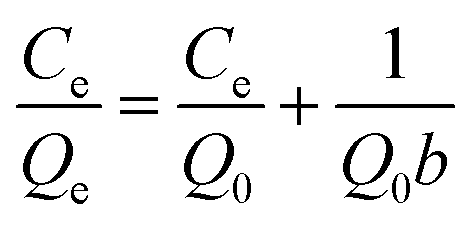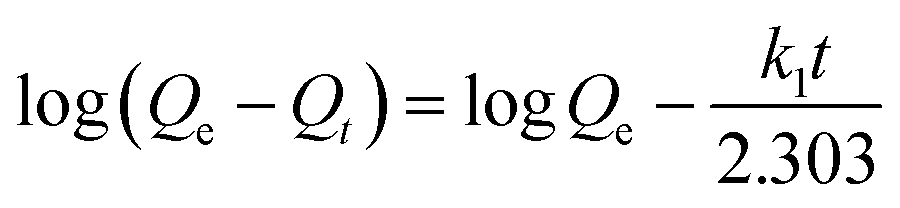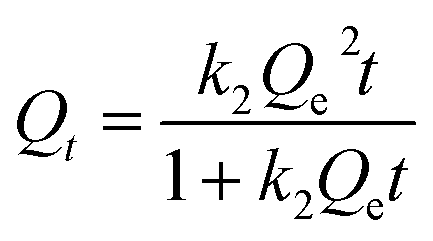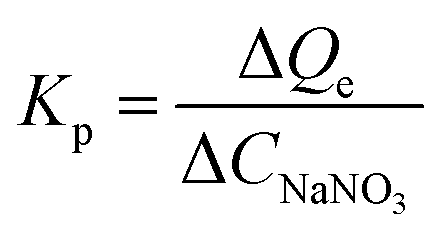DOI:
10.1039/D0RA02034F
(Paper)
RSC Adv., 2020,
10, 19943-19951
Highly efficient removal of Cu(II) by novel dendritic polyamine–pyridine-grafted chitosan beads from complicated salty and acidic wastewaters†
Received
3rd March 2020
, Accepted 30th April 2020
First published on 27th May 2020
Abstract
In this study, dendritic polyamine chitosan beads with and without 2-aminomethyl pyridine were facilely prepared and characterized. Compared to CN (without the pyridine function), more adsorption active sites, larger pores, higher nitrogen content, higher specific surface area, and higher strength could be obtained for CNP (with the pyridine function). CNP microspheres afforded a larger adsorption capacity than those obtained by CN for different pH values; further, the uptake amounts of Cu(II) were 0.84 and 1.12 mmol g−1 for CN and CNP beads, respectively, at pH 5. The CNP microspheres could scavenge Cu(II) from highly acidic and salty solutions: the maximum simulated uptake amount of 1.93 mmol g−1 at pH 5 could be achieved. Due to the strong bonding ability and weakly basic property of pyridine groups, the adsorption capacity of Cu(II) at pH 1 was 0.75 mmol g−1 in highly salty solutions, which was comparative to those obtained from the commercial pyridine chelating resin M4195 (QCu(II) = 0.78 mmol g−1 at pH 1). In addition, a distinct salt-promotion effect could be observed for CNP beads at both pH 5 and 1. Therefore, the prepared adsorbent CNP beads can have promising potential applications in the selective capturing of heavy metals in complex solutions with higher concentrations of H+ and inorganic salts, such as wastewaters from electroplating liquid and battery industries.
1. Introduction
Nowadays, the threat of heavy metal ions (HMIs) has become an urgent issue with regard to serious aqueous pollution.1–3 Residual HMIs can get enriched in living organisms, resulting in organ and tissue damage.4,5 Therefore, it is significant to completely remove HMIs from water. However, HMIs usually coexist with huge amounts of alkali or alkaline-earth metal salts and high-concentration H+ in the effluents from mining, electroplating, and battery manufacturing industries,6,7 resulting in ineffective removal by traditional technologies such as coagulation, ion exchange, and so on.8–10
Recently, biomass-derived adsorbents, such as lignin, cellulose, chitosan, biochar, and so on, have shown promising potential in environmental domains mainly because of their low cost and biodegradability.11,12 They usually exhibit large volume, micropores, mesopores, large specific surface area, and abundant functional groups; because of this, they have an obviously higher surface charge and they are more conducive toward the adsorption of various pollutants in water.13,14 Biomass-derived adsorbents can efficiently capture HMIs and organic pollutants from wastewaters via the chelating action, hydrogen bonding, and ion exchange.15,16 In particular, chitosan—a natural polysaccharide composed of flexible hydrophilic structures and abundant adsorption sites—has been utilized to decontaminate hazardous wastewaters.17,18 However, depending only on the original hydroxyl (–OH) and amino (–NH2) groups of raw chitosan molecules, the adsorption capacity and selectivity toward HMIs via complexation reactions are fairly insufficient. According to earlier studies, adsorption capacities partly rely on the density of the active sites as well as the structural deformation of the introduced functional moieties.19,20 Polyethylenimine (PEI), a dendritic polymer possessing abundant amine groups, is an excellent grafting group for functionalizing chitosan molecules.21,22 Modification with PEI can yield a superior structure with porous texture, leading to favorable adsorption of HMIs via the chelating reaction.23,24 However, PEI amines (primary, secondary, and tertiary amine groups) are susceptible to protonation and can be occupied by H+ in acidic systems, leading to a substantial decrease in the adsorption performance toward HMIs.25
To extend of the sorption pH values and enhance the adsorption ability of PEI-grafted chitosan, the introduction of pyridine groups is a worthwhile choice.26 Owing to the strong electron-withdrawing effect of the aromatic ring, the nitrogen atom of the pyridine group exhibits lower pKa (pKa NH+ → N < 1) and it can coordinate with HMIs in acidic solutions.27–29 Moreover, additional bond strength can be obtained via π antibonding from the populated d orbitals of the HMIs to the π* orbitals of the pyridine aromatic system.30 Further, it is envisioned that by the grafting of rigid pyridine groups, the applicability and stability of adsorbents toward HMIs can show distinct improvements.
In this study, dendritic PEI-grafted chitosan (CN) beads and pyridine–PEI-grafted chitosan (CNP) beads were facilely prepared and characterized. The adsorption behaviors of CNP and CN beads toward Cu(II) were explored: microspheric CNPs exhibited stronger bonding ability and wider sorption pH. The adsorption mechanism was also investigated. The adsorption capacity of CNP toward Cu(II) was comparative to that obtainable from commercial pyridine chelating resins (M4195). Furthermore, the bis(2-pyridylmethyl)amine reagent used for preparing M4195 is costly, while 2-chloromethylpyridine hydrochloride reagent (used in CNP production) is much cheaper.28 Therefore, this research is expected to open up newer avenues with regard to the facile preparation of high-performance green adsorbents.
2. Experimental section
2.1 Materials
All the organic reagents were purchased from Shanghai Meryer Chemical Technology Co., Ltd. (Shanghai, PR China). All the inorganic analytical reagents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, PR China). All these reagents were used as purchased without any further treatment.
2.2 Preparation of CN and CNP beads
The synthesis routes of CN and CNP beads used in this study is outlined in Scheme 1. Firstly, the pyridine function was performed by reacting PEI with 2-chloromethylpyridine hydrochloride, yielding an orange viscous liquid. Secondly, the above orange liquid and chitosan were preliminarily crosslinked by dropping epichlorohydrin. After preliminary crosslinking, the homogeneous solution was pumped into an alkaline medium to form microspheres. Finally, to improve the strength of the microspheres, secondary crosslinking was carried out in a sodium hydroxide aqueous solution by slowly dripping epoxy chloropropane. Similarly, milky white CN beads were prepared only by omitting the process of the pyridine function of PEI and straightforwardly mixing PEI with the chitosan mother solution.31
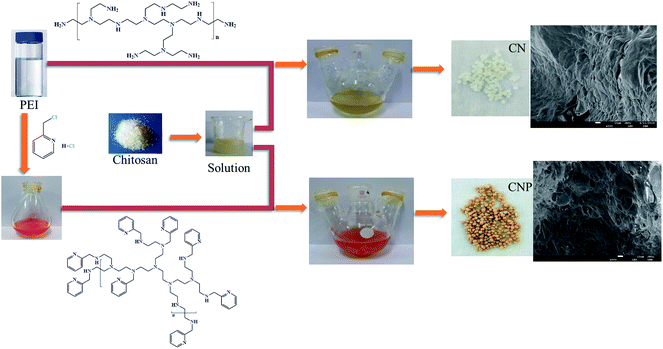 |
| | Scheme 1 Preparation of CNP and CN beads. | |
2.2.1 Pyridine function of PEI. An ethanol solution (30 mL) containing PEI (0.90 g), 2-chloromethylpyridine hydrochloride (1.5 g), and sodium carbonate (1.94 g) was heated at 85 °C for 2 h. After the reaction, the solvent was evaporated in a vacuum, and an orange viscous liquid was obtained for subsequent crosslinking.
2.2.2 Preliminarily crosslinking and formation of microspheres. Here, 0.90 g chitosan was dissolved in an acetic acid solution (0.60 mL acetic acid in 30.00 mL distilled water) at room temperature under continuous stirring at 780 rpm for 15 min. Under mechanical stirring, the orange viscous liquid (PEI modified with pyridine groups) was added into the chitosan mother solution in a three-necked flask at 60 °C. Then, epichlorohydrin (0.30 mL) was added dropwise as a crosslinking agent; preliminary crosslinking was accomplished after 2 h. The obtained homogeneous solution was pumped into a medium containing anhydrous sodium sulfate (13 g), sodium hydroxide (16 g) in 300 mL distilled water, and 2 mL ethanol; composite microspheres were formed and filtered out.
2.2.3 Secondary crosslinking. To improve the strength of the microspheres, secondary crosslinking was performed. The microspheres were put in a 100 mL aqueous solution containing sodium hydroxide (pH 13), and 0.50 mL epoxy chloropropane was added dropwise. The crosslinking reaction was heated at 60 °C for 2 h. The microspheres were separated, extracted with ethanol, and washed with distilled water to become neutral (pH 6–8). Finally, the prepared brownish yellow beads (CNP) were utilized for the efficient removal of HMIs from acidic and salty solutions.
2.3 Characterizations of CN and CNP beads
Elemental analysis (EA, Vario MICRO Analyzer, Germany) and Fourier-transform infrared (FT-IR) spectroscopy (NEXUS 870, America) were used to analyze the chelating functional groups of the beads. The specific surface area and porous texture of the adsorbents were determined from the N2 isotherms data at 77 K using the ASAP 2020 adsorption analyzer (Micromeritics Instrument Co., USA). The surface chemical states of the beads before and after the interaction with metal ions were further analyzed by X-ray photoelectron spectrometry (XPS) (ESCALAB-2, Great Britain) and by using the XPSPEAK41 software. The zeta potential measurements were performed by using a Zeta Plus 4 analyzer (Brookhaven Co., USA). The thermogravimetric analysis (TGA) characterization was conducted to investigate the thermal properties of the CN and CNP beads using a thermal analyzer (PerkinElmer, USA).
2.4 Adsorption features of CN and CNP beads
2.4.1 Static adsorption experiments. To evaluate the effect of solution pH on the adsorption performance, static experiments were carried out in the batch mode and in triplicate by mixing 0.025 g beads with 50 mL solution containing various defined initial concentrations of Cu(II) at pH values of 1, 2, 3, 4, and 5. Further, the adsorption isotherm experiments were conducted at the best pH value with the initial concentrations of Cu(II) within 1–5 mmol L−1. In different salty systems, 0.025 g beads were added into 50 mL solutions containing corresponding metal ions in 100 mL conical flasks, and a series of inorganic salts with various concentration ratios were introduced. Then, the conical flasks were shaken in an incubator shaker at 298 K and 120 rpm for 24 h, and the residual concentrations of the metal ions were analyzed.Furthermore, Langmuir and Freundlich models were applied to describe the isotherm data in single systems with the following forms.
| |
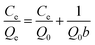 | (1) |
where
b (L mmol
−1) is the affinity parameter or Langmuir adsorption constant, which reflects the free energy of adsorption;
Kf and
n are constant isotherm parameters; and
Q0 (mmol g
−1) is the capacity parameter.
2.4.2 Kinetic adsorption experiments. The kinetic adsorption experiments were conducted in a single Cu(II) system. The concentration of each HMI was 1 mmol L−1 in all the systems. Briefly, 500 mL solution with 0.25 g CNP was agitated at 120 rpm and 298 K in an incubator shaker, during which they were sampled at predefined time intervals. The pseudo-first-order rate equation (eqn (3)) and pseudo-second-order rate equation (eqn (4)) expressed in the following nonlinear forms were both used to describe the kinetic adsorption process.| |
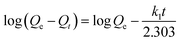 | (3) |
| |
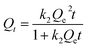 | (4) |
where Qt is the adsorption capacity (mmol g−1) at time t; k1 and k2 are the rate constants of the pseudo-first-order (min−1) and pseudo-second-order (g mmol−1 min−1) expressions, respectively.
3. Results and discussion
3.1 Characterizations of CN and CNP beads
As shown in Scheme 1, after grafting 2-aminomethyl pyridine groups, the color of CNP microspheres turned to brownish yellow. As compared to milky white CN beads, CNP microspheres shrank and became smaller in size. After adding the pyridine function, the hydrophobic–hydrophobic interactions and steric effects increased inside the microspheres due to the introduction of aromatic rings; expectedly, the moisture content decreased and pore volume increased significantly. The moisture content of the microspheres decreased from 95.10% for CN to 91.21% for CNP beads, and the mechanical strength of the microspheres increased after the addition of the pyridine function. Further, the surfaces of CNP beads were rougher and more porous relative to those of the CN beads, as confirmed by the SEM images (Scheme 1); this facilitated the access of adsorbates. Accordingly, the specific surface area, mean pore size, and pore volume increased after modification with pyridine (Table 1 and Fig. S1†). For CNP beads, the mean pore size was 20.1124 nm, which was larger than that of the radius of the hydrated metal ion, allowing the efficient diffusion of hydrated metal ions into the internal space of the microspheres. Due to the same skeleton of microspheres, similar thermal stability was exhibited by both CN and CNP beads; no distinct decomposition occurred until ∼310 °C (Fig. S2†).
Table 1 Structural parameters of CN and CNP beads
| Adsorbents |
BET specific area (m2 g−1) |
Pore volume (cm3 g−1) |
BJH average adsorption aperture (nm) |
| CN |
1.2116 |
0.001121 |
— |
| CNP |
2.043 |
0.011143 |
20.1124 |
| Adsorbents |
C (%) |
H (%) |
N (%) |
| CN |
42.265 |
7.759 |
8.905 |
| CNP |
56.568 |
6.108 |
12.774 |
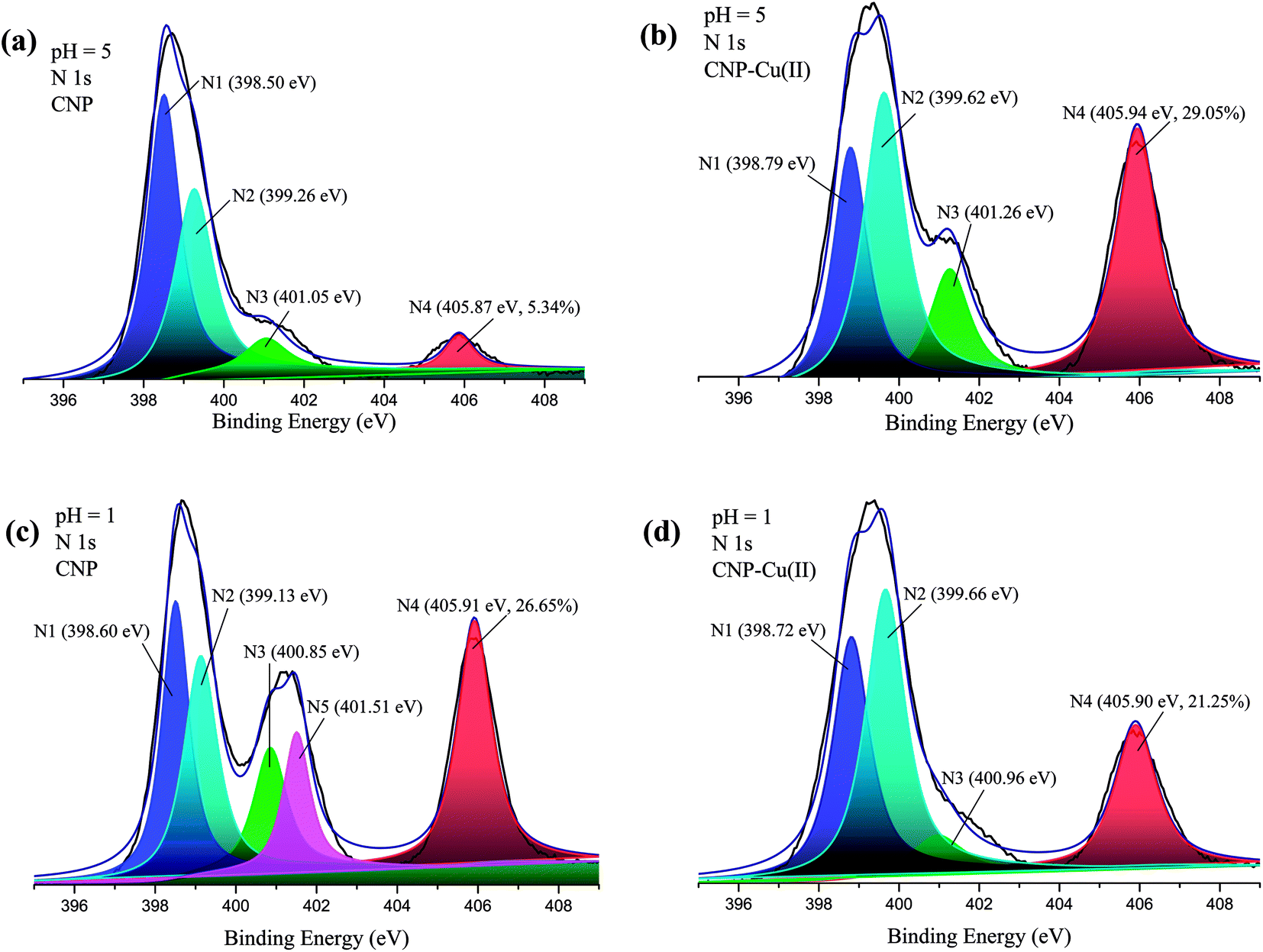
After pyridine functionalization, a new absorption peak associated with the stretching vibration of pyridine at ∼1548 cm−1 was observed in the IR spectra (Fig. S3†);28 further, the nitrogen content increased significantly from 8.91% for CN to 12.70% for CNP (Table 1), indicating the successful introduction of the pyridine functional group. As amine groups were susceptible to protonation, the surfaces of the microspheres were supposed to be positive.32 Correspondingly, the zeta potentials of both CN and CNP beads were largely positive at pH values between 2.0 and 10.0. The zeta potential measurements showed that the zero electric points of both CN and CNP beads were located at ∼10 (Fig. S4†). The surface chemical compositions of the CN and CNP beads were explored by XPS analysis. The binding energy peak of O1s at 531.90 eV was attributed to the oxygen atoms of –OH. The N1s core-level XPS spectra could be deconvoluted into 4 peaks at binding energies of 398.50, 399.13, 401.05, and 405.87 eV, which could be attributed to the nitrogen atoms in neutral amine (N1: C–N or –NH), pyridine (N2: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), protonated amine (N3: –NH+), and nitrate ions (N4: NO3−), respectively (Fig. S5†).28
N), protonated amine (N3: –NH+), and nitrate ions (N4: NO3−), respectively (Fig. S5†).28
3.2 Effect of pH on adsorption
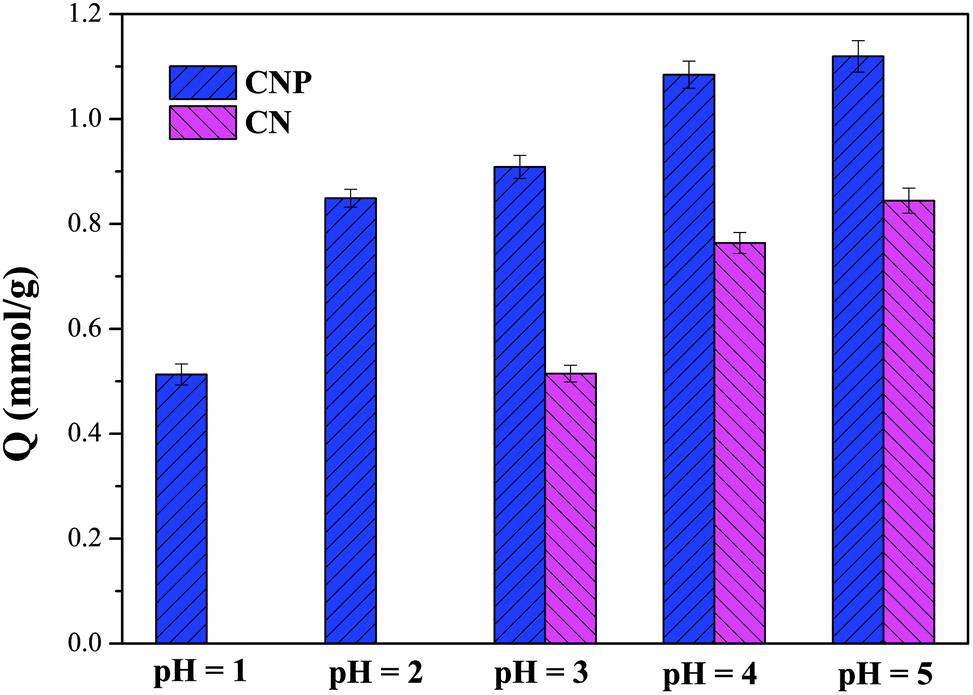
Solution pH is one of the most important factors for the adsorption of HMIs, because it influences the ionization state of the functional groups.33,34 The pH effect on the adsorption behaviors of CN and CNP beads is shown in Fig. 1. Due to the high bonding affinity of Cu(II) (intermediate partial hard acids) with nitrogen atoms (intermediate partial hard bases), both CN and CNP beads exhibited a large adsorption capacity to Cu(II). At pH 5, the uptake amounts of Cu(II) were 0.84 and 1.12 mmol g−1 for CN and CNP beads, respectively. With an increase in H+ concentration, the adsorption capacity decreased. Owing to the protonation of nitrogen atoms, the amine groups on the CN beads were unavailable to trap Cu(II) at pH 2.32 However, the removal capacity of CNP beads toward Cu(II) was 0.51 mmol g−1 at pH 1, which was comparable to M4195 (QCu(II) = 0.78 mmol g−1 at pH 1).35 It can be inferred that the introduction of pyridine groups can elevate the uptake amount and improve acid resistance. Therefore, in the subsequent investigation, the discussion mainly focuses on CNP beads, and the adsorption of Cu(II) in both weakly and highly acidic solutions (pH 5 and 1, respectively) is discussed.
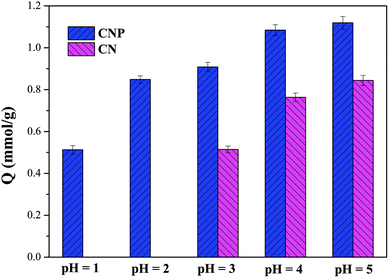 |
| | Fig. 1 Effect of pH on the adsorption of CN and CNP beads. | |
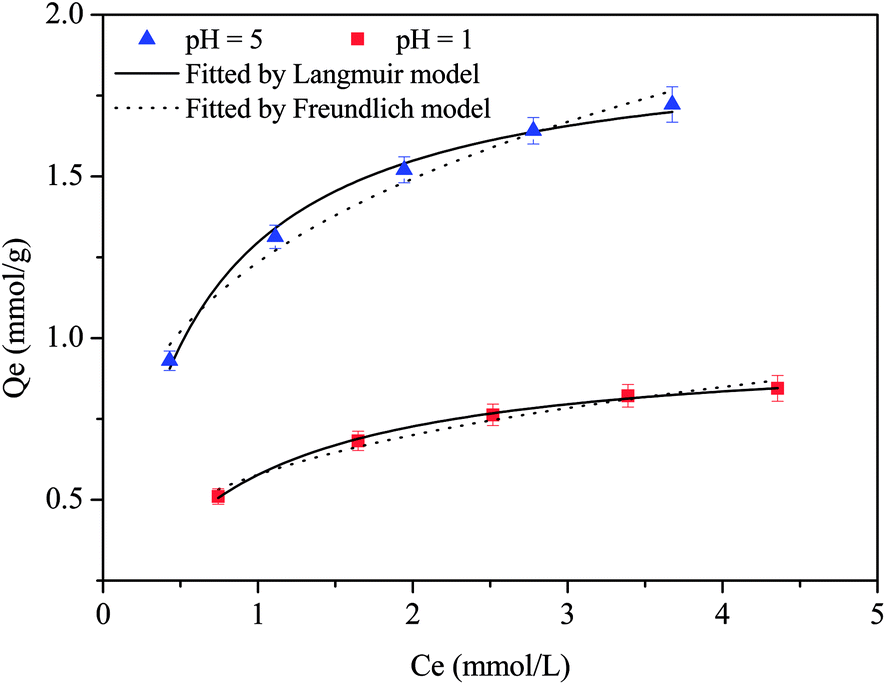
3.3 Adsorption isotherms
The adsorption isotherms of Cu(II) at pH 5 and 1 were described by means of the Langmuir and Freundlich models (Fig. 2 and Table S1†); the data could be better fitted by the Langmuir model, which yielded a higher correlation coefficient. This indicated the presence of a monolayer adsorption process.36 The maximum simulated adsorption capacities at pH 5 and 1 were 1.93 and 0.98 mmol g−1, respectively, which exceeded those of most of the reported polyamine adsorbents (Table S2†). Commonly, the adsorption isotherms of amine-functionalized adsorbents toward Cu(II) were investigated in weakly acidic solutions. At pH 4 or 5, the maximum uptake capacities of D001-PEI,37 amine-functionalized SBA-15,38 silica-supported 2-aminomethyl pyridine adsorbent,30 aminothiazole-functionalized adsorbent,39 dual-primary-amine chelating resin,40 tannic-acid-grafted PEI encapsulated in alginate beads,41 nanofibrillated cellulose/PEI aerogels,23 polyethylene polyamine@tannic acid-encapsulated MgAl-layered double hydroxide adsorbents,42 and polystyrene-supported 2-aminomethylpyridine chelating resin43 were 1.55 mmol g−1, 0.54 mmol g−1, 0.78 mmol g−1, 1.35 mmol g−1, 1.74 mmol g−1, 1.36 mmol g−1, 2.74 mmol g−1, 0.92 mmol g−1 and 1.53 mmol g−1, respectively. The scavenging of Cu(II) was mainly accomplished by the formation of coordination bonds between the nitrogen atoms and Cu(II) ions.38–40 Although some pyridine-functionalized adsorbents exhibited removing ability toward Cu(II) at pH 1 or 2, the uptake capacity was less than 0.3 mmol g−1 due to the inadequate number of pyridine groups.30,43
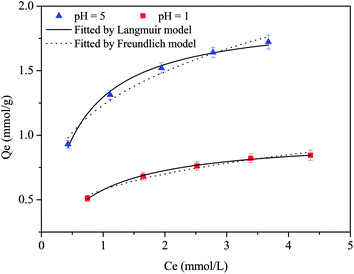 |
| | Fig. 2 Adsorption isotherms of CNP at pH 5 and 1. | |
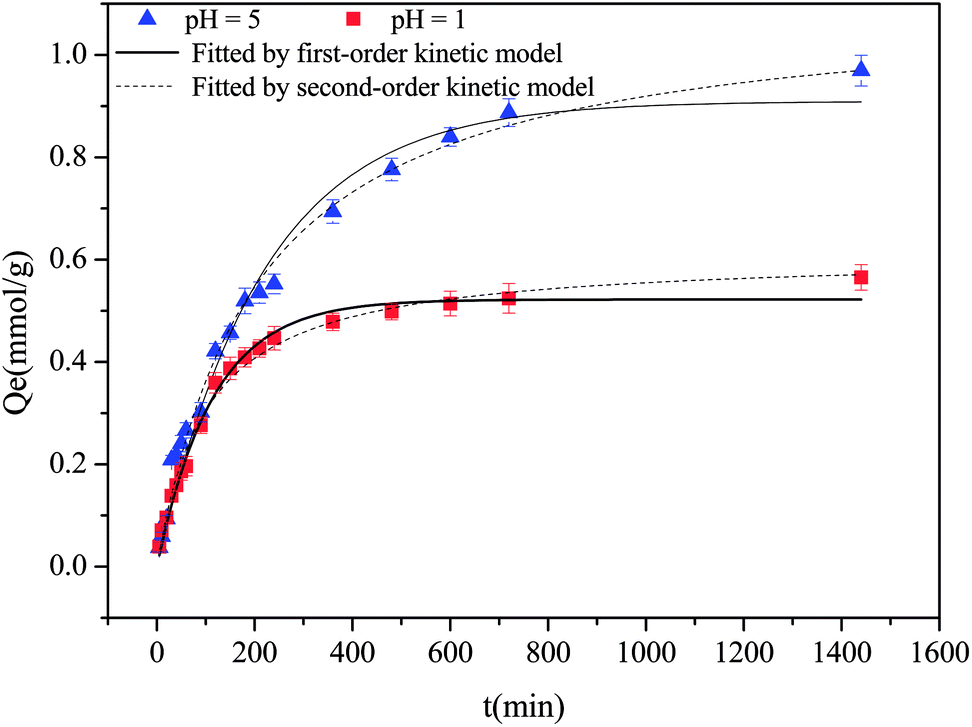
3.4 Adsorption kinetics
The adsorption kinetics of Cu(II) at pH 5 and 1 are shown in Fig. 3. Evidently, over 80% adsorption occurred within 5 h and equilibrium was attained after 24 h. The kinetic data were simulated by pseudo-first- and pseudo-second-order models with the fitting data, as listed in Table S3;† the pseudo-second-order models could more effectively describe the kinetic adsorption process, indicating the presence of a rate-controlling chemisorption process.44
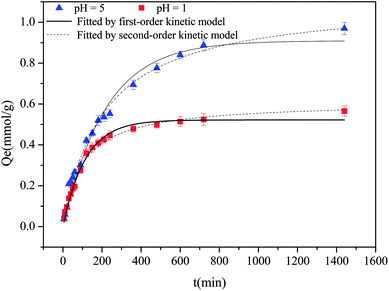 |
| | Fig. 3 Kinetic adsorption behaviors of Cu(II) at pH 5 and 1. | |
3.5 Effect of inorganic salts on adsorption
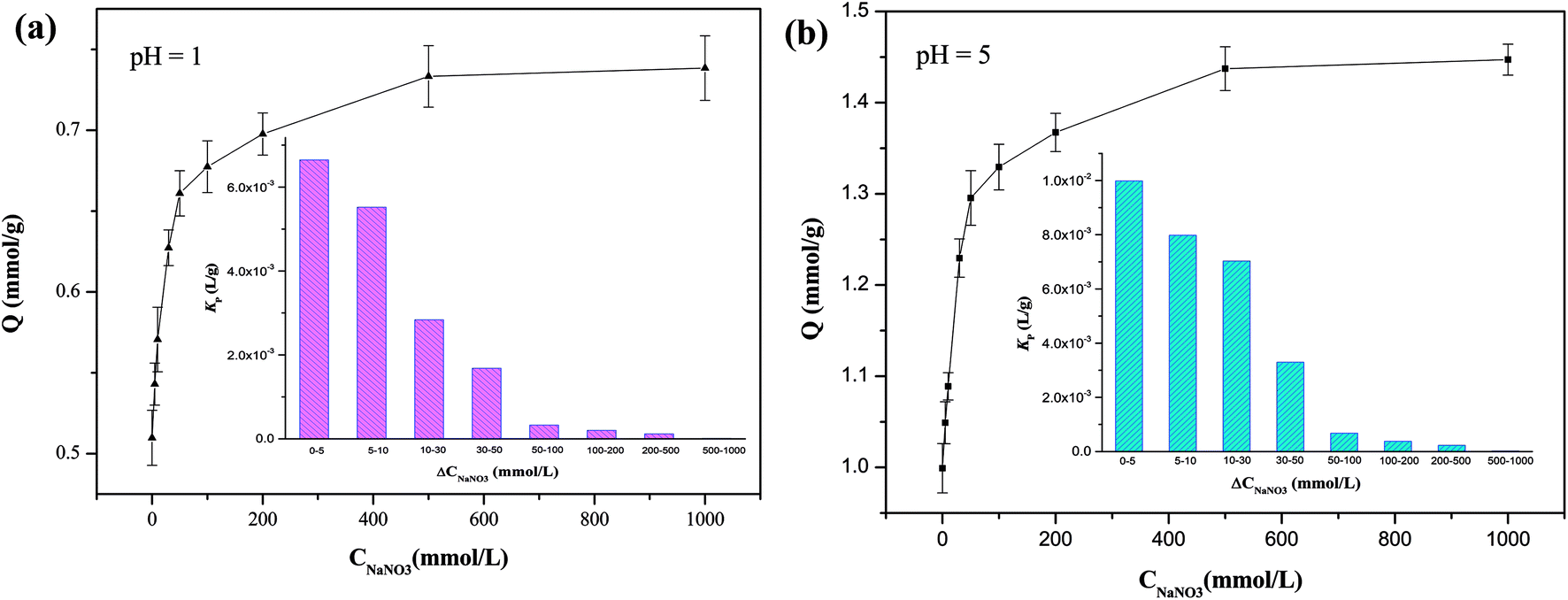
Huge amounts of inorganic salts are present in wastewaters from mining, electroplating, and novel materials manufacturing industries.32,45,46 The copresence of salts could either consume the active sites or inhibit the adsorption of target HMIs by changing the form of HMIs.47 For instance, Cu(II) can react with Cl− to form CuCl2 molecules as well as CuCl3− and CuCl42− anions for higher concentrations of Cl−. In a salty solution (CNaNO3 = 1000 mmol L−1), the adsorption capacity of Cu(II) was 0.75 mmol g−1 in a highly acidic solution (pH 1) and almost approached the saturated adsorption capacity, demonstrating that most of the active adsorption sites could be accessible by Cu(II) ions, yielding abundant NaNO3 (Fig. 4). In addition, the uptake amount of Cu(II) reached 1.45 mmol g−1 at pH 5. The enhancement ability of NaNO3 can be described by the promotion index Kp (L g−1) (eqn (5)) as follows:40| |
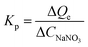 | (5) |
where ΔQe (mmol g−1) is the increment in adsorption capacity with an increase in the initial concentration of NaNO3, ΔCNaNO3 (mmol L−1). In an acidic solution at pH 1, in the low-salt region (0–10 mmol L−1), Kp was 6.0 × 10−3 L g−1, indicating a strong salt-promotion effect. However, this effect was obviously weakened in the high-salt region (500–1000 mmol L−1), as evidenced by the Kp value of 1.0 × 10−5 L g−1, implying a marginal impact of the salt-promotion effect (Fig. 4). A similar tendency for Kp was observed at pH 5.
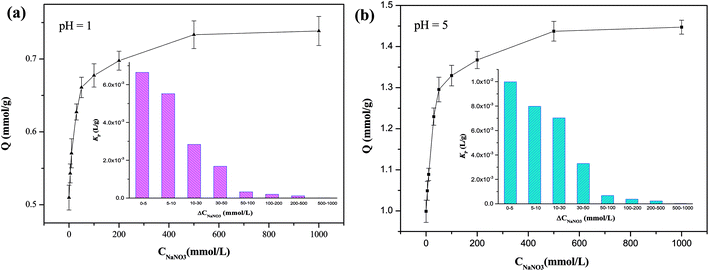 |
| | Fig. 4 Adsorption amounts and Kp values of Cu(II) for different concentrations of NaNO3. | |
The effect of other Na+ salts on adsorption was further explored, and all the salts exhibited a promotion effect with adsorption amounts in the range from 0.71 to 0.76 mmol g−1 at pH 1 and that from 1.35 to 1.52 mmol g−1 at pH 5 due to electrostatic screening exerted by the anions (Fig. S6†).39 Chloride ions exerted a stronger salt-promotion effect than that by NO3−, which could be attributed to the complexation between Cl− ion and Cu(II) to form negatively charged CuCl3− and CuCl42− and subsequently an ion-exchange effect with protonated N atoms in addition to the chelating effect.48,49 Therefore, more adsorption sites were available and the uptake amount was enhanced (Q = 0.78 mmol g−1 for pH 1; Q = 1.64 mmol g−1 for pH 5; CNaNO3 = 500 mmol L−1). However, the adsorption capacity decreased in solutions with higher concentrations of Cl− (>500 mmol L−1) owing to the serious competition effect from Cl− anions. The promotion effect in SO42−-bearing systems is slightly lower than those in NO3−- and Cl−-bearing systems due to the large steric hindrance from SO42−.50,51
3.6 Regeneration and reusability
The regeneration and reusability of the adsorbent are crucially important in wastewater treatment processes. The exhausted CNP with Cu(II) was subject to regeneration with 3 M HCl (∼10% HCl) in static desorption.35 The obtained result showed that preloaded Cu(II) could be effectively extracted with desorption efficiency higher than 98.5%. Then, the desorbed microspheres were thoroughly rinsed with distilled water until the effluent was neutral. Five continuous cycles of adsorption and regeneration were performed to evaluate the reusability, and the uptake amount of Cu(II) decreased by only 5% (Fig. S7†), indicating that CNP beads could be effectively reused without any significant capacity loss.
3.7 Adsorption mechanism
The inner adsorption mechanism of CNP beads was investigated by examining the FT-IR and XPS data. The FT-IR spectra of the CNP beads before and after loading with HMIs at pH 5 are shown in Fig. S8.† The stretching vibrations of pyridine at 1548 cm−1 and those of C–N at 1463 cm−1 were significantly weakened, and they almost disappeared after adsorption, indicating that pyridine and aliphatic amines were involved in the chelating interaction with HMIs. In addition, the appearance of new peaks at 1608 and 1384 cm−1 could be attributed to the stretching vibrations of CN and NO3−, respectively.28,30,43 NO3− anions were simultaneously adsorbed along with HMIs for charge balance. The stretching vibration of C–O at 1070 cm−1 remained unchanged, implying the absence of oxygen atoms in the adsorption of HMIs.
To further distinguish the kinds of functional groups and confirm the coordinating interaction between the nitrogen atoms and HMIs, the XPS characterizations of CNP before and after capturing HMIs were carried out (Fig. 5). The N1s core-level XPS spectra of CNP (pH 5) were deconvoluted into 4 peaks, namely, 398.50, 399.26, 401.05, and 405.87 eV, which could be ascribed to the nitrogen atoms in neutral amine (N1: C–N or –NH), pyridine (N2: CN), protonated amine (N3: –NH+), and nitrate ions (N4: NO3−), respectively.28,43,52 After the adsorption of Cu(II) at pH 5, it was expected that the 2 peaks involving neutral nitrogen (N1) and pyridine nitrogen (N2) showed obvious shifts to 398.79 and 399.62 eV, confirming the coordination effect between the lone pair of electrons in the nitrogen atoms and empty orbitals of Cu(II). The molar ratio of NO3− significantly increased from 5.34% in CNP to 29.05% in CNP–Cu(II), confirming the synchronous adsorption of NO3− counterions along with Cu(II) for maintaining electric neutrality. In addition, the binding energy of O1s remained almost unchanged before and after adsorbing Cu(II) ions (Fig. S9†), indicating that oxygen atoms were hardly involved in the formation of the coordination bond.
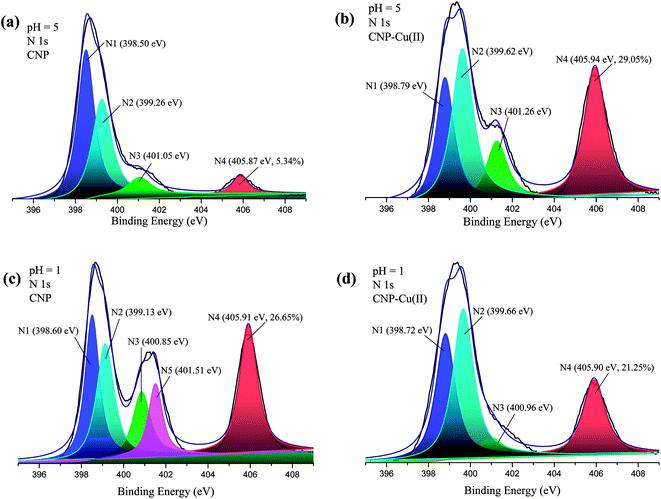 |
| | Fig. 5 XPS spectra (N1s) of CNP beads before and after adsorbing Cu(II) at pH 5 and 1. | |
The core-level spectra of N1s of CNP and CNP–Cu at pH 1 were also investigated, and the spectra could be fitted by 5 peaks (Fig. 5). The peak at 401.51 eV corresponded to nitrogen atoms in protonated pyridine (N5: CN+).28,43,52 The peak of N2 was obviously shifted to higher energies due to the formation of a shared bond with Cu(II). The characteristic peak associated with protonated pyridine (N5) almost disappeared after adsorption, indicating that H+ ions of protonated pyridine were dislodged by Cu(II). Meanwhile, the peaks corresponding to N4 and N3 did not exhibit any distinct shifts before and after adsorption. Therefore, it could be inferred that only nitrogen atoms of the pyridine groups exhibited chelating effects with Cu(II) at pH 1, which was further verified by the FT-IR spectra according to which the stretching vibrations of pyridine at 1523 cm−1 exhibited distinctly reduced intensity after adsorption (Fig. S8†).
According to the literature, the pKa values of bis(2-pyridylmethyl) amine in sulfate solutions were 1.5, 2.7, and 3.5; therefore, amine and pyridyl nitrogen atoms could be protonated at pH 1.27,28,35 In this case, the adsorption of HMIs in a low-pH solution would have to cater to competition from H+. As expected, the adsorption of Cu(II) in a solution at pH 1 showed that H+ ions were firstly adsorbed rapidly by the nitrogen atoms, resulting in a slight increase in pH. Then, the adsorbed H+ was displaced by Cu(II) and dislodged into the bulk solution. Therefore, a decline in pH would be detected during the latter half (Fig. S10†).53
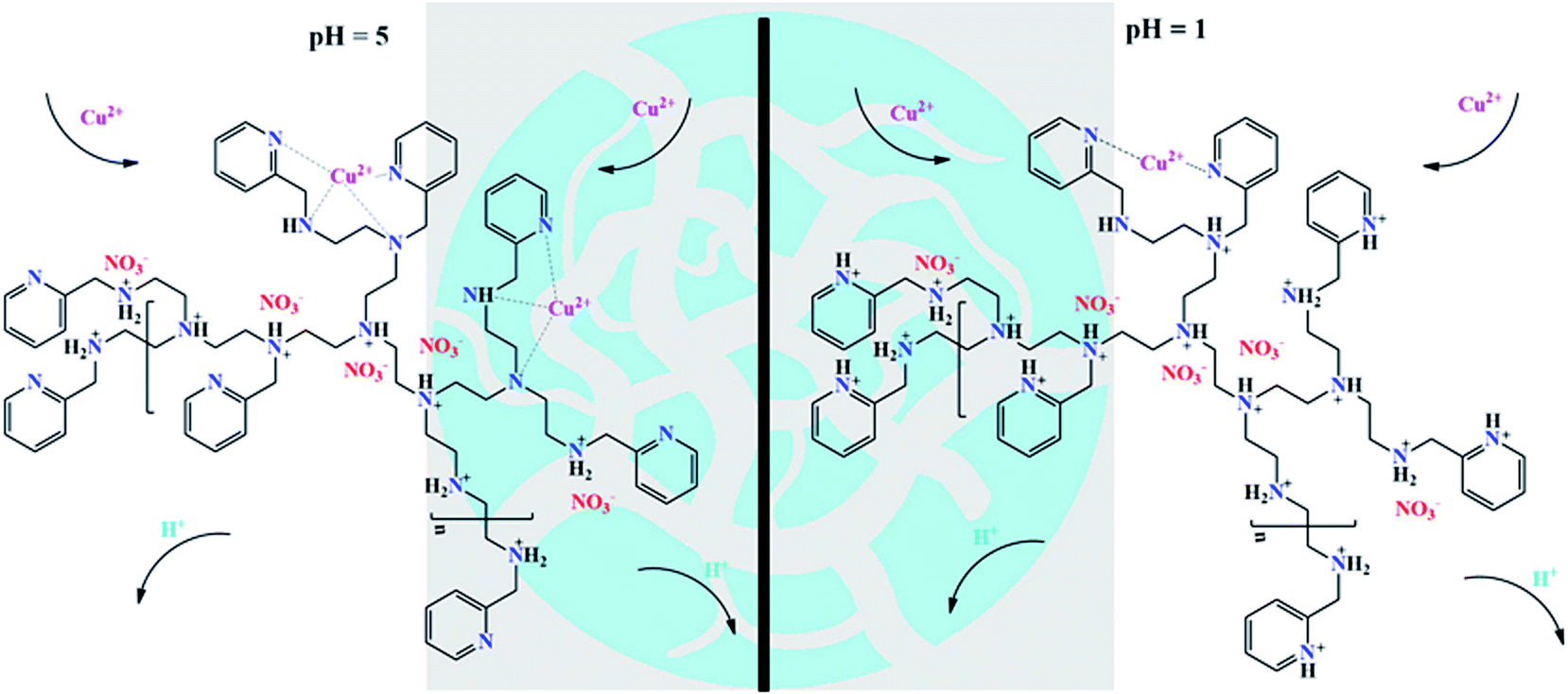
As shown in Fig. 6, the adsorbent CNP beads were capable of capturing HMIs through a coordination effect between the aliphatic amine, pyridine, and HMIs, while only nitrogen atoms of pyridine participated in the chelating effect at both pH 1 and 5.28,30,54 This result was consistent with the reported studies according to which the polyamine adsorbent lost its adsorption ability below pH 3,32 but pyridine-functionalized beads could selectively recycle HMIs at pH 1. Due to the weakly basic characteristic of the aliphatic amines and pyridine rings, competitive adsorption between the Cu(II) ions and H+ could be observed even at low pH. Therefore, the nitrogen atoms of the active adsorption sites would be partly protonated, leading to lower adsorption capacities. With an increase in the pH values and decrease in H+ concentration, fewer active adsorption sites could be seen in their protonated forms, thereby exhibiting higher affinity toward Cu(II) ions.28,30,43,52 In salty systems, more active sites could be released owing to the electrostatic shielding effect exerted by the abundant anions, thereby leading to a significant promotion in the adsorption capacity.
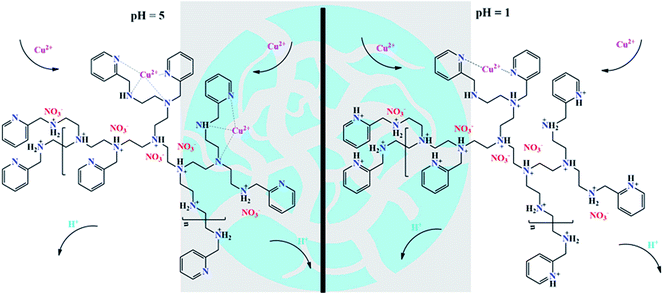 |
| | Fig. 6 Possible mechanism of the adsorption process. | |
4. Conclusions
In summary, dendritic polyamine chitosan beads with (CNP) and without (CN) the grafting of pyridine groups were prepared for the subsequent removal of Cu(II) ions. CNP beads showed better properties in terms of mechanical strength and adsorption behaviors relative to those of CN beads. At pH 5, the uptake amounts of Cu(II) were 0.84 and 1.12 mmol g−1 for CN and CNP beads, respectively. In acidic solutions (pH 1), the removal capacity toward Cu(II) of CNP beads still remained 0.51 mmol g−1, while the adsorption of CN beads was marginal and could be neglected. Both amine and pyridine groups contributed toward the adsorption of HMIs at pH 5, with maximum simulated adsorption capacities of QCu(II) = 1.93 mmol g−1 for CNP beads. However, only pyridine groups were responsible for adsorption at pH 1. The adsorption mechanism was verified by means of the FT-IR and XPS spectra. In salty solutions, due to the electrostatic shielding effect, the introduction of inorganic salts exerted a significant positive impact on the adsorption characteristics, and the uptake amount of Cu(II) was enhanced by 47.1% at pH 1 and 45.5% at pH 5. The exhausted CNP beads could be effectively regenerated by 3 M HCl with the desorption efficiency higher than 98.5%, and the uptake amount of Cu(II) was decreased only by 5% after 5 cycles of adsorption and regeneration. In brief, we provided the preparation of low-cost, efficient, and environmentally friendly adsorbents with potential applications in the selective removal of HMIs from acidic and salty solutions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Natural Science Foundation of Hainan Province (No. ZDYF2019140, 219MS041), National Natural Science Foundation of China (No. 21961009, 51708281).
Notes and references
- J. Chen, Y. S. Liu, J. N. Zhang, Y. Q. Yang, L. X. Hu, Y. Y. Yang, J. L. Zhao, F. R. Chen and G. G. Ying, Bioresour. Technol., 2017, 238, 70–77 CrossRef CAS PubMed.
- S. Bolisetty, M. Peydayesh and R. Mezzenga, Chem. Soc. Rev., 2019, 48, 463–487 RSC.
- H. Wang, Z. Wang, R. Yue, F. Gao, R. Ren, J. Wei, X. Wang and Z. Kong, Chem. Eng. J., 2020, 383, 123107 CrossRef.
- Y. Niu, X. Jiang, K. Wang, J. Xia, W. Jiao, Y. Niu and H. Yu, Sci. Total Environ., 2020, 700, 134509 CrossRef CAS PubMed.
- Q. Lu, S. Wang, X. Bai, F. Liu, C. Li, Y. Deng and S. Tian, Hum. Ecol. Risk Assess., 2020 DOI:10.1080/10807039.2019.1710811.
- X. Qiu, H. Hu, J. Yang, C. Wang and Z. Cheng, Hydrometallurgy, 2018, 180, 121–131 CrossRef CAS.
- Z. M. M. A. Tawila, S. Ismail, S. S. A. Amr and E. K. A. Elkhair, RSC Adv., 2019, 9, 27825–27834 RSC.
- L. K. Lautz, G. D. Hoke, Z. Lu, D. I. Siegel, K. Christian, J. D. Kessler and N. G. Teale, Environ. Sci. Technol., 2014, 48, 9061–9069 CrossRef CAS PubMed.
- G. Zeng, Y. Liu, L. Tang, G. Yang, Y. Pang, Y. Zhang, Y. Zhou, Z. Li, M. Li, M. Lai, X. He and Y. He, Chem. Eng. J., 2015, 259, 153–160 CrossRef CAS.
- A. Bashir, L. A. Malik, S. Ahad, T. Manzoor, M. A. Bhat, G. N. Dar and A. H. Pandith, Environ. Chem. Lett., 2018, 17, 729–754 CrossRef.
- Y. Dong, D. Sang, C. He, X. Sheng and L. Lei, RSC Adv., 2019, 9, 29015–29022 RSC.
- B. Wang, Z. Bai, H. Jiang, P. Prinsen, R. Luque, S. Zhao and J. Xuan, J. Hazard. Mater., 2019, 364, 192–205 CrossRef CAS.
- H. N. Tran, H. C. Nguyen, S. H. Woo, T. V. Nguyen, S. Vigneswaran, A. Hosseini-Bandegharaei, J. Rinklebe, A. K. Sarmah, A. Ivanets, G. L. Dotto, T. T. Bui, R.-S. Juang and H.-P. Chao, Crit. Rev. Environ. Sci. Technol., 2019, 49, 2155–2219 CrossRef CAS.
- D. Mohan, A. Sarswat, Y. S. Ok and C. U. Pittman Jr, Bioresour. Technol., 2014, 160, 191–202 CrossRef CAS.
- M. Bilal, J. A. Shah, T. Ashfaq, S. M. H. Gardazi, A. A. Tahir, A. Pervez, H. Haroon and Q. Mahmood, J. Hazard. Mater., 2013, 263, 322–333 CrossRef CAS PubMed.
- A. L. Cazetta, O. Pezoti, K. C. Bedin, T. L. Silva, A. P. Junior, T. Asefa and V. C. Almeida, ACS Sustainable Chem. Eng., 2016, 4, 1058–1068 CrossRef CAS.
- D. Zhang, L. Wang, H. Zeng, B. Rhimi and C. Wang, Environ. Sci.: Nano, 2020, 7, 793–802 RSC.
- M. Vakili, S. Deng, G. Cagnetta, W. Wang, P. Meng, D. Liu and G. Yu, Sep. Purif. Technol., 2019, 224, 373–387 CrossRef CAS.
- X. Yang, Y. Wan, Y. Zheng, F. He, Z. Yu, J. Huang, H. Wang, Y. S. Ok, Y. Jiang and B. Gao, Chem. Eng. J., 2019, 366, 608–621 CrossRef CAS.
- Y. Wu, H. Pang, Y. Liu, X. Wang, S. Yu, D. Fu, J. Chen and X. Wang, Environ. Pollut., 2019, 246, 608–620 CrossRef CAS.
- A. Yasin, Y. Chen, Y. Liu, L. Zhang, X. Zan and Y. Zhang, Polym. Chem., 2020, 11, 810–819 RSC.
- C. Liu, R.-N. Jin, X.-k. Ouyang and Y.-G. Wang, Appl. Surf. Sci., 2017, 408, 77–87 CrossRef CAS.
- J. Li, K. Zuo, W. Wu, Z. Xu, Y. Yi, Y. Jing, H. Dai and G. Fang, Carbohydr. Polym., 2018, 196, 376–384 CrossRef CAS PubMed.
- Q. Meng, J. Liu, Y. Jiang and Q. Teng, J. Chem. Eng. Data, 2019, 64, 2618–2626 CrossRef CAS.
- J. Chen, X. Liu, S. Wang, A. Wang, Z. Wang, Q. Zeng, Z. Li and L. Zhang, Polymer, 2019, 175, 71–80 CrossRef CAS.
- H. Rezania, V. Vatanpour, E. Salehi, N. Gavari, A. Shockravi and M. Ehsani, J. Polym. Environ., 2019, 27, 1790–1800 CrossRef CAS.
- A. Wołowicz and Z. Hubicki, Chem. Eng. J., 2012, 197, 493–508 CrossRef.
- L. Zong, F. Liu, D. Chen, X. Zhang, C. Ling and A. Li, Chem. Eng. J., 2018, 334, 995–1005 CrossRef CAS.
- P. Mech, M. Bogunia, A. Nowacki and M. Makowski, J. Phys. Chem. A, 2020, 124, 538–551 CrossRef CAS PubMed.
- L. Bai, H. Hu, W. Zhang, J. Fu, Z. Lu, M. Liu, H. Jiang, L. Zhang, Q. Chen and P. Tan, J. Mater. Chem., 2012, 22, 17293–17301 RSC.
- C. Ling, Y. Zhao, Z. Ren, J. Han, C. Zhu and F.-Q. Liu, Chin. Chem. Lett., 2019, 30, 2196–2200 CrossRef CAS.
- X.-P. Zhang, F.-Q. Liu, C.-Q. Zhu, C. Xu, D. Chen, M.-M. Wei, J. Liu, C.-H. Li, C. Ling, A.-M. Li and X.-Z. You, RSC Adv., 2015, 5, 75985–75997 RSC.
- W. Zhan, C. Xu, G. Qian, G. Huang, X. Tang and B. Lin, RSC Adv., 2018, 8, 18723–18733 RSC.
- L. Xie, Z. Yu, S. M. Islam, K. Shi, Y. Cheng, M. Yuan, J. Zhao, G. Sun, H. Li, S. Ma and M. G. Kanatzidis, Adv. Funct. Mater., 2018, 28, 1800502 CrossRef.
- D. Kołodyńska, W. Sofińska-Chmiel, E. Mendyk and Z. Hubicki, Sep. Sci. Technol., 2014, 49, 2003–2015 CrossRef.
- B. Song, G. Zeng, J. Gong, P. Zhang, J. Deng, C. Deng, J. Yan, P. Xu, C. Lai, C. Zhang and M. Cheng, Chemosphere, 2017, 172, 449–458 CrossRef CAS.
- Y. Chen, B. Pan, H. Li, W. Zhang, L. Lv and J. Wu, Environ. Sci. Technol., 2010, 44, 3508–3513 CrossRef CAS.
- E. Da'na and A. Sayari, Chem. Eng. J., 2011, 166, 445–453 CrossRef.
- C. Xu, F. Q. Liu, J. Gao, L. J. Li, Z. P. Bai, C. Ling, C. Q. Zhu, D. Chen and A. M. Li, J. Hazard. Mater., 2014, 280, 1–11 CrossRef CAS PubMed.
- C. Zhu, F. Liu, C. Xu, J. Gao, D. Chen and A. Li, J. Hazard. Mater., 2015, 287, 234–242 CrossRef CAS PubMed.
- C. Bertagnolli, A. Grishin and T. Vincent, et al., Sep. Sci. Technol., 2015, 50, 2879–2906 Search PubMed.
- Q. Huang, J. Zhao and M. Liu, et al., J. Taiwan Inst. Chem. Eng., 2018, 82, 92–101 CrossRef CAS.
- X. Qiu, H. Hu, J. Yang, C. Wang, Z. Cheng and G. Ji, Chem. Pap., 2018, 72, 2071–2085 CrossRef CAS.
- W. Peng, Z. Xie, G. Cheng, L. Shi and Y. Zhang, J. Hazard. Mater., 2015, 294, 9–16 CrossRef CAS.
- X. Huang, J. Yang, J. Wang, J. Bi, C. Xie and H. Hao, Chemosphere, 2018, 206, 513–521 CrossRef CAS PubMed.
- L. Xu, D. Wu, W. Liu, X. Xu and G. Cao, J. Environ. Manage., 2019, 237, 495–503 CrossRef CAS PubMed.
- Y. Zhang, C. Zhu, F. Liu, Y. Yuan, H. Wu and A. Li, Sci. Total Environ., 2019, 646, 265–279 CrossRef CAS PubMed.
- R. R. Navarro, K. Tatsumi, K. Sumi and M. Matsumura, Water Res., 2001, 35, 2724–2730 CrossRef CAS PubMed.
- W. Chouyyok, Y. Shin, J. Davidson, W. D. Samuels, N. H. LaFemina, R. D. Rutledge, G. E. Fryxell, T. Sangvanich and W. Yantasee, Environ. Sci. Technol., 2010, 44, 6390–6395 CrossRef CAS PubMed.
- C. Liu, R. Bai and L. Hong, J. Colloid Interface Sci., 2006, 303, 99–108 CrossRef CAS PubMed.
- J. He, H.-X. Xue, C.-W. Lü, Q.-Y. Fan, Y. Liang, Y. Sun, L.-L. Shen and S. Bai, Environ. Geol., 2009, 58, 1499–1508 CrossRef CAS.
- M. X. Tan, Y. N. Sum, J. Y. Ying and Y. Zhang, Energy Environ. Sci., 2013, 6, 3254–3259 RSC.
- C. Liu, R. Bai and Q. S. Ly, Water Res., 2008, 42, 1511–1522 CrossRef CAS.
- S. Radi, S. Tighadouini, M. Bacquet, S. Degoutin, L. Janusc and Y. N. Mabkhotd, RSC Adv., 2016, 6, 82505–82514 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02034f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Chen Lia,
Xiao-Peng Zhang
a,
Chen Lia,
Xiao-Peng Zhang