L. Mueller and K. Matyjaszewski, Macromolecules, 2008, 41, 1067–1069 CrossRef CAS.
- J. Rzayev and J. Penelle, Macromolecules, 2002, 35, 1489–1490 CrossRef CAS.
- J. Rzayev and J. Penelle, Angew. Chem., Int. Ed., 2004, 43, 1691–1694 CrossRef CAS.
- P. Ye, P.-F. Cao, Z. Su and R. Advincula, Polym. Int., 2017, 66, 1252–1258 CrossRef CAS.
- T. Arita, M. Buback, O. Janssen and P. Vana, Macromol. Rapid Commun., 2004, 25, 1376–1381 CrossRef CAS.
- M. J. Monteiro, R. Bussels, S. Beuermann and M. Buback, Aust. J. Chem., 2002, 55, 433–437 CrossRef CAS.
- Y. Ogo, Journal of Macromolecular Science: Part C: Polymer Reviews, 1984, 24, 1–48 CrossRef.
- M. Buback and C. Kowollik, Macromolecules, 1999, 32, 1445–1452 CrossRef CAS.
- P. Maksym, M. Tarnacka, A. Dzienia, K. Erfurt, A. Brzęczek-Szafran, A. Chrobok, A. Zięba, K. Kaminski and M. Paluch, Polymer, 2018, 140, 158–166 CrossRef CAS.
- P. Maksym, M. Tarnacka, A. Dzienia, K. Wolnica, M. Dulski, K. Erfurt, A. Chrobok, A. Zięba, A. Brzózka, G. Sulka, R. Bielas, K. Kaminski and M. Paluch, RSC Adv., 2019, 9, 6396–6408 RSC.
- P. Maksym, M. Tarnacka, A. Dzienia, K. Erfurt, A. Chrobok, A. Zięba, K. Wolnica, K. Kaminski and M. Paluch, Polym. Chem., 2017, 8, 5433–5443 RSC.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- R. Devasia, R. L. Bindu, R. Borsali, N. Mougin and Y. Gnanou, Macromol. Symp., 2005, 229, 8–17 CrossRef CAS.
- O. Rodriguez-Gonzalez, R. Buckow, T. Koutchma and V. M. Balasubramaniam, Compr. Rev. Food Sci. Food Saf., 2015, 14, 536–554 CrossRef.
- D. Wan, K. Satoh, M. Kamigaito and Y. Okamoto, Macromolecules, 2005, 38, 10397–10405 CrossRef CAS.
- V. P. Torchilin, T. S. Levchenko, K. R. Whiteman, A. A. Yaroslavov, A. M. Tsatsakis, A. K. Rizos, E. V. Michailova and M. I. Shtilman, Biomaterials, 2001, 22, 3035–3044 CrossRef CAS.
- A. Benahmed, M. Ranger and J. C. Leroux, Pharm. Res., 2002, 18, 323–328 CrossRef PubMed.
- T. W. Chung, K. Y. Cho, H. C. Lee, J. W. Nah, J. H. Yeo, T. Akaike and Ch. S. Cho, Polymer, 2004, 45, 1591–1597 CrossRef CAS.
- F. Kremer and A. Schonhals, Broadband Dielectric Spectroscopy, Springer, New York, 2003 Search PubMed.
- O. Oparaji, S. Narayanan, A. Sandy, S. Ramakrishnan and D. Hallinan, Macromolecules, 2018, 51, 2591–2603 CrossRef CAS.
- T. Nicolai and G. Floudas, Macromolecules, 1998, 31, 2578–2585 CrossRef CAS.
- Z. Wojnarowska, H. Feng, Y. Fu, S. Cheng, B. Carroll, R. Kumar, V. N. Novikov, A. Kisliuk, M. T. Saito, N. G. Kang, J. W. Mays, A. P. Sokolov and V. Bocharova, Macromolecules, 2017, 50, 6710–6721 CrossRef CAS.
- H. Vogel, Phys. Z., 1921, 22, 645–646 CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- G. Tammann and W. Hesse, Z. Anorg. Allg. Chem., 1926, 156, 245–257 CrossRef CAS.
- T. G. Fox Jr and P. J. Flory, J. Appl. Phys., 1950, 21, 581–591 CrossRef.
- F. Haaf, A. Sanner and F. Straub, Polym. J., 1985, 17, 143–152 CrossRef CAS.
- M. Teodorescu and M. Bercea, Polym.-Plast. Technol. Eng., 2015, 54, 923–943 CrossRef CAS.
- B. Vatsha, J. C. Ngila and R. M. Moutloali, Phys. Chem. Earth, 2013, 67–69, 125–131 Search PubMed.
- W. Albrecht, J. Schauer, T. Weigel, K. Richau, T. Groth and A. Lendlein, J. Membr. Sci., 2007, 291, 10–18 CrossRef CAS.
- S. S. Madaeni, N. Arast, F. Rahimpour and Y. Arast, Desalination, 2011, 280, 305–312 CrossRef CAS.
- J. Barzin, S. S. Madaeni, H. Mirzadeh and M. Mehrabzadeh, J. Appl. Polym. Sci., 2004, 92, 3804–3813 CrossRef CAS.
- R. Verma, Int. J. Pharm., 2003, 263, 9–24 CrossRef CAS PubMed.
- M.-K. Chun, C.-S. Cho and H.-K. Choi, J. Appl. Polym. Sci., 2004, 94, 2390–2394 CrossRef CAS.
- V. Bühler, Polyvinylpyrrolidone Excipients for Pharmaceuticals, Springer, Berlin, Heidelberg, 2005, p. 88 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02246b |
| This journal is © The Royal Society of Chemistry 2020 |