
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew fused conjugated molecules with fused thiophene and pyran units for organic electronic materials†
Daoliang Chenab,
Danlei Zhuab,
Gaobo Linab,
Mingxu Dua,
Dandan Shiab,
Qian Peng a,
Lang Jianga,
Zitong Liua,
Guanxin Zhanga and
Deqing Zhang*ab
a,
Lang Jianga,
Zitong Liua,
Guanxin Zhanga and
Deqing Zhang*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Organic Solids, CAS Center of Excellence of Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 26th March 2020
Abstract
Rigid and planar conjugated molecules have substantial significance due to their potential applications in organic electronics. Herein we report two highly fused ladder type conjugated molecules, TTCTTC and TTTCTTTC, with up to 10 fused rings in which the fused-thiophene rings are fused to the chromeno[6,5,4-def]chromene unit. Both molecules show high HOMO levels and accordingly they can be oxidized into their radical cations with absorptions extending to 1300 nm in the presence of trifluoroacetic acid. Thin films of TTCTTC and TTTCTTTC exhibit p-type semiconductor properties with hole mobilities up to 0.39 cm2 V−1 s−1. Moreover, TTCTTC shows a high fluorescence quantum yield of up to 16.5% in the solid state.
Introduction
Fused conjugated molecules such as arene analogues with heteroatoms including sulfur and nitrogen have been intensively studied,1 with the aim of improving the stabilities of arene-based conjugated molecules, tune their HOMO/LUMO energies,2 modulate the intermolecular interactions and thus improve their semiconducting properties.3 In comparison, examples of arene-analogues with oxygen remain seldom. Chi, Zeng and their coworkers separately reported bis(anthraoxa)quinodimethanes with nine and ten fused six-membered rings and oxygen-embedded quinoidal pentacene and nonacene as arene-analogues with formal pyran units.4 Extended O-doped polycyclic aromatic hydrocarbons and molecular ribbons have been synthesized by Bonifazi and coworkers.5 You and coworkers have recently devised a [4 + 2] synthetic strategy6 to synthesize O-containing porphyrins and investigated their absorption and electrochemical properties.7 Formal pyran units have also been incorporated into conjugated molecules with radical character.8 In fact, conjugated molecules incorporating oxygen atoms in their backbones have been investigated less for organic electronic materials comparing with their sulfur and nitrogen atom counterparts.9 In this regard, the series of studies by Bnedikov and coworkers have aroused interest in oligofurans10 which possess many of the properties which enable them to be interesting as organic electronic materials.10,11 Nakamura and coworkers devised a new synthetic method for polycyclic furans and investigated their emissive and semiconducting properties in the solid state,12 demonstrating the high potential of fused polycyclic furans as organic optoelectronic materials.9,12,13Considering the fact that thiophene, in particular fused-thiophene rings containing fused conjugated molecules such as BTBT and its analogues,14 show outstanding semiconducting properties, it is interesting to integrate a pyran moiety with fused-thiophene rings to form newly fused conjugated molecules as potential organic electronic materials. In this paper, we report two highly fused conjugated molecules, thieno[2′′,3′′:4′,5′] thieno[3′,2′:2,3] chromeno[6,5,4-def] thieno[2′,3′:4,5] thieno[3,2-b] chromene (TTCTTC) and thieno[2′′′,3′′′:4′′,5′′] thieno[2′′,3′′:4′,5′] thieno [3′,2′:2,3] chromeno[6, 5,4-def] thieno[2′′,3′′:4′,5’] thieno[2′,3′:4,5] thieno[3,2-b] chromene (TTTCTTTC) (Scheme 1), with two formal pyran units. Two alkyl chains are attached to both TTCTTC (R = n-octadecyl) and TTTCTTTC (R = 2-ethylhexyl) to ensure their good solubility in organic solvents. Both TTCTTC and TTTCTTTC can be synthesized in 3 steps with acceptable yields. The results reveal that they have high HOMO energy levels, up to −4.69 eV. Thin films of these two compounds exhibit p-type semiconducting properties with hole mobility up to 0.39 cm2 V−1 s−1. In particular, the thin film of TTCTTC is emissive with a quantum yield of 16.5% in the solid state.
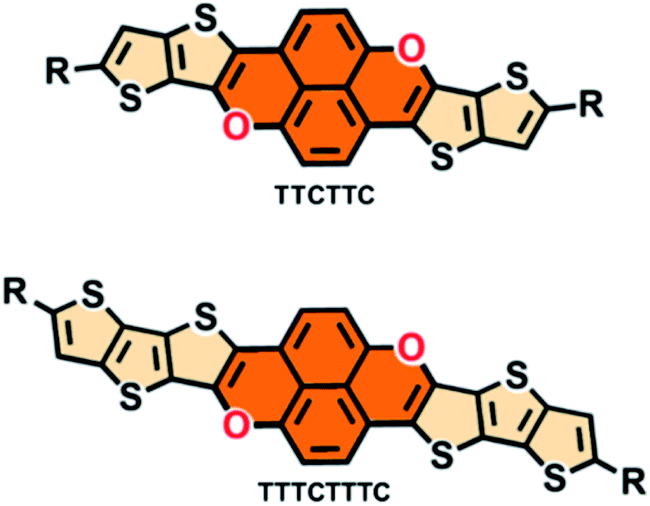 | ||
| Scheme 1 Chemical structures of TTCTTC and TTTCTTTC. | ||
Results and discussion
The synthetic approaches are outlined in Scheme 2. The synthesis started with compound 1 which underwent a halogen–lithium exchange reaction to afford zinc reagent 2. The selective Negishi coupling of 2 with 3-bromo-2-iodothieno[3,2-b]thiophene and 3-bromo-2-iododithieno[3,2-b:2′,3′-d]thiophene in the presence of PdCl2(PPh3)2 yielded 3a and 4a in 49.8% and 52.3% yields, respectively. The respective demethylation of 3a and 4a by BBr3 afforded 3b and 4b. The intramolecular C–O coupling reaction within 3b and 4b, which was catalyzed by CuI/1,10-phenloine, yielded the target molecules TTCTTC and TTTCTTTC in 61.5% and 58.3% yields, respectively. The chemical structures of TTCTTC and TTTCTTTC were characterized with NMR spectra and MS data, and their purities were checked with elemental analysis (see ESI†). Thermal decomposition was not observed for TTCTTC and TTTCTTTC when the temperatures were below 282 °C and 316 °C, respectively, based on the thermogravimetric analysis data (see Fig. S1†).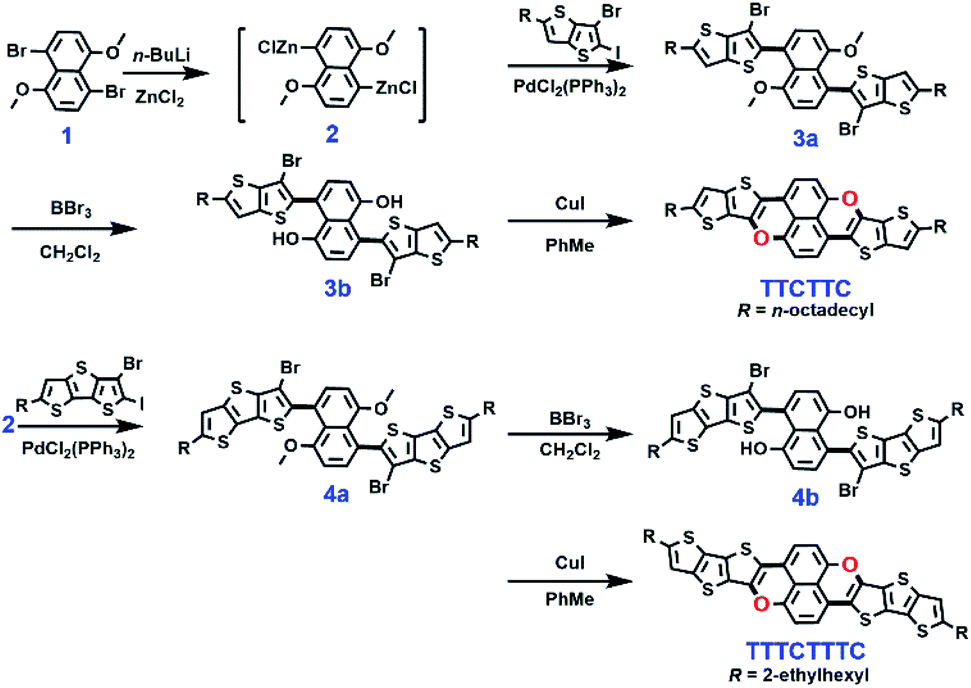 | ||
| Scheme 2 The synthesis of TTCTTC and TTTCTTTC. | ||
Single crystals of TTCTTC and TTTCTTTC suitable for X-ray analysis were obtained by crystallization from the respective solutions in toluene, and their crystal structures were successfully determined (see ESI†). Fig. 1 shows their molecular structures and intermolecular arrangements. Obviously, the conjugated cores of both TTCTTC and TTTCTTTC are fully planar. All bond lengths (see Tables S2 and S3†) are in the normal ranges. Molecules of TTCTTC interact to form layers via short interatomic contacts (C⋯H short contacts, 2.39 Å; S⋯S short contacts, 3.42 Å). The molecular layers are further assembled to form columns via π–π interactions (3.46 Å). For TTTCTTTC, molecules interact to form layers via interatomic S⋯S contacts (3.70 Å), and the conjugated cores are further assembled by intermolecular π–π interactions (3.50 Å) as depicted in Fig. 1. Such intermolecular interactions and arrangements are beneficial for intermolecular charge transport as discussed below.
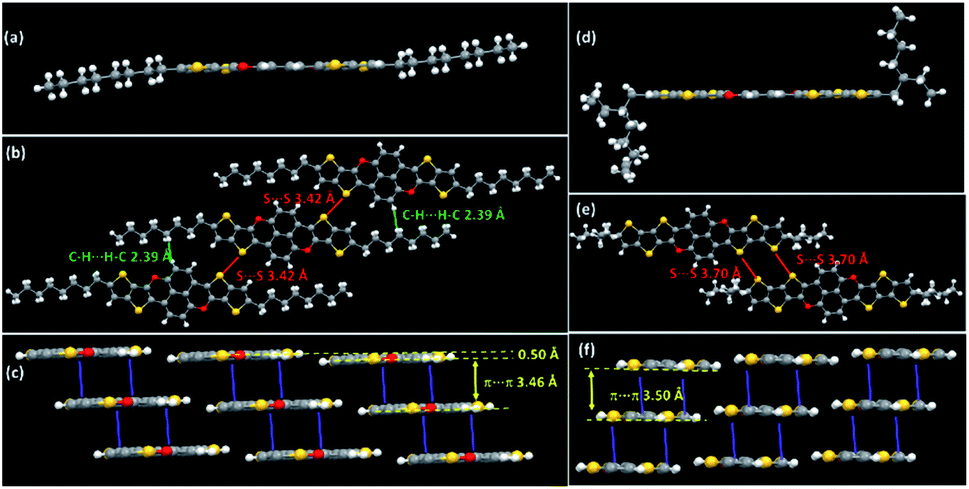 | ||
| Fig. 1 Molecular structures and intermolecular interactions and arrangements of TTCTTC (a–c) and TTTCTTTC (d–f) crystal lattices. | ||
The cyclic voltammograms of TTCTTC and TTTCTTTC were measured and are shown in Fig. 2. As shown in Fig. 2, two reversible oxidation waves were detected at Eox11/2 = −0.01 V (vs. Fc/Fc+) and Eox21/2 = 0.41 V (vs. Fc/Fc+) for TTCTTC, whereas TTTCTTTC displayed two oxidation waves at Eox11/2 = 0.02 V (vs. Fc/Fc+) and Eox21/2 = 0.41 V (vs. Fc/Fc+). Based on the respective onset oxidation potentials, HOMO energies were estimated to be −4.69 eV for TTCTTC and −4.71 eV for TTTCTTTC. As discussed below, the optical band gaps (Egopt) of TTTCTTTC and TTCTTC were estimated to be 2.58 eV and 2.48 eV, respectively. Accordingly, the LUMO energies of TTCTTC and TTTCTTTC were calculated to be −2.11 eV and −2.23 eV, respectively, using the equation: LUMO = HOMO + Egopt. On the basis of the high lying HOMO energy levels, which are comparable with those of TTF15 and pentacene,16 thin films of TTCTTC and TTTCTTTC are expected to show p-type semiconducting behavior. The high HOMO energy levels are also supported by the DFT calculations. Fig. S5† shows the calculated HOMO and LUMO orbitals of TTCTTC and TTTCTTTC. It is clear that the HOMO orbitals are mainly distributed on the central chromeno[6,5,4-def]chromene cores for both molecules, while their LUMO orbitals are evenly distributed across the whole conjugated backbone. Therefore, the resultant excited states of the two molecules exhibit charge transfer features from the central conjugated core to the fused-thiophenes. The calculated HOMO energies of TTCTTC and TTTCTTTC were −4.45 eV and −4.48 eV, respectively, whereas the respective calculated LUMO energies were −1.34 eV and −1.54 eV. The calculated HOMO energies of TTCTTC and TTTCTTTC are close as determined with cyclic voltammetry as discussed above.
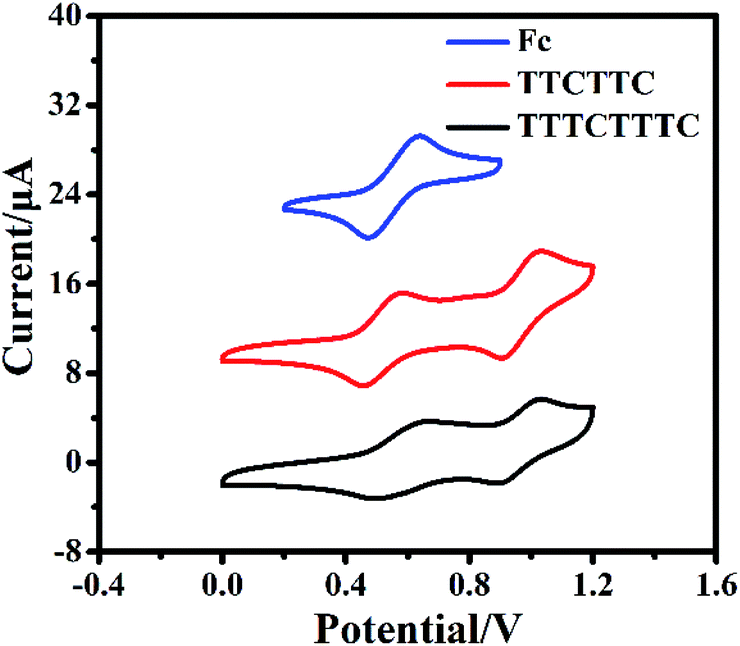 | ||
| Fig. 2 Cyclic voltammograms of TTCTTC (0.1 mM) and TTTCTTTC (0.1 mM) in o-dichlorobenzene at a scan rate of 100 mV s−1, with Pt as the working and counter electrodes, and Ag/AgCl (saturated KCl) as the reference electrode, and n-Bu4NPF6 (0.1 M) as the supporting electrolyte. The cyclic voltammogram of ferrocene was measured under the same conditions. | ||
Fig. 3 shows the absorption spectra of TTCTTC and TTTCTTTC in solution and solid state. The solution of TTCTTC shows strong absorption at 295 nm, 406 nm, 436 nm, and 463 nm. In comparison, the absorption peaks of TTTCTTTC in solution are red-shifted to 319 nm, 334 nm, 422 nm, 450 nm, and 478 nm. Such absorption red-shift can be attributed to the elongation of conjugation length of TTTCTTTC compared with TTCTTC. This agrees with the calculated electron absorptions based on their optimized geometries. As shown in Table S4,† the calculated absorption peaks are at 461.1 nm (S0 → S2) and 313.8 nm (S0 → S6) for TTCTTC in solution and those for TTTCTTC are at 490.9 nm (S0 → S2) and 349.3 nm (S0 → S4). These are in good agreement with the respective two dominant absorption bands of TTCTTC and TTTCTTTC. The other absorption peaks may be caused by electron-vibration coupling, which is usually observed for planar conjugated molecules.
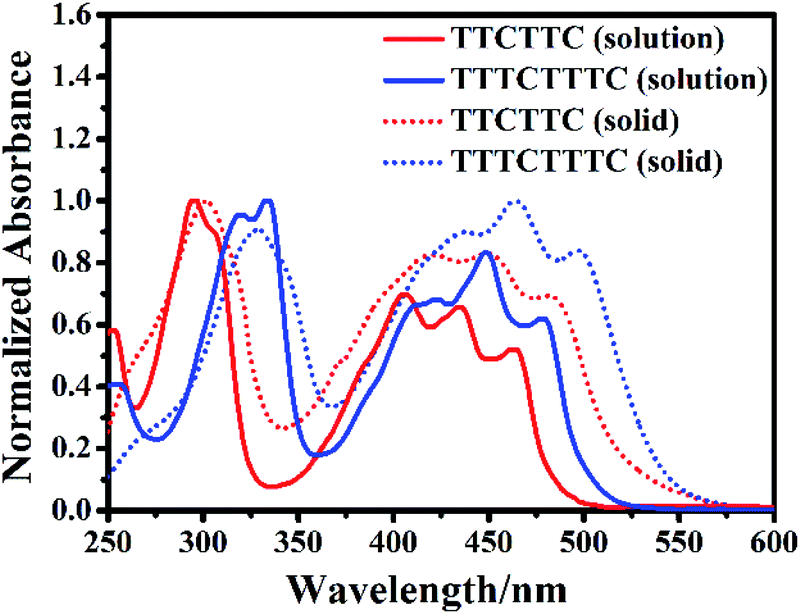 | ||
| Fig. 3 Normalized UV-vis absorption spectra of the dichloromethane (10 μM) solutions of TTCTTC and TTTCTTTC and their solid samples. | ||
The absorption spectra of both molecules are red-shifted in their solid states compared with the respective absorption in solution (see Fig. 3). For instance, the solid sample of TTCTTC shows absorption at 301 nm, 420 nm, 450 nm and 482 nm which are red-shifted by 6 nm, 14 nm, 14 nm and 19 nm, respectively, in comparison with those in solution. On the basis of the respective onset absorption in the solid state, the optical bandgaps of TTCTTC and TTTCTTTC were estimated to be 2.33 eV and 2.31 eV, respectively.
The emission spectra in solution and solid state were measured (see Fig. S6†) and their emission maxima, quantum yields and fluorescence lifetimes are listed in Table S5.† To our delight, both solutions of TTCTTC and TTTCTTTC are emissive with quantum yields of 13.8% and 13.6%, respectively. Furthermore, TTCTTC is also emissive around 571 nm in the solid state with a quantum yield of 16.5%, while TTTCTTTC shows rather weak emission in the solid state (Table S5 and Fig. S7†).
Considering that TTCTTC and TTTCTTTC possess high HOMO energy levels, we investigated their absorption spectra after oxidation. It was reported that electron donors with high HOMO energy levels such as N–N linked bicarbazole or biacridine derivatives,17 and tetrabenzoporphyrin,18 can be oxidized by acids such CF3COOH. As shown in Fig. 4, new absorption peaks around 650 nm and 980 nm were observed after the addition of CF3COOH (1% wt) into the CH2Cl2 solution of TTCTTC. Similarly, new absorption peaks around 680 nm and 1150 nm emerged after the addition of CF3COOH (1% wt) into the CH2Cl2 solution of TTTCTTTC. Furthermore, strong ESR signals with g = 2.0039 and 2.0040 were observed for both solutions of TTCTTC and TTTCTTTC after addition of CF3COOH, respectively (see Fig. S8†). We also measured the absorption spectra of TTCTTC and TTTCTTTC (see Fig. S9†) after electrochemical oxidation by applying oxidation potentials at 0.6 V (vs. Ag/AgCl). Clearly, the absorption after addition of CF3COOH matches well with the respective ones after electrochemical oxidation. These results show that both TTCTTC and TTTCTTTC can be oxidized by CF3COOH. This could be attributed to their high HOMO energy levels.
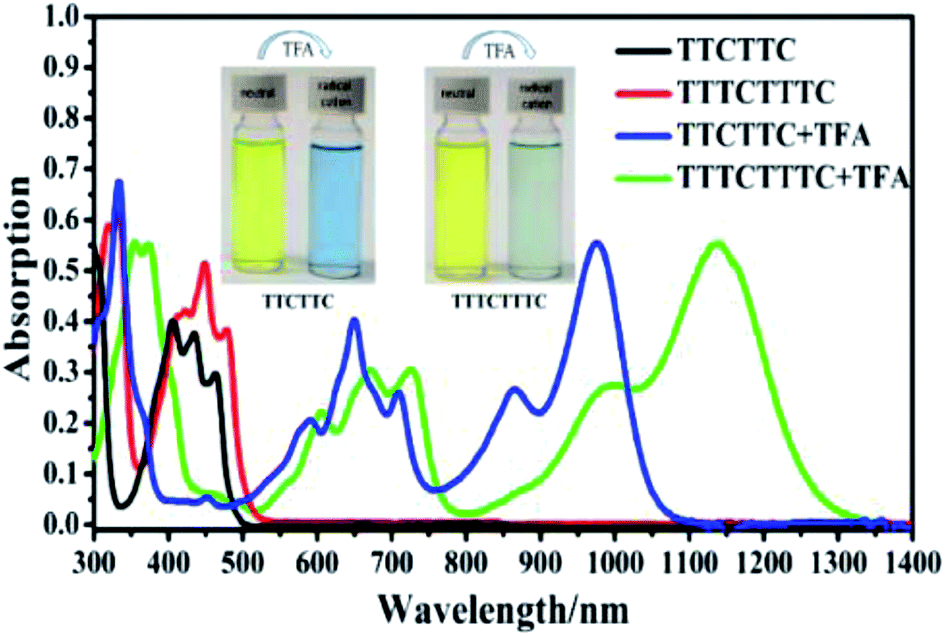 | ||
| Fig. 4 UV-vis-NIR absorption spectra of CH2Cl2 solutions (10 μM) of TTCTTC and TTTCTTTC and related UV-vis-NIR absorption spectra after the addition of CF3COOH (1% wt). | ||
In order to evaluate the charge transporting properties, organic field-effect transistor (OFET) devices with bottom-gate/bottom-contact (BGBC) structures were fabricated by vacuum deposition of the thin films on the octadecyltrichlorosilane (OTS)-modified SiO2 (300 nm)/Si substrates with a pre-patterned gold source and drain electrodes. The devices were measured under an inert atmosphere. Fig. 5, S10 and S11† show the transfer and output curves, from which the semiconducting performance data was extracted and summarized in Table S6.† In the transfer curves for OFETs with thin films of TTCTTC and TTTCTTTC, IDS increases by applying the negative VGS (see Fig. 5, S10 and S11†). Thus, both thin-films exhibit p-type semiconducting behavior. This is in good agreement with the high HOMO energy levels of TTCTTC and TTTCTTTC as discussed above. The maxima and average hole mobilities of devices with thin films of TTCTTC deposited on the substrates at 25 °C were measured to be 0.31 and 0.19 cm2 V−1 s−1, respectively, with high Ion/off ratios. The average hole mobility for a thin film of TTCTTC slightly increased to 0.25 cm2 V−1 s−1 by increasing the substrate temperature to 50 °C. But, the hole mobility started to decrease by further increasing the substrate temperature to 90 °C as listed in Table S6.† In comparison, the maxima hole mobility is only 0.016 cm2 V−1 s−1 for the thin film of TTTCTTTC deposited on the substrate at 25 °C (see Table S6†). Only a small enhancement of charge mobility was observed by increasing the substrate temperature for the thin film of TTTCTTTC. This can be attributed to the poor crystallinity of the thin film of TTTCTTTC (Fig. S12–S14†). The intermolecular transfer integrals were calculated on the basis of the crystal structures of TTCTTC and TTTCTTTC. As shown in Fig. S4,† TTCTTC shows multiple charge transfer pathways with transfer integrals of 25.97 and −36.19 meV, which are larger than those of TTTCTTTC (14.34 meV and 1.35 meV). This is in good agreement with the observation that the thin film of TTCTTC exhibits higher hole mobility than that of TTTCTTTC.
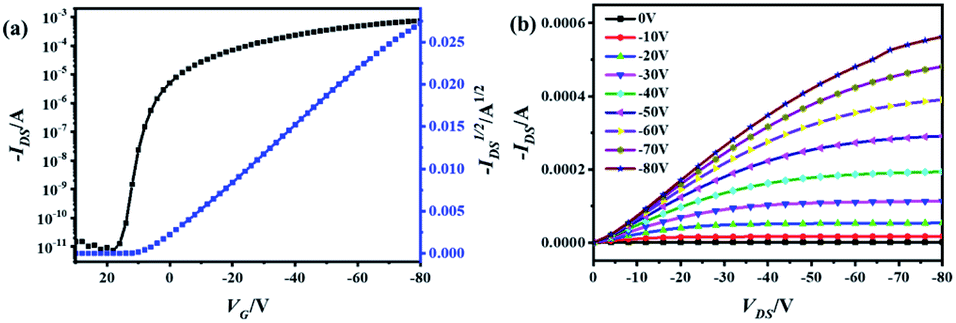 | ||
| Fig. 5 Transfer (a) and output (b) characteristics of OFETs with thin films of TTCTTC deposited on OTS-modified SiO2/Si substrate. | ||
Conclusions
In conclusion, we report the synthesis, characterization and semiconducting properties of two fused conjugated molecules, TTCTTC and TTTCTTTC, entailing pyran and fused thiophene moieties. The crystal structures show that molecules of TTCTTC interact via C⋯H and S⋯S interactions to form layers which are further connected via π–π stacking with a distance of 3.46 Å. Intermolecular π–π interactions were also observed for TTTCTTTC in the crystal lattice. Both electrochemical measurements and DFT calculations show that both TTCTTC and TTTCTTTC possess high HOMO energy levels. As a result, they can be oxidized to their radical cations by CF3COOH. The resulting radical cations show strong absorption in the near-infrared light region up to 1300 nm. Both conjugated molecules are emissive in solution, with fluorescence quantum yields up to 13.8%. But, only TTCTTC shows relatively strong emission in the solid state. Both thin films of TTCTTC and TTTCTTTC exhibit p-type semiconducting properties. The thin film of TTCTTC shows relatively high hole mobility of up to 0.39 cm2 V−1 s−1, whereas the average hole mobility of a thin film of TTTCTTTC is only 0.017 cm2 V−1 s−1. This can be attributed to the fact that the thin film of TTCTTC shows high crystallinity based on the XRD studies, while the thin film of TTTCTTTC is of low crystallinity. Further elongation of the conjugation lengths and incorporation of functional groups to these fused and expanded conjugated molecules are underway.Experimental section
Synthesis of 3a
Under a nitrogen atmosphere, 2.7 ml of n-butyl lithium (1.6 M) was added to 1 (0.67 g, 1.95 mmol) in dry THF solution (20 ml) at −78 °C. The resulting solution was stirred at this temperature for 10 min. When the reaction system was slowly warmed up to 0 °C, 2.5 ml of ZnCl2 (2 M) in dry THF was added to the system. After the reaction mixture was stirred for 30 min at 0 °C, 3-bromo-2-iodo-5-octylthieno[3,2-b]thiophene19 (3.0 g, 6.6 mmol) and PdCl2(PPh3)2 (50 mg, 0.07 mmol) were added. The reaction system was then heated to 70 °C and stirred for 12 h under a nitrogen atmosphere. After removal of the THF, 50 ml of water was added to the mixture which was then extracted with dichloromethane and dried over Na2SO4. After removal of dichloromethane, the crude product was purified using silica gel column chromatography with petroleum ether (60–90 °C)/dichloromethane (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to give 3a (820 mg, 0.97 mmol) in 49.8% yield. 1H NMR (300 MHz, CDCl3, 25 °C) δ = 7.42 (d, J = 9 Hz, 2H), 7.00 (s, 2H), 6.88 (d, J = 9 Hz, 2H), 3.62 (s, 6H), 2.92 (t, J = 7.5 Hz, 4H), 1.75 (d, J = 6 Hz, 4H), 1.43–1.30 (m, 20H), 0.90 (t, J = 6 Hz, 6H). 13C NMR (75 MHz, CDCl3, 25 °C) δ = 157.22, 146.87, 141.62, 137.70, 135.02, 132.02, 125.95, 121.03, 117.00, 105.89, 101.55, 56.14, 31.90, 31.67, 31.22, 29.38, 29.26, 29.14, 22.71, 14.15. HRMS (MALDI-TOF): m/z calcd for C40H46Br2O2S4: 844.0747 [M]+; found: 844.0758.
:1, v/v) to give 3a (820 mg, 0.97 mmol) in 49.8% yield. 1H NMR (300 MHz, CDCl3, 25 °C) δ = 7.42 (d, J = 9 Hz, 2H), 7.00 (s, 2H), 6.88 (d, J = 9 Hz, 2H), 3.62 (s, 6H), 2.92 (t, J = 7.5 Hz, 4H), 1.75 (d, J = 6 Hz, 4H), 1.43–1.30 (m, 20H), 0.90 (t, J = 6 Hz, 6H). 13C NMR (75 MHz, CDCl3, 25 °C) δ = 157.22, 146.87, 141.62, 137.70, 135.02, 132.02, 125.95, 121.03, 117.00, 105.89, 101.55, 56.14, 31.90, 31.67, 31.22, 29.38, 29.26, 29.14, 22.71, 14.15. HRMS (MALDI-TOF): m/z calcd for C40H46Br2O2S4: 844.0747 [M]+; found: 844.0758.
Synthesis of 3b
Under a nitrogen atmosphere, 3a (265 mg, 0.31 mmol) was dissolved in 20 ml of dry dichloromethane. The solution was cooled to −78 °C, and 1 ml of BBr3 (1 M) solution in dichloromethane was added slowly. Then, it was allowed to increase to room temperature and stirred for 12 h. 50 ml of water was added to the mixture which was then extracted with dichloromethane and dried over Na2SO4. After removal of dichloromethane, the mixture was purified using silica gel column chromatography with petroleum ether (60–90 °C)/dichloromethane (1:1, v/v) to give 3b (188 mg, 0.23 mmol) in 74.2% yield. 1H NMR (400 MHz, [D8]THF, 25 °C) δ = 8.75 (s, 2H), 7.26 (d, J = 8 Hz, 2H), 7.11 (s, 2H), 6.77 (d, J = 8 Hz, 2H), 2.97 (t, J = 8 Hz, 4H), 1.86–1.73 (m, 4H), 1.58–1.24 (m, 20H), 0.95 (t, J = 6 Hz, 6H). 13C NMR (100 MHz, [D8]THF, 25 °C) δ = 155.28, 146.15, 142.04, 137.64, 135.37, 131.14, 125.50, 119.82, 117.12, 109.47, 101.78, 31.87, 31.73, 30.78, 29.34, 29.25, 28.98, 22.57, 13.46. HRMS (ESI): m/z calcd for C38H42Br2O2S4 + Br−: 898.9576 [M + Br]−; found: 898.9583.
Synthesis of TTCTTC
3b (106 mg, 0.13 mmol) and K2CO3 (300 mg, 2.17 mmol) were added to 30 ml of dry toluene in a Schlenk tube. After nitrogen gas was bubbled through the mixture for 10 min, 1,10-phenanthroline (20 mg, 0.11 mmol) and CuI (10 mg, 0.05 mmol) was added under a nitrogen atmosphere. It was then heated to 100 °C and stirred for 24 h. Then, after cooling to room temperature, the reaction mixture was directly subjected to separation using basic Al2O3 column chromatography with toluene as the eluent to give TTCTTC (52 mg) in 61.5% yield. 1H NMR (400 MHz, [D8]THF/CS2 v/v = 1:4, 25 °C) δ = 6.80 (s, 2H), 6.53 (d, J = 8.0 Hz, 2H), 6.50 (d, J = 8.0 Hz, 2H), 2.83 (t, J = 7.6 Hz, 4H), 1.35–1.22 (m, 24H), 0.86 (m, 6H). 13C NMR (100 MHz, [D8]THF/CS2 v/v = 1:4, 25 °C) δ = 150.28, 149.01, 143.45, 135.36, 126.69, 123.50, 121.78, 117.32, 117.01, 114.30, 110.02, 32.32, 31.92, 31.45, 29.83, 29.75, 29.58, 23.27, 14.45. HRMS (MALDI-TOF) m/z calcd for C38H40O2S4: 656.1911 [M]+; found 656.1892; elemental analysis calcd (%) for C38H40O2S4: C, 69.47; H, 6.14; S, 19.52; found: C 69.74; H 6.09; S 19.37.
Synthesis of 4a
Under a nitrogen atmosphere, 3.0 ml of n-butyl lithium (1.6 M) was added to 1 (0.74 g, 2.14 mmol) in dry THF solution (20 ml) at −78 °C. The resulting solution was stirred at this temperature for another 10 min. Then, the reaction system was warmed to 0 °C, and 3.5 ml ZnCl2 (2 M) in THF was added slowly. The reaction was stirred at this temperature for 30 min, then 3-bromo-6-(2-ethylhexyl)-2-iododithieno[3,2-b:2′,3′-d]thiophene19 (3.0 g, 5.85 mmol) and PdCl2(PPh3)2 (30 mg, 0.04 mmol) were added to the reaction system. The reaction was heated to 70 °C and stirred at this temperature for 12 h. Then, the mixture was cooled and evaporated to remove the excess THF. 50 ml of water was added to the mixture which was then extracted with dichloromethane and dried over Na2SO4. After removal of dichloromethane, the crude product was purified using silica gel column chromatography with petroleum ether (60–90 °C)/dichloromethane (2:1, v/v) to give 4a (1.08 g) in 52.3% yield. 1H NMR (400 MHz, CDCl3, 25 °C) δ = 7.46 (d, J = 8.0 Hz, 2H), 7.03 (s, 2H), 6.91 (d, J = 8 Hz, 2H), 3.64 (s, 6H), 2.87 (d, J = 4 Hz, 4H), 1.67 (m, 2H), 1.50–1.24 (m, 16H), 0.95–0.91 (m, 12H). 13C NMR (100 MHz, CDCl3, 25 °C) δ = 157.31, 146.01, 141.13, 138.67, 132.24, 129.67, 128.09, 125.93, 120.77, 118.57, 105.91, 105.87, 102.60, 56.15, 41.54, 35.22, 32.44, 28.94, 25.55, 23.05, 14.20, 10.89. HRMS (MALDI-TOF) m/z calcd for C44H46O2S6: 956.0188 [M]+; found 956.0120.
Synthesis of 4b
Under a nitrogen atmosphere 4a (420 mg, 0.44 mmol) was dissolved in 20 ml of dry dichloromethane. The mixture was then cooled to −78 °C, and 1 ml of BBr3 (1 M) solution in dichloromethane was added slowly at this temperature. Then, it was allowed to increase to room temperature and stirred for 12 h. 50 ml of water was added to the mixture which was then extracted with dichloromethane and dried over Na2SO4. After removal of the solvents, the crude product was purified using silica gel column chromatography with petroleum ether (60–90 °C)/dichloromethane (1:1, v/v) to give 4b (294 mg) in 72.7% yield. 1H NMR (400 MHz, [D8]THF, 25 °C) δ = 7.17 (d, J = 8 Hz, 2H), 7.02 (s, 2H), 6.67 (d, J = 8 Hz, 2H), 2.79 (d, J = 8 Hz, 4H), 1.57 (m, 2H), 1.31–1.23 (m, 16H), 0.85–0.79 (m, 12H). 13C NMR (125 MHz, [D8]THF, 25 °C) δ = 155.48, 145.52, 141.77, 140.25, 138.56, 131.39, 129.55, 128.01, 125.56, 119.52, 118.81, 109.60, 102.23, 41.65, 34.83, 32.38, 28.84, 25.47, 22.90, 13.47, 10.21. HRMS (ESI) m/z calcd for C42H42Br2O4S6 + Br−: 1010.9018 [M + Br]−; found 1010.9071.
Synthesis of TTTCTTTC
4b (114 mg, 0.12 mmol), K2CO3 (300 mg, 2.17 mmol) and 60 ml of toluene were added to a Schlenk tube. After nitrogen gas was bubbled through the solution for 10 min, CuI (10 mg, 0.05 mmol) and 1,10-phenanthroline (20 mg, 0.11 mmol) were added. The reaction mixture was heated to 100 °C and stirred for 24 h. Then, the reaction mixture was separated directly using basic Al2O3 column chromatography using toluene as the eluent to give TTTCTTTC (51 mg) in 58.3% yield as a yellow solid. 1H NMR (400 MHz, [D8]THF/CS2 v/v = 1:4, 25 °C) δ = 6.91 (s, 2H), 6.60 (d, J = 8 Hz, 2H), 6.56 (d, J = 8 Hz, 2H), 2.81 (d, J = 8 Hz, 4H), 1.62 (m, 2H), 1.37–1.22 (m, 16H), 0.92–0.88 (m, 12H). 13C NMR (100 MHz, [D8]THF/CS2 v/v = 1:4, 25 °C) δ = 150.26, 147.11, 143.89, 141.05, 129.58, 129.15, 127.67, 123.44, 121.51, 118.77, 116.69, 114.36, 110.12, 41.73, 35.36, 32.73, 29.23, 25.94, 23.52, 14.34, 10.97. HRMS (MALDI-TOF) m/z calcd for C42H40O2S6: 768.1352 [M]+; found 768.1348; elemental analysis calcd (%) for C42H40O2S6: C, 65.59; H, 5.24; S, 25.01; found: C 65.38; H 5.21; S 24.98.
Fabrication of OFET devices
OFET devices for TTCTTC and TTTCTTTC were fabricated in a BGBC configuration. A layer of 300 nm thick SiO2 was used as a gate dielectric layer and a heavily doped n-type silicon wafer was chosen as the gate electrode. The gold drain/source (D/S) electrodes with thickness of 30 nm were adopted and deposited on the substrates by photo-lithography. Mixed solutions of hydrogen peroxide and sulfuric acid were used to treat the substrates and were soaked for about 0.5 h. Then they were cleaned using deionized water and rinsed in isopropanol. The obtained Si/SiO2 substrates were then put into a vacuum oven and treated with OTS at 125 °C for 4 h. The OTS modified substrates were then washed with chloroform, n-hexane and isopropanol, respectively. Molecules of TTCTTC and TTTCTTTC were slowly deposited (0.1 Å s−1) onto the OTS modified substrates to form the thin films (about 40 nm in thickness) at different substrate temperatures (25, 50, and 90 °C). The measurements were conducted under a nitrogen atmosphere using a Keithley 4200 SCS analyzer. The mobilities were calculated with the equation: ID = μCi(W/2L)(VG − Vth)2, in which ID is the drain current, Ci is capacitance per unit area of the insulating layer (11.5 nF cm−2), W is channel width, L is channel length, and Vth is the threshold voltage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support of NSFC (21790363, 21871271, 21661132006).References
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef CAS; X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491 CrossRef; M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036 CrossRef; M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291 CrossRef PubMed; L. Qiu, C. Yu, N. Zhao, W. Chen, Y. Guo, X. Wan, R. Yang and Y. Liu, Chem. Commun., 2012, 48, 12225 RSC; S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Chem. Commun., 2015, 51, 4528 RSC; H. Yamada, Y. Yamaguchi, R. Katoh, T. Motoyama, T. Aotake, D. Kuzuhara, M. Suzuki, T. Okujima, H. Uno, N. Aratani and K.-i. Nakayama, Chem. Commun., 2013, 49, 11638 RSC.
- T. Zheng, Z. Cai, R. Ho-Wu, S. H. Yau, V. Shaparov, T. Goodson and L. Yu, J. Am. Chem. Soc., 2016, 138, 868 CrossRef CAS; Y. Huang, D. Wu, J. Huang, Q. Guo, J. Li and J. You, Angew. Chem., Int. Ed., 2014, 53, 12158 CrossRef; S.-L. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428 CrossRef PubMed; S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Chem. Commun., 2011, 47, 11394 RSC; H. J. Son, F. He, B. Carsten and L. Yu, J. Mater. Chem., 2011, 21, 18934 RSC; J. Cao, C. Zuo, B. Du, X. Qiu and L. Ding, Chem. Commun., 2015, 51, 12122 RSC.
- H. Dong, C. Wang and W. Hu, Chem. Commun., 2010, 46, 5211 RSC; H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397 CrossRef CAS PubMed; S. Vegiraju, D.-Y. Huang, P. Priyanka, Y.-S. Li, X.-L. Luo, S.-H. Hong, J.-S. Ni, S.-H. Tung, C.-L. Wang, W.-C. Lien, S. L. Yau, C.-L. Liu and M.-C. Chen, Chem. Commun., 2017, 53, 5898 RSC; D. Wang, X. Qiao, G. Ouyang, H. Wu and H. Li, Chem. Commun., 2019, 55, 6253 RSC; M. Melucci, M. Zambianchi, L. Favaretto, M. Gazzano, A. Zanelli, M. Monari, R. Capelli, S. Troisi, S. Toffanin and M. Muccini, Chem. Commun., 2011, 47, 11840 RSC; X. Shi, J. Chang and C. Chi, Chem. Commun., 2013, 49, 7135 RSC.
- S. Dong, T. Y. Gopalakrishna, Y. Han, H. Phan, T. Tao, Y. Ni, G. Liu and C. Chi, J. Am. Chem. Soc., 2019, 141, 62 CrossRef CAS PubMed; Y. Wang, S. Qiu, S. Xie, L. Zhou, Y. Hong, J. Chang, J. Wu and Z. Zeng, J. Am. Chem. Soc., 2019, 141, 2169 CrossRef.
- A. Berezin, N. Biot, T. Battisti and D. Bonifazi, Angew. Chem., Int. Ed., 2018, 57, 8942 CrossRef CAS; S. Daphné, D. Nicola and B. Davide, Angew. Chem., Int. Ed., 2016, 55, 5947 CrossRef.
- J. Yin, M. Tan, D. Wu, R. Jiang, C. Li and J. You, Angew. Chem., Int. Ed., 2017, 56, 13094 CrossRef CAS.
- C. Li, L. Zhu, W. Liang, R. Su, J. Yin, Y. Hu, Y. Lan, D. Wu and J. You, Chem. Sci., 2019, 10, 7274 RSC.
- K. P. Rao, T. Kusamoto, F. Toshimitsu, K. Inayoshi, S. Kume, R. Sakamoto and H. Nishihara, J. Am. Chem. Soc., 2010, 132, 12472 CrossRef CAS; C. M. Wehrmann, R. T. Charlton and M. S. Chen, J. Am. Chem. Soc., 2019, 141, 3240 CrossRef PubMed; O. Anamimoghadam, M. D. Symes, D.-L. Long, S. Sproules, L. Cronin and G. Bucher, J. Am. Chem. Soc., 2015, 137, 14944 CrossRef.
- Z. Zhao, H. Nie, C. Ge, Y. Cai, Y. Xiong, J. Qi, W. Wu, R. T. K. Kwok, X. Gao, A. Qin, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2017, 4, 1700005 CrossRef CAS; H. Tsuji and E. Nakamura, Acc. Chem. Res., 2017, 50, 396 CrossRef PubMed.
- O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546 CrossRef CAS PubMed.
- Y. Xiong, J. Tao, R. Wang, X. Qiao, X. Yang, D. Wang, H. Wu and H. Li, Adv. Mater., 2016, 28, 5949 CrossRef CAS PubMed.
- C. Mitsui, J. Soeda, K. Miwa, H. Tsuji, J. Takeya and E. Nakamura, J. Am. Chem. Soc., 2012, 134, 5448 CrossRef CAS PubMed; H. Tsuji, C. Mitsui, L. Ilies, Y. Sato and E. Nakamura, J. Am. Chem. Soc., 2007, 129, 11902 CrossRef.
- D. Chen, J. Li, W. Ma, B. Li, Y. Zhen, X. Zhu, W. Hu, H. Tsuji and E. Nakamura, Asian J. Org. Chem., 2018, 7, 2228 CrossRef CAS; K. Nakanishi, D. Fukatsu, K. Takaishi, T. Tsuji, K. Uenaka, K. Kuramochi, T. Kawabata and K. Tsubaki, J. Am. Chem. Soc., 2014, 136, 7101 CrossRef.
- T. Mori, T. Nishimura, T. Yamamoto, I. Doi, E. Miyazaki, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 13900 CrossRef CAS PubMed; K. Niimi, S. Shinamura, I. Osaka, E. Miyazaki and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 8732 CrossRef PubMed.
- S. Adeel, M. E. Abdelhamid, A. Nafady, Q. Li, L. L. Martin and A. M. Bond, RSC Adv., 2015, 5, 18384 RSC.
- O. L. Griffith, J. E. Anthony, A. G. Jones and D. L. Lichtenberger, J. Am. Chem. Soc., 2010, 132, 580 CrossRef CAS PubMed.
- P. Pandit, K. Yamamoto, T. Nakamura, K. Nishimura, Y. Kurashige, T. Yanai, G. Nakamura, S. Masaoka, K. Furukawa, Y. Yakiyama, M. Kawano and S. Higashibayashi, Chem. Sci., 2015, 6, 4160 RSC.
- Y. Zhen, K. Inoue, Z. Wang, T. Kusamoto, K. Nakabayashi, S.-i. Ohkoshi, W. Hu, Y. Guo, K. Harano and E. Nakamura, J. Am. Chem. Soc., 2018, 140, 62 CrossRef CAS PubMed.
- H.-Y. Chen, G. Schweicher, M. Planells, S. M. Ryno, K. Broch, A. J. P. White, D. Simatos, M. Little, C. Jellett, S. J. Cryer, A. Marks, M. Hurhangee, J.-L. Brédas, H. Sirringhaus and I. McCulloch, Chem. Mater., 2018, 30, 7587 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1963694 and 1975874. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01984d |
| This journal is © The Royal Society of Chemistry 2020 |