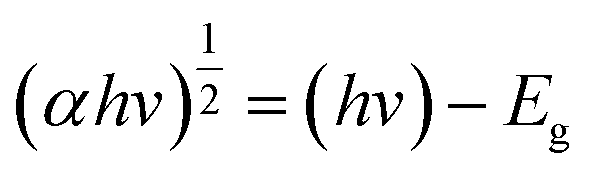DOI:
10.1039/D0RA04726K
(Paper)
RSC Adv., 2020,
10, 29432-29440
Lateral size reduction of graphene oxide preserving its electronic properties and chemical functionality†
Received
28th May 2020
, Accepted 4th August 2020
First published on 10th August 2020
Abstract
Graphene oxide (GO) is widely considered as a graphene precursor when chemically reduced. Nevertheless, through the precise control of two parameters: lateral size and oxidation degree, GO can be useful in many applications as modified graphene oxide or functional reduced graphene oxide. Commonly, the decrease in GO lateral size, involves a change in the C/O ratio and therefore a modification in a large number of characteristics. Here, a simple but effective approach to synthesize GO with lateral dimensions below 100 nm and without modification of its chemical, optical and electronic features is presented. The use of a sonifier at low temperature allows to rapidly reduce the lateral size in ∼82% while preserving the C/O ratio and consequently the chemical stability, the band gap, the electronic energy levels and the functionality. This method will allow several applications from biomedicine to energy, where reliable reduced size of GO is required.
1. Introduction
Graphene holds a myriad of exceptional characteristics, becoming one of the most promising two-dimensional materials.1 One of the most explored approaches to overcome the major challenges of graphene's synthesis and processing is through graphene oxide (GO). Basically, GO is a highly oxidized graphene, exhibiting oxygen groups such as hydroxyl, carboxyl, epoxy and carbonyl, described by the Lerf–Klinowski model.2 As expected, after its chemical reduction, GO is frequently perceived as a precursor for graphene, rather than chemically derived graphene3,4 or reduced graphene oxide5 (rGO). In this context, the rGO synthesis through GO as an intermediate has several advantages, being the two most important: scalability3 and simple further chemical modification ability of GO to obtain functional rGO,6,7 for even wider range of properties and applications than pure graphene.8
In this regard, it is also important to point out the two most significant GO parameters such as: (i) lateral size, and (ii) oxidation degree (C/O ratio). It has been reported that GO lateral dimensions around 100 nm improves significantly its stability.9,10 Moreover, the lateral size is especially important in biomedical applications,11–14 as well for the optical and electronic properties.15–17 Several studies have been conducted to synthesize GO with lateral dimensions around 100 nm using different techniques, such as ultrasonication and milling,10,11,14,18,19 oxidation,20,21 ultracentrifugation,12,16,22,23 circular flow fractionation,24 oxidation/centrifugation15 and filtration.24
A challenge arises when the lateral size of GO is intended to decrease without modification of its chemical, optical and electronic features. The second parameter, C/O ratio, is related to the total number of functional groups in GO and therefore, is considered as the oxidation degree. The C/O ratio is the most crucial parameter because it simultaneously controls two properties: electronic (i.e., band gap) and further chemical functionality.5,7,25 When the lateral size reduction process of GO is prepared without considering a possible chemical reduction,16,18,26 it may lead into a different C/O ratio. Thus, rGO with low solubility in a wide range of solvents is obtained, because of the coexistence of remaining oxygen groups and the sp2 recovery in the structure.5 As the aforementioned oxidation degree is carefully taken into consideration in a lateral size reduction process, allows further fine tuning when GO is chemically reduced, i.e., rGO final surface chemistry and characteristics through simple chemical techniques.6,7
When both parameters are accurately controlled, is possible to use GO not only as graphene precursor, but also in different applications like biomedicine,11,18,23,27 catalysis,22 polymer nanocomposites,28,29 adsorption,24 electronics,25 organic solar cells,30,31 and specially in energy storage such as supercapacitors, where if the precise C/O ratio and size can be controlled, the small lateral size GO could be used in hierarchical structures.32–34 Based on that, is crucial to control both parameters, because if a lateral size reduction leads into a C/O ratio modification on GO, it will affect the electronic properties, its solubility and further chemical functionalisation. Herein, we report a simple but, highly effective approach to synthesize GO around 100 nm in lateral size and high concentration by means of ultrasound and low temperature conditions, without change in the electronic properties and excellent solubility in water.
2. Experimental
2.1 Materials
Graphite nanoparticles (GNPs) from ACS materials were used as a starting material. Potassium permanganate (KMnO4), potassium nitrate (KNO3), hydrochloric acid (HCl 37.3%) and sulfuric acid (H2SO4 98%) were purchased from CTR Scientific. Ethanol, hydrogen peroxide (H2O2 30%) and methanol were purchased from J.T. Baker. All the chemicals were used as received. Deionized water (DI H2O) was employed for the complete process.
2.2 Graphene oxide synthesis
The GO synthesis was performed from GNPs by a modified Hummers' method as reported elsewhere.5,8,35 Briefly, 0.5 g of GNPs were oxidized in 30 mL of H2SO4 and 0.295 g of KNO3 with 3 g of KMnO4 to achieve formation of Mn2O7 complex. The reaction was carried out for 6 hours, stirring the solution at 450 rpm controlling the temperature below 15 °C. The reaction was quenched with 100 mL of DI H2O and 6 mL of H2O2 30%. Next, the obtained bright yellow dispersion (graphite oxide) was centrifuged at 3500 rpm for 10 minutes to remove the supernatant and 100 mL of HCl 10% were added to the precipitate and centrifuged under the same conditions. The washing process was repeated 3 times with DI H2O. Afterwards, 80 mL of DI H2O were added, and the dispersion was placed in the ultrasonic bath VWR Symphony Ultrasonic Cleanser Model 97043-944 for 1 hour for exfoliation. Finally, the supernatant in water dispersion was recovered, stored and labelled as GO-0.
2.3 Graphene oxide lateral size reduction
The lateral size decrease of GO was achieved by a Branson sonifier model SFX250, applying a 40% amplitude and a controlled temperature of 18 °C. The procedure was performed as follows: the starting GO dispersion (GO-0) was diluted at [3 mg mL−1] and gauged to 10 mL followed by sonication. Every 30 minutes for a lapse of 240 minutes, 1 mL of the GO dispersion was taken and gauged at 10 mL in centrifuge vials named as GO-30, GO-60, GO-90, GO-120 and GO-240, referring to the time in the ultrasonic probe. Later, the dispersions were centrifuged at 3500 rpm for 1 hour to remove the less stable GO, then the supernatant was analysed. The resulting yield after the size reduction and centrifugation was determinate to be 90%.
2.4 Characterisation
The Scanning Electron Microscopy in Scanning Transmission Electron Microscopy (STEM) mode was used for morphology characterisation using a FEI Nova NanoSem200 equipment. Diluted [0.1 mg mL−1] water dispersions were analysed by Zeta-potential and Dynamic Light Scattering (DLS) Malvern Zetasizer Nano S to determine stability and size distribution, respectively.
A dried sample by vacuum filtration at room temperature was analysed by X-ray photoelectron spectroscopy (XPS), analyses were carried out with a Thermo Scientific Escalab 250Xi instrument. The base pressure during analysis was ∼10−10 mbar and the photoelectrons were generated with the AlKα (1486.68 eV) X-ray source with monochromator and a spot size of 650 μm. The X-ray voltage and power were 14 kV and 350 W, respectively. The acquisition conditions for the high-resolution spectra were 20 eV pass energy, 45° take-off angle and 0.1 eV per step. Selected high resolution region spectra were recorded covering the C1s and O1s signals. The recorded photoelectrons peaks were analysed with the Avantage software V 5.41.
A drop casting sample on a glass substrate was prepared from the GO dispersion, in order to be analysed by Raman spectroscopy which was performed on a confocal Raman microscope alpha300R from WITec. A 50× objective was used for all measurements and the system was used unpolarized with a 2.33 eV laser. The laser power was reduced to avoid sample damage. All spectra were gathered over 2 s exposures and 10 accumulations.
UV-Vis-NIR Cary 5000 was used for optical measurements in diluted [0.1 mg mL−1] water dispersions. An Electrochemical Workstation CH-Instruments 650A was used to determine the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels. The cyclic voltammetry (CV) was carried out using a three-electrode cell with a platinum wire as working electrode, as well as counter electrode and Ag/Ag+ as the reference electrode, prepared with an AgNO3 [0.1 M] aqueous solution, calibrated with ferrocene (Fc/Fc+). A solution of tetrabutylammonium hexafluorophosphate (Bu4NPF6) [0.1 M] in anhydrous acetonitrile was used as the support electrolyte. Prior to each measurement, N2 was bubbled in the cell for 15 minutes to remove the oxygen. Afterwards, the needle was placed over the solution and kept it there during the measurements.
3. Theory: proposed lateral size reduction mechanism
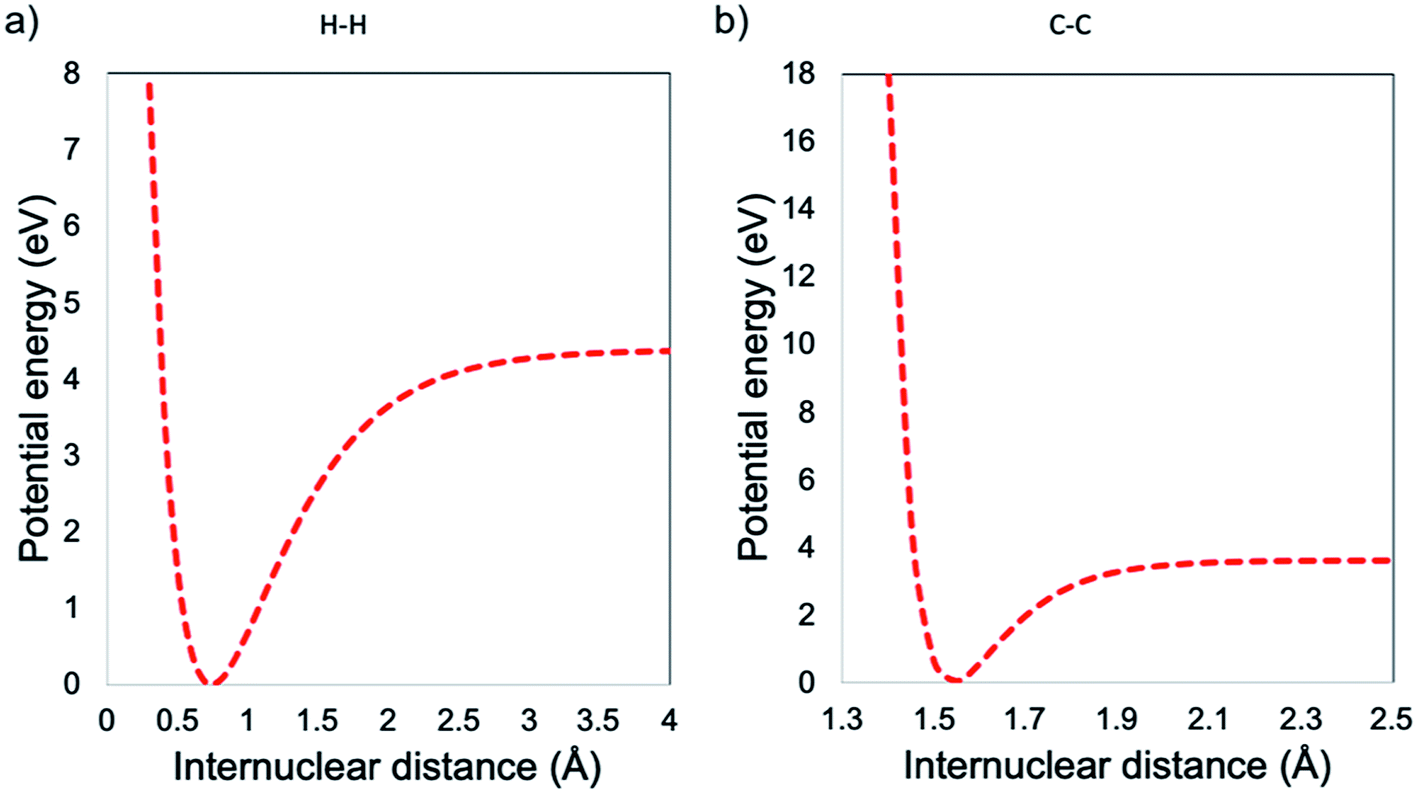
In this work, it was proposed to synthesize GO and afterwards decrease the lateral size without compromising the C/O ratio and therefore without affecting its chemical functionality and electronic properties. In this regard, we propose that the mechanism must be different from the hot spot,18 where the final product is reduced both chemically and in lateral size. The hot spot theory is associated to an atomic reduction of GO by heat, in other words, a chemical–thermal reduction, leading to a rGO and not to GO.18 In the present work, the mechanical anharmonicity behaviour, was explored. Hereof, for a simple molecule, the homolytic cleavage of a bond that defines the Bond Dissociation Energy (BDE) is described in the Morse potentials, i.e., a mathematical function that describes how the potential energy changes along the coordinate for the internal motion. All these functions derive from the bonding forces that hold the atoms together in the molecule, by assuming that vibrational motion obeys Hooke's law. The Morse potential describes bond-stretching vibrations: in the y-axis, the potential energy in eV versus the internuclear distance in the x-axis see Fig. 1.36,37
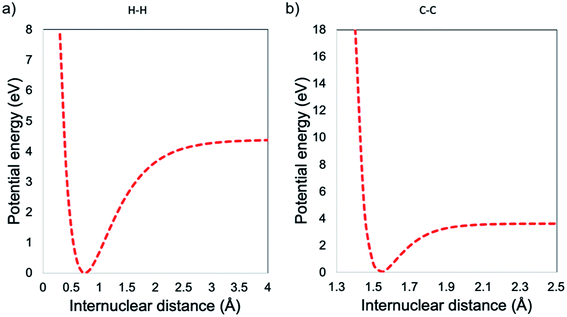 |
| | Fig. 1 Morse potential energy curve for (a) H–H bonding and (b) C–C bonding. | |
At small values of internuclear distance, the positive charges on the two nuclei cause mutual repulsion, which progressively increases the potential energy, preventing their approaching. At large values of internuclear distance the molecule dissociates: two neutral atoms are formed and, since they do not influence each other, the force constant is zero and internuclear distance can then be increased to infinity with no further change of the potential energy. As one can see from the examples in Fig. 1, the H–H bond can handle a higher distortion before the breakage, when compared with the C–C bond. In this context, will be expected that the decrease in lateral size is carried out by the breakage within the GO structure of the sp3 bonding, i.e., the lower BDE compared with the sp2. Nevertheless, by the control of the low temperature, it is possible to avoid the modification in the average C/O ratio, i.e., preventing the thermal–chemical reduction, as seen in Fig. 2.
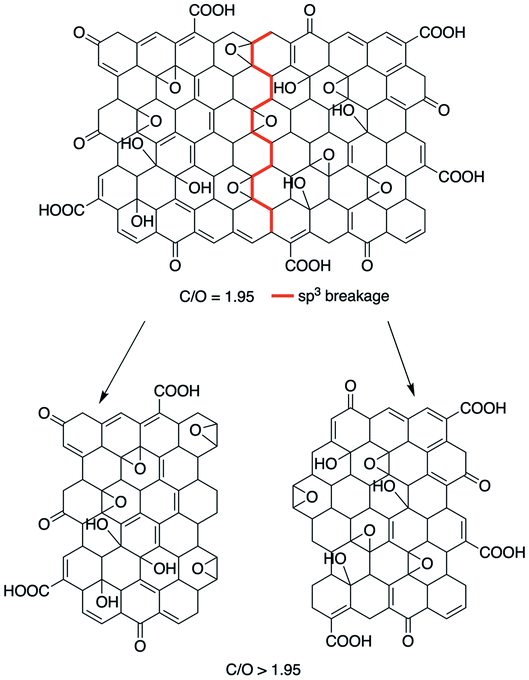 |
| | Fig. 2 Schematic representation of the size reduction mechanism in the x–y plane, without modification of the C/O ratio. | |
4. Results and discussion
In order to analyse the lateral size decrease, SEM in STEM mode was used as well as DLS. Afterwards, to confirm that lateral size modification did not lead to a chemical reduction, XPS was performed to all the samples in order to compare the chemical state and modifications of GO at the surface. Optical characteristics were analysed by UV-Vis and refined by Tauc plot. The electronic features such as the HOMO, LUMO and electrical band gap was analysed by cyclic voltammetry.
4.1 Lateral size determination
The STEM micrographs are presented in Fig. 3 for GO-0 and GO-240, corresponding to the initial and the final samples, see all pictures (at low and high magnifications) in Fig. S1 in the ESI.† As one can see, the lateral size acquired from the STEM are similar from the values obtained by DLS, presented in Table 1, which is in agreement from previous works.8,12,14,29
 |
| | Fig. 3 STEM micrographs of (a) the starting GO and (b) after 240 minutes treatment. | |
Table 1 DLS average size-time dependent
| Sample |
Time (min) |
Size (nm) |
| GO 0 |
0 |
500 |
| GO 30 |
30 |
144 |
| GO 60 |
60 |
118 |
| GO 90 |
90 |
108 |
| GO 120 |
120 |
98 |
| GO 240 |
240 |
86 |
The average size obtained by DLS, is shown in the Table 1, and the complete information from the technique can be found in Fig. S2 in the ESI.† Also, stability measurements by Zeta-potential are presented in Fig. S3 in the ESI.†
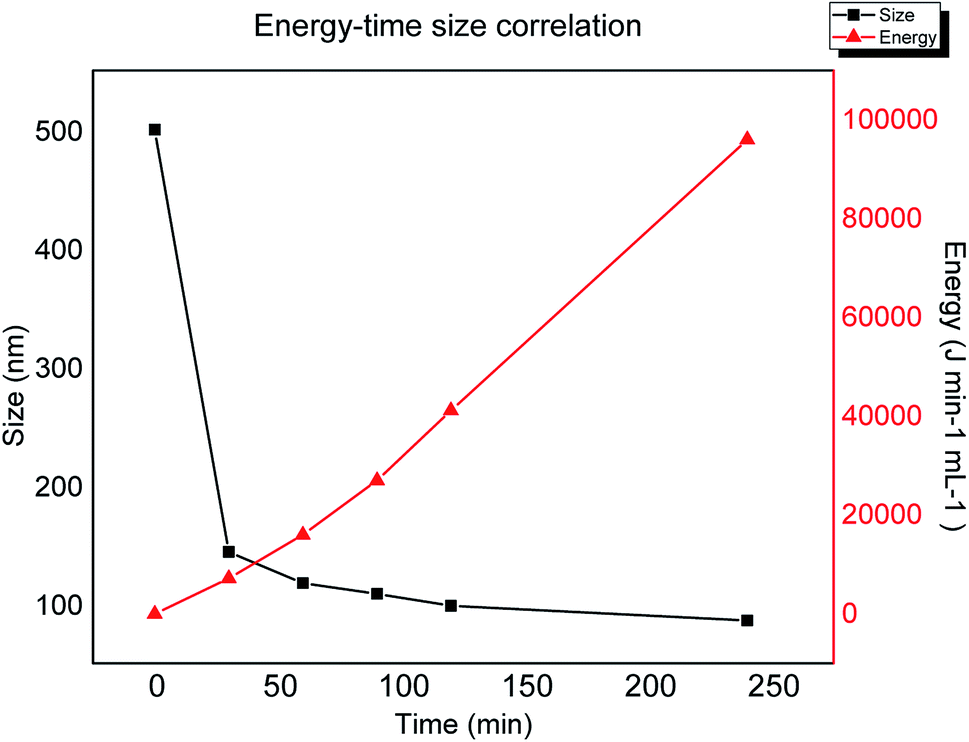
In order to analyse the relation between lateral size against the applied energy during sonication, the plot in Fig. 4 shows the progression in size reduction depending on the energy during the time of the experiment. It is clear that the major change in size is achieved during the first stage of the experiment (first two hours), however, based on the plot one can interpolate or extrapolate the required energy in order to obtain a specific desired size. This plateau behaviour could be explained by the depletion of sp3 breakage areas, confirming that the mechanism is related to the lowest BDE by mechanical vibration.
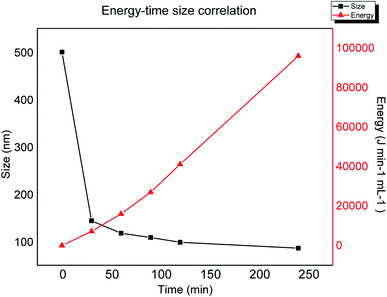 |
| | Fig. 4 Lateral size obtained by dynamic light scattering versus applied energy during sonication. | |
4.2 XPS analysis
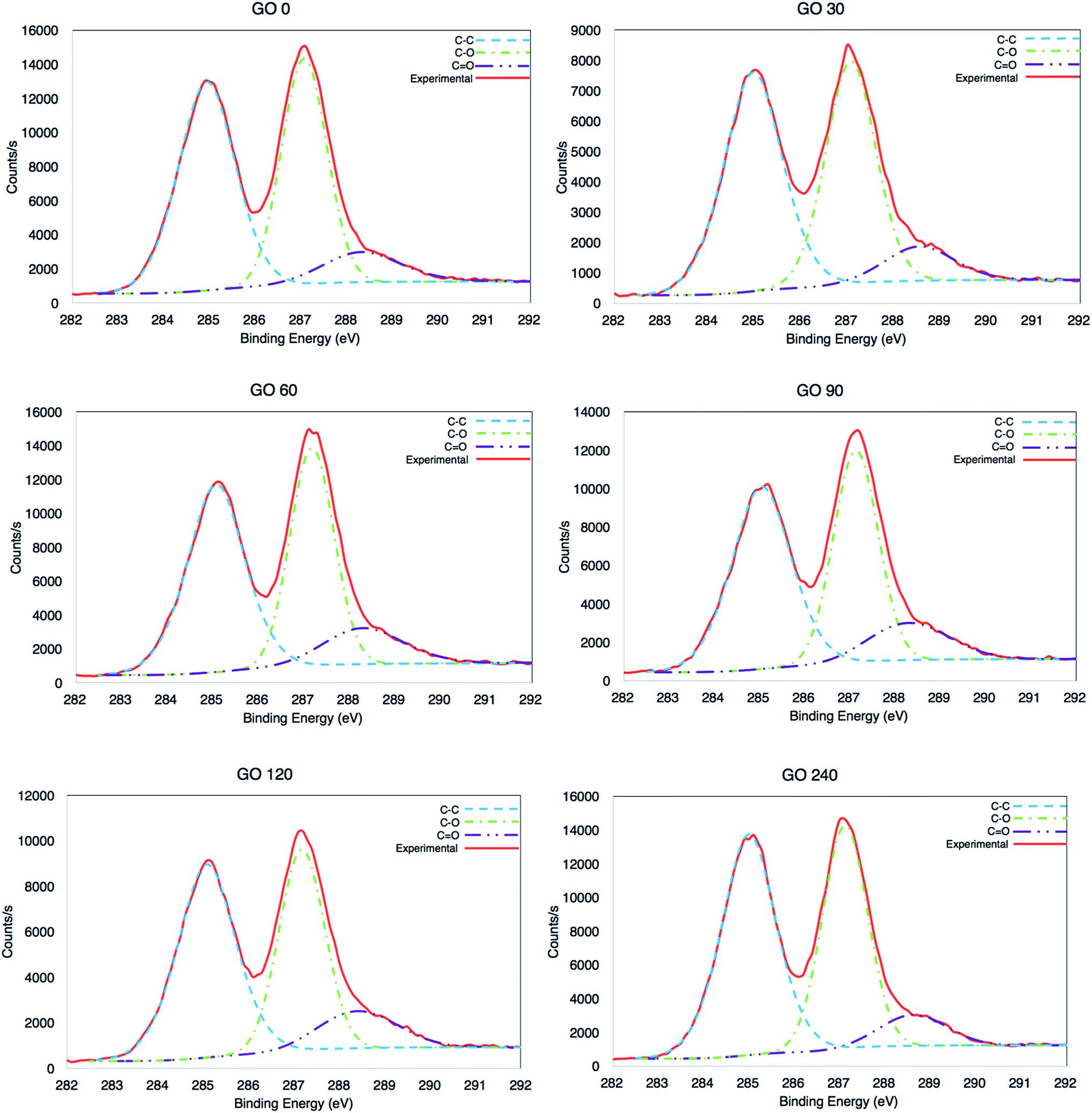
The chemical state of the synthesized materials was analysed by X-ray photoelectron spectroscopy (XPS). The high resolution C1s were recorded from 279 eV to 298 eV and are presented in Fig. 5. The signal coming from all the GOs is characterised of highly oxidized material, produced by C–C at ∼284.5 eV, but also C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at ∼286.7 eV and ∼288.7 eV, respectively.5,38
O at ∼286.7 eV and ∼288.7 eV, respectively.5,38
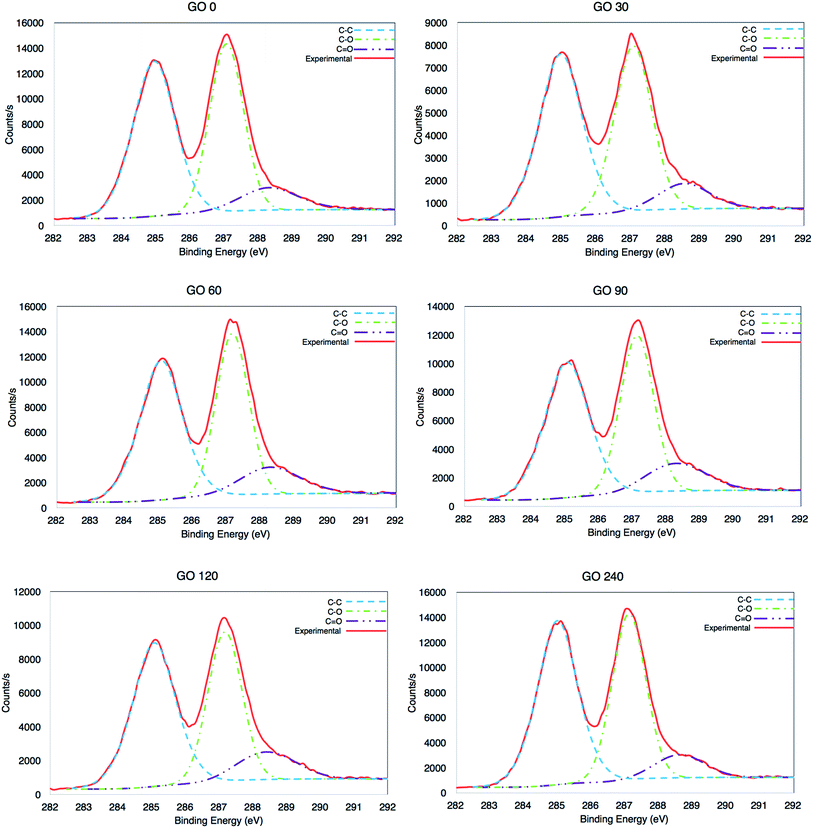 |
| | Fig. 5 High resolution XPS C1s analysis of GOs at 0, 30, 60, 90, 120 and 240 minutes. | |
It is clear that there was a minor variation between the samples (±3%), as seen in Table 2, where the average value is 1.94, which corresponds to a highly oxidized material. The C/O ratio was calculated adding the area in the high-resolution C1s, from the signals for the three species (C–C, C–O and CO) and then divided it by the area of C–O and CO. However, also the carbon and oxygen content per atomic percentage from the survey is also included, see surveys in Fig. S4 in ESI.†
Table 2 C/O ratio from high-resolution XPS results and carbon–oxygen content from survey
| Sample |
Time (min) |
C/O ratio |
Carbon (atomic%) |
Oxygen (atomic%) |
| GO-0 |
0 |
1.95 |
67.76 |
32.24 |
| GO-30 |
30 |
1.96 |
68.4 |
31.6 |
| GO-60 |
60 |
1.89 |
67.54 |
32.46 |
| GO-90 |
90 |
1.89 |
66.45 |
33.55 |
| GO-120 |
120 |
1.95 |
67.88 |
32.12 |
| GO-240 |
240 |
2.01 |
68.36 |
31.64 |
4.3 Structural study by Raman spectroscopy
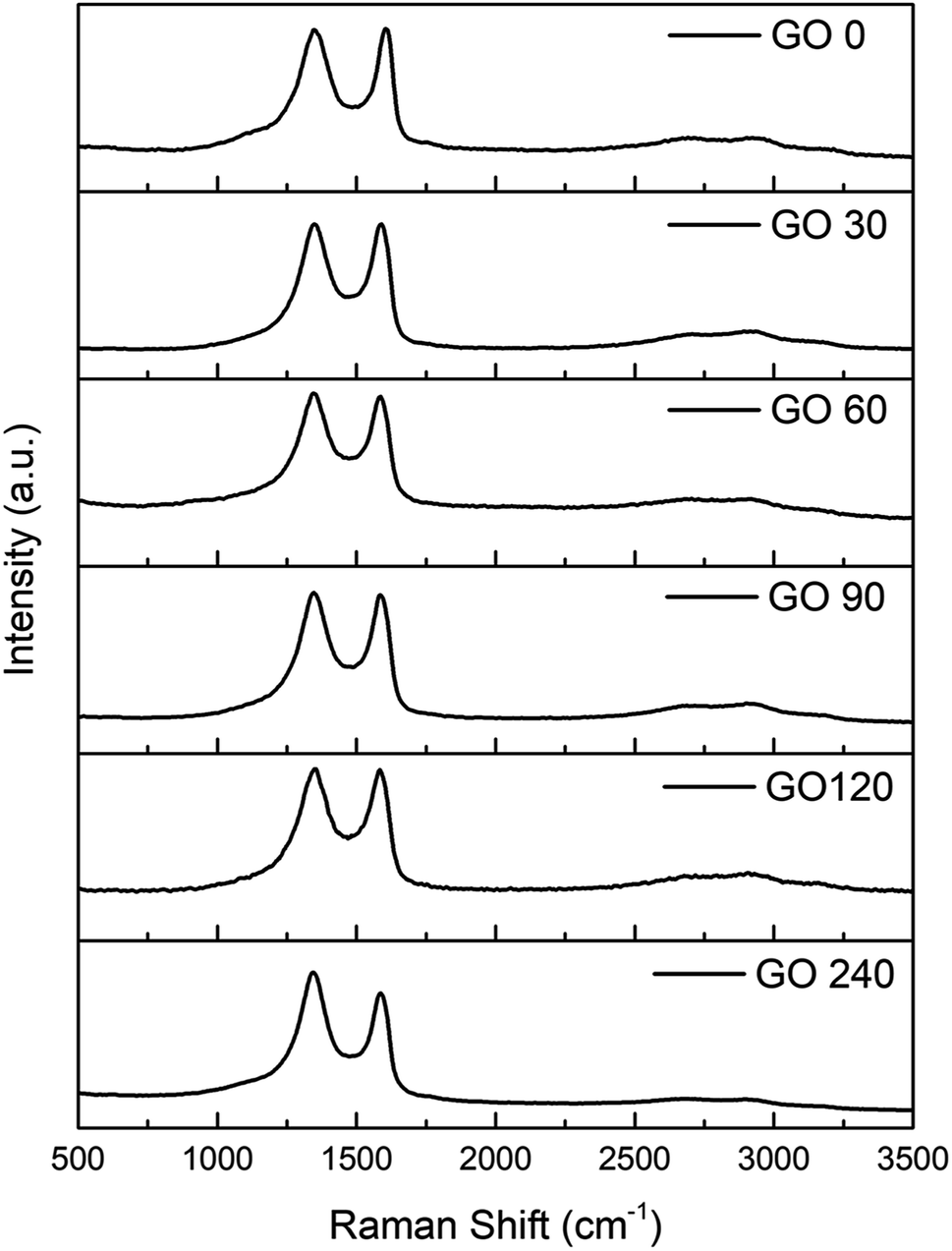
In order to compare the structural quality of the starting material against the synthesized one, Raman spectra is presented in Fig. 6. It is well known that the particular dispersion of π electrons in graphene offers a powerful and efficient insight of their electronic properties, and therefore its structural crystallinity. It can be noticed that all spectra exhibit an intense band from 1450–1660 cm−1 corresponding to the G band, due vibrational E2g degenerative mode observed in sp2 carbons. Furthermore, an equal intense band is observed at 1260–1400 cm−1, assigned to the D band an is related to the A1g mode.39 The D peak is originated due the interaction between phonons and defects, such as in-plane substitution heteroatoms, vacancies, or grain boundaries. In this study, the ratio between the D and G band are basically the same except for the last sample, GO-240, which can be explained by an increment in the number of grain boundaries attributed to the smaller size.
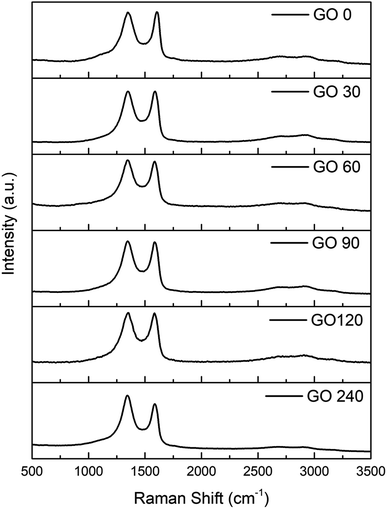 |
| | Fig. 6 Raman spectra of GO at 0, 30, 60, 90, 120 and 240 minutes. | |
4.4 Optical properties and optical band gap
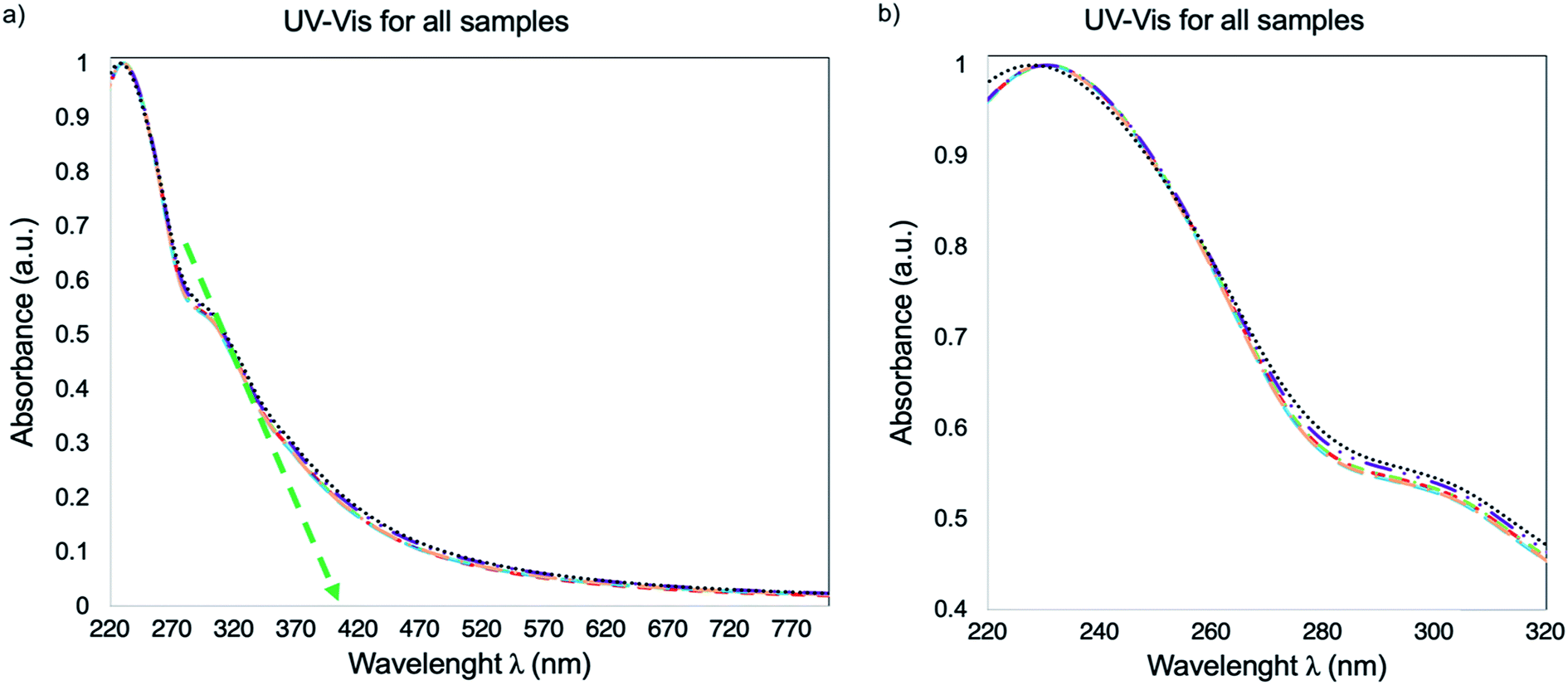
The UV-Vis spectra for the DI H2O dispersions of GO-0, GO-30, GO-60, GO-90, GO-120 and GO-240 is presented in Fig. 7. As one can see, the spectrum is the same for all the samples, with the typical5 transitions π → π* and n → π* at 232 and 305 nm indicating the absence of a chemical reduction process, despite the fact that in previous reports18,24,26 the size reduction is associated to a chemical reduction process. The optical band gap (Eoptg) stablished from the spectra in Fig. 7 (green dotted arrow) is calculated with an onset of 412 nm, which corresponds to 3 eV.
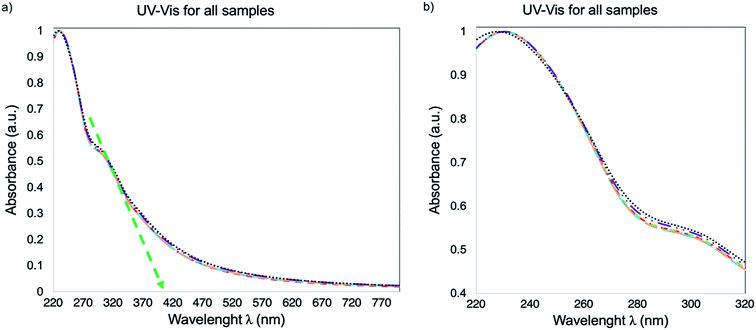 |
| | Fig. 7 UV-Vis spectra of GO-0, GO-30, GO-60, GO-90, GO-120 and GO-240. (a) Spectra from 220 to 800 nm, the green dotted arrow points out a 412 nm onset. (b) The spectra are a zoom between 220 nm to 320 nm for a better appreciation of the overlap between each sample. | |
In order to analyse the indirect Eoptg values with a more precise approach, Tauc plots were performed using the UV-Vis data which are shown in Fig. 5. The Tauc's relation8,40,41 is written below:
| |
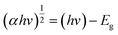 | (1) |
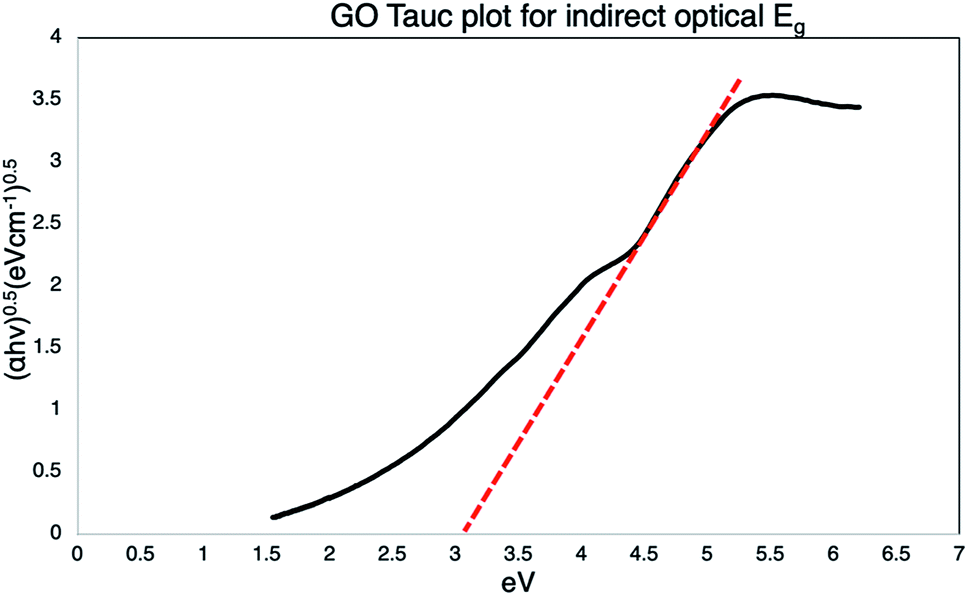
The Tauc's plot is constructed to determine the Eoptg value, extrapolating a tangent line (red dotted) to achieve intersection with the x axis (Eg in eV), as shown in Fig. 8, obtaining a value of 3 eV.
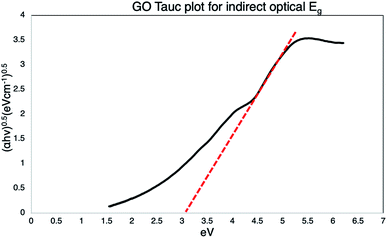 |
| | Fig. 8 Tauc plot of GO, the extrapolation of the dotted tangent line, intersect the x-axis defining the Eoptg to 3 eV. | |
4.5 Electronic features: HOMO–LUMO levels and electronic band gap
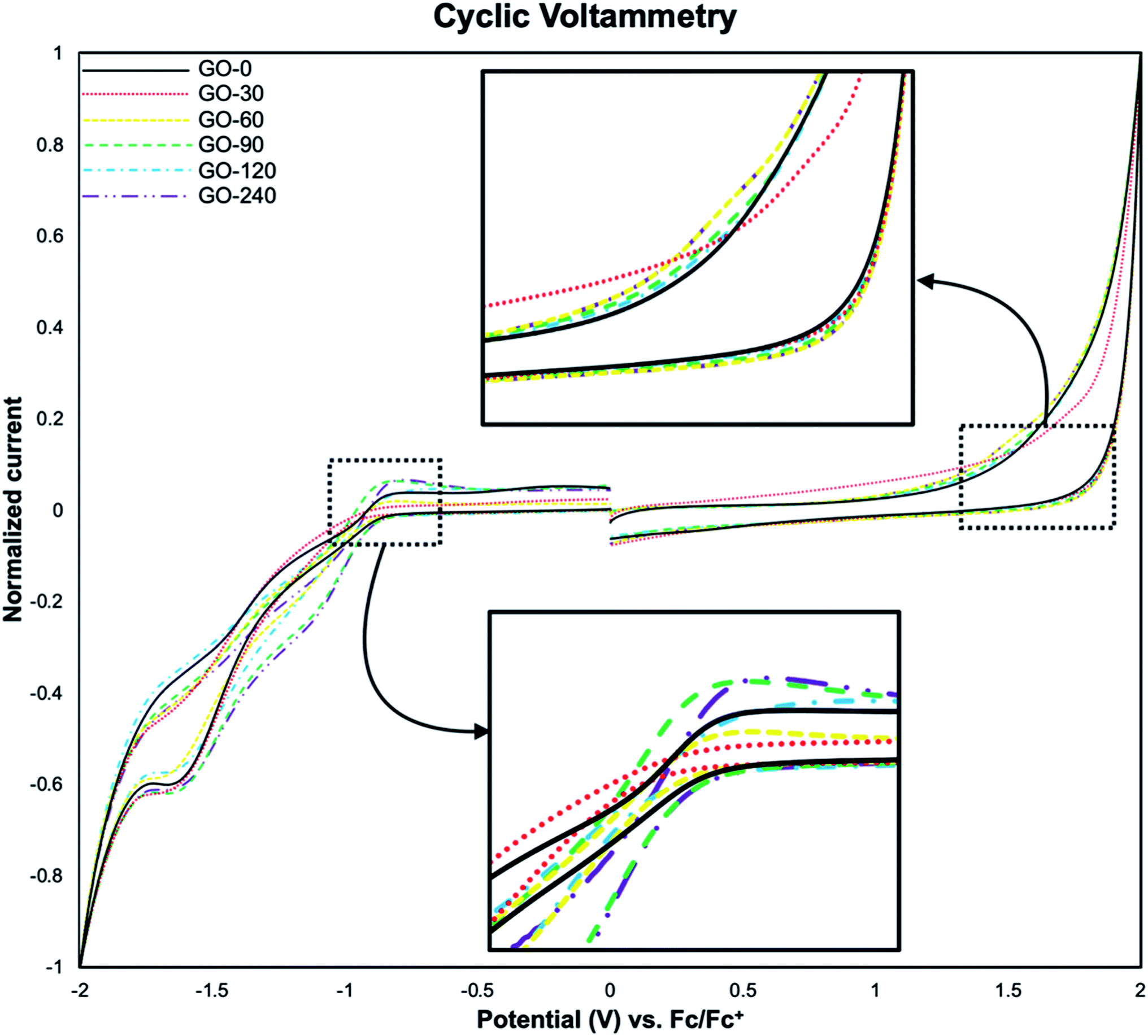
The analysis of energy levels corresponding to the HOMO and the LUMO was performed by cyclic voltammetry, using the oxidation onset (Eox) and reduction onset (Ered) values obtained from the voltamperograms. In Fig. 9 it is illustrated the voltamperogram for all the samples. It is worth mentioning that, the discontinuities at 0 V are inherent to the technique. First, a swept from 0 V to 2 V and back to 0 V is done, followed by a swept from 0 V to −2 V and back to 0 V. The current is in the order of μA, so when it is normalized the effect on the discontinuities become larger. However, the inflections on the plots (pointed by black arrows in Fig. 9), are the more relevant section. The HOMO and LUMO values were calculated with reference against ferrocene using the following equations:8,42| | |
EHOMO = −(Eox + 5.13) eV
| (2) |
| | |
ELUMO = −(Ered + 5.13) eV
| (3) |
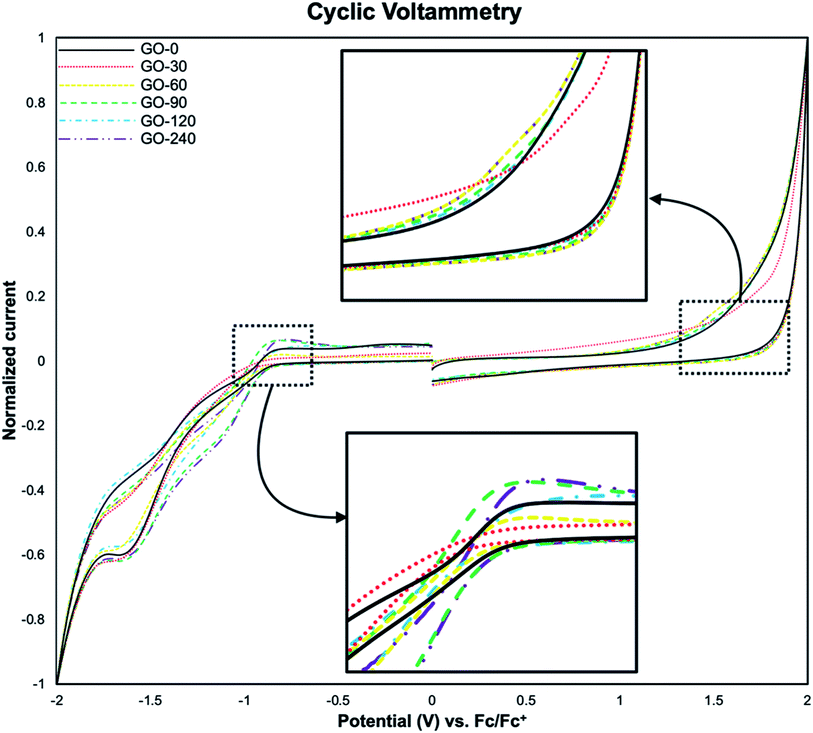 |
| | Fig. 9 Voltamperograms of GO-0, GO-30, GO-60, GO-90, GO-120 and GO-240. The insets show the inflection points used as oxidation and reduction onset. | |
The HOMO–LUMO values obtained from eqn (2) and (3), are −6.6 eV and −4.2 eV respectively, being 2.5 eV the electronic band gap (ECVg), in accordance to previously reported values.8,30,31,40,43 These values measured by CV show differences compared to the Eoptg obtained from UV-Vis data. This difference has been attributed to the intrinsic nature of the methods per se used for the measurements, as previously reported,42 Table 3 summarise the HOMO–LUMO estimated values.
Table 3 HOMO–LUMO values for all the samples
| Sample |
HOMO (eV) |
LUMO (eV) |
| GO-0 |
−6.69 |
−4.25 |
| GO-30 |
−6.68 |
−4.20 |
| GO-60 |
−6.67 |
−4.22 |
| GO-90 |
−6.67 |
−4.23 |
| GO-120 |
−6.66 |
−4.20 |
| GO-240 |
−6.67 |
−4.23 |
5. Conclusions
The graphene oxide lateral size decreases without compromising other relevant parameters, such as C/O ratio, crystallinity and band gap was achieved. This easy method of synthesis opens the opportunity to produce not only chemical derived graphene, but also to synthesize specific functionalised graphene oxide and reduced graphene oxide with a desired size for a specific biomedical, films, sensors or photovoltaic application. Moreover, the highly stable values for HOMO–LUMO levels, band gap, structural and C/O ratio are highly desirable, since facilitate further chemical modifications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Authors are thankful for the technical support and facilities at CIMAV Campus Monterrey and Chalmers University of Technology, Department of Chemistry and Chemical Engineering, as well as the Mexican National Research Council (CONACyT) for the scholarship (U. A. M.-R.). We would also like to thank the technical staff of CIMAV Campus Monterrey: Lilia Magdalena Bautista Carrillo for DLS and Zeta-potential measurements, Luis Gerardo Silva Vidaurri for XPS measurements and Nayeli Pineda for STEM micrographs.
References
- R. Garg, S. Elmas, T. Nann and M. R. Andersson, Adv. Energy Mater., 2017, 7, 1601393 CrossRef.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS PubMed.
- Q. Ke and J. Wang, Journal of Materiomics, 2016, 2, 37–54 CrossRef.
- M. A. Velasco-Soto, S. A. Pérez-García, J. Alvarez-Quintana, Y. Cao, L. Nyborg and L. Licea-Jiménez, Carbon, 2015, 93, 967–973 CrossRef CAS.
- S. Bai, X. Shen, G. Zhu, Z. Xu and Y. Liu, Carbon, 2011, 49, 4563–4570 CrossRef CAS.
- U. A. Méndez-Romero, M. A. Velasco-Soto, L. Licea-Jiménez, J. Álvarez-Quintana and S. A. Pérez-García, in Graphene Materials-Structure, Properties and Modifications, InTech, 2017 Search PubMed.
- U. A. Méndez-Romero, S. A. Pérez-García, X. Xu, E. Wang and L. Licea-Jiménez, Carbon, 2019, 146, 491–502 CrossRef.
- J. Luo, L. J. Cote, V. C. Tung, A. T. L. Tan, P. E. Goins, J. Wu and J. Huang, J. Am. Chem. Soc., 2010, 132, 17667–17669 CrossRef CAS PubMed.
- T. J. Nacken, C. E. Halbig, S. E. Wawra, C. Damm, S. Romeis, J. Walter, M. J. Tehrani, Y. Hu, Y. Ishii, S. Eigler and W. Peukert, Carbon, 2017, 125, 360–369 CrossRef CAS.
- R. Artur Filipe, N. Leon, L. Neus, P. M. Sourav, F. Bengt, B. Cyrill and K. Kostas, 2D Materials, 2018, 5, 035020 CrossRef.
- E. Bidram, A. Sulistio, A. Amini, Q. Fu, G. G. Qiao, A. Stewart and D. E. Dunstan, Carbon, 2016, 103, 363–371 CrossRef CAS.
- Z. Liu, S. M. Tabakman, Z. Chen and H. Dai, Nat. Protoc., 2009, 4, 1372 CrossRef CAS PubMed.
- J. Wang, Y. Cao, Q. Li, L. Liu and M. Dong, Chem.–Eur J., 2015, 21, 9632–9637 CrossRef CAS PubMed.
- J. Zhao, S. Pei, W. Ren, L. Gao and H.-M. Cheng, ACS Nano, 2010, 4, 5245–5252 CrossRef CAS PubMed.
- S. Pandit and M. De, ChemistrySelect, 2017, 2, 10004–10009 CrossRef CAS.
- S. G. Kim, S. H. Wang, C. M. Ok, S. Y. Jeong and H. S. Lee, Carbon, 2017, 125, 280–288 CrossRef CAS.
- G. Gonçalves, M. Vila, I. Bdikin, A. de Andrés, N. Emami, R. A. S. Ferreira, L. D. Carlos, J. Grácio and P. A. A. P. Marques, Sci. Rep., 2014, 4, 6735 CrossRef PubMed.
- C. E. Halbig, T. J. Nacken, J. Walter, C. Damm, S. Eigler and W. Peukert, Carbon, 2016, 96, 897–903 CrossRef CAS.
- C.-Y. Su, Y. Xu, W. Zhang, J. Zhao, X. Tang, C.-H. Tsai and L.-J. Li, Chem. Mater., 2009, 21, 5674–5680 CrossRef CAS.
- L. Zhang, J. Liang, Y. Huang, Y. Ma, Y. Wang and Y. Chen, Carbon, 2009, 47, 3365–3368 CrossRef CAS.
- Y. Noh, Y. Kim, S. Lee, E. J. Lim, J. G. Kim, S. M. Choi, M. H. Seo and W. B. Kim, Nanoscale, 2015, 7, 9438–9442 RSC.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. Sanchez Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- S. Zhang, Y. Li, J. Sun, J. Wang, C. Qin and L. Dai, RSC Adv., 2016, 6, 74053–74060 RSC.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394–3398 CrossRef CAS PubMed.
- J. Chen, Y. Li, L. Huang, N. Jia, C. Li and G. Shi, Adv. Mater., 2015, 27, 3654–3660 CrossRef CAS PubMed.
- G. Reina, N. D. Q. Chau, Y. Nishina and A. Bianco, Nanoscale, 2018, 10, 5965–5974 RSC.
- P. Sriram, R. Nutenki, V. R. Mandapati, M. Karuppiah and S. I. Kattimuttathu, Polym. Compos., 2017, 38, 852–862 CrossRef CAS.
- M. Huskić, S. Bolka, A. Vesel, M. Mozetič, A. Anžlovar, A. Vizintin and E. Žagar, Eur. Polym. J., 2018, 101, 211–217 CrossRef.
- F. Bonaccorso, N. Balis, M. M. Stylianakis, M. Savarese, C. Adamo, M. Gemmi, V. Pellegrini, E. Stratakis and E. Kymakis, Adv. Funct. Mater., 2015, 25, 3870–3880 CrossRef CAS.
- M. M. Stylianakis, M. Sygletou, K. Savva, G. Kakavelakis, E. Kymakis and E. Stratakis, Adv. Opt. Mater., 2014, 3, 658–666 CrossRef.
- W. Wei, W. Chen, L. Ding, S. Cui and L. Mi, Nano Res., 2017, 10, 3726–3742 CrossRef CAS.
- W. Wei, J. Wu, S. Cui, Y. Zhao, W. Chen and L. Mi, Nanoscale, 2019, 11, 6243–6253 RSC.
- W. Wei, W. Ye, J. Wang, C. Huang, J.-B. Xiong, H. Qiao, S. Cui, W. Chen, L. Mi and P. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 32269–32281 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- E. V. Anslyn and D. A. Dougherty, Modern physical organic chemistry, University science books, 2006 Search PubMed.
- J. M. Hollas, Modern spectroscopy, John Wiley & Sons, 2004 Search PubMed.
- C. K. Chua and M. Pumera, Small, 2015, 11, 1266–1272 CrossRef CAS PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- F. Zheng, W.-L. Xu, H.-D. Jin, X.-T. Hao and K. P. Ghiggino, RSC Adv., 2015, 5, 89515–89520 RSC.
- A. Mathkar, D. Tozier, P. Cox, P. Ong, C. Galande, K. Balakrishnan, A. Leela Mohana Reddy and P. M. Ajayan, J. Phys. Chem. Lett., 2012, 3, 986–991 CrossRef CAS PubMed.
- D. Gedefaw, M. Tessarolo, M. Bolognesi, M. Prosa, R. Kroon, W. Zhuang, P. Henriksson, K. Bini, E. Wang, M. Muccini, M. Seri and M. R. Andersson, Beilstein J. Org. Chem., 2016, 12, 1629–1637 CrossRef CAS PubMed.
- B. Zhang, Y.-L. Liu, Y. Chen, K.-G. Neoh, Y.-X. Li, C.-X. Zhu, E.-S. Tok and E.-T. Kang, Chem.–Eur J., 2011, 17, 10304–10311 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra04726k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac,
Qunping Fan
*ac,
Qunping Fan