
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEthanol vs. water: influence of the terminal functional group of the alkyl chain and environment of the self-assembly process on electron transport through the thiol layer
Agata Kowalczyk *ab and
Cong Yu*bc
*ab and
Cong Yu*bc
aFaculty of Chemistry, University of Warsaw, Pasteura St. 1, PL-02-093 Warsaw, Poland. E-mail: akowalczyk@chem.uw.edu.pl; congyu@ciac.ac.cn
bState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, PR China
cUniversity of Science and Technology of China, Hefei, 230026, China
First published on 5th June 2020
Abstract
Self-assembly of alkanethiol chains on metallic surfaces is a spontaneous process which leads to the formation of highly ordered layers. However, the organization of the thiol chains on the surface strongly depends on the intermolecular interactions between the terminal groups in the chain. The solution environment also plays an important role. In this paper we present the effect of solution solvent (water and ethanol) and the presence of various hydrophilic terminal groups (–OH, –NH2 and –COOH) on the quality and electrochemical properties of the formed alkanethiol layers. In the studies we applied voltammetry, atomic force microscopy and quartz crystal microbalance to characterize the morphology, packing density and ability to electron exchange through the thiol layer. The blocking properties of the formed SAMs expressed as the electron-transfer rate constant as well as their organization have been examined using a model electrochemical probe, Fe(CN)63−. With the increase in the polarity of the terminal functional group the regularity of the thiol layer decreased.
Introduction
The surface properties are of great importance to biomolecule-surface interactions. By adjusting the abundance, type, and spatial distribution of surface modifiers chemical and structural properties of surfaces such as stability, surface charge, wettability and topography can be controlled.1–3 That is why the different types of modifying layers have attracted considerable attention for molecular-level surface design in e.g. catalysis,4,5 membrane transport,6,7 molecular recognition,8,9 cell adhesion,10,11 biosensing,12–14 and protein-surface interactions.15,16 There are many examples of modifying layers, which allow change and control of nanoscale surface properties, such as self-assembled alkanethiol monolayers (SAMs), polymers, reduced diazonium salts, reduced graphene oxide etc.17–21Self-assembled alkanethiol monolayers are very popular because they are considered to be stable, easy in preparation, well-organized and, due to the variety of thiols with various terminal groups, can mimic surfaces with different charge and extreme different properties. From the applicability point of view of such layers the functionalization process is desired to facilitate the surface interactions. The high stability of thiol monolayer self-assembled on gold surface is related to the fact that the alkanethiol reacts strongly with gold and forms a bond with covalent character.22 Additionally, intermolecular and surface forces such as van der Waals forces, electrostatic interactions and hydrogen bonding ensure the compact packing of the SAM, stabilize the structure and play key role in a wide variety of chemicals and biochemical surface interactions.23–25 The quality of the formed layers strongly depends on many factors; the most important are thiol chain length, presence of functional terminal groups in thiol chain, and environment solvent of the adsorption process.26 The number of carbon atoms in the thiol chain is a key factor in the layer packing density and its organization (uniformity) that is reflected in the rate of electron exchange through the layer. This rate is a very important parameter in many applications. Taking into account the electron tunneling mechanism, the electron-transfer rate constant is expected to decrease exponentially as SAM thickness increases.27,28 The way to improve electron exchange through a SAM layer is the application of short chain alkanethiols. Nevertheless, only alkanethiols with the sufficiently high number of carbon atoms in the alkyl chain form a well-organized, densely packed crystalline-like monolayers,29 the SAM layers of short chain alkanethiols are not as well defined and uniform as the layers of long chain alkanethiols. However, the permeability of the SAM layers depends not only on layer thickness,30–32 but also on electrode potential,31,33–35 presence of terminal group, and, to a large extent, on solvent used as the adsorption medium.36,37 Since the layer is a barrier for ions and molecules, the thicker is the layer (longer alkanethiol chain) the worst its permeability is observed. As mentioned above, thiol layers of short alkanethiols are not well organized which is conducive to good permeability of the layer. Due to the fact, that the van der Waals forces stabilizing the interactions between carbon chains in the layer are weaker in the case of short alkanethiols, such layers contain more defects compared to those formed from longer alkanethiol chains.38,39 However, the packing of the SAM layer depends not only on the van der Waals forces but also is influenced by the terminal group of the alkanethiol chains. In the case of small terminal groups (–SH, –COOH, –OH, –NH2) the structure of the SAM layer is still controlled by van der Waals interactions and the SAM layers are still quite compact. But in the case of bulky terminal groups, e.g. the carboxylic group, the ordering in SAMs decrease.40–43 The SAM layers are considered to be impermeable to ions and molecules in a specific potential range.35 The potential-induced changes in alkanethiol layer organization are related to the values of the potential applied to the electrode. Generally, both extremely negative and positive potential values are not conducive to layer stability due to the reductive or oxidative electrodesorption of alkanethiol chains. They induce structural changes in the monolayer, lead to appearance of defects in SAM, and in consequence increase permeability of the SAM layer. According to the literature data the alkanethiol layers are stable and impermeable in the potential window from −0.5 to 0.5 V vs. Ag/AgCl for Au/–S–(CH2)nR; n ≤ 15, where R is the terminal group, e.g. –CH3, –Cl, –OH, –COOH, –CN.35,41,44–46 The values of critical potentials given above depend on chain length; the longer alkanethiol chain the wider potential window of stability is. At potentials more positive or negative than the appropriate critical potential the structure of the SAM layer changes. It may involve ill-defined structural transformation or the reductive or oxidize electrodesorption of entire alkanethiol chains. In consequence the potential-induced defects appear and the layer loses its insulating ability.47–52 Furthermore, during the formation of the SAM layer in an aqueous solution the unique phenomenon occurs: the hydrophobic alkanethiol chains associate and reduce the aqua contact area.53 The alkanethiol film repels water molecules adjacent to it and in consequence a gap between the monolayer film and water arises.36 The water content in the solution environment is very important in the formation process of well-organized and uniform SAM layers, which depends on how the clustering of SAMs due to the van der Waals interactions is controlled by surrounded water structures. The clustering of thiols in the surrounding water changes with water content in the solution environment; the randomness of the packing of the alkane chains increases with a decrease in water content. In consequence, the arising local disorder leads to an increase in layer roughness. So, it can be concluded that the quality of self-assembled monolayer is strongly dependent on surrounding water structure.
Taking into account the literature data related to the formation of SAM layers the influence of solution environment on the quality of the formed thiol layers should be an interesting aspect especially in the case of formation of thiol layers terminated with polar groups (–OH, –NH2, –COOH). Herein, we report how the presence of polar terminal groups influence on the morphology, thickness, spatial distribution of alkyl chains, and in consequence on the electron transport through the layer self-assembled in aqueous and ethanolic environments. The studies were performed with the use of cyclic voltammetry (CV), quartz crystal microbalance (QCM), atomic force microscopy (AFM), and contact angle measurements. The obtained data indicate that from the electrochemical applicability point of view the nature and quality of the formed layers strongly depend on the terminal group of thiol chains and solvent used.
Experimental section
Chemicals
All chemicals were of the highest purity available. Sodium hydroxide, potassium dihydrogen phosphate, dibasic potassium phosphate, potassium sulfate, potassium ferricyanide, 1-octanethiol (HS–C8H17), 8-mercapto-1-octanol (HS–C8H16–OH), 8-amino-1-octanethiol (HS–C8H16–NH2) and 8-mercaptooctanoic acid (HS–C7H14–COOH) were purchased from Sigma. All measurements were made in 0.1 M phosphate buffer (PB) of pH 7.0 containing 0.15 M K2SO4. Ultrapure water (Milli-Q, Millipore, conductivity of 0.056 μS cm−1) was used to prepare all solutions.Voltammetric measurements
Cyclic voltammetry experiments were performed using a CHI-1040C electrochemical workstation (CH Instrument) in the three-electrode system. A gold disc electrode (Au; ϕ = 1.6 mm, BAS Instruments) was used as the working electrode, a Ag/AgCl/3 M KCl as the reference electrode and a gold wire served as the auxiliary electrode. Before each measurement the surface of the working electrode was polished with 1 μm Al2O3 powder on a wet pad. After each polishing, the electrode surface was rinsed with direct stream of ultrapure water to completely remove alumina from the electrode surface, and dried with argon. Next, the gold electrodes were electrochemically cleaned by cycling them in 0.1 M H2SO4 solution in a potential range from −0.3 to 1.5 V (vs. Ag/AgCl/3 M KCl) with scan rate 0.05 V s−1, until a stable voltammogram typical for a bare gold electrode was obtained.The double-layer capacitance measurements for bare and SAMs coated gold electrodes were performed in 0.1 M deoxygenated phosphate buffer (PB) of pH 7.0 containing 150 mM K2SO4 in the potential range from −0.1 to 0.5 V (vs. Ag/AgCl/3 M KCl) at scan rate 1 V s−1.
Atomic force microscopy and contact angle measurements
All AFM images were collected in Peak Force Tapping mode using ScanAsyst Fluid + probes (Bruker) with nominal spring constant of 0.4 N m−1. Each probe was carefully calibrated using thermal tune module. The images were recorded in a 0.1 M PB of pH = 7.0 aqueous solution of. Thickness of the monolayer was estimated using the AFM-based nanolithography technique. Namely, a given area of the film was scanned using silicon nitride probe under relatively high load in the range of 50–60 nN. Due to the high forces applied, the molecules were removed from the substrate by the scanning probe. Next, the applied load was decreased down to 1 nN, and the scanning area was increased to get the topography of the shaved region and the surrounding intact monolayer. Such images were subjected to cross-sectional analysis, which allowed measurements of the height differences between bare and monolayer-coated substrate. Such differences corresponded to the monolayer thickness measured under 1 nN load applied to the AFM tip.Contact angle measurements were done after modification of the gold surface with appropriate SAM monolayer. The measurements were performed using a Hetta Lite Optical Fensiometer, model TL100.
QCM experiments
Quartz crystal microbalance experiments were performed using an Autolab model with a 6 MHz Au/TiO2 quartz crystal resonator. The resonant frequency of the quartz crystal lattice vibrations in a thin quartz crystal wafer was measured as a function of the mass attached to the crystal surface. For thin rigid films, the interfacial mass change, Δm, is linearly related to the change in the resonance oscillation frequency, Δf, of the QCM. This relation is given by the Sauerbrey equation.54,55 The piezoelectrically active (geometrical) surface area of the working Au electrode was 0.353 cm2 and the real surface area was A = 0.527 cm2 (the roughness factor, R equaled 1.5). Before the use the Au-QCM electrodes were electrochemically cleaned by voltammetric cycling in 0.5 M NaOH solution in a potential range from 0 to 0.8 V (with a 10 s scan stop at 0.8 V) vs. Ag/AgCl/3 M KCl with scan rate 50 mV s−1. Next, the electrodes were cycled in 0.1 M H2SO4 solution in a potential range from −0.3 to 1.5 V (vs. Ag/AgCl/3 M KCl) until a stable voltammogram, typical for a bare gold electrode, was observed. The final step of the electrochemical cleaning procedure was the activation step in 0.1 M perchloric acid solution. It was done through the cycling in a potential range from −0.65 to 0.95 V (vs. Ag/AgCl/3 M KCl) at a high scan rate (>1 V s−1).Preparation of thiols layers
All thiols layers were formed on freshly cleaned bare gold electrode from 1 mM aqueous and ethanolic (99.8%) solutions of appropriate thiol. The self-assembly process lasted circa 12 h. Next, the electrodes with self-assembled thiol layers were delicately rinsed with water or ethanol respectively, to remove physically adsorbed thiol molecules, and gently dried with argon.Results and discussion
AFM and contact angle characteristic
To get the information about the morphology of the formed thiol layers, the AFM and contact angle measurements were performed. The main consequence of the presence of –OH, –NH2 and –COOH terminal groups in the alkanethiol chain is the change in wettability of the modified surface (contact angle measurements); this is shown in Table 1. With increasing the polarity of the terminal group in the thiol chains a decrease in the contact angle value was observed. The introduction of the 8-mercaptooctanoic acid, the most polar studied alkanethiol, onto the electrode surface resulted in more than twice lower contact angle value compared to 1-octanethiol for both aqueous and ethanolic solutions.| Cdl [μF cm−2] | dCV [nm] | dAFM [nm] | Em [V] | ks [cm s−1] | Contact angle | |
|---|---|---|---|---|---|---|
| a This value may corresponds to the electrode process based on penetration of the thiol layer by the redox probe molecules. | ||||||
| Bare | 34.2 ± 5.4 | — | — | 0.188 | (2.7 ± 0.2) × 10−2 | 80° ± 0.3° |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||
| Aqueous solutions | ||||||
| Au/–S–C8H17 | 1.2 ± 0.2 | 1.7 ± 0.4 | 1.0 ± 0.3 | 0.196 | (0.9 ± 0.3) × 10−4a | 95° ± 1.3° |
| Au/–S–C8H16–OH | 1.1 ± 0.2 | 1.8 ± 0.5 | 1.2 ± 0.3 | 0.145 | (2.6 ± 0.6) × 10−4 | 72° ± 2.3° |
| Au/–S–C8H16–NH2 | 1.2 ± 0.1 | 1.7 ± 0.5 | 1.3 ± 0.5 | 0.188 | (1.1 ± 0.2) × 10−2 | 56° ± 2.5° |
| Au/–S–C7H14–COOH | 0.9 ± 0.1 | 2.5 ± 0.8 | 1.3 ± 0.6 | 0.235 | (0.5 ± 0.1) × 10−2 | 41° ± 1.8° |
|
||||||
| Ethanolic solutions | ||||||
| Au/–S–C8H17 | 2.2 ± 0.3 | 0.9 ± 0.4 | 1.0 ± 0.2 | — | — | 98° ± 0.5° |
| Au/–S–C8H16–OH | 1.9 ± 0.1 | 1.1 ± 0.4 | 1.2 ± 0.3 | 0.125 | (0.6 ± 0.1) × 10−4 | 68° ± 0.8° |
| Au/–S–C8H16–NH2 | 0.9 ± 0.1 | 2.4 ± 0.9 | 1.3 ± 0.4 | 0.184 | (1.2 ± 0.3) × 10−2 | 60° ± 0.9° |
| Au/–S–C7H14–COOH | 0.9 ± 0.1 | 2.3 ± 0.5 | 1.4 ± 0.5 | 0.238 | (0.7 ± 0.1) × 10−2 | 46° ± 1.3° |
Moreover, the AFM data (see Fig. 1) showed that the terminal groups affected the ordering of the SAMs. The regularity of Au/–S–C8H16–OH layer was very similar to the Au/–S–C8H17 layer regardless of thiol adsorption environment. In turn, the introduction of the –NH2 and –COOH terminal groups to alkanethiol chains led to an increase in the irregularity of the layer. This effect is a consequence of the greater participation of the intermolecular hydrogen bonds between terminal groups at the lost of cohesion of the hydrocarbon chains. The surface roughness increased at least 1.5 times (see in Fig. 1). The presence of these groups could favour the local formation of a multilayer, wherein the formed adlayer was definitely less uniform. However, the estimated layer thicknesses are quite similar to each other, regardless the type of the terminal group of the alkanethiol and solution environment (Table 1, AFM data).
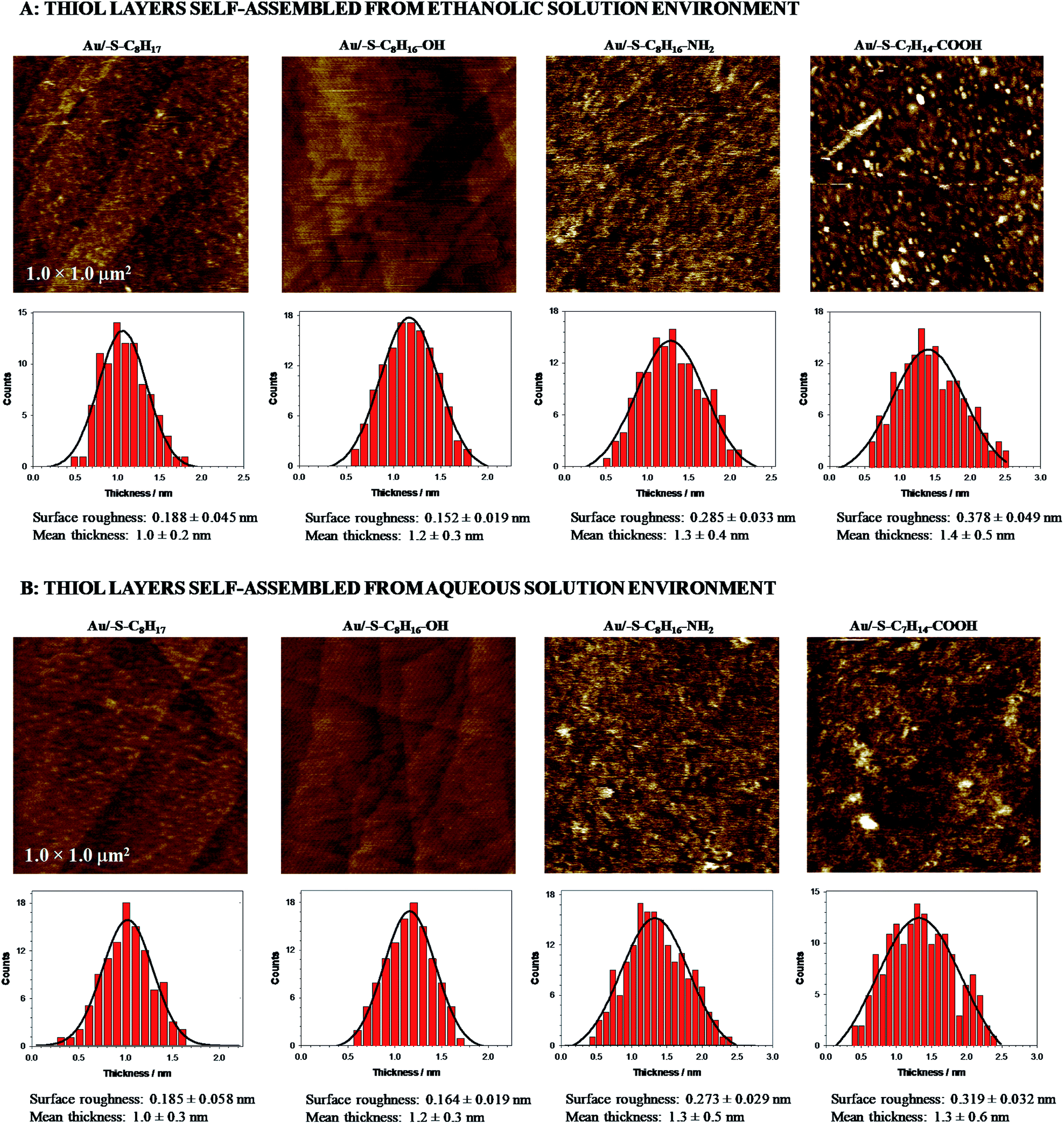 | ||
| Fig. 1 AFM micrographs and histograms obtained for gold electrode modified with 1-octanethiol, 8-amino-1-octanethiol, 8-mercapto-1-octanol and 8-mercaptooctanoic acid self-assembled in ethanolic (A) and aqueous (B) solution environment. | ||
The carbon chains in alkanethiol monolayers on gold substrate are inclined at an angle of circa 30° relative to the normal to the surface of the substrate, which causes the real thickness of the monolayer to be smaller than the length of the particles forming it.56 It should be emphasized that the presence of the terminal groups affects the inclination of the alkanethiol chain versus the electrode surface; the chains are increasingly inclined towards the electrode surface. The increase in the value of the angle between the carbon chain and the normal to the surface of the substrate from 30° to 40° leads to a decrease in the layer thickness by ca. 0.1 nm (calculations were done for the length of 1-octanethiol chain). It should be also noted that the exchange of the –CH3 terminal group for such highly polar group as –COOH will cause a change in this angle by more than 10°. Taking into account the above statement it can be concluded that the lack of significant changes in the layer thickness determined from the AFM data, absolutely do not rule out the formation of the multilayer. These considerations are additionally supported by double increase in the roughness factor for Au/–S–C7H14–COOH compared to Au/–S–C8H17. Additionally, the thiol layer containing the –COOH groups was very sensitive to the solution environment where the self-assembled process was performed. Only for the Au/–S–C7H14–COOH the surface roughness was significantly higher in the case when the layer was formed in ethanolic solution. The ethanolic solution environment changed the clustering of thiols and in consequence the randomness of the packing density of alkyl chains was higher. Further, the local disorder in the layer took place, which was proved by an increase in the roughness factor of the tested layers. In the aqueous environment the alkyl chains associated via van der Waals and hydrophobic interactions; this could influence the organization of the layer. Large roughness of the –NH2 and –COOH terminated films, compared to 1-octanethiol, could be also related to the formation of the aggregates/dimers.57 Chidsey and Loiacono have shown that the SAM layer containing terminal carboxylic groups are quasi-liquid, while those containing –OH and –NH2 groups are quasi-crystalline.58
Voltammetric characteristic
Quasi-liquid monolayers exhibit the highest permeability for ions and water molecules, which was directly demonstrated by the electrochemical properties of the SAMs. One basic electrochemical parameter, which characterises the quality of the thiol layer is double-layer capacitance (Cdl). On the basis of Cdl values it is possible to estimate layer thickness and get the information about the morphology and tightness of the formed layer. Theoretically the capacitance value for unmodified, bare gold electrode should be circa 20 μF cm−2,59 and for polycrystalline gold electrodes it can be even 2–3 times higher, whereas for electrodes modified with well organised thiol monolayers, the capacitance value should be circa 1–2 μF cm−2. The capacitance values between 2 and 20 μF cm−2 suggest the presence of the defects in the thiol layer. For all tested thiol layers, regardless of the used solvent environment, the values of Cdl (calculated at E = 0.1 V as Ctotal = 0.5(|Ia| + |Ic|)(vA)−1) were at least one order of magnitude lower compared to bare gold electrode. Such low capacitance values, see Table 1, indicated that the formed thiol layers were well organised, homogenous and tight. It is known, that the formation of the well organised SAM layer for short alkanethiols is problematic. The organisation of the monolayer is better for the longer alkyl chains.56 Unfortunately, the longer alkyl chain the more hindered is the electron transport. It should be noted that the mechanism of electron transfer across alkanethiol SAMs is attributed to tunnelling and in some cases may also occurs through layer defects.60,61 The blocking properties of the formed thiol monolayers have been checked against the model electrochemical system, Fe(CN)63−. The typical cyclic voltammograms obtained with electrodes modified with thiol layers and are presented in Fig. 2. The layers were adsorbed in aqueous and ethanolic solutions and the alkanethiols were correspondingly modified. In the case of 1-octanethiol layer, formed in ethanolic solution, the current signals related to the reduction and oxidation processes of selected redox probe (Fe(CN)63−) were not observed (see the left curve in Fig. 2B). Such result indicates hindered electron transfer through the 1-octanethiol layer, which means that the formed layer was well-organized and tight. To detect the presence of possible defects in the –SC8H17 layer and to see whether Fe(CN)63− species can penetrate the layer, the modified electrode was immersed in 5 mM Fe(CN)63− solution overnight. As it is shown in Fig. 3 the current signals of the redox probe were barely visible for all scan rates. Such behaviour indicates that octanethiol layer is very regular and practically free of defects. However, such effect was observed only in the case when the layer was formed in ethanolic solution environment. In the case of aqueous environment the current signals of redox probe were visible just after placing the electrode in the redox probe solution (see right curve in Fig. 2B). However, the intensity of the observed current signals was much lower and the separation between anodic and cathodic peaks was much bigger compared to the bare electrode. The presence of redox probe current signals indicates more randomness in alkyl chains orientation in the layer compared to the layer formed in ethanolic solution. The randomness in the alkanethiol chains arrangement in the layer led to more layer defects and an increase in the rate of the electron exchange between the redox probe and the electrode surface.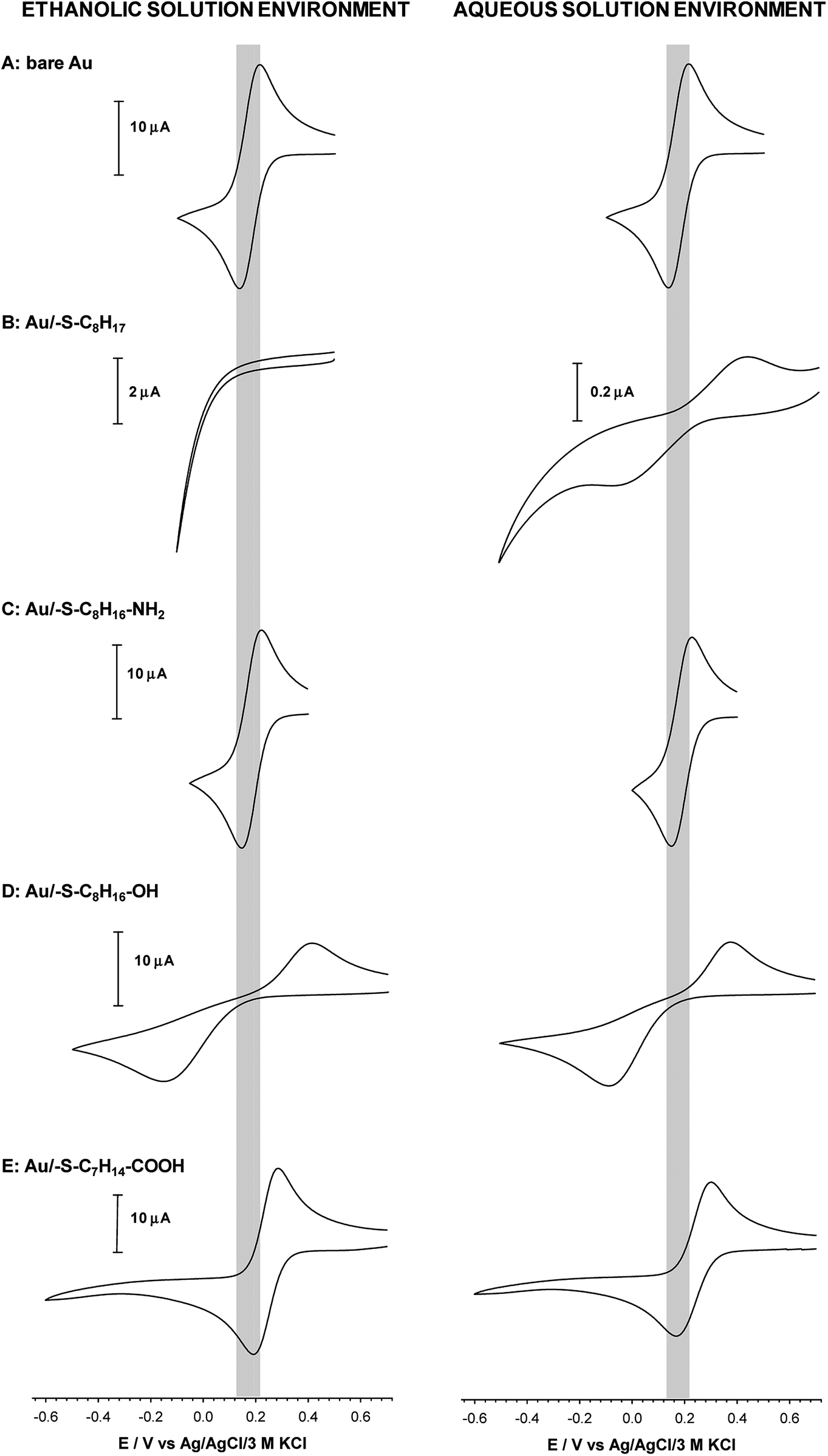 | ||
| Fig. 2 Cyclic voltammograms of Fe(CN)63− recorded at bare (A) and modified with: 1-octanethiol (B), 8-amino-1-octanethiol (C), 8-mercapto-1-octanol (D) and 8-mercaptooctanoic acid (E) gold disc electrodes. Experimental conditions: 5 mM Fe(CN)63− in 0.1 M phosphate buffer with addition 150 mM K2SO4 (pH 7.0); scan rate 0.05 V s−1. Thiol layers were formed in 1 mM ethanolic and aqueous solution. | ||
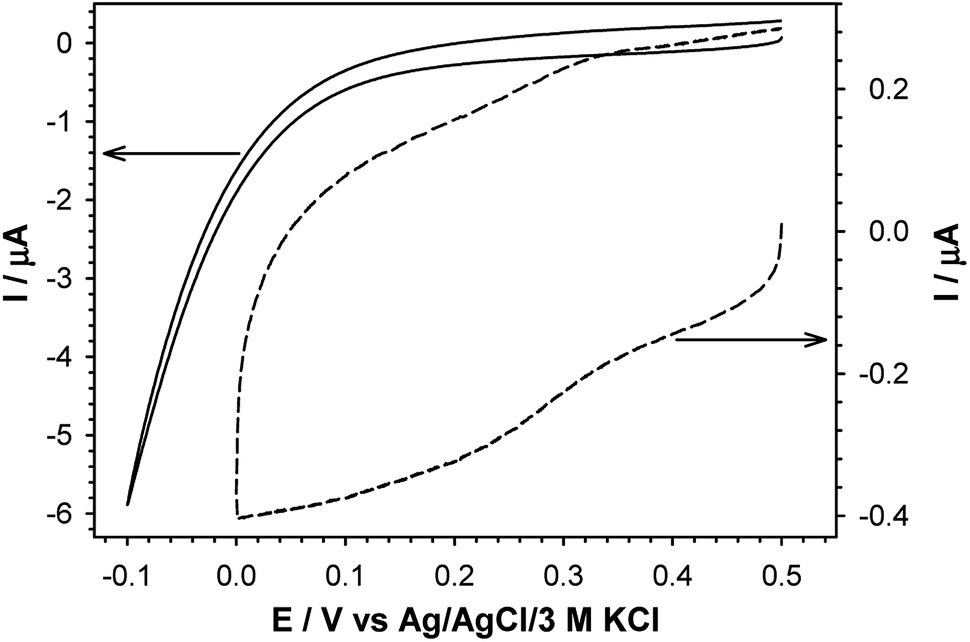 | ||
| Fig. 3 Cyclic voltammograms of 5 mM Fe(CN)63− in 0.1 M phosphate buffer containing 150 mM K2SO4, pH 7.0, recorded at gold disc electrode modified with 1-octanethiol layer (solid line) and Fe(CN)63− entrapped in 1-octanethiol layer after 12 h soaking (dashed line) in 0.1 M phosphate buffer containing 150 mM K2SO4, pH 7.0. | ||
The presence of polar terminal groups in the thiol chains may result in a change in ion permeability as well as a change in the ordering of the layer. In the experimental conditions (pH 7.0) the amine groups were protonated, which led to the electrostatic attraction of negatively charged redox probe, and in consequence no changes in the voltammograms obtained with the electrode modified with thiol containing terminal amine group, compared to bare electrode, were observed (see Fig. 2C). In this case the influence of the solution environment (ethanol/water) on the CV characteristic was negligible.
The exchange of –NH2 terminal group for –OH led to a decrease in current intensity and on increase in peaks separation (see Fig. 2D). However, in the case of the Au/–SC8H16–OH layer formed in ethanolic solution the oxidation and reduction processes of the redox probe were shifted by circa 50 mV towards more positive and more negative potentials, respectively. This fact suggests that the obtained layer was more uniform, compared to the layer formed in the aqueous environment. In the case of –S–C7H14–COOH layer the positions and intensities of current signals were strongly dependent on the number of consecutively recorded voltammetric curves, regardless of the solution environment. The registration of subsequent scans at the same scan rate led to a decrease in peak separation and improvement of the signal shape, see Fig. 4. Such behaviour proved the dependence of the layer stability on applied potential; the alkanethiol chains were prone to the changes in orientation and tilt during the voltammetric cycling. The susceptibility of thiol chains on reorientation strongly depended on the potential range. In position of more positive potential led to faster layer reorganization which was directly connected with the water content in the layer. Therefore, before the voltammetric experiments the –S–C7H14–COOH layers were first cycled between 0.72 and −0.65 V in 5 mM Fe(CN)63− (0.1 M phosphate buffer containing 150 mM K2SO4, pH 7.0) at scan rate 0.05 V s−1 until a stable voltammogram was obtained. In the case of other studied thiol layers the separation as well as the intensities of the current signals were independent of the number of cycles, so the stabilization step was not necessary. Those layers were stable in the applied potential window.
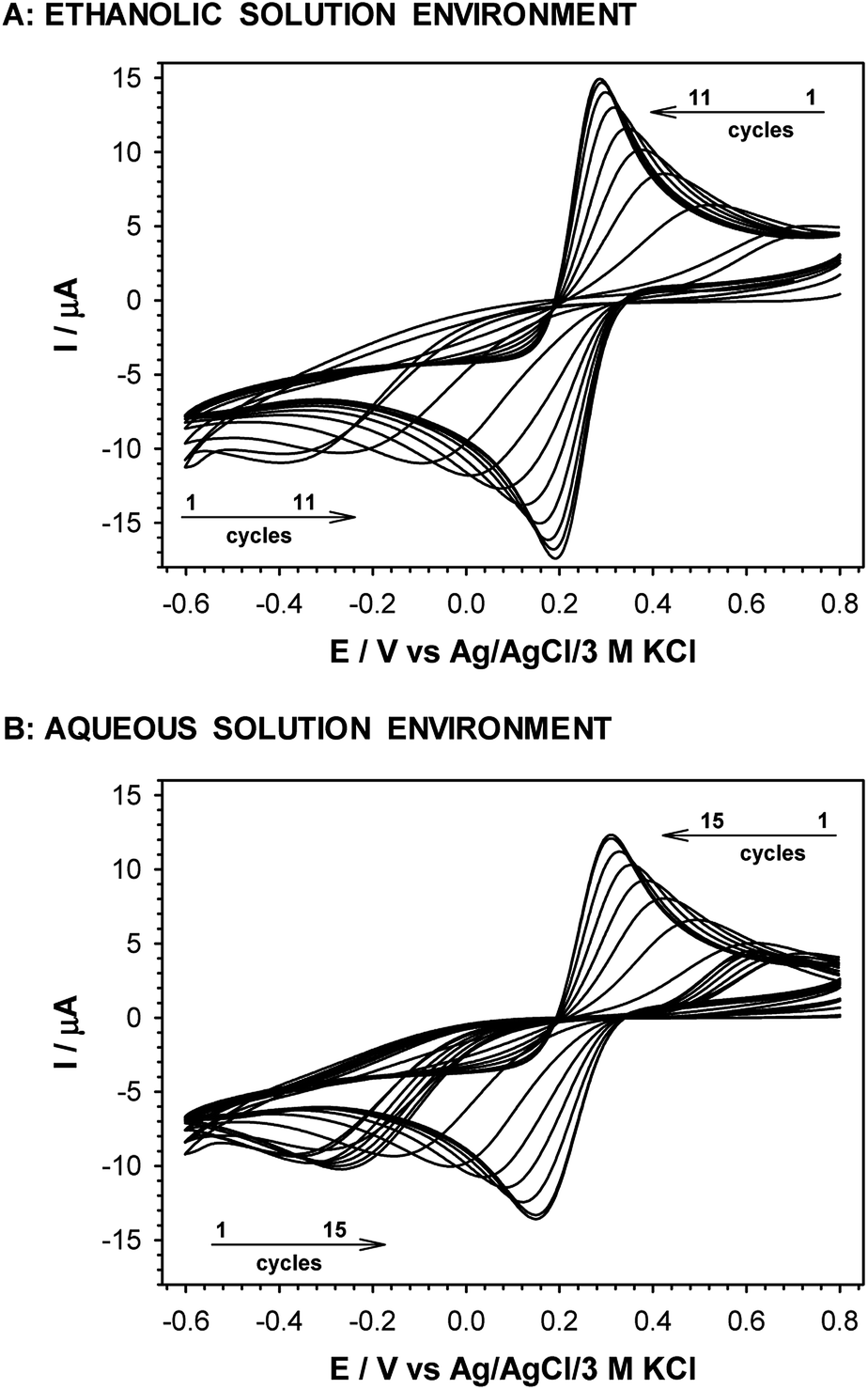 | ||
| Fig. 4 Subsequent cyclic voltammograms obtained with gold disc electrode modified with 8-mercaptooctanoic acid. 5 mM Fe(CN)63− in 0.1 M phosphate buffer containing 150 mM K2SO4, pH 7.0, scan rate 0.05 V s−1. Thiol layers were formed in 1 mM thiol ethanolic and aqueous solutions. | ||
The cyclic voltammograms of Fe(CN)63− obtained with all tested thiol modified electrodes at various scan rates are presented in Fig. 5. The dependences of oxidation and reduction peak currents versus square root of scan rate are presented in the insets in Fig. 5. They give the information that the redox process is diffusional. Such behavior was observed in the case of all tested thiol layers except for 1-octanethiol layer formed in ethanolic solution; then the redox signal was not seen at all. The presence of the layer on the surface of the electrode forces the electron transport between the surface of the electrode and the redox probe present in the solution through the layer by tunneling.
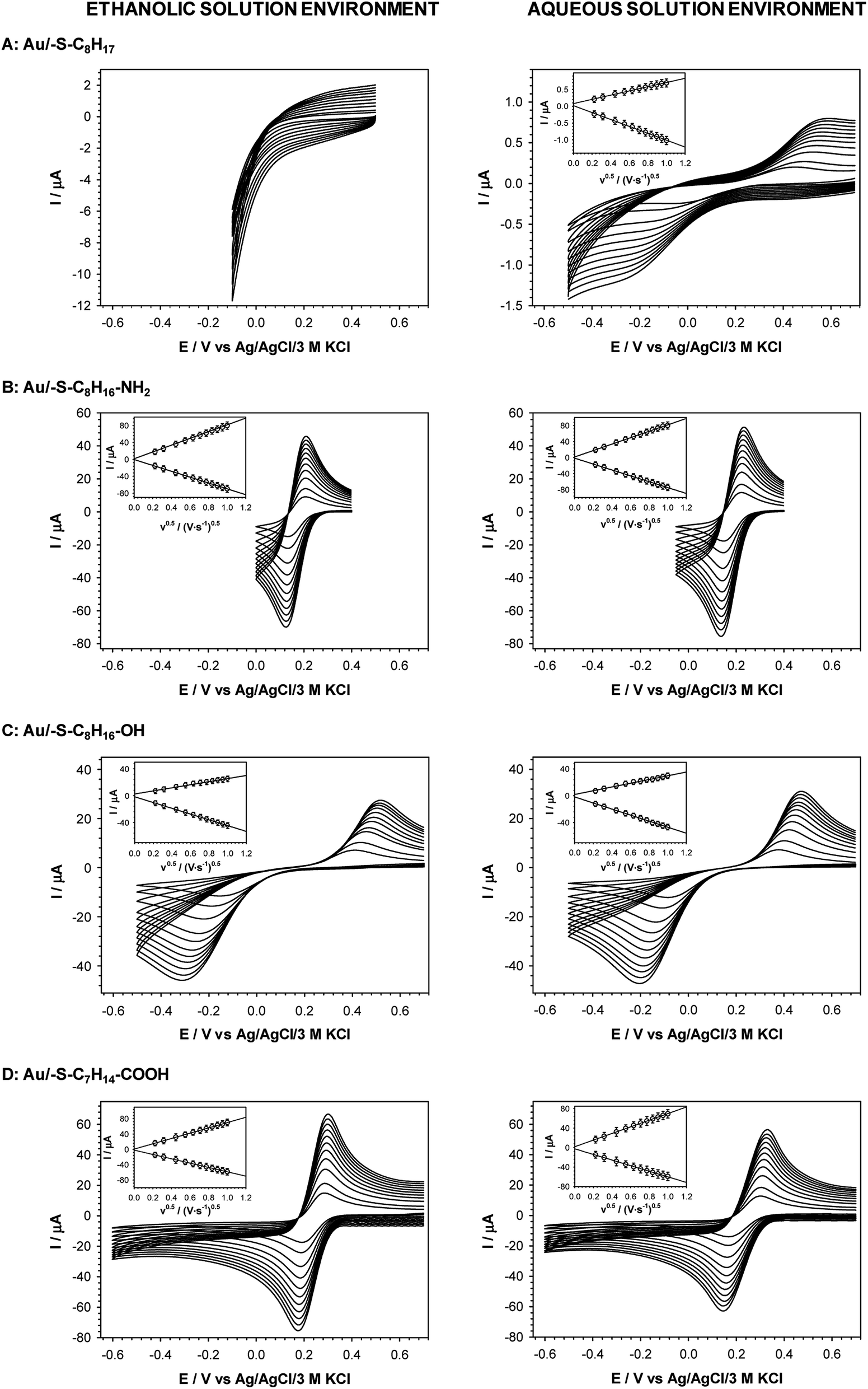 | ||
| Fig. 5 Cyclic voltammograms of 5 mM Fe(CN)63− recorded for various scan rates at gold electrodes modified with: 1-octanethiol (A), 8-amino-1-octanethiol (B), 8-mercapto-1-octanol (C) and 8-mercaptooctanoic acid (D). Insets: plots of the anodic and cathodic peak currents versus square root of scan rate. Experimental conditions: 5 mM Fe(CN)63− in 0.1 M phosphate buffer with addition 150 mM K2SO4 (pH 7.0). Thiol layers were formed in 1 mM ethanolic and aqueous solution. | ||
The efficiencies of the electron exchange can be very well illustrated by appropriate electron-transfer rate constants (ks). To determine the ks values for the terminal groups in alkanethiol chain the cyclic voltammograms at increasing scan rates were recorded. For an irreversible charge transfer process, peak separation should be much higher than 300 mV, and then the ks value can be determined from the intercept of the dependence ln(Ip) versus (Ep − E0′), where Ip is given by eqn (1):62,63
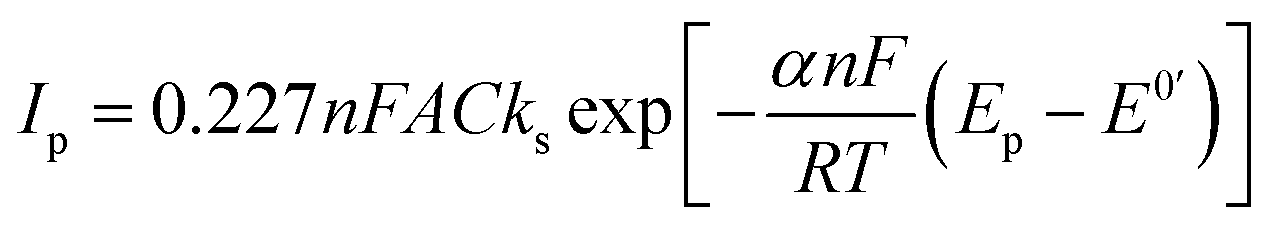 | (1) |
In eqn (1) Ip is peak current value, n is number of exchanged electrons, F is Faraday constant (9.649 × 105 C mol−1), A is surface area, C is concentration of redox probe, Ep is peak potential, E0′ is formal potential, R is gas constant (8.31 J mol−1 K−1), T is temperature, α is transmission coefficient. Conditions for using eqn (1) took place in the case of –S–C8H16–OH layers (regardless of the solution environment of the self-assembly process) and –S–C8H17 layer formed in the aqueous solution. For a quasi-reversible process (Au/–S–C8H16–NH2 and Au/–S–C7H14–COOH) the ks value should be calculated from the slope of the dependence Ψ = f(ν−0.5):64
| Ψ = ks[πDnFv/RT]−0.5 | (2) |
To get some information about influence of the studied SAM layers on the thermodynamics of the redox probe the voltammetric midpoint potentials (Em) were also calculated (see Table 1). The midpoint potential is the potential at which the concentrations of the redox probe species are equal so it should be close to the formal potential. The obtained data clearly showed that the Em value depends on the type of the SAM layer; the observed Em changes were in the range 4–60 mV. The obtained results confirmed the presence of interactions between the redox probe and the terminal groups in the thiol chains.
QCM data
The maximal surface area occupied by one molecule of thiol is 21 × 10−16 cm2,67 and for the real surface area of the electrode (0.527 cm2) the theoretical mass increase obtained for a monolayer of –SC8H17 is ca. 60 ng. The apparent mass increase (mthiol = 4.3Δf) related to the observed experimental frequency shifts for accumulation of –SC8H17, –SC8H16–OH, –SC8H16–NH2 and –SC7H14–COOH in the ethanolic solution, are: 66.6; 71.4; 77 and 81.7 ng, respectively. It means that the deposition of thiol layers containing –CH3 and –OH groups from the anhydrous environment leads to the formation of a monolayer. In the case of the –NH2 and –COOH terminal groups the determined mass increases calculated from the QCM data was ca. 22% higher than for 1-octanethiol. Additionally, it should be noted that the presence of above mentioned groups leads to a significant increase of the thiol chain tilt from the normal to the surface and in consequence to a smaller amount of the thiol molecules in the monolayer. The QCM data confirmed the aggregation process of thiol chains with the –NH2 and –COOH terminal groups. The resonant frequency shifts −Δf (corresponding to the mass accumulation) for the self-assembly process of the studied thiols with various terminal groups were different when the process was performed in the ethanolic and aqueous solutions, see Fig. 6.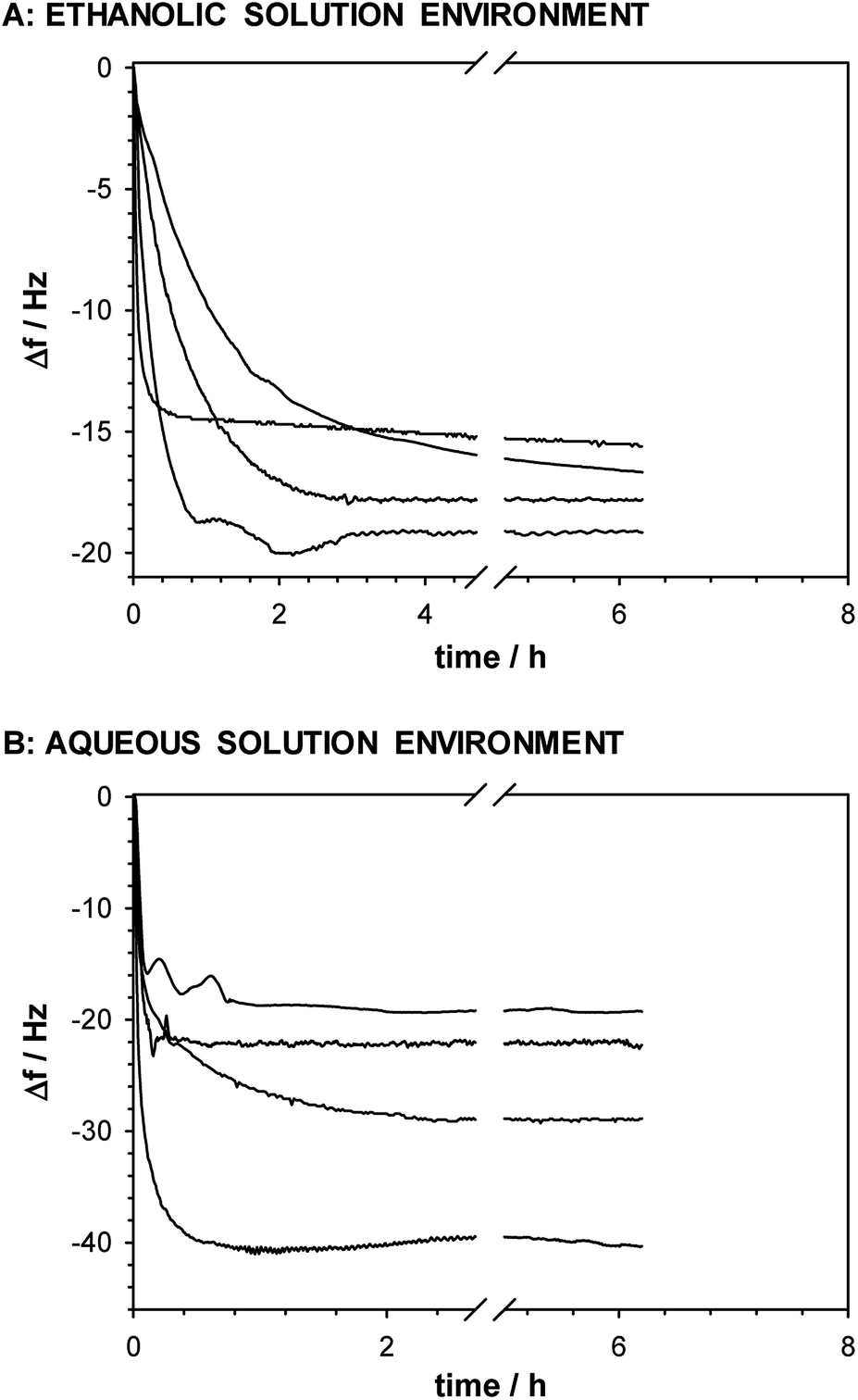 | ||
| Fig. 6 Frequency shifts recorded during self-assembly process of appropriate thiols in ethanolic (A) and aqueous (B) solutions. | ||
The higher frequency shifts, especially for the thiols with the –NH2 and –COOH terminal groups, were observed for the layer formation in the aqueous solution environment. Such effect is a consequence of layer hydration and possible aggregation process. The QCM data are in very good agreement with the AFM data.
Conclusions
The results presented in this paper showed that the quality and thickness of the formed thiol layer on the gold surface are strongly dependent upon the functional terminal group in the thiol chain and on the solution layer formation; also the water content is a crucial point. The quality of the obtained layer was evaluated using several techniques. AFM and QCM results allowed us to conclude that randomness of alkyl chains is higher in the case of the thiols containing the –NH2 and –COOH groups regardless of the solution for the self-assembly process. Moreover, the permeability of such layers for ions and water molecules increased compared to the hydrophobic thiol chain. The presence of functional terminal groups in the thiol chains influences the electron transport through the layer. SAMs of the thiol molecules without the functional group blocked completely the electron exchange between the redox probe dissolved in the solution and the electrode surface. However, such situation took place only when the layer was formed in the ethanolic solution. In the case of the aqueous solution the electron exchange was only hampered. The electron transport was also hindered in the case of the thiol layer containing the –OH group, while SAMs with the –NH2 and –COOH groups, due to the presence of layer defects practically weren't a barrier for the electron transport. Generally, the peak current values scaled up linearly with the square root of the scan rate indicating semi-infinite diffusional electron transport through the thiol layer. Based on the voltammetric studies, it turned out that the stability of –SC7H14–COOH layer was dependent on the applied potential. The –SC7H14–COOH layer was unstable and easily reorganized during the cyclization step, when the potential-induced defects arose.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CAS President's International Fellowship Initiative [grant number 2018VBC0008], the National Natural Science Foundation of China [grant numbers 51761145102, 21874128], and the Jilin Provincial Science and Technology Development Project [grant number 20190201069JC]. Agata Kowalczyk thanks Prof. Sławomir Sęk for AFM experiments.Notes and references
- J. H. Park, Z. Schwartz, R. Olivares-Navarrete, B. D. Boyan and R. Tannenbaum, Langmuir, 2011, 27, 5976 CrossRef CAS PubMed.
- Z. P. Aguilar, Nanomaterials for medical applications, Elsevier, Oxford, 2013 Search PubMed.
- B. D. Malhotra and A. Ali, Nanomaterials for biosensors. Fundamentals and applications, Elsevier, Amsterdam, 2018 Search PubMed.
- B. J. O'Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804 CrossRef.
- A. Weingarten, Nat. Rev. Chem., 2018, 2, 0136 CrossRef.
- S. Matosevic and B. M. Paegel, Nat. Chem., 2013, 5, 958 CrossRef CAS.
- S. Abdu, M.-C. Martí-Calatayud, J. E. Wong, M. García-Gabaldón and M. Wessling, ACS Appl. Mater. Interfaces, 2014, 6, 1843 CrossRef CAS.
- T. Ogoshi, S. Takashima and T. Yamagishi, J. Am. Chem. Soc., 2015, 137, 10962 CrossRef CAS.
- M. Park, BioChip J., 2019, 13, 82 CrossRef CAS.
- S. Guo, X. Zhu and X. J. Loh, Mater. Sci. Eng., C, 2017, 70, 1163 CrossRef CAS PubMed.
- K. Ishihara, T. Kitagawa and Y. Inoue, ACS Biomater. Sci. Eng., 2015, 1, 103 CrossRef CAS.
- Y. Si, J. W. Park, S. Jung, G. S. Hwang, E. Goh and H. J. Lee, Biosens. Bioelectron., 2018, 121, 265 CrossRef CAS PubMed.
- E. Matysiak-Brynda, J. P. Sęk, A. Kasprzak, A. Królikowska, M. Donten, M. Patrzalek, M. Poplawska and A. M. Nowicka, Biosens. Bioelectron., 2019, 128, 23 CrossRef CAS.
- M. Stobiecka and M. Hepel, Biosens. Bioelectron., 2011, 26, 3524 CrossRef CAS PubMed.
- M. Kulkarni, A. Mazare, J. Park, E. Gongadze, M. S. Killian, S. Kralj, K. von der Mark, A. Igliča and P. Schmuki, Acta Biomater., 2016, 45, 357 CrossRef CAS PubMed.
- M. Stobiecka, A. Chalupa and B. Dworakowska, Biosens. Bioelectron., 2016, 84, 37 CrossRef CAS PubMed.
- M. Hepel and C.-J. Zhong, ACS Symposium Series, American Chemical Society, Washington, DC, 2012, vol. 1112, ch. 11, p. 293 Search PubMed.
- A. Tiwari, H. K. Patra and A. P. F. Turner, Advanced bioelectronic materials, John Wiley & Sons, New Jersey, 2015 Search PubMed.
- A. Kowalczyk, J. P. Sęk, A. Kasprzak, M. Poplawska, I. P. Grudzinski and A. M. Nowicka, Biosens. Bioelectron., 2018, 117, 232 CrossRef CAS PubMed.
- A. M. Nowicka, M. Fau, A. Kowalczyk, M. Strawski and Z. Stojek, Electrochim. Acta, 2014, 126, 11 CrossRef CAS.
- E. Matysiak-Brynda, B. Wagner, M. Bystrzejewski, I. P. Grudzinski and A. M. Nowicka, Biosens. Bioelectron., 2018, 109, 83 CrossRef CAS PubMed.
- C. A. Widrig, C. Chung and M. Porter, J. Electroanal. Chem., 1991, 310, 335 CrossRef CAS.
- J. Dai, Z. Li, J. Jin, J. Cheng, J. Kong and S. Bi, J. Electroanal. Chem., 2008, 624, 315 CrossRef CAS.
- E. Pensa, E. Cortes, G. Corthey, P. Carro, C. Vericat, M. H. Fonticelli, G. Benitez, A. A. Rubert and R. C. Salvarezza, Acc. Chem. Res., 2012, 45, 1183 CrossRef CAS PubMed.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CrossRef CAS PubMed.
- Z. Yang, A. Gonzalez-Cortes, G. Jourquin, J. Viré, J. Kauffmann and J. Delplancke, Biosens. Bioelectron., 1995, 10, 789 CrossRef CAS.
- C. E. D. Chidsey, Science, 1991, 251, 919 CrossRef CAS.
- D. E. Khoshtariya, T. D. Dolidze, M. Shushanyan, K. L. Davis, D. H. Waldeck and R. van Eldik, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2757 CrossRef CAS PubMed.
- R. F. DeBono, G. D. Loucks, D. Della Manna and U. J. Krull, Can. J. Chem., 1996, 74, 677 CrossRef CAS.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559 CrossRef CAS.
- E. Boubour and R. B. Lennox, J. Phys. Chem. B, 2000, 104, 9004 CrossRef CAS.
- V. A. Nikitina, A. V. Rudnev, G. A. Tsirlina and T. Wandlowski, J. Phys. Chem. C, 2014, 118, 15970 CrossRef CAS.
- N. Darwish, P. K. Eggers, S. Ciampi, Y. Zhang, Y. Tong, S. Ye, M. N. Paddon-Row and J. J. Gooding, Electrochem. Commun., 2011, 13, 387 CrossRef CAS.
- B. O'Brien, H. Sahalov and P. C. Searson, Appl. Phys. Lett., 2010, 97, 043110 CrossRef.
- E. Boubour and R. B. Lennox, Langmuir, 2000, 16, 7464 CrossRef CAS.
- U. Kumar Sur and V. Lakshminarayanan, J. Colloid Interface Sci., 2002, 254, 410 CrossRef PubMed.
- U. Kumar Sur and V. Lakshminarayanan, J. Electroanal. Chem., 2004, 565, 343 CrossRef.
- C. Vericat, M. E. Vela, G. Benitez, P. Carrob and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805 RSC.
- S. A. Kislenkoa, V. A. Nikitinab and R. R. Nazmutdinov, High Energy Chem., 2015, 49, 341 CrossRef.
- A. Ulman, J. E. Eilers and N. Tillman, Langmuir, 1989, 5, 1147 CrossRef CAS.
- C. E. D. Chidsey and D. N. Loiacono, Langmuir, 1990, 6, 682 CrossRef CAS.
- M. M. Walczak, D. D. Popenoe, R. S. Deinhammer, B. D. Lamp, C. Chung and M. D. Porter, Langmuir, 1991, 7, 2687 CrossRef CAS.
- O. Azzaroni, M. E. Vela, H. Martin, A. Hernandez Creus, G. Andreasen and R. C. Salvarezza, Langmuir, 2001, 17, 6647 CrossRef CAS.
- R. G. Nuzzo, L. H. Dubois and D. L. Allara, J. Am. Chem. Soc., 1990, 112, 558 CrossRef CAS.
- L. H. Dubois, B. R. Zegarski and R. G. Nuzzo, J. Am. Chem. Soc., 1990, 112, 570 CrossRef CAS.
- J. A. M. Sondag-Huethorst and L. G. J. Fokkink, Langmuir, 1995, 11, 2237 CrossRef CAS.
- M. M. Walczak, C. A. Alves, B. D. Lamp and M. D. Porter, J. Electroanal. Chem., 1995, 396, 103 CrossRef.
- D.-F. Yang, C. P. Wilde and M. Morin, Langmuir, 1997, 13, 243 CrossRef CAS.
- D.-F. Yang, C. P. Wilde and M. Morin, Langmuir, 1996, 12, 6570 CrossRef CAS.
- S.-I. Imabayashi, M. Iida, D. Hobara, Z. Q. Feng, K. Niki and T. Kakiuchi, J. Electroanal. Chem., 1997, 428, 33 CrossRef CAS.
- A. Badia, S. Arnold, V. Scheumann, M. Zizlsperger, J. Mack, G. Jung and W. Knoll, Sens. Actuators, B, 1999, 54, 145 CrossRef CAS.
- M. Nishizawa, T. Sunagawa and H. Yoneyama, J. Electroanal. Chem., 1997, 436, 213 CrossRef CAS.
- A. Matsuzaki, M. Nagoshi and N. Hara, ISIJ Int., 2011, 51, 108 CrossRef CAS.
- S. Bruckenstein and M. Shay, Electrochim. Acta, 1985, 30, 1295 CrossRef CAS.
- A. Wieckowski, Interfacial electrochemistry. theory, experiment, and application, Marcel Dekker, New York, 1999, ch. 34, p. 599 Search PubMed.
- M. D. Porter, T. B. Bright, D. L. Allara and C. E. D. Chidsey, J. Am. Chem. Soc., 1987, 109, 3559 CrossRef CAS.
- H. Wang, S. Chen, L. Li and S. Jiang, Langmuir, 2005, 21, 2633 CrossRef CAS PubMed.
- C. E. D. Chidsey and D. N. Loiacono, Langmuir, 1990, 6, 709 CrossRef.
- A. Świetłow, M. Skoog and G. Johansson, Electroanalysis, 1992, 4, 921 CrossRef.
- G. Benitez, C. Vericat, S. Tanco, F. Remes Lenicov, M. F. Castez, M. E. Vela and R. C. Salvarezza, Langmuir, 2004, 20, 5030 CrossRef CAS PubMed.
- C. Vericat, F. Remes Lenicov, S. Tanco, G. Andreasen, M. E. Vela and R. C. Salvarezza, J. Phys. Chem. B, 2002, 106, 9114 CrossRef CAS.
- A. Y. Gokhshtein and Y. P. Gokhshtein, Dokl. Akad. Nauk SSSR, 1960, 131, 601 CAS.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 6, 706 CrossRef.
- I. Lavagnini, R. Antiochia and F. Magno, Electroanalysis, 2004, 16, 505 CrossRef CAS.
- D. R. Lide and H. P. R. Frederikse, CRC Handbook of Chemistry and Physics, CRC Press, New York, 1997, pp. 5–94 Search PubMed.
- A. Haran, D. H. Waldeck, R. Naaman, E. Moons and D. Cahen, Science, 1994, 263, 948 CrossRef CAS PubMed.
- G. E. Poirier and E. D. Pylant, Science, 1996, 272, 1145 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |