
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstructing Rh–Rh3+ modified Ta2O5@TaON@Ta3N5 with special double n–n mutant heterojunctions for enhanced photocatalytic H2-evolution
Wenli Zhangab,
Hongquan Jiang *ab,
Wei Zhangab and
Shuying Zangc
*ab,
Wei Zhangab and
Shuying Zangc
aKey Laboratory of Photonic and Electronic Bandgap Materials, Ministry of Education, Harbin Normal University, Harbin 150025, P. R. China. E-mail: h.q.jiang1119@163.com
bKey Laboratory of Synthesis of Functional Materials and Green Catalysis, Colleges of Heilongjiang Province, Harbin Normal University, Harbin 150025, P. R. China
cKey Laboratory of Remote Sensing Monitoring of Geographic Environment, Colleges of Heilongjiang Province, Harbin Normal University, Harbin 150025, P. R. China
First published on 10th August 2020
Abstract
A multiple core–shell heterostructure Rh–Rh3+ modified Ta2O5@TaON@Ta3N5 nanophotocatalyst was successfully constructed through nitriding Rh3+-doped Ta2O5 nanoparticles, which exhibited a much higher carrier separation efficiency about one order of magnitude higher than the Ta2O5@Ta3N5 precursor, and thus an excellent visible light photocatalytic H2-evolution activity (83.64 μmol g−1 h−1), much superior to that of Rh anchored Ta2O5@TaON (39.41 μmol g−1 h−1), and improved stability due to the residual Rh–O/N in the Ta3N5 shell layer. Rh-modifying significantly extended light absorption to the overall visible region. Localized built-in electric fields with hierarchical potential gradients at the multiple interfaces including a Rh/Ta3N5 Schottky junction and double n–n Ta3N5/TaON/Ta2O5 mutant heterojunctions, drove charge carriers to directionally transfer from inside to outside, and efficiently separate. Enhanced photoactivity was ascribed to a synergetic effect of improved light absorption ability, increased carrier separation efficiency, and accelerated surface reaction. A promising strategy of developing excellent Ta3N5-based photocatalysts for solar energy conversion is provided by constructing double n–n mutant heterojunctions.
Introduction
In the 21st century, in order to solve the serious problems of fossil energy shortage and environment deterioration, solar driven overall water splitting using particulate photocatalysts provides an ideal way to massively convert ultimate renewable solar energy into clean hydrogen energy, because of the simplicity and the most cost-effective approach.1–4 Tantalum nitride (Ta3N5), as a promising visible light responsive photocatalyst, has attracted great attention due to a suitable band gap of about 2.1 eV in regard to the solar spectrum and adaptive conduction band and valence band position for overall water splitting. However, its intrinsic drawbacks including low charge carrier separation efficiency, poor carrier transportation, and easy self-oxidation by photogenerated holes have impeded its commercial application.5–7Constructing heterojunction has been proved to be a convenient and effective method driving efficient carrier separation in bulk phase, fast transportation, and fast reaction at the semiconductor/liquid interfaces for Ta-based photocatalysts.6–12 To date, many kinds of effective heterojunctions have been developed, such as semiconductor/semiconductor heterojunctions (p–n junction Ag3PO4/Ta3N5 and Ta3N5/BaTaO2N, n–n junction CoOx/Ta3N5, Z-scheme junction TaON/Bi2O3 and Ta3N5/Bi2O3),6–9 cocatalyst/semiconductor heterojunction (Ru/TaON),10 and non-metal/semiconductor heterojunction (Ta3N5/g-C3N4).11 Previously, we constructed Rh/TaON/Ta2O5 heterojunctions by coupling Rh/TaON Schottky junction and n–n TaON/Ta2O5 mutant heterojunction to synergetically enhance photocatalytic H2-evolution activity.12 However, most previous research for Ta3N5-based photocatalysts focused on the charge carrier separation and utilization in two or three component heterojunctions, and little attention has been paid on designing multiple core–shell heterostructures.
Moreover, solar spectrum responses and redox capabilities of tantalum (oxy)nitrides can be effectively tuned by the composition and crystal structure.13 As reported by Domen et al., the valence band positions of tantalum (oxy)nitrides decreased in the order of Ta3N5 > TaON > Ta2O5, while their conduction position were very close.14 Specially, the morphology control of (oxy)nitrides can be achieved from topotactic transformation of metal oxide precursors during nitridation process.15 Inspired by above works, we expected to construct Ta2O5@TaON@Ta3N5 multiple core–shell hetero-junctions by accurate controlling nitridation of Ta2O5 nanoparticles, which would present hierarchical potential gradients, thereby forming internal built-in electric fields to facilitate photogenerated carrier separation and inside-out directional transfer.
Herein, we report the controllable synthesis of multiple core–shell heterostructure Rh–Rh3+ modified Ta2O5@TaON@Ta3N5 nano-photocatalyst using Rh-doped Ta2O5 as precursor through one-step high-temperature nitridation reduction process, which exhibits a much higher separation efficiency of photogenerated carrier and photocatalytic H2-evolution activity than the reported Rh anchored Ta2O5@TaON. To our knowledge, special double n–n mutant heterojunctions in as-prepared multiple core–shell heterostructures are reported for the first time. Furthermore, intimate interfaces of heterostructures are achieved by the controllable nitridation process. This work provides a new strategy to develop efficient core–shell Ta3N5-based photocatalysts for solar energy conversion by constructing the double n–n mutant heterojunctions.
Experimental
Synthesis of multiple core–shell heterostructure Rh–Rh3+ modified Ta2O5@TaON@Ta3N5 nanophotocatalyst
Rh-doped Ta2O5 nanoparticles precursor was prepared through hydrolysis of TaCl5 in ethanol solution via the hydrothermal process at 160 °C for 3 h, followed by a calcination in a muffle furnace at 700 °C for 3 h, as reported in the cite.12 The multiple core–shell heterostructure Rh–Rh3+ modified Ta2O5@TaON@Ta3N5 nanophotocatalyst was controllably synthesized by nitriding the Rh-doped Ta2O5 precursor under NH3 gas flow with the flow rate of 50 mL min−1 in a tube furnace at 850 °C for 6 h, denoted as Rh-Ta-6. For comparison, the other Rh-modified samples were synthesized by nitriding the Rh-doped Ta2O5 precursor under the same ammonolysis conditions except for nitriding time (3 h, 9 h, and 12 h, respectively). As-prepared samples were denoted as Rh-Ta-3, Rh-Ta-9, and Rh-Ta-12, respectively. The synthetic routes of above Rh-modified samples are shown in Scheme 1. In addition, the Ta2O5@Ta3N5 nanophotocatalyst was obtained by nitriding Ta2O5 nanoparticles at 850 °C for 3 h, as reported in the cite.14 Ta3N5 nanophotocatalyst was obtained by nitriding Ta2O5 nanoparticles at 850 °C for 24 h.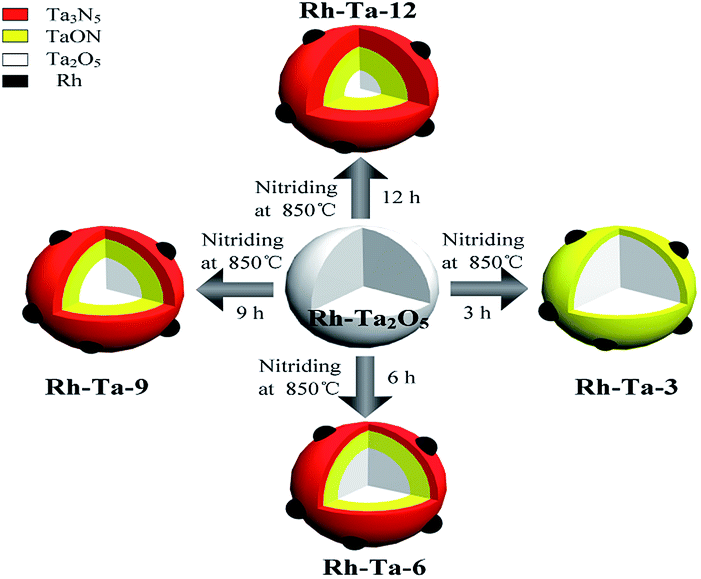 | ||
| Scheme 1 Schematic illustration of the synthetic processes of multiple core–shell heterostructure Rh modified samples. | ||
Characterization
A Japan Shimadzu XRD-6000 X-ray diffractometer was used to collect the XRD patterns of the samples using Cu Kα radiation (λ = 1.5418 Å) in the range of 10–90° (2θ). A FEI Tecnai G2TF20 transmission electron microscope (TEM) was used to investigate the morphologies and microstructures of the samples at an accelerating voltage of 200 KV. X-ray photoelectron spectroscopy (XPS) measure was carried out on a PHI-5400 ESCA System instrument with Al Kα radiation (hν = 1486.6 eV), and the binding energy of C 1s at 284.6 eV was used as a reference. Depth profiling was performed by monatomic Ar ion sputtering for 10 s, followed by XPS acquisition. Fourier-transform infrared (FTIR) spectra were obtained on a VERTEX 80 FTIR spectrometer using KBr as diluent. UV-vis diffuse reflectance spectra (DRS) were collected on a Shimadzu UV-2550 spectrophotometer using BaSO4 as reference standard. Ultraviolet photoelectron spectroscopy (UPS) was recorded with XPS instrument using He I excitation (21.2 eV) and a constant pass energy of 5 eV in the ultrahigh vacuum chamber.Electrochemical measurements
Photocurrent responses and Mott–Schottky curves were carried out in a three-electrode electrochemical system with a quartz window, using a CHI660E electrochemical workstation. A platinum electrode was used as counter electrode, and an Ag/AgCl electrode as the reference electrode. The photocatalyst was loaded on an FTO conducting glass, which was used as the working electrode. 0.5 mol L−1 Na2SO4 aqueous solution was used as the electrolyte for the photocurrent measurement, a 350 W Xe lamp was served as the light source. 1 mol L−1 NaOH solution was used as the electrolyte for the Mott–Schottky curve measurement. The applied potential window was −1.0 V to 1.0 V, and the applied frequency was 1 kHz.Photocatalytic H2-evolution tests
Photocatalytic H2-evolution tests of as-prepared samples were performed under visible light irradiation (300 W xenon lamp, λ > 420 nm), using CEL-SPH2N photocatalytic water splitting into H2 system, as reported in our previous report.12Results and discussion
Structures and morphologies
The phase structures of as-prepared Rh-modified samples were investigated by the X-ray diffraction (XRD) patterns shown in Fig. 1. As observed, after nitriding for 3 h, main diffraction peaks at 29.06°, 32.6°, 35.1°, 35.64°, 36.68°, 39.9°, 42.14°, 50.9°, 52.1°, and 57.6° correspond to (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11), (111), (002), (020), (200), (021), (
11), (111), (002), (020), (200), (021), (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 11), (022), (22), and (113) lattice planes of monoclinic TaON (JCPDS no. 70-1193),12,15 while very weak peaks at 22.8° corresponding to (001) crystal planes of Ta2O5 should result from Ta2O5 cores.15 The results imply that Ta2O5 in the shell layer of Rh-Ta2O5 nanoparticles could topochemically transform into TaON through nitriding at 850 °C for 3 h. After nitriding for 6 h, new peaks at 24.46°, 31.34°, 36.05°, and 46.66° corresponding to (110), (023), (113), and (025) crystal planes of monoclinic Ta3N5 (JCPDS 79-1533),15,16 respectively, are detected due to the formation of Ta3N5 shell layers, confirming the coexistence of Ta3N5, TaON, and Ta2O5 in the Rh-Ta-6 sample. Furthermore, the intensities of the diffraction peaks for Ta3N5 further increase with prolonging the nitridation time, such as typically changes shown at 36.05°, indicating more and more Ta3N5 are generated from topotactic transformation of TaON in the shell layers. After nitriding at 850 °C for 24 h, only peaks for monoclinic Ta3N5 are detected, confirming that Ta2O5 has been nitrided through TaON completely into Ta3N5. However, no peaks corresponding to Rh are detected for all Rh-modified samples, due to a much lower Rh-modifying amount than the XRD detection limit.
11), (022), (22), and (113) lattice planes of monoclinic TaON (JCPDS no. 70-1193),12,15 while very weak peaks at 22.8° corresponding to (001) crystal planes of Ta2O5 should result from Ta2O5 cores.15 The results imply that Ta2O5 in the shell layer of Rh-Ta2O5 nanoparticles could topochemically transform into TaON through nitriding at 850 °C for 3 h. After nitriding for 6 h, new peaks at 24.46°, 31.34°, 36.05°, and 46.66° corresponding to (110), (023), (113), and (025) crystal planes of monoclinic Ta3N5 (JCPDS 79-1533),15,16 respectively, are detected due to the formation of Ta3N5 shell layers, confirming the coexistence of Ta3N5, TaON, and Ta2O5 in the Rh-Ta-6 sample. Furthermore, the intensities of the diffraction peaks for Ta3N5 further increase with prolonging the nitridation time, such as typically changes shown at 36.05°, indicating more and more Ta3N5 are generated from topotactic transformation of TaON in the shell layers. After nitriding at 850 °C for 24 h, only peaks for monoclinic Ta3N5 are detected, confirming that Ta2O5 has been nitrided through TaON completely into Ta3N5. However, no peaks corresponding to Rh are detected for all Rh-modified samples, due to a much lower Rh-modifying amount than the XRD detection limit.
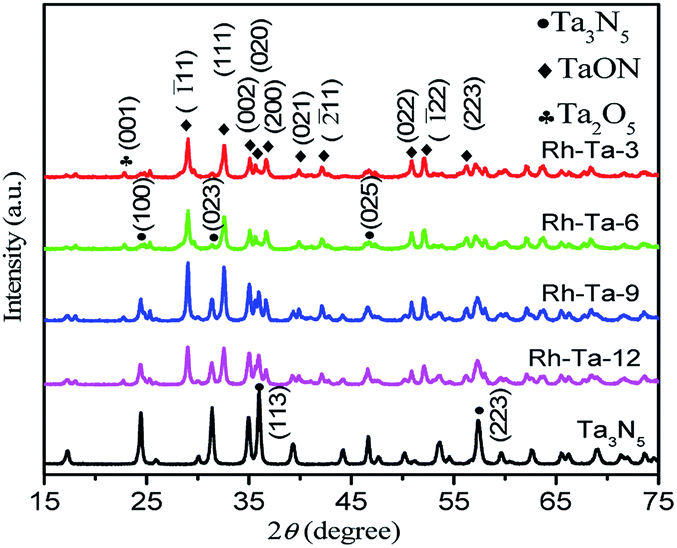 | ||
| Fig. 1 X-ray diffraction patterns of Rh-Ta-3, Rh-Ta-6, Rh-Ta-9, Rh-Ta-12, and Ta3N5. | ||
To confirm the morphologies and microstructures of as-prepared Rh-modified samples with different nitridation times, the transmission electron micrograph (TEM) images are shown in Fig. 2a–d. All Rh-modified samples are irregular nanoparticles with sizes of 50–60 nm, and many small Rh nano hemispheres with diameters of 6–10 nm uniformly distribute on the sample surfaces, which will play a significant role in electron transfer.17 Accordingly, their high-resolution transmission electron micrograph (HR-TEM) images are displayed in Fig. 2e–h. The lattice spacing of 0.22 nm corresponding to the (111) planes of Rh confirms the presence of metallic Rh in all Rh-modified samples (JCPDS no. 88-2334).18 After nitriding at 850 °C for 3 h, the outer layer displays clear fringe spacing of 0.27 nm (Fig. 2e), which is assigned to the (111) crystal planes of TaON (JCPDS no. 70-1193),12,15 confirming the formation of Rh/TaON heterojunction. As the nitridation time increasing to 6 h, a thin outermost layer with lattice spacing of 0.36 nm corresponding to the (110) crystal plane of Ta3N5 appears between Rh nanospheres and TaON layer (Fig. 2f),15,16 constructing the Rh/Ta3N5/TaON heterojunctions. As observed in Fig. 2g, upon nitriding for 9 h, the outer layer with Ta3N5 (110) crystal planes obviously thickens, and its crystallinity increases, and meanwhile, the preferential plane of the TaON layer transforms from (111) crystal planes to (11) crystal planes. As the nitridation time up to 12 h, the lattice spacing of 0.28 nm corresponds to the (112) crystal planes of Ta3N5 (Fig. 2h). Unfortunately, the lattice fringes of Ta2O5 cores, which has been detected in the above XRD patterns, are not observed probably due to the masking effect of outer Ta3N5 or/and TaON layers.
 | ||
| Fig. 2 TEM images of Rh-Ta-3 (a), Rh-Ta-6 (b), Rh-Ta-9 (c), Rh-Ta-12 (d) and HR-TEM images of Rh-Ta-3 (e), Rh-Ta-6 (f), Rh-Ta-9 (g), and Rh-Ta-12 (h). | ||
Moreover, the EDX mapping for the Rh-Ta-6 sample further confirms the existence of Rh, O, Ta, and N elements (Fig. 3). The vast majority of Rh element aggregates are distributed discretely on the surface, but Ta and N elements are highly dispersed in the sample. A concentration gradient of dispersed O element is also observed in the sample, further revealing the presence of shell–core structure.
 | ||
| Fig. 3 (a) TEM, and EDX mapping of (b) Rh, (c) O, (d) Ta, and (e) N elements of Rh-Ta-6. | ||
Chemical compositions and surface functional groups
The surface compositions and chemical states of the Rh-Ta-6 sample were investigated by XPS. The survey XPS spectrum of the Rh-Ta-6 sample demonstrates the presence of Rh, Ta, O, and N elements. As shown in Fig. 4a, the Rh 3d peaks fitted into two pairs of peaks with the binding energies for Rh 3d5/2 peaks at 306.45 eV and 308.49 eV are attributed to Rh0 and Rh3+, respectively.19,20 The contributions of Rh0 and Rh3+ are 91.05% and 8.95%, respectively, confirming the presence of a little Rh3+, except for most Rh0. Therefore, under the nitridation condition at 850 °C, a majority of doped-Rh3+ ions were reduced to Rh0, and extracted from the Ta2O5 lattices, then aggregating into nanospheres on the surfaces. However, a small amount of Rh3+ ions still remained in the lattices of the Ta-based (oxy)nitrides, which played a key role on keeping their topological structures, otherwise Ta2O5 nanoparticles would be transformed into Ta3N5 nanosheets. In addition, it was also found that the Rh-modifying strongly suppressed the phase transformation from TaON to Ta3N5 under the nitridation condition at 850 °C.12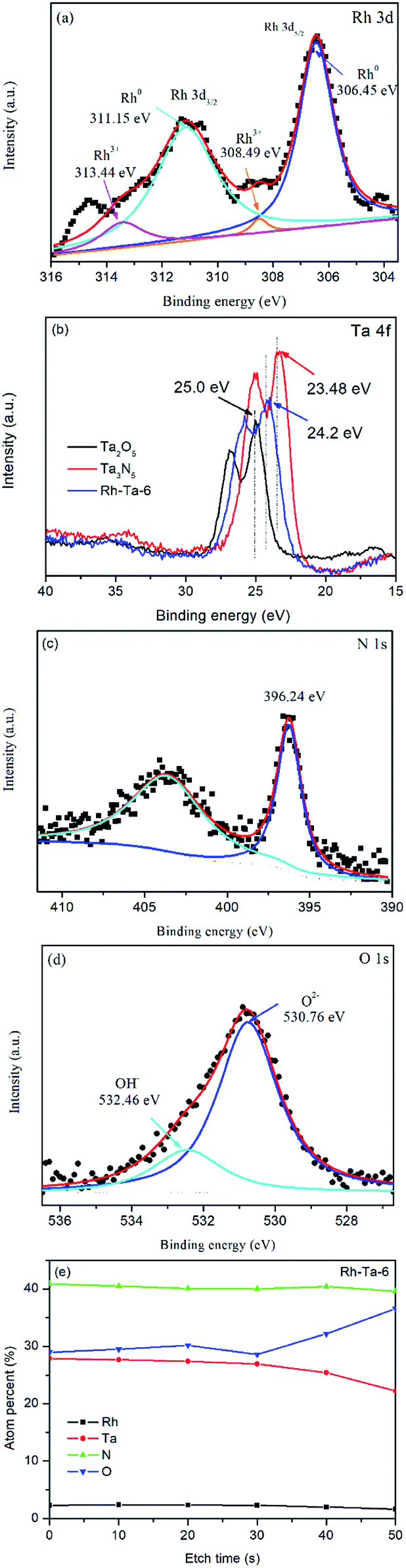 | ||
| Fig. 4 (a) XPS spectra of Rh 3d for Rh-Ta-6; (b) XPS spectra of Ta 4f for Ta2O5, Rh-Ta-6, and Ta3N5; (c) high resolution XPS spectra of N 1s (d) and O 1s (e) for Rh-Ta-6; the atomic percentages of the etched layers for Rh-Ta-6 changing with Ar ion etching time. | ||
As seen in Fig. 4b, the binding energies of Ta 4f peaks for the Rh-Ta-6 sample appear at 24.2 eV and 26.1 eV with a spin orbital separation of 1.9 eV, corresponding to Ta 4f7/2 and Ta 4f5/2,21 respectively, confirm the existence of Ta5+, which are a little lower than the Ta 4f7/2 (25.0 eV) and Ta 4f5/2 (26.84 eV) in pure Ta2O5, but a little higher than the Ta 4f7/2 (23.48 eV) and Ta 4f5/2 (25.3 eV) in pure Ta3N5, due to slight changes in the electron density of Ta5+ by partly substitution of a lower electronegativity N for O forming layer Ta3N5 and inter layer TaON. In addition, as shown in Fig. 4c, the peak at 396.24 eV corresponds to the N 1s binding energy of N3−, attributing to the N–Ta bond resulted from the substitution of the nitrogen ion (N3−) for oxygen ion (O2−) in the Ta2O5 crystal lattices.12 As observed in Fig. 4d, the O 1s peak is fitted into two peaks at 530.76 eV corresponding to the lattice oxygen in Ta–O linkages and at 532.49 eV associated with the chemisorbed O species.22
Furthermore, the surface atomic ratio of N/Ta evaluated by the elemental sensitive factor method is 1.328, higher than that of TaON (1.0), while its O/Ta is 0.967, lower than that of TaON (1.0), further confirming the formation of multiple shell–core heterostructure Ta2O5@TaON@Ta3N5 in Rh-Ta-6 sample. In order to further prove indeed such a kind multiple core–shell heterostructure, XPS depth profiling for the Rh-Ta-6 sample was also performed using monatomic Ar ion sputtering for 10 s, to confirm the subsurface information.23 However, the XPS acquisition was collected not from single Rh-Ta-6 nanoparticle but from Rh-Ta-6 tableting, so only statistical result could be obtained. As seen in Fig. 4e, the atomic percentages of Ta, N, and Rh gradually decreased, and the atomic percentage of O stepped up with Ar ion etching. This result is in accord with the multiple shell–core heterostructures.
To verify the surface bonding, the FTIR spectra of Rh-Ta2O5, Ta2O5@Ta3N5, and as-prepared Rh-modified samples are given in Fig. 5. The peaks at 897, 590, 543, 492, 461, and 436 cm−1 are assigned to the vibrational modes relative to Ta2O5 phonon bands.24 The main peak at 590 cm−1 in Rh-Ta2O5, which is not observed in the other samples, is related to the bending vibration of TaO6 octahedral units.25 For Ta2O5@ Ta3N5 and Rh-Ta2O5 samples, the peaks at 1600 cm−1 and 1667 cm−1 are attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O adsorbed and chemisorbed on the surface,26,27 respectively, and the peaks at 1122 cm−1 to the C–O–C symmetric stretching vibration,28 and the peaks at 1037 cm−1 to the bridged bidentate carbonate,29 which result from the residual organic groups on the surfaces. In contrast, they are not found in our target materials, indicating that the most surface residues were eliminated by the high temperature nitridation process. However, for all samples, the peaks at 1634 cm−1 are assigned to the O–H bending vibration of surface adsorbed water. Furthermore, a new peak appears at 1385 cm−1, corresponding to the surface carbonate species.30
O adsorbed and chemisorbed on the surface,26,27 respectively, and the peaks at 1122 cm−1 to the C–O–C symmetric stretching vibration,28 and the peaks at 1037 cm−1 to the bridged bidentate carbonate,29 which result from the residual organic groups on the surfaces. In contrast, they are not found in our target materials, indicating that the most surface residues were eliminated by the high temperature nitridation process. However, for all samples, the peaks at 1634 cm−1 are assigned to the O–H bending vibration of surface adsorbed water. Furthermore, a new peak appears at 1385 cm−1, corresponding to the surface carbonate species.30
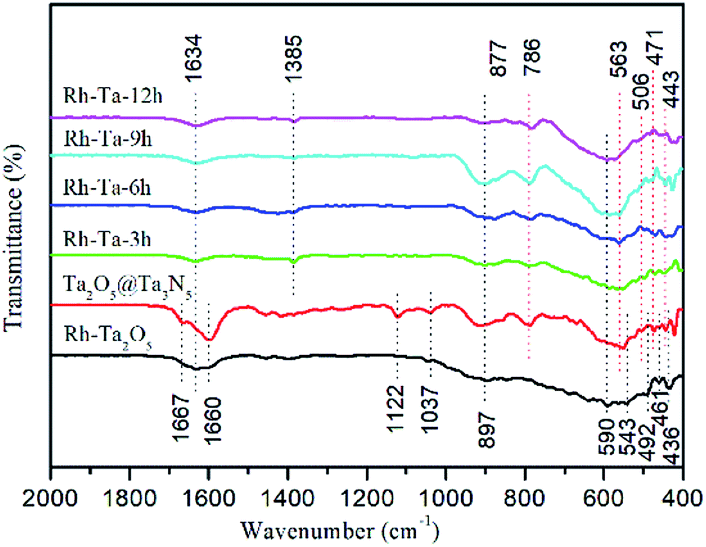 | ||
| Fig. 5 FTIR spectra of Rh-Ta2O5, Ta2O5@Ta3N5, Rh-Ta-3, Rh-Ta-6, Rh-Ta-9, and Rh-Ta-12. | ||
For all nitridated samples, the peaks at 877, 786, 563, 506, 471, and 443 cm−1 should be assigned to Ta–N bonds.12 Unfortunately, no peaks at 575 cm−1 corresponding to the Rh–O stretching can be observed.31 Interestingly, as compared with the absorption at 506–443 cm−1 in Ta2O5@Ta3N5, the absorption of Ta–N bonds in all Rh-modified samples have been shifted significantly to high wave number about 3–8 cm−1, which should be ascribed to the residual Rh–O/N bonds in the crystal lattices of the Ta-based (oxy)nitrides.
UV-vis absorption
The UV-vis diffuse reflectance spectra (DRS) of pure Ta2O5, Ta3N5 and as-prepared Rh-modified samples with different nitridation times are shown in Fig. 6. As observed, pure Ta2O5 can only absorb the UV light of wavelength less than 321 nm (about 3.86 eV), while Ta3N5 obtained from nitriding Ta2O5 at 850 °C for 24 h exhibits strong absorption the visible light of wavelength less than 608 nm (about 2.04 eV).12 Compared with pure Ta2O5 and Ta3N5, all Rh-modified samples present very strong light absorption from the ultraviolet to the whole visible region, mainly resulted from the localized surface plasmon resonance (LSPR) effects of modified Rh metal nanoparticles on their surfaces. In addition, sub-band-gap introduced by the residual Rh3+ (0.75 Å for the ionic radius of Rh3+) in the shell layer lattices (0.7 Å for Ta5+) and nitrogen vacancies (VN) also contribute to absorb longer wavelength visible light.32 For Rh-modified samples, as the nitridation time more than 3 h, their light adsorption capacities present a little decrease owing to both the decrease of residual Rh3+ in the lattices of the shell layer and the agglomeration of surface Rh extracted from the Ta2O5 lattices, as well as their adsorption edges near 600 nm in relate to electron excitation from the valence band the conduction band Ta3N5 are more distinct accordingly, due to gradual thickening of the Ta3N5 shell layer with prolonging the nitridation time. Therefore, the improved light harvesting abilities for Rh-modified samples should contribute to enhancing their photocatalytic H2-evolution.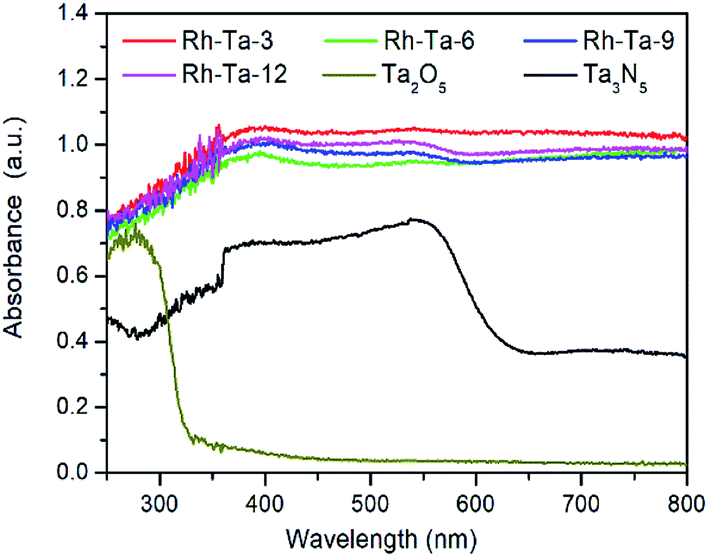 | ||
| Fig. 6 UV-vis absorption spectra of Ta2O5, Rh-Ta-3, Rh-Ta-6, Rh-Ta-9, Rh-Ta-12, and Ta3N5. | ||
Charge separation, photocatalytic H2-evolution activity and proposed charge separation mechanism
Photocurrent measurement was used to qualitatively investigate the separation and transfer of photogenerated charge carriers, and a higher photocurrent intensity generally means a higher charge separation efficiency, thus a higher photoactivity.12 Fig. 7a presents the photocurrent action spectra of unmodified Ta2O5@Ta3N5 and Rh-modified samples with different nitridation times with 0.2 V bias vs. Ag/AgCl for several on–off irradiation cycles under a 350 W Xe lamp. Clearly, their photocurrent signals increase in the following order: Ta2O5@Ta3N5 < Rh-Ta-12 < Rh-Ta-9 < Rh-Ta-3 < Rh-Ta-6, which is not the same as the order of their light harvesting abilities, but well consistent with their photocatalytic H2-evolution activities from aqueous solution containing 20 vol% methanol as a sacrificial agent (Fig. 7b), further confirming that the charge carrier separation efficiency plays a decisive role in enhancing the photocatalytic H2-evolution performance for the Rh-modified samples. Moreover, Rh-modifying significantly improves the photocurrent responses, as well as the photocatalytic H2-evolution activities. More interestingly, the Rh-Ta-6 sample exhibits the highest photocurrent response about one order magnitude higher than that of Ta2O5@Ta3N5, and excellent photocatalytic H2-evolution activity (83.64 μmol g−1 h−1), much superior to Ta2O5@Ta3N5 (21.75 μmol g−1 h−1) and Rh-anchored Ta2O5@TaON (39.41 μmol g−1 h−1).12 In particular, the photocurrent signal for the Rh-Ta-6 sample also exhibits a better stability than all other Rh-modified samples, which should be ascribed to the residual Rh–O/N in the Ta3N5 shell layer. Further research for Rh-modified Ta3N5-based photocatalysts is needed to improve photocatalytic performance for practical application.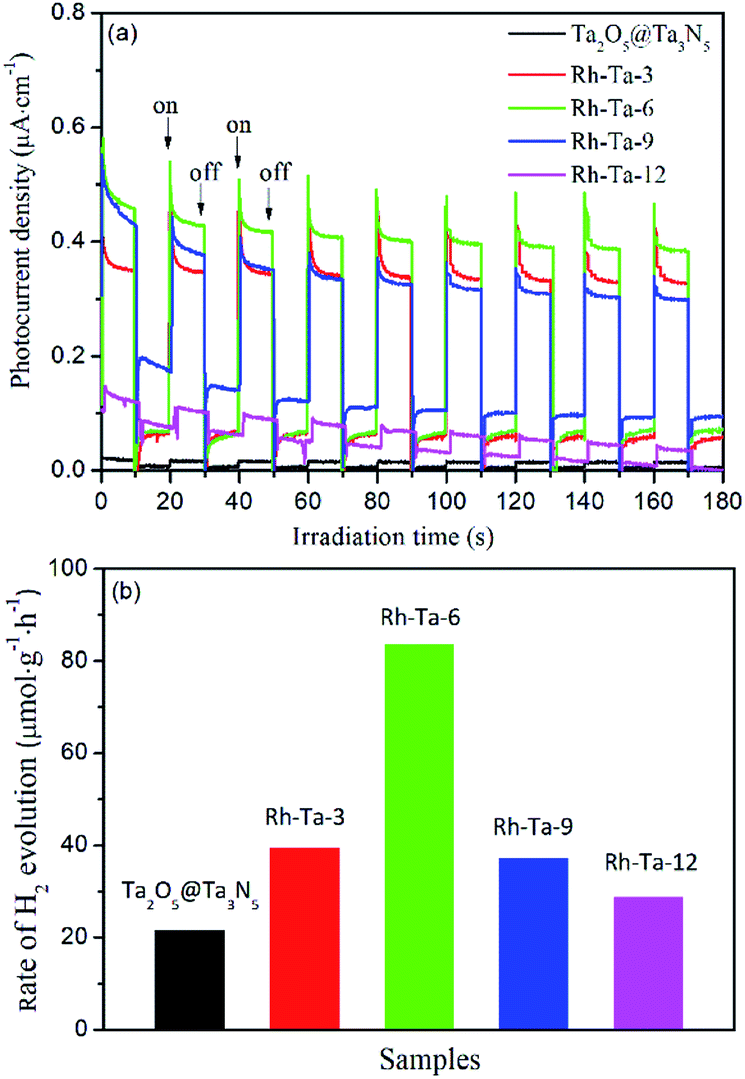 | ||
| Fig. 7 (a) Photocurrent action spectra and (b) photocatalytic H2-evolution activities of Ta2O5@Ta3N5, Rh-Ta-3, Rh-Ta-6, Rh-Ta-9, and Rh-Ta-12. | ||
Additionally, Mott–Schottky plots and VB-XPS spectra were determined to investigate their relative conduction band (CB) and valence band (VB) levels, respectively. Fig. 8a presents the Mott–Schottky plots of Rh-Ta2O5, Rh-Ta-3, and Rh-Ta-6 samples. As observed in Fig. 8a, all the samples exhibit positive slopes in the liner Mott–Schottky regions, confirming that they are n-type conductive semiconductors. Their flat-band (FB) potentials are obtained by the intercept of the extrapolation from the Mott–Schottky plots on the potential axis to be −0.74 V, −0.76 V, and −0.84 V, without further correction based on the pH values of their isoelectric points. Generally speaking, the CB bottom of n-type semiconductor is more negative by ca. 0.2 V than the FB potential.7,14 Consequently, it can be deduced that the relative band alignment of CB is Ta2O5 > TaON > Ta3N5. Moreover, as seen in Fig. 8b, the VB positions based on linear extrapolation are 2.64 eV, 2.52 eV, and 1.45 eV for Rh-Ta2O5, Rh-Ta-3, and Rh-Ta-6 samples, respectively, confirming that the VB position shifts to lower binding energies with prolonging the nitridation time. Thus it can be obtained that the relative band alignment of VB is Ta2O5 > TaON > Ta3N5.
 | ||
| Fig. 8 (a) Mott–Schottky plots and (b) VB-XPS spectra of Rh-Ta2O5, Rh-Ta-3, and Rh-Ta-6 samples. | ||
The above results are well consistent with the reported absolute band edge positions in the vacuum level, the higher energy of N 2p orbital can push the VB top upward in the order of Ta2O5 > TaON > Ta3N5,14 whereas the less repulsive electrostatic potential at the Ta sites (29.77 V for Ta2O5, 28.80 V for TaON, and 27.11 V for Ta3N5) by the electrons in the CB bottom push down the CB bottom in the order of Ta2O5 > TaON > Ta3N5 (Scheme 2a).36
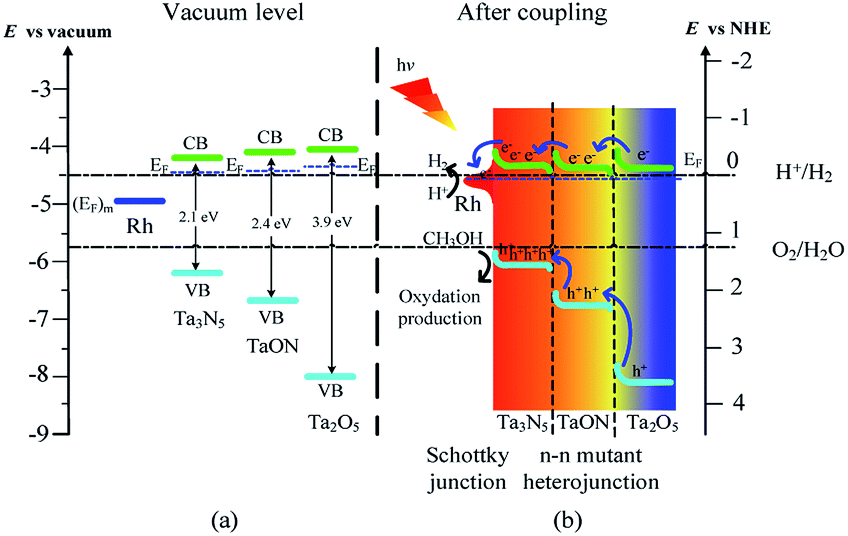 | ||
| Scheme 2 Schematic illustration of (a) band positions of Rh, Ta3N5, TaON, and Ta2O5 in vacuum level, (b) directional transfer of charge carriers in Rh/Ta3N5 Schottky junction and double n–n Ta3N5/TaON/Ta2O5 mutant heterojunctions for Rh-Ta-6. | ||
How are the charge carrier separation and transfer driven by internal built-in fields within the Rh-Ta-6 sample so as to improve the photocatalytic water splitting into H2? Based on the above analysis, we propose a probable mechanism illustrated in Scheme 2. Firstly, Rh-modifying obviously improve the light absorption abilities from ultraviolet to whole visible region. Specially, Rh can initiate the resonant energy transfer under longer wavelength irradiation, leading to surface plasmon resonance (SPR) induced electron transportation from Rh to Ta3N5, which contributes to the improved photocurrent and photoactivity.33 Furthermore, Rh with a higher work function (4.98 eV)34 than that of Ta3N5 (4.5 eV)35 serves as an electron sink to facilitate the photogenerated charge transfer from Ta3N5 under shorter wavelength irradiation, then an upward band bending is produced in the Ta3N5 side, thus a Schottky-type barrier is formed in the Rh/Ta3N5 interface, which accelerates the photogenerated electron–hole separation.12 On the other hand, double n–n Ta3N5/TaON/Ta2O5 mutant heterojunctions are formed in the triple core–shell heterostructures. Upon coupling, the photogenerated electrons transfer from Ta2O5 core with the highest Fermi energy level, through TaON interlayer to Ta3N5 shell with the lowest Fermi energy level in turn for achieving thermodynamic equilibrium, and lead to forming an accumulated charge layer in the Ta3N5 side and a depletion layer in the TaON side at the Ta3N5/TaON interface, as well as an accumulated charge layer in the TaON side and a depletion layer in the Ta2O5 side at the TaON/Ta2O5 interface. As a consequence, localized built-in electric fields are formed in the negative and positive space charge regions at the multiple hierarchical interfaces including the Rh/Ta3N5 Schottky junction and n–n Ta3N5/TaON/Ta2O5 mutant heterojunctions, the directions of which are inside-out consistently, promoting the fast directional transfer and efficient separation of photogenerated charge carriers within the Rh-Ta-6 sample, and then improving its photoactivity for water splitting into H2 (Scheme 2b).
In addition, Rh anchored on the surface may extract the photogenerated electrons from the VB of the Ta3N5 shell layer and subsequently transfer them into the solution due to the very low overpotential.34,37 Therefore, the superior photocatalytic H2 evolution performance for the Rh-Ta-6 sample can be ascribed to a synergetic effect of above factors.
Conclusions
A novel multiple core–shell heterostructure Rh–Rh3+ modified Ta2O5@TaON@Ta3N5 nano-photocatalyst with hierarchical potential gradients was controllably synthesized via nitriding at 850 °C for 6 h from Rh-doped Ta2O5 precursor, which exhibited an excellent photoactivity for water splitting into H2 (83.64 μmol g−1 h−1), much superior to previously reported Rh anchored Ta2O5@TaON (39.41 μmol g−1 h−1). Stability was also improved probably due to residual Rh–O/N in Ta3N5 shell layer. Rh-modifying significantly improved the light absorption ability from ultraviolet to overall visible region, mainly due to LSPR effects of the surface-modified Rh and sub-band-gap behavior of residual Rh3+ in the shell layer lattices and surface N-vacation. Internal built-in electric fields were formed at the multi-hierarchical interfaces including the Rh/Ta3N5 Schottky junction and double n–n Ta3N5/TaON/Ta2O5 mutant heterojunctions, which driven the rapid directional transfer from inside to outside and efficient separation of photogenerated charge carriers. By comparison, the superior photocatalytic H2 evolution activity could be ascribed to the synergetic effect of such positive factors including improved light absorption abilities, enhanced charge carrier separation efficiency within the multi-hierarchical heterojunctions, as well as accelerated surface reaction. Constructing multiple shell–core heterostructure Ta2O5@TaON@Ta3N5 with double n–n mutant heterojunctions was proved to be a promising strategy for further developing efficient and stable Ta3N5-based photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Key Project of Innovative Scientific Research for Postgraduate students of Harbin Normal University (No. HSDSSCX2018-17).References
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- S. Khan, S. R. Teixeira and M. J. L. Santos, RSC Adv., 2015, 5, 103284–103291 RSC.
- S. S. Zhu and D. W. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- T. Jing, Y. Dai, X. C. Ma, W. Wei and B. B. Huang, RSC Adv., 2015, 5, 59390–59397 RSC.
- L. Wang, A. Mazare, I. Hwang, S. So, N. T. Nguyen and P. Schmuki, Chem. Commun., 2017, 53, 11763–11766 RSC.
- W. Wang, H. B. Fang, Y. Z. Zheng, Y. K. Che, X. Tao and J. F. Chen, RSC Adv., 2015, 5, 62519–62526 RSC.
- B. B. Dong, J. Y. Cui, Y. Y. Gao, Y. Qi, F. X. Zhang and C. Li, Adv. Mater., 2019, 31, 1808185 CrossRef.
- E. Nurlaela, H. Wang, T. Shinagawa, S. Flanagan, S. Ould-Chikh, M. Qureshi, Z. Mics, P. Sautet, T. L. Bahers, E. Cánovas, M. Bonn and K. Takanabe, ACS Catal., 2016, 6, 4117–4126 CrossRef CAS.
- S. P. Adhikari, Z. D. Hood, K. L. More, I. Ivanov, L. F. Zhang, M. Gross and A. Lachgar, RSC Adv., 2015, 5, 54998–55005 RSC.
- M. Hara, J. Nunoshige, T. Takata, J. N. Kondo and K. Domen, Chem. Commun., 2003, 3000–3001 RSC.
- Y. H. Jiang, P. P. Liu, Y. C. Chen, Z. Z. Zhou, H. J. Yang, Y. Z. Hong, F. Li, L. Ni, Y. S. Yana and D. H. Gregoryc, Appl. Surf. Sci., 2017, 391, 392–403 CrossRef CAS.
- H. Q. Jiang, W. Zhang, S. Y. Zang and W. L. Zhang, Int. J. Hydrogen Energy, 2019, 44, 24218–24227 CrossRef CAS.
- Q. L. Liu, Z. Y. Zhao and J. H. Yi, Phys. Chem. Chem. Phys., 2018, 20, 12005–12015 RSC.
- W. J. Chun, A. Ishikawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1798–1803 CrossRef CAS.
- D. Abeysinghe and S. E. Skrabalak, ACS Energy Lett., 2018, 3, 1331–1344 CrossRef CAS.
- S. M. Wang, Z. Y. Li, Y. Guan, L. Lu, Z. Shi, P. Wang, S. C. Yan and Z. G. Zou, Appl. Catal., B, 2019, 245, 220–226 CrossRef CAS.
- B. Niu and Z. M. Xu, J. Catal., 2019, 371, 175–184 CrossRef CAS.
- S. J. Li, X. F. Shen, J. S. Liu and L. S. Zhang, Environ. Sci.: Nano, 2017, 4, 1155–1167 RSC.
- S. S. Chen, Y. Qi, Q. Ding, Z. Li, J. Y. Cui, F. X. Zhang and C. Li, J. Catal., 2016, 339, 77–83 CrossRef CAS.
- E. Grabowskaa, M. Diak, T. Klimczuk, W. Lisowski and A. Z. Medynska, Mol. Catal., 2017, 434, 154–166 CrossRef CAS.
- S. Khan, M. J. L. Santos, C. Malfatti, J. Dupont and S. R. Teixeira, Chem.–Eur. J., 2016, 22, 18501–18511 CrossRef CAS PubMed.
- J. Y. Cui, Y. P. Luo, B. B. Dong, Y. Qi, M. J. Jia, F. X. Zhang and C. Li, Sol. RRL, 2019, 1900445 CrossRef.
- B. W. Boote, L. Men, H. P. Andaraarachchi, U. Bhattacharjee, J. W. Petrich, J. Vela and E. A. Smith, Chem. Mater., 2017, 29, 6012–6021 CrossRef CAS.
- J. L. Ferrari, K. O. Lima, L. J. Q. Maia, S. J. L. Ribeiro and R. R. Goncalves, J. Am. Ceram. Soc., 2011, 94, 1230–1237 CrossRef CAS.
- G. M. D. Pietro, C. Pereira, R. R. Goncalves, S. J. L. Ribeiro, C. D. Freschi, F. C. Cassanjes and G. Poirier, J. Am. Ceram. Soc., 2015, 98, 2086–2093 CrossRef.
- N. Hérault, L. Olivet, L. Pirault-Roy, C. Especel, M. A. Vicerich, C. L. Pieck and F. Epron, Appl. Catal., A, 2016, 517, 81–90 CrossRef.
- Q. S. Gao, C. Giordano and M. Antonietti, Small, 2011, 7, 3334–3340 CrossRef CAS PubMed.
- J. Cao, L. Ren, N. Li, C. W. Hu and M. H. Cao, Chem.–Eur. J., 2013, 19, 12619–12623 CrossRef CAS PubMed.
- C. D. DiGiulio, V. G. Komvokis and M. D. Amiridis, Catal. Today, 2012, 184, 8–19 CrossRef CAS.
- J. Múnera, B. Faroldi, E. Frutis, E. Lombardo, L. Cornaglia and S. G. Carrazán, Appl. Catal., A, 2014, 474, 114–124 CrossRef.
- C. I. Cabello, M. Muñoz, I. L. Botto and E. Payen, Thermochim. Acta, 2006, 447, 22–29 CrossRef CAS.
- J. J. Wang, Z. S. Li, J. H. Jiang, J. Y. Feng and Z. G. Zou, Phys. Chem. Chem. Phys., 2015, 17, 8166–8171 RSC.
- D. W. Wang, S. C. Pillai, S. H. Ho, J. B. Zeng, Y. Li and D. D. Dionysiou, Appl. Catal., B, 2018, 237, 721–741 CrossRef CAS.
- Q. Wang, M. Ming, S. Niu, Y. Zhang, G. Y. Fan and J. S. Hu, Adv. Energy Mater., 2018, 8, 1801698 CrossRef.
- S. M. Wang, Y. Guan, L. Lu, Z. Shi, S. C. Yan and Z. G. Zou, Appl. Catal., B, 2018, 224, 10–16 CrossRef CAS.
- Z. H. Cui and H. Jiang, J. Phys. Chem. C, 2017, 121, 3241–3251 CrossRef CAS.
- M. Sheikhzadeh, S. Hejazi, S. Mohajernia, O. Tomanec, M. Mokhtar, A. Alshehri, S. Sanjabi, R. Zboril and P. Schmuki, ChemCatChem, 2019, 11, 6258–6262 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |