
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbamoyl-N-aryl-imine-urea: a new framework to obtain a putative leishmanicidal drug-candidate†
Marina A. Alves‡
ab,
Aline C. de Queiroz‡c,
Anderson Brandão Leitec,
Felipe T. Martins d,
Antonio C. Doriguettod,
Eliezer J. Barreiroab,
Magna S. Alexandre-Moreira*c and
Lídia M. Lima*ab
d,
Antonio C. Doriguettod,
Eliezer J. Barreiroab,
Magna S. Alexandre-Moreira*c and
Lídia M. Lima*ab
aInstituto Nacional de Ciência e Tecnologia de Fármacos e Medicamentos (INCT-INOFAR), Laboratório de Avaliação e Síntese de Substâncias Bioativas (LASSBio, ®), Universidade Federal do Rio de Janeiro (UFRJ), CCS, PO Box 68023, Cidade Universitária, 21941-902, Rio de Janeiro, RJ, Brazil Web: http://www.inct-inofar.ccs.ufrj.br Web: http://www.lassbio.icb.ufrj.br
bPrograma de Pós-graduação em Química, Instituto de Química, UFRJ, 21941-909, Rio de Janeiro, RJ, Brazil
cLaboratório de Farmacologia e Imunologia, Instituto de Ciências Biológicas e da Saúde, Universidade Federal de Alagoas (UFAL), 57072-900, Maceió, AL, Brazil. E-mail: suzana.magna@gmail.com; Tel: +55 82 3214 1528
dInstituto de Química, Universidade Federal de Goiás (UFG), Campus Samambaia, CP 131, Goiânia, GO 74001-970, Brazil
First published on 26th March 2020
Abstract
Leishmaniasis is a neglected parasitic disease, and current treatment includes limitations of toxicity, variable efficacy, high costs and inconvenient doses and treatment schedules. Therefore, new leishmanicidal drugs are still an unquestionable medical need. In this paper we described the design conception of a new framework, the carbamoyl-N-aryl-imine-urea, to obtain putative leishmanicidal drug-candidates. Compounds 9a–e and 10a–e were designed and synthesized and their leishmanicidal activity was studied in comparison to pentamidine, miltefosine and meglumine antimoniate. The conformational profile of the new carbamoyl-N-aryl-imine-urea framework was investigated by X-ray diffraction studies, using compound 9a as a model. The plasma stability of this putative peptide mimetic subunit was studied for compound 10e (LASSBio-1736). Among the congeneric series, LASSBio-1736 was identified as a new antileishmanial drug-candidate, displaying plasma stability, cytotoxicity against amastigote forms of L. amazonensis and L. braziliensis, and leishmanicidal activity in a cutaneous leishmaniasis murine model, without preliminary evidence of hepatic or renal toxicity.
Introduction
Leishmaniasis is a parasitic disease caused by about 20 Leishmania species and is transmitted to humans by more than 30 different species of phlebotomine sandflies. The parasitosis is clinically classified as (i) cutaneous leishmaniasis (CL), (ii) mucocutaneous leishmaniasis (ML), and (iii) visceral leishmaniasis (VL), the most severe form. Collectively they comprise one of the most prevalent neglected disease, with more than 2 million new cases occurring annually.1 Although leishmaniasis had been associated with risk factors such as socio-economic status, demographic area and human behaviors, an increase in its worldwide incidence has been attributed to migration, travel to endemic areas, deforestation, urbanization and organ transplant.2 The most common form of leishmaniasis is CL, which represents over 90% of cases distributed across three main endemic regions: (i) Afghanistan, Iran, Saudi Arabia and Syria; (ii) Algeria and Tunisia; and (iii) Brazil and Peru.3 It is mainly caused by L. major and L. tropica in the Old World, while L. amazonensis, L. braziliensis and L. guyanensis are responsible for the most cases of infection in the new world.A promising strategy to treat parasitic infections is to inhibit parasite proteases. Also known as peptidases or peptide hydrolases, those enzymes are responsible by the catabolism of proteins and polypeptides through the cleavage of peptide bonds. Among peptidases subtypes, cysteinyl proteases (CPs) have been considered druggable targets for the design and development of new leishmanicidal drugs, since they play vital roles in the life cycle of Leishmania species.4–6
Numerous inhibitors of CPs have been described in the literature. They are designed considering the catalytic mechanism of cysteinyl proteases that predict the formation of a tetrahedral intermediate, generated from the nucleophilic addition of the cysteine residue to an electrophilic peptide carbonyl group.7 Two types of CPs inhibitors have been reported: peptide analogues and peptide mimetic derivatives. Their common feature is the presence of an electrophilic functionality that can be attacked by the catalytic cysteine thiolate in the active site. The peptide mimetic approach has been considered more advantageous, since it can overcome the undesirable drug-likeness properties associated with peptide structures, such as low solubility, poor bioavailability and metabolic instability.8,9
Over the years, several chemotypes have been explored as a possible core to form transient covalent bond with sulfur atom of an essential cysteine residue from cysteinyl proteases, subsequently, inhibiting the catalytic function of those enzymes. Amide (1),10 acylhydrazone (2–4),11–13 hydrazone (5),14 thiosemicarbazone (6)15 and semicarbazone (7)16 are amongst the most prevalent chemotypes (Fig. 1).
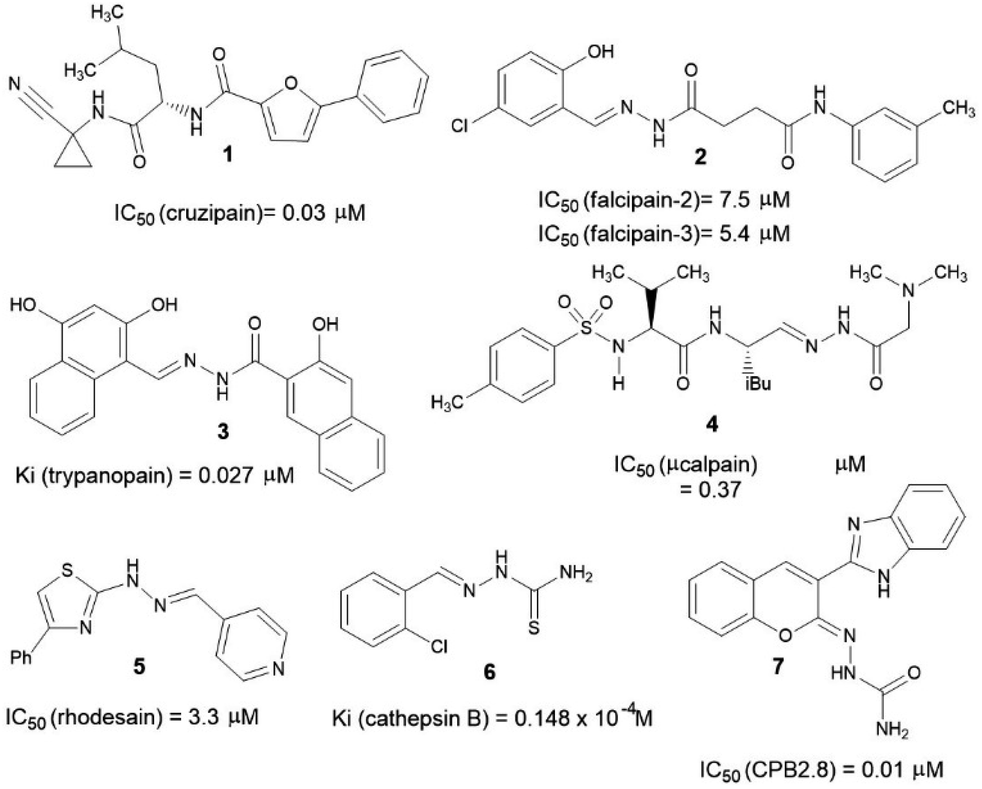 | ||
| Fig. 1 Examples of cysteine proteases inhibitors bearing a peptide mimetic framework. | ||
In this paper we describe the design, synthesis, plasma stability and leishmanicidal activity of new compounds bearing a novel putative peptide mimetic framework: carbamoyl-N-aryl-imine-urea.
Adopting the classical ligand-based drug design approach, the new carbamoyl-N-aryl-imine-ureas 9a–e and 10a–e were planned by molecular modifications at the prototype 8. The design conception considered the hybridization among the amide (RCONHR) and semicarbazone (RNHCONHN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR) fragments of the prototypes 1 and 8, respectively (Fig. 2). From this approach, compound 9a was designed holding a new framework as a linker to connect the aromatic rings, presented in the lead-compound 8. In view of the toxicophoric profile of 5-nitrofuranyl subunit,17 it was further replaced by phenyl ring. Substitution on the phenyl ring by electron donating and electron withdrawing groups allowing the conception of compounds 9b–e. To investigate the ortho-effect on the activity of compounds 9a–e, their analogues containing the chloro atom introduced in the position 2 of the phenyl ring linked to carbonyl subunit (10a–e) were also proposed (Fig. 2).
CHR) fragments of the prototypes 1 and 8, respectively (Fig. 2). From this approach, compound 9a was designed holding a new framework as a linker to connect the aromatic rings, presented in the lead-compound 8. In view of the toxicophoric profile of 5-nitrofuranyl subunit,17 it was further replaced by phenyl ring. Substitution on the phenyl ring by electron donating and electron withdrawing groups allowing the conception of compounds 9b–e. To investigate the ortho-effect on the activity of compounds 9a–e, their analogues containing the chloro atom introduced in the position 2 of the phenyl ring linked to carbonyl subunit (10a–e) were also proposed (Fig. 2).
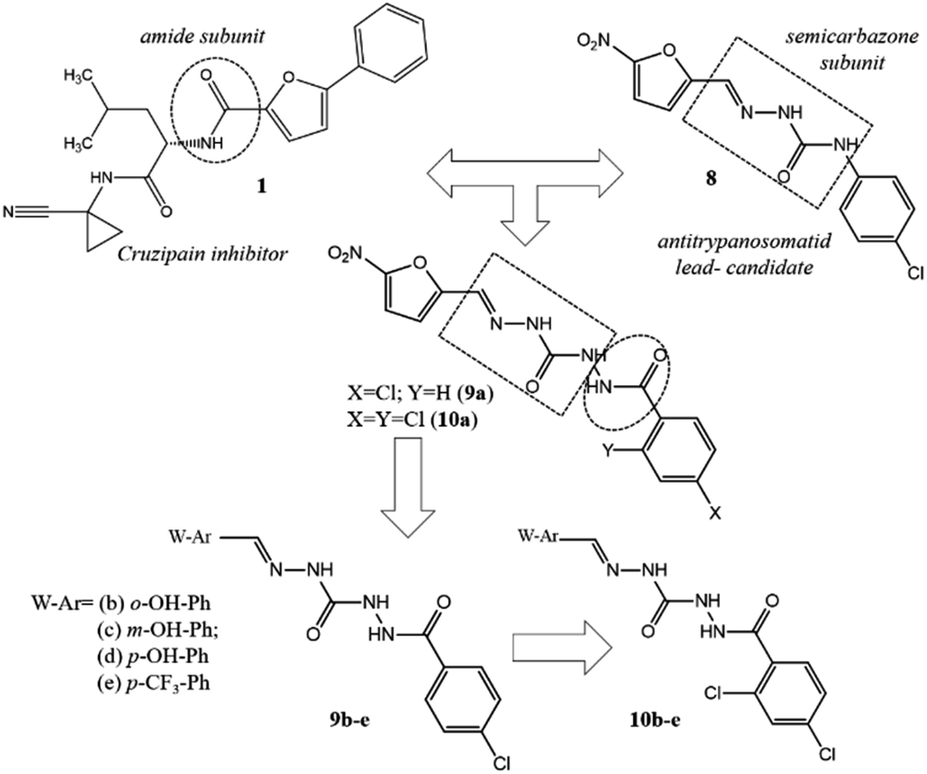 | ||
| Fig. 2 Design conception of compounds 9a–e and 10a–e bearing a new peptide mimetic framework – the carbamoyl-N-aryl-imine-urea. | ||
Results and discussion
Synthesis
Compounds 9a–e and 10a–e were synthetized in three linear steps from the hydrazides 11 and 12, obtained commercially (Fig. 3). First, the phenyl carbamates (13 and 14) were prepared from compounds 11 and 12 by condensation reaction with phenyl chloroformate, using chloroform as solvent at room temperature.18 The key intermediates 15 and 16 were obtained in good yield from the carbamates 13 and 14 (respectively), exploring hydrazinolysis step in the presence of hydrazine hydrate and ethanol as solvent at room temperature.19 The synthesis of the target compounds 9a–e and 10a–e was concluded by condensation of the key-intermediates (15 and 16) with functionalized aldehydes, in the presence of ethanol at room temperature.19 Compounds 9a–e and 10a–e were obtained in overall yield ranging from 53 to 95%.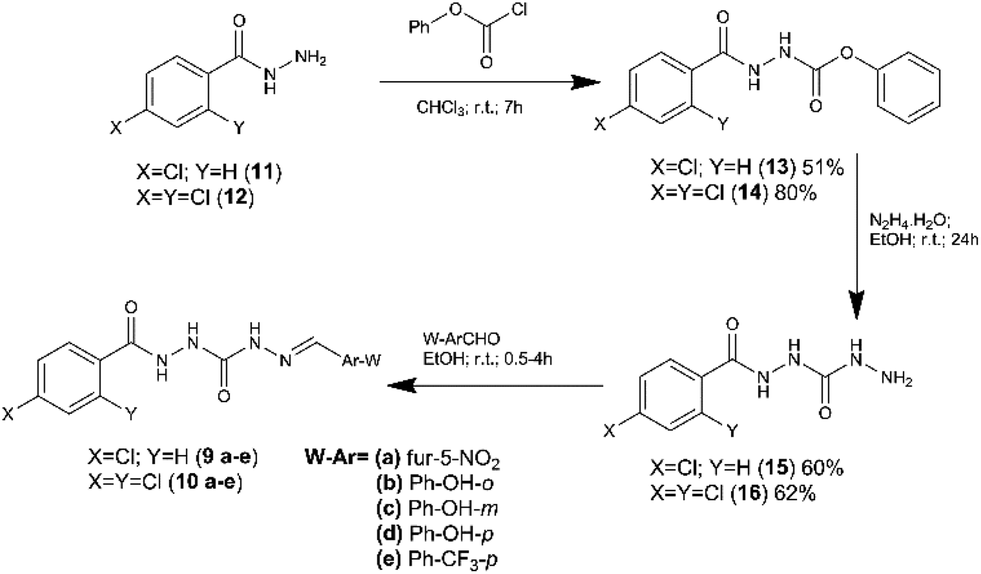 | ||
| Fig. 3 Synthetic route to obtain the target compounds 9a–e and 10a–e. | ||
The chemical structure of compounds 9a–e and 10a–e was elucidated by 1H and 13C NMR, IR and mass spectrometry. The analysis of those spectra data has revealed that all compounds were synthetized as a single diastereoisomer (Z or E).
Single crystal structure determination
To determine unequivocally the relative stereochemistry of the imine double bond (NCH) and the molecular structure of the new peptide mimetic derivatives, X-ray diffraction studies have been proposed. However, considering the difficulty of getting compounds 9a–e and 10a–e in crystalline form, the X-ray crystallography assays have been performed only with derivative 9a, obtained as crystal solid.
As depicted in Fig. 4, compound 9a (LASSBio-1491) has crystallized in the centrosymmetric monoclinic space group P21/c. Crystal data and refinement results are listed in Table S1.† There were two molecules of compound 9a in the asymmetric unit (Fig. 4), which were labeled as A and B (atom labels ending with the corresponding capital letter). The occurrence of these two molecules features is a typical case of conformerism around only one rotatable single bond (Fig. 5). These two conformers differ only for a rotation of ca. 180° around the C4–C5 bond axis connecting the 5-nitrofuran ring to acylhydrazone group. The O3–C4–C5–N2 and C3–C4–C5–N2 torsions differ for 177.7 (10)° and 178.0 (13)° between the two conformers (O3–C4–C5–N2 = 175.8 (4)° in A and −1.9 (6)° in B; C3–C4–C5–N2 = −2.3 (8)° in A and 175.7 (5)° in B). This rotation changes the relative orientation of the heterocyclic O3 furan and the O4 carbonyl oxygens in the conformers. While these oxygens are on the same side of the open chain encompassing them in conformer A, they are on opposite sides in conformer B. Except for this conformational difference, the remaining backbones of conformers A and B is similar [root mean square deviation (r.m.s.d.) of 0.156 Å for all overlaid corresponding non-hydrogen atoms from both conformers, excluding nitrofuran atoms O1–O3, N1, and C1–C3; see Fig. 5]. These equivalent backbones can be described by two molecular planes which are almost bent perpendicularly. This twist is gotten on the N4–N5 hydrazine bond (C6–N4–N5–C7 = −70.7 (5)° in A and −82.7 (5)° in B), which gives rise to one plane adjusted from N4 to nitro group (r.m.s.d. of the fourteen fitted non-hydrogen atoms is 0.116 Å in A and 0.0245 Å in B) and another one from N5 to Cl1 (r.m.s.d. of the ten fitted non-hydrogen atoms is 0.145 Å in A and 0.164 Å in B). These two molecular planes are bent by 69.88 (7)° and 78.32 (7)° in molecules A and B, respectively (Fig. 5). A slight twist is also found in both conformers around the C7–C8 bond axis connecting the 4-chlorobenzene ring to carbonyl moiety (O5–C7–C8–C9 = 16.0 (6)° in A and 17.1 (6)° in B).
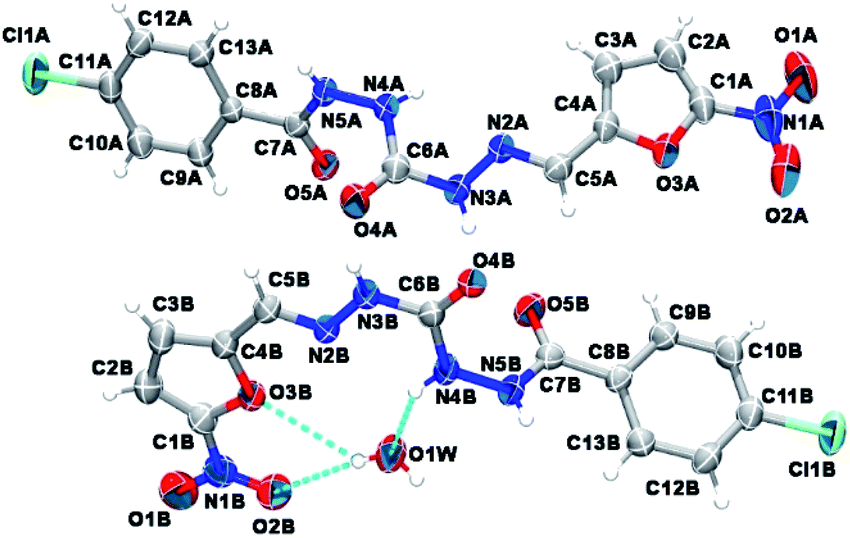 | ||
| Fig. 4 Asymmetric unit of compound 9a (LASSBio-1491) elucidated in this study. Non-hydrogen atoms are represented as 30% probability ellipsoids and randomly labeled, while hydrogens are shown as arbitrary radius spheres. Cyan dashed lines draw hydrogen bonds. | ||
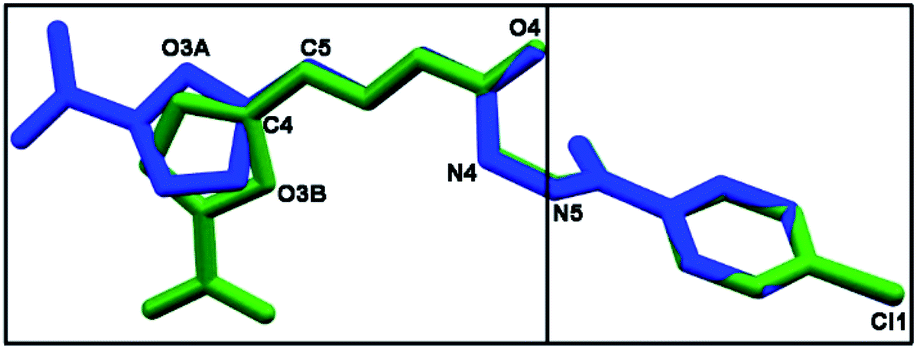 | ||
| Fig. 5 A molecular superposition of conformers A (blue) and B (green) of compound 9a (LASSBio-1491). Hydrogen atoms were omitted for clarity. Some key atoms were labeled to identify molecular fragments. The two molecular planes crossing through both conformers were split over two frames (left and right). | ||
Besides the two conformers, there is also one water molecule in the asymmetric unit of compound 9a, revealing that this compound has crystallized as a hemihydrate. Water molecule selectively interacts only with conformer B, being even responsible for the conformational change found between the two conformers. The whole water molecules lie onto the molecular plane adjusted from N4 to nitro group of molecule B, where it acts as a hydrogen bonding acceptor from N4B–H4B and as a bifurcated hydrogen bonding donor to the oxygens O2B (heterocyclic furan oxygen) and O3B (from nitro group) (Fig. 4). These last contacts enforce the rotation about C4–C5, introducing conformerism in the crystal structure of the studied compound.
Biological assays
| Compound | IC50a |
|---|---|
| a Concentration required to give 50% death of cells (IC50) was calculated by linear regression analysis from the culture growth constant (Kc) values at employed concentrations (100, 10, 1 and 0.1 μM).23 This constant corresponds to the slope resulting from plotting the log of the growth measurement versus time for each drug concentration. | |
| Miltefosine | >100 μM |
| Pentamidine | 35.5 ± 3.4 μM |
| LASSBio-1491 (9a) | >100 μM |
| LASSBio-1704 (9b) | >100 μM |
| LASSBio-1709 (9c) | 85.2 ± 2.3 μM |
| LASSBio-1706 (9d) | 86.2 ± 8.7 μM |
| LASSBio-1737 (9e) | >100 μM |
| LASSBio-1703 (10a) | >100 μM |
| LASSBio-1705 (10b) | 78.1 ± 4.5 μM |
| LASSBio-1708 (10c) | >100 μM |
| LASSBio-1707 (10d) | >100 μM |
| LASSBio-1736 (10e) | >100 μM |
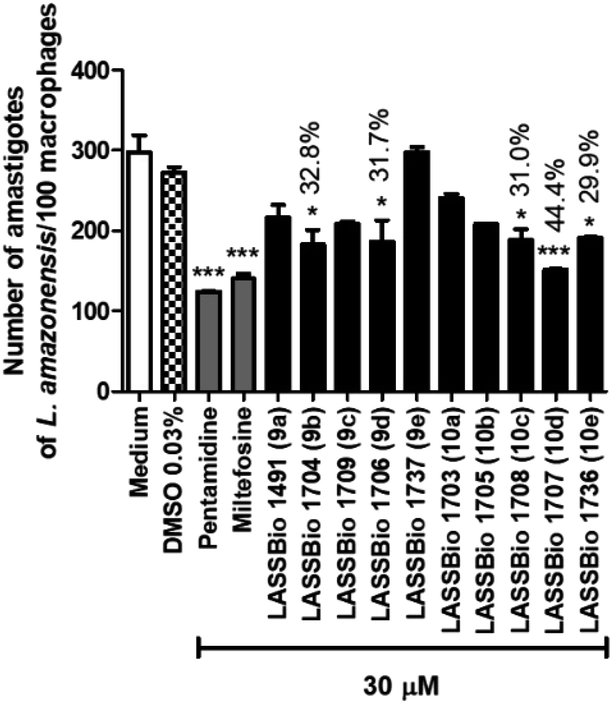 | ||
| Fig. 6 Effect of pentamidine and carbamoyl-N-aryl-imine-urea derivatives (9a–e and 10a–e) against intracellular amastigote forms of L. amazonensis at concentration of 30 μM. | ||
| Compound | L. amazonensis | L. braziliensis | ||
|---|---|---|---|---|
| IC50a (μM) | Maximum effectb (% ± S.E.M.) | IC50a (μM) | Maximum effectb (% ± S.E.M.) | |
| a IC50 is the concentration required to give 50% death of parasites, calculated by linear regression analysis from the Kc values at employed concentrations (100, 10, 1, 10−1, 10−2 and 10−3 μM).24 This constant corresponds to the slope resulting from plotting the log of the growth measurement versus time for each drug concentration.b Maximum effect (ME) is expressed as mean ± standard error of maximum toxicity average of triplicates of a representative experiment. The values of maximum effect were considered significant when *p < 0.05, **p < 0.01 compared to the 0.1% DMSO group. | ||||
| Miltefosine | 22.0 ± 1.8 | 58.1 ± 4.9** | 32.1 ± 1.1 μM | 84.0 ± 1.5** |
| Pentamidine | 32.8 ± 4.6 | 59.1 ± 5.6** | 78.4 ± 4.7 μM | 61.7 ± 2.2** |
| LASSBio-1704 (9b) | 85.3 ± 5.0 | 55.6 ± 1.4** | >100 μM | 48.1 ± 4.5** |
| LASSBio-1706 (9d) | >100 | NA | 3.6 ± 0.6 μM | 70.4 ± 1.9** |
| LASSBio-1708 (10c) | >100 | 31.0 ± 1.7** | >100 μM | NA |
| LASSBio-1707 (10d) | >100 | 44.4 ± 0.2** | >100 μM | NA |
| LASSBio-1736 (10e) | 84.0 ± 0.3 | 57.6 ± 0.2** | 5.3 ± 2.6 μM | 57.2 ± 5.2** |
Further, the leishmanicidal activity against intracellular amastigotes forms of L. braziliensis were also investigated (Table 2). The carbamoyl-N-arylimine-ureas 9d (LASSBio-1706) and 10e (LASSBio-1736) showed anti-amastigote activity with IC50 values of 3.6 μM and 5.3 μM, respectively. These results revealed the higher potency of 9d and 10e in comparison with the standard drugs miltefosine (IC50 = 32.1 μM) and pentamidine (IC50 = 78.4 μM).
As shown in Table 2, significant variations in activity of carbamoyl-N-arylimine-ureas were observed against the intracellular amastigote of L. amazonensis and of L. braziliensis, which can be owing the variances among both Leishmania spp. or due the differences in the mechanism of action of those compounds when compared to the standards pentamidine and miltefosine.
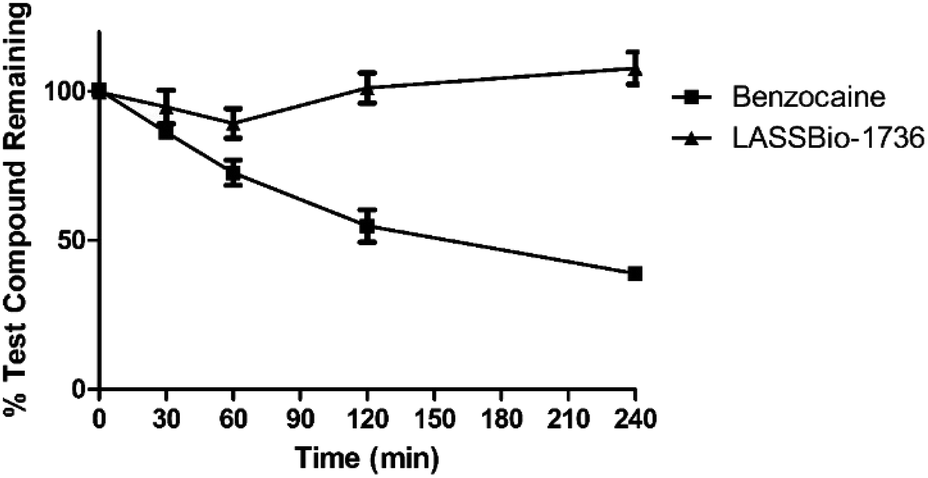 | ||
| Fig. 7 Plasma stability of 10e (LASSBio-1736) ▲ and standard benzocaine ■. Data were obtained through the compound recovery percentage analyzed by UPLC-MS/MS. | ||
So, BALB/c mice infected with L. amazonensis were treated during 28 days with compound 10e (30 μmol per kg per day, i.p.) or with the standards miltefosine (30 μmol per kg per day, p.o.) and meglumine antimoniate (30 μmol per kg per day, i.p.). As demonstrated in Fig. 8 and 9, the carbamoyl-N-arylimine-urea 10e (LASSBio-1736) has reduced the lesion sizes of infected ear like miltefosine, and both have shown better efficacy than meglumine antimoniate.
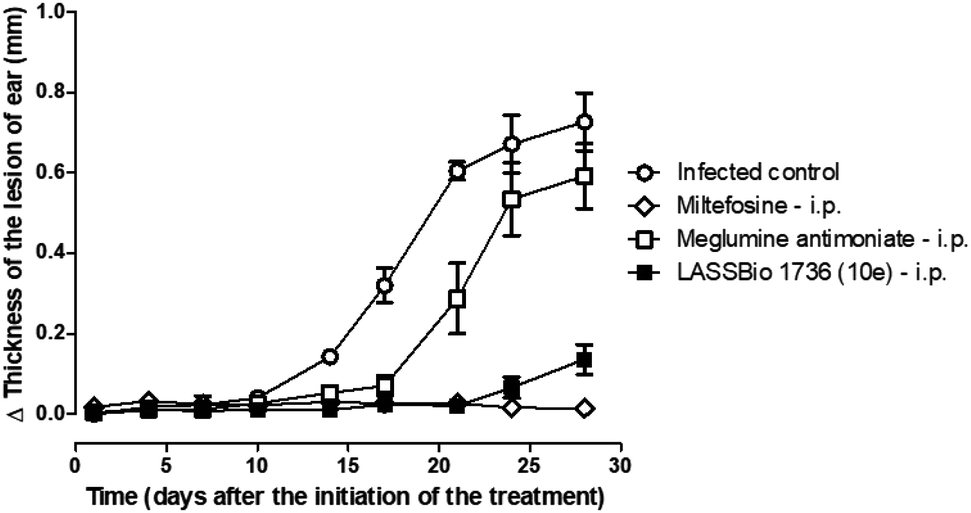 | ||
| Fig. 8 In vivo efficacy of meglumine antimoniate, miltefosine and 10e (LASSBio-1736) treatments (30 μmol per kg per day × 28 days) in BALB/c mice infected with L. amazonensis. Lesion sizes were monitored weekly. Values are the mean of lesion sizes in five mice in each group and bars represent the standard error of the mean. | ||
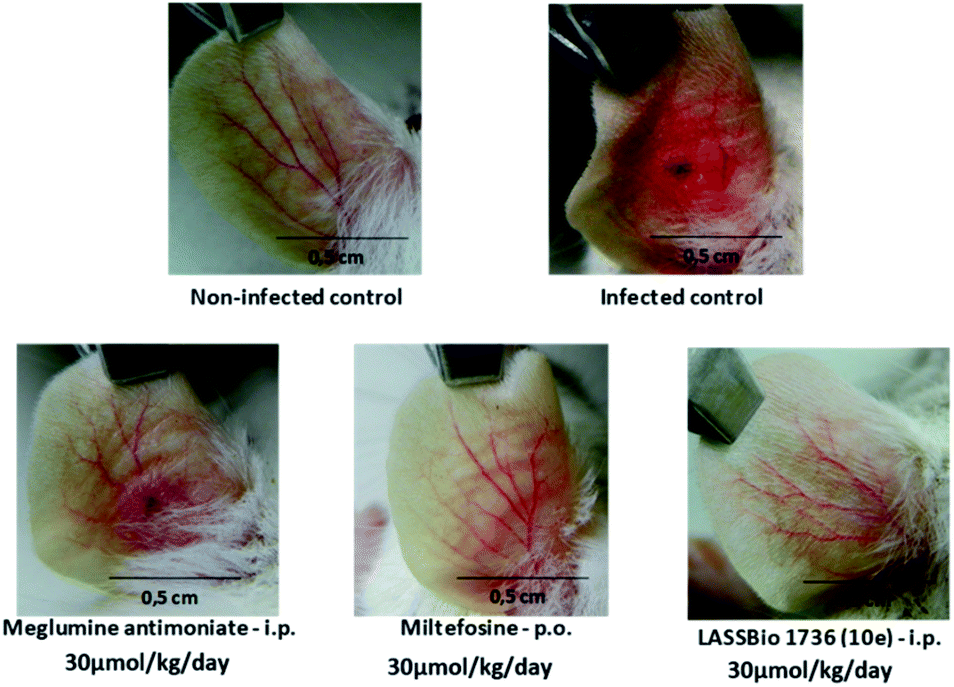 | ||
| Fig. 9 Images of lesions after of treatment of BALB/c mice infected with L. amazonensis untreated and treated with meglumine antimoniate, miltefosine and 10e (LASSBio-1736) at dose 30 μmol per kg per day × 28 days. Photographs were taken on the first day after end of treatment. In the control groups, infected control, and meglumine antimoniate, the lesions showed an intense swelling and ulcerated by after treatment cessation. In groups treated with miltefosine and 10e (LASSBio-1736), the photographs revealed a complete healing of the nodules and ulcers. | ||
Regarding the parasite burden, 10e has decreased parasite load in infected ear as miltefosine, but it has not reduced the L. amazonensis titer in draining lymph node (Fig. 10).
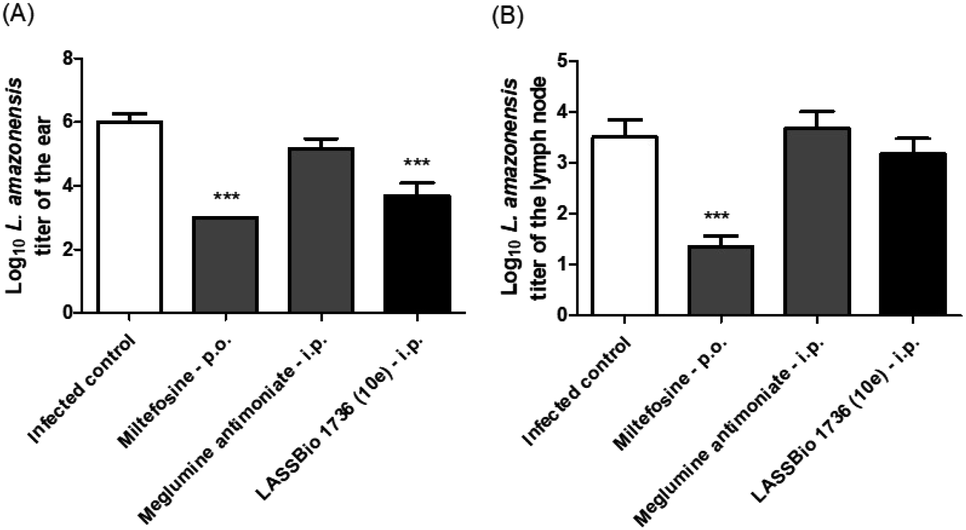 | ||
| Fig. 10 Parasite burden throughout the course of miltefosine (30 μmol per kg per day × 28 days, p.o.), meglumine antimoniate and LASSBio-1736 (10e) (30 μmol per kg per day × 28 days, i.p.) treatments in BALB/c mice infected with L. amazonensis. (A) log10 of parasites loads of the infected ear. (B) log10 of parasites loads of the draining lymph node. The parasite loads of infected ears and draining lymph nodes were determined using a quantitative limiting-dilution assay. Values are the mean of parasites loads in five mice in each group and bars represent the standard error of the mean. ***P < 0.001 vs. control. | ||
The survival of BALB/c mice infected with L. amazonensis untreated and treated during 28 days with the daily dose of 30 μmol kg−1 with meglumine antimoniate, miltefosine and 10e (LASSBio-1736) was analyzed. Animal mortality was not observed for the groups treated with 10e and miltefosine, while 20% of death was identified from the 25th day of treatment with meglumine antimoniate (Fig. 11). After the end of treatment, the animal's toxicity was investigated looking for any alterations on spleen weight and on alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine (CREA) and urea serum levels. As shown in Fig. 12 and 13 compound 10e (LASSBio-1736) did not significantly interfere with serum levels of these biochemical markers, suggesting no hepato- and nephrotoxic activities. Moreover, like the standards drugs, 10e (LASSBio-1736) did not alter animal's spleen weight.
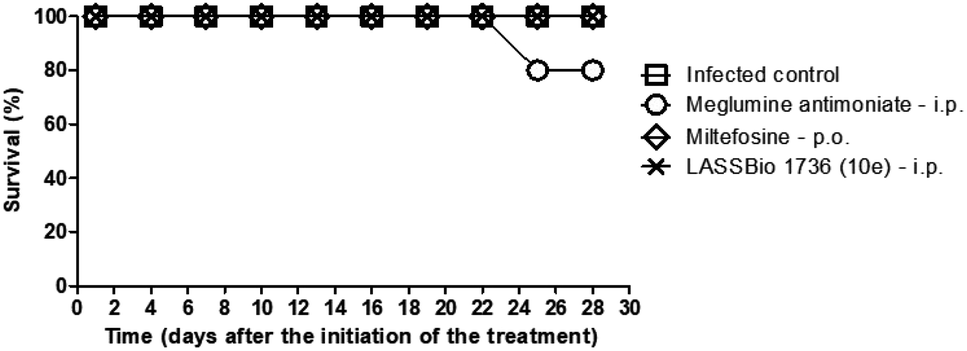 | ||
| Fig. 11 Survival of BALB/c mice infected with L. amazonensis untreated and treated with meglumine antimoniate, miltefosine, and LASSBio-1736 (10e) at dose 30 μmol per kg per day × 28 days. Data are from representative experiments, n = 5 mice per group. Treatment with meglumine antimoniate induced 20% of mortality in the group and other treatments presented survival rate of 100%. | ||
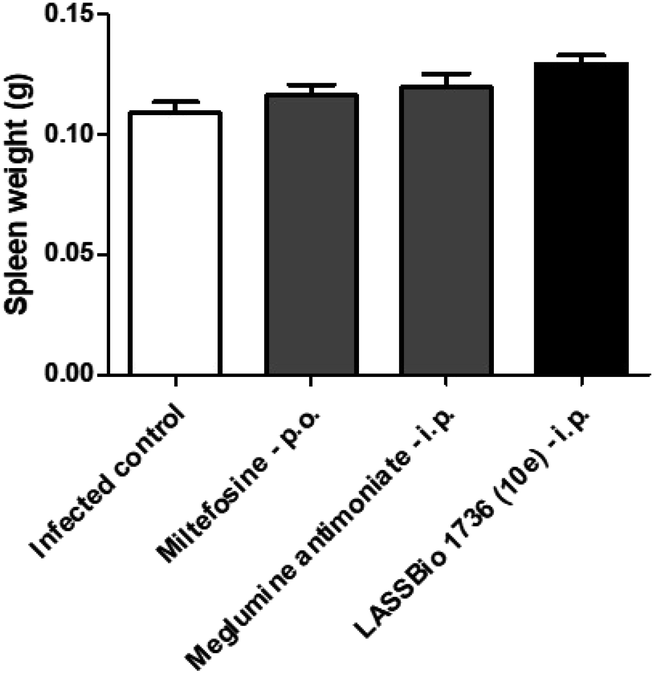 | ||
| Fig. 12 In vivo effect of miltefosine (30 μmol per kg per day × 28 days, p.o.), meglumine antimoniate and LASSBio-1736 (10e) (30 μmol per kg per day × 28 days, i.p.) in spleen weight of BALB/c mice infected with L. amazonensis. Spleen weight was verified in last day of treatment. Values are the mean of the spleen weight in five mice in each group and bars represent the standard error of the mean. *P < 0.05 vs. control. | ||
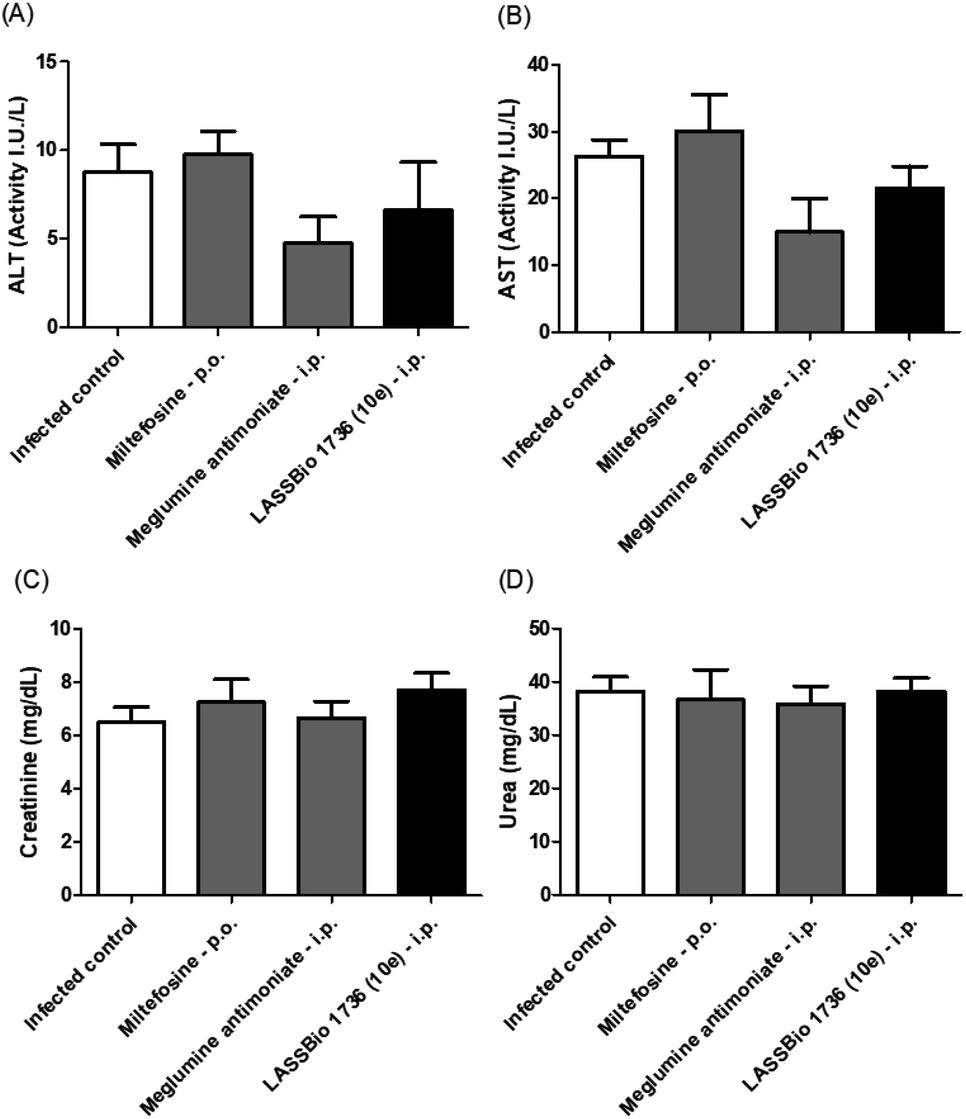 | ||
| Fig. 13 In vivo effect of miltefosine (30 μmol per kg per day × 28 days, p.o.), meglumine antimoniate and LASSBio-1736 (10e) (30 μmol per kg per day × 28 days, i.p.) treatments on serum ALT (A), AST (B), creatinine (C) and urea (D) levels of BALB/c mice infected with L. amazonensis. Values are the mean of the plasma levels of biochemical parameters in five mice in each group and bars represent the standard error of the mean. *P < 0.05, **P < 0.01 vs. control. | ||
Experimental section
Chemistry
Reagents and solvents were purchased from commercial suppliers. The reactions were monitored by thin layer chromatography, which was performed on aluminum sheets pre-coated with silica gel 60 (HF-254, Merck) to a thickness of 0.25 mm. The chromatograms were viewed under ultraviolet light (254–265 nm). 1H NMR and 13C NMR spectra were determined in deuterated dimethyl sulfoxide using a Bruker DPX-200 at 200 MHz, Varian Mercury-300 (300 MHz), Varian MR-400 (400 MHz). Signal multiplicities are represented by: s (singlet), d (doublet), t (triplet), q (quadruplet), m (multiplet) and br (broad signal). Infrared (IR) spectra were obtained with a FTLA 2000-100 spectrophotometer using potassium bromide plates. Melting points of final products were determined with a Quimis 340 apparatus and are uncorrected. The purity of compounds was determined by HPLC (>95%) using the Shimadzu LC20AD apparatus, a Kromasil 100-5C18 (4.6 mm × 250 mm) column and the SPD-M20A detector (Diode Array) at 220–310 nm for quantification of analyte in a 1 mL min−1 constant flux. The injector was programmed to inject a volume of 20 μL. The mobile phases used were: CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 1:1; 4:1; 6:4 and 7:3. The results of elemental analysis were obtained FlashEA 1112 Series instrument (Thermo Scientific, Waltham, USA) from samples previously dried under vacuum. The HRMS analysis was performed using a QExactive™ Hybrid Quadrupole Orbitrap Mass Spectrometer (Thermo Fisher Scientific, Waltham, USA) using electrospray ionization (ESI). Standard working solutions of compounds 9a–e and 10a–e (1 μg mL−1) were prepared with methanol/water 7:3 and fortified with 0.1% formic acid and 5 mM NH4COOH (ammonium formate), and the solutions were used by direct infusion.
:methanol 5%). Then, 10 mL of hexane was added to the reaction, keeping it under stirring for 10 minutes. Following, it was vacuum filtered and washed with hexane to give the product 13 or 14.
:H2O 1:1; 4:1; 6:4 and 7:3. The results of elemental analysis were obtained FlashEA 1112 Series instrument (Thermo Scientific, Waltham, USA) from samples previously dried under vacuum. The HRMS analysis was performed using a QExactive™ Hybrid Quadrupole Orbitrap Mass Spectrometer (Thermo Fisher Scientific, Waltham, USA) using electrospray ionization (ESI). Standard working solutions of compounds 9a–e and 10a–e (1 μg mL−1) were prepared with methanol/water 7:3 and fortified with 0.1% formic acid and 5 mM NH4COOH (ammonium formate), and the solutions were used by direct infusion.
:methanol 5%). Then, 10 mL of hexane was added to the reaction, keeping it under stirring for 10 minutes. Following, it was vacuum filtered and washed with hexane to give the product 13 or 14.
5.1. Preparation of the 4-chloro-phenylcarbamoyl-o-phenylcarbamate (compound 13; LASSBio-1702). Compound 13 was obtained in 70% of yield, as a white solid with mp = 182–185 °C. IR (KBr) (cm−1): 3277 (νNH); 1730 (νC
O); 1661 (νCO); 1010 (δ Ar-Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.62 (s, 1H, Ar-CONH); 9.92 (s, 1H, NHCO); 7.90 (d, 2H, H2, H6, J = 10 Hz); 7.60 (d, 2H, H3, H5, J = 10 Hz); 7.44 (d, 2H, H3′, H5′, J = 8 Hz); 7.26 (t, 1H, H4, J = 6 Hz); 7.18 (d, 2H, H2′, H6′, J = 8 Hz); 97% purity in HPLC (R.T. = 3.6 min, CH3CN:H2O (6:1)); HRMS calculated for C14H11ClN2O3: [M + H]+ = 291.05309, found: m/z 291.05241.
5.2. Preparation of 2,4-dichloro-phenylcarbamoyl-o-phenylcarbamate (compound 14; LASSBio-1492). Compound 14 was obtained in 80% of yield, as a white solid with mp = 180–184 °C; IR (KBr) (cm−1): 3247 (νNH); 1741 (νC
O); 1661 (νCO); 1033 (νAr-Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.49 (s, 1H, ArCONH); 10.05 (s, 1H, NHNHCO), 7.74 (d, 1H, H5, J = 2 Hz); 7.58–7.14 (m, 7H, H3, H6, H2′, H3′, H4′, H5′, H6′); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 163.1 (ArCONH), 154.2 (NHNHCO); 150.6 (C1′); 135.4 (C4), 133.1 (C1); 131.7 (C2); 130.5 (C4′); 129.5 (C3′, C5′); 129.4 (C2′, C6′); 127.4 (C3); 125.4 (C5); 121.5 (C6); 98% purity in HPLC (R.T. = 4.25 min, CH3CN:H2O (7:1)); HRMS calculated for C14H10Cl2N2O3: [M + H]+ = 325.01412, found: m/z 325.01385.
:methanol 5%). The reaction volume was reduced in vacuum system and after addition of ice occurred precipitation of the product, which was filtered under vacuum.
6.1. Preparation of intermediate 4-chloro-phenylcarbamoyl-N-amine-urea (compound 15; LASSBio-1710). Yield: 64%, white solid, mp 187–190 °C; IR (KBr) (cm−1): 3328 (νNH); 1676 (νC
O); 1597 (νCO); 1013 (δ Ar-Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.24 (s, 1H, Ar-CONH); 8.17 (s, 1H, NHCONH); 7.89 (d, H2, H6, J = 8); 7.63 (s, 1H, NHCONH); 7.55 (d, 2H, H3, H5, J = 8); 4.17 (s, 2H, NH2); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.7 (Ar-CO); 160.3 (NHCONH); 136.9 (C4); 132.0 (C1); 129.9 (C2, C6); 128.9 (C3, C5); 97% purity in HPLC (R.T. = 3.6 min, CH3CN:H2O (6:1)); HRMS calculated for C8H9ClN4O2: [M + H]+ = 229.04867, found: m/z 229.04852.
6.2. Preparation of intermediate 2,4-dichloro-phenylcarbamoyl-N-amine-urea (compound 16; LASSBio-1493). Yield: 62%, white solid, mp 190–194 °C; IR (KBr) (cm−1): 3297 (νNH); 1698, (νC
O); 1651 (νCO); 1041 (νC–Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.17 (s, 1H, Ar-CONH); 8.24 (s, 1H, NHCONH); 7.70 (d, 1H, H5, J = 2 Hz); 7.60–7.50 (m, 3H, H3, H6, NHCONH); 4.15 (s, 2H, NH2); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.0 (Ar-CO); 159.3 (NHCONH); 135.0 (C4); 133.5 (C1); 131.7 (C2); 130.8 (C3); 128.3 (C5); 127.2 (C6); 97% purity in HPLC (R.T. = 3.6 min, CH3CN:H2O (6:1)); HRMS calculated for C8H8Cl2N4O2: [M + H]+ = 263.00970, found: m/z 263.00932.
:5–10% methanol) indicated the completion of reaction. The volume of the reaction was reduced under reduced pressure (2/3 of volume), and after addition of ice was observed precipitation of the product which was vacuum filtered and washed with cold water.
7.1. Preparation of compound 4-chloro-phenylcarbamoyl-N-5-nitrofuranimine-urea (compound 9a; LASSBio-1491). Yield: 87%, yellow solid, mp 217–220 °C; IR (KBr) (cm−1): 3246 (νNH); 1659 (νC
O); 1504 (νCO); 1596 and 1351 (δ Ar-NO2); 1015 (δ Ar-Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 11.24 (s, 1H, Ar-CONH); 10.39 (s, 1H, NHCONH); 9.12 (s, 1H, NHCONH); 7.93–7.89 (m, 3H, NCH, H2, H6); 7.80 (d, 1H, H3, J = 4 Hz); 7.58 (d, 2H, H3, H5, J = 8 Hz); 7.31 (d, 1H, H4, J = 4 Hz); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.3 (Ar-CONH); 154.5 (C1′); 152.8 (C2′); 151.4 (NHCONH); 136.6 (CNH); 131.3 (C4); 129.3 (C2, C6); 129.2 (C1); 128.6 (C3, C5); 115.1 (C3′); 112.6 (C4′); 99% purity in HPLC (R.T. = 4.98 min, CH3CN:H2O (1:1), λ = 370 nm); HRMS calculated for C13H10ClN5O5: [M + H]+ = 352.04432, found: m/z = 352.04417.
7.2. Preparation of compound 4-chloro-phenylcarbamoyl-N-2-hydroxyphenylimine-urea (compound 9b; LASSBio-1704). Yield: 53%, white solid, mp >250 °C; IR (KBr) (cm−1): 3309 (νNH); 1701 and 1545 (νC
O); 1664 and 1484 (δ N–H); 1011 (δ Ar-Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.65 (s, 1H, Ar-CONH); 10.30 (s, 1H, NHCONH); 10.02 (s, 1H, OH); 8.98 (s, 1H, NHCONH); 8.22 (s, 1H, NCH); 7.89 (t, 3H, H2, H6, J = 8 Hz, H2′); 7.57 (d, 2H, H3, H5, J = 8 Hz); 7.17 (t, 1H, H4′, J = 6 Hz); 6.88–6.81 (m, 2H, H3′, H5′); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.2 (Ar-CONH), 156.0 (C6′); 155.1 (NHCONH); 139.3 (CNH); 136.5 (C4); 131.5 (C1); 130.4 (C4′); 129.3 (C2, C6); 128.5 (C3, C5); 127.1 (C2′); 120.2 (C1′); 119.1 (C3′), 116.0 (C5′); 99% purity in HPLC (R.T. = 4.86 min, CH3CN:H2O (4:6), λ = 320 nm). HRMS calculated for C15H13ClN4O3: [M + H]+ = 333.07489, found: m/z = 333.07432.
7.3. Preparation of compound 4-chloro-phenylcarbamoyl-N-3-hydroxyphenylimine-urea (compound 9c; LASSBio-1709). Yield: 91%, white solid, mp 225–227 °C; IR (KBr) (cm−1): 3342 (νNH); 1692 (νC
O); 1655 (νCO); 1015 (νAr-Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.62 (s, 1H, Ar-CONH); 10.28 (s, 1H, NHCONH); 9.47 (s, 1H, OH); 8.97 (s, 1H, NHCONH); 7.90 (d, 2H, H2, H6, J = 4 Hz); 7.80 (s, 1H, NCH); 7.57 (d, 2H, H3, H5, J = 4 Hz); 7.17 (m, 3H, H2′, H3′, H6′); 6.77 (d, 1H, H4′, J = 4 Hz); 98% purity in HPLC (R.T. = 4.24 min, CH3CN:H2O (1:1), λ = 279 nm). HRMS calculated for C15H13ClN4O3: [M + H]+ = 333.07489, found: m/z = 333.07407.
7.4. Preparation of compound 4-chloro-phenylcarbamoyl-N-4-hydroxyphenylimine-urea (compound 9d; LASSBio-1706). Yield: 86%, white solid, mp 223–225 °C; IR (KBr) (cm−1): 3369 (νNH); 1701 (νC
O); 1651 (νCO); 1014 (νAr-Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.49 (s, 1H, Ar-CONH); 10.28 (s, 1H, NHCONH); 9.77 (s, 1H, OH); 8.92 (s, 1H, NHCONH); 7.90 (d, 2H, H2, H6, J = 8 Hz); 7.79 (s, 1H, NCH); 7.59 (m, 4H, H3, H5, H2′, H6′); 6.77 (d, 2H, H3′, H5′, J = 8 Hz); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.2 (Ar-CONH); 158.7 (C4′); 155.3 (NHCONH); 141.2 (CNH); 136.5 (C4); 131.5 (C1); 129.3 (C2, C6); 128.5 (C3, C5, C2′, C6′); 125.6 (C1′); 115.4 (C3′, C5′); 99% purity in HPLC (R.T. = 2.95 min, CH3CN:H2O (7:1), λ = 287 nm); HRMS calculated for C15H13ClN4O3: [M + H]+ = 333.07489, found: m/z = 333.07455.
7.5. Preparation of compound 4-chloro-phenylcarbamoyl-N-4-trifluoromethylphenylimine-urea (compound 9e; LASSBio-1737). Yield: 85%, white solid, mp 201–203 °C; IR (KBr) (cm−1): 3338 (νNH); 1701 (νC
O); 1663 (νCO); 1015 (νAr-Cl); 1131 and 1100 (C–F); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.95 (s, 1H, Ar-CONH); 10.34 (s, 1H, NHCONH); 9.24 (s, 1H, NHCONH); 8.05 (d, 2H, H2, H6, J = 8 Hz); 7.96 (d, 2H, H2′, H6′, J = 8 Hz); 7.90 (s, 1H, NCH); 7.75 (d, 2H, H3, H5, J = 8 Hz); 7.60 (d, 2H, H3′, H5′, J = 8 Hz); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.2 (Ar-CONH); 155.0 (NHCONH); 139.1 (NCH); 138.5 (C4); 136.5 (C1′); 131.4 (C4′); 129.3 (C2, C6, C2′, C6′); 128.5 (C3, C5, C1); 127.3 (C3′, C5′); 125.3 (CF3); 99% purity in HPLC (R.T. = 5.95 min, CH3CN:H2O (6:1), λ = 320 nm). HRMS calculated for C16H12ClF3N4O2: [M + H]+ = 385.06736, found: m/z = 385.06695.
7.6. Preparation of compound 2,4-dichloro-phenylcarbamoyl-N-5-nitrofuranimine-urea (compound 10a; LASSBio-1703). Yield: 87%, yellow solid, mp 222–225 °C; IR (KBr) (cm−1): 3387 (νNH); 1727 (νC
O); 1667 (νCO); 1015 (νC–Cl); 1244 and 1281 (NO); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 11.22 (s, 1H, Ar-CONH); 10.22 (s, 1H, NHCONH); 9.18 (s, 1H, NHCONH); 7.86 (s, 1H, NCH); 7.79 (d, 1H, H3′, J = 4 Hz); 7.71 (s, 1H, H5); 7.57 (m, 2H, H3, H6); 7.32 (d, 1H, H4′, J = 4 Hz); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.2 (Ar-CONH); 154.0 (NHCONH); 152.7 (C2′); 151.2 (C1′); 135.1 (CNH); 133.3 (C4); 131.7 (C1); 130.7 (C2); 129.3 (C5); 129.1 (C6); 127.2 (C3); 115.0 (C3′); 112.5 (C4′); 99% purity in HPLC (R.T. = 3.41 min, CH3CN:H2O (7:1), λ = 370 nm); HRMS calculated for C13H9Cl2N5O5: [M + H]+ = 386.00535, found: m/z = 386.00500.
7.7. Preparation of compound 2,4-dichloro-phenylcarbamoyl-N-2-hydroxyphenylimine-urea (compound 10b; LASSBio-1705). Yield: 78%, white solid, mp 210–212 °C; IR (KBr) (cm−1): 3436–1852 (νOH); 3232 (νNH); 1708 (νC
O); 1655 (νCO); 1046 (νC–Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.66 (s, 1H, Ar-CONH); 10.16 (s, 1H, NHCONH); 10.03 (s, 1H, OH); 9.11 (s, 1H, NHCONH); 8.23 (s, 1H, NCH); 7.92 (d, 1H, H2′, J = 8 Hz); 7.73 (d, 1H, H5); 7.60 (m, 2H, H3, H6); 7.20 (t, 1H, H4′, J = 6 Hz); 6.86–6.79 (m, 2H, H3′, H5′); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.3 (Ar-CO); 155.9 (C6′); 154.8 (NHCONH); 139.1 (NCH); 135.1 (C4); 133.5 (C1); 131.7 (C2); 130.8 (C4′); 130.5 (C3); 129.4 (C5); 127.3 (C6); 127.1 (C2′); 120.3 (C1′); 119.1 (C3′); 116.0 (C5′); 99% purity in HPLC (R.T. = 3.52 min, CH3CN:H2O (7:1), λ = 276 nm); HRMS calculated for C15H12Cl2N4O3: [M + H]+ = 367.03592, found: m/z = 367.03557.
7.8. Preparation of compound 2,4-dichloro-phenylcarbamoyl-N-3-hydroxyphenylimine-urea (compound 10c; LASSBio-1708). Yield: 96%, white solid, mp 223–225 °C; 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.62 (s, 1H, Ar-CONH); 10.12 (s, 1H, NHCONH); 9.48 (s, 1H, OH); 9.05 (s, 1H, NHCONH); 7.80 (s, 1H, N
CH); 7.71 (s, 1H, H5); 7.70–7.50 (m, 2H, H3, H6); 7.19–16 (m, 3H, H2′, H3′, H6′); 6.76 (d, 1H, H4′, J = 8 Hz); 98% purity in HPLC (R.T. = 3.12 min, CH3CN:H2O (7:1), λ = 279 nm). HRMS calculated for C15H12Cl2N4O3: [M + H]+ = 367.03592, found: m/z = 367.03550.
7.9. Preparation of compound 2,4-dichloro-phenylcarbamoyl-N-4-hydroxyphenylimine-urea (compound 10d; LASSBio-1707). Yield: 90%, white solid, mp 225–227 °C; IR (KBr) (cm−1): 3362 (νNH); 1694 (νC
O); 1651 (νCO); 1098 (νC–Cl); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.53 (s, 1H, Ar-CONH); 10.16 (s, 1H, NHCONH); 9.83 (s, 1H, OH); 9.05 (s, 1H, NHCONH); 7.84 (s, 1H, NCH); 7.76 (s, 1H, H5); 7.67 (d, 2H, H2′, H6′, J = 10 Hz); 7.58–7.53 (m, 2H, H5, H6); 6.82 (d, 2H, H3′, H5′, J = 10 Hz); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.3 (Ar-CONH); 158.7 (C4′); 155.0 (NHCONH); 141.2 (NCH); 135.1 (C4); 133.6 (C1); 131.7 (C2); 130.8 (C3); 129.4 (C5); 128.5 (C6); 127.3 (C1′); 125.5 (C2′, C6′); 115.4 (C3′, C5′); 99% purity in HPLC (R.T. = 3.05 min, CH3CN:H2O (7:1), λ = 287 nm); HRMS calculated for C15H12Cl2N4O3: [M + H]+ = 367.03592, found: m/z = 367.03563.
7.10. Preparation of compound 2,4-dichloro-phenylcarbamoyl-N-4-trifluoromethylphenylimine-urea (compound 10e; LASSBio-1736). Yield: 60%, white solid, mp 213–215 °C; IR (KBr) (cm−1): 3555 (νNH); 1703 (νC
O); 1655 (νCO); 1015 (νC–Cl); 1164–1105 (C–F); 1H-NMR (200 MHz, DMSO-d6) δ (ppm): 10.95 (s, 1H, Ar-CONH); 10.17 (s, 1H, NHCONH); 9.33 (s, 1H, NHCONH); 8.09 (d, 2H, H2′, H6′, J = 10 Hz); 7.97 (s, 1H, CNH); 7.77–7.74 (m, 3H, H3, H5, H6); 7.55 (m, 2H, H3′, H5′); 13C-NMR (50 MHz. DMSO-d6) δ (ppm): 165.3 (Ar-CONH); 154.7 (NHCONH); 139.1 (NCH); 138.5 (C1′); 135.2 (C4); 133.5 (C1); 131.7 (C2′, C6′); 130.80 (C2); 129.4 (C3′, C5′); 129.3 (C5); 127.4 (C6, C3); 125.3 (C4′); 121.4 (CF3); 99% purity in HPLC (R.T. = 5.82 min, CH3CN:H2O (6:1), λ = 340 nm); HRMS calculated for C16H11Cl2F3N4O2: [M + H]+ = 419.02839, found: m/z = 419.02792.
000 ng mL−1 solubilized in DMSO) was added in 195 μL of rat plasma solution (1:1) diluted with PBS (pH 7.4). After vortexing the mixture was placed in a shaker at 37 °C under vigorous stirring for 0, 30, 60, 120 and 240 minutes. After each reaction time, 600 μL of cold methanol containing 100 ng mL−1 of ketoconazole (internal standard) were added to the wells to stop the reaction. The solution was mixed and centrifuged at 13000 rpm for 15 min. The supernatant (400 μL) were placed in 2 mL vials, and analyzed by UPLC-MS/MS.The analysis was performed using a Thermo Scientific Dionex UltiMate 3000 UHPLC liquid chromatography system (Thermo Fisher Scientific, Waltham, USA) coupled to a TSQ Quantiva Triple-Stage Quadrupole Mass Spectrometer, using electrospray ionization (ESI) and equipped with a degasser and TriPlus RSH autosampler (Thermo Fisher Scientific, Waltham, USA). A Syncronis column (Thermo, Waltham, USA; C8, 2.1 × 50 mm and 1.7 μM particle size) was employed and maintained at 40 °C. The mobile phase was prepared with Milli-Q water containing 0.1% formic acid and 5 mM ammonium formate (A) and with methanol containing 0.1% formic acid (B), and an 8 μL injection volume was used. Gradient elution at a flow rate of 0.3 mL min−1 was performed as follows: 1 min isocratic 90% A, decreased to 20% A at 10 min, and reduced to 0% A at 12.0 min through 14 min. The percentage of A was then increased to the initial condition of 90% at 14.1 min and was maintained until 16 min to equilibrate the column before the next injection. The total analysis time per sample was 16.0 min.
Detection in single reaction monitoring (SRM) mode was used for determination of the LASSBio-1736 (10e) m/z 419.1 → 205.0, benzocaine m/z 166.1 → 137.9 and ketoconazole (IS) 532.1 → 490.3, with a tolerance of mass deviation of 6 ppm, operating in positive ionization mode. The data were acquired using TraceFinder 3.2. software (Thermo Fisher Scientific, Waltham, USA), and analyzed using GraphPad Prism 5 program.
Biology
To assess the activity of the test derivatives against the amastigote stage of L. amazonensis and L. braziliensis the model of infection in coverglass was realized.31 The murine macrophages (J774.A1 cell line) were prepared in 24-well cell culture plate (flat bottom, clear, Corning) at 2 × 105 adherent cells per well, infected with 2 × 106 promastigotes in glass coverslips placed inside 1 mL medium culture. The cultures were cultured or not with the test derivatives or reference drugs, and kept for 24 h at 37 °C, 5% CO2. After 24 h of incubation, the coverslips were washed, stained with Giemsa–May–Grünwald, and intracellular amastigotes were counted in 100 macrophages, using an optical microscope, on the objective of 100×. Thus, during the counting of 100 macrophages, the amount of amastigotes present within these defence cells was also determined. Data obtained from in vitro experiments were expressed as the mean ± S.E.M. of duplicate cultures of representative assays. Statistical differences between the treated and the control groups were evaluated by ANOVA and Dunnett hoc tests. Differences with a p value < 0.05 or lower were considered significant.
Experimental conclusions
A unique peptide mimetic framework was proposed for the first time based on molecular hybridization between amide and semicarbazone subunits. From this framework, a series of carbamoyl-N-aryl-imine-ureas were prepared and evaluated. Compound 10e (LASSBio-1736) was identified as a new antileishmanial drug candidate, displaying plasma stability, cytotoxicity against amastigote forms of L. amazonensis and L. braziliensis, and leishmanicidal activity in a cutaneous leishmaniasis murine model, without preliminary evidence of hepatic or renal toxicity.These data also highlight perspectives for investigating the potential of 10e (LASSBio-1736) as a cysteine protease inhibitor, as well as encouraging the extension of these studies to determine the mechanism of induction of parasite death and perform LASSBio-1736 (10e) structure optimization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Leishmania collection of the Oswaldo Cruz Institute, Dr Eduardo Caio Torres dos Santos at Oswaldo Cruz Institute – Fiocruz and Dr Valéria de Matos Borges at Gonçalo Moniz Research Center – Fiocruz for having provided Leishmania species. We also thanks CNPQ (BR), CAPES (BR), FAPERJ (RJ) and FAPEAL (AL) by financial support.References
- WHO, Epidemiological situation, available at: https://www.who.int/leishmaniasis/burden/en/, accessed 7 June 2019 Search PubMed.
- A. Oryan and M. Akbari, Asian Pac. J. Trop. Med., 2016, 9, 925–932 CrossRef CAS PubMed
.
- M. Akhoundi, T. Downing, J. Votýpka, K. Kuhls, J. Lukeš, A. Cannet, C. Ravel, P. Marty, P. Delaunay, M. Kasbari, B. Granouillac, L. Gradoni and D. Sereno, Mol. Aspects Med., 2017, 57, 1–29 CrossRef CAS PubMed
- J. H. McKerrow, J. C. Engel and C. R. Caffrey, Bioorg. Med. Chem., 1999, 7, 639–644 CrossRef CAS
- M. Sajid and J. H. McKerrow, Mol. Biochem. Parasitol., 2002, 120, 1–21 CrossRef CAS
- H. J. Atkinson, P. C. Babbitt and M. Sajid, Trends Parasitol., 2009, 25, 573–581 CrossRef CAS PubMed
- R. W. W. Hooft, Front. Pharmacol., 1998, 7, 107–119 Search PubMed
- G. J. Moore, Trends Pharmacol. Sci., 1994, 15, 124–129 CrossRef CAS
- R. W. W. Hooft, Med. Chem., 1998, 6, 189–194 Search PubMed
- E. Salas-Sarduy, L. U. Landaburu, J. X. Karpiak, K. P. Madauss, J. J. Cazzulo, F. Agüero and V. E. Alvarez, Sci. Rep., 2017, 7, 12073–12085 CrossRef PubMed
- P. V. Desai, A. Patny, Y. Sabnis, B. Tekwani, J. Gut, P. Rosenthal, A. Srivastava and M. Avery, J. Med. Chem., 2004, 47, 6609–6615 CrossRef CAS PubMed
- M. Nakamura and J. Inoue, Bioorg. Med. Chem. Lett., 2002, 12, 1603–1606 CrossRef CAS PubMed
- R. W. W. Hooft, Mol. Med., 1998, 6, 660–669 Search PubMed
- N. C. Fonseca, L. F. da Cruz, F. da Silva Villela, G. A. do Nascimento Pereira, J. L. de Siqueira-Neto, D. Kellar, B. M. Suzuki, D. Ray, T. B. de Souza, R. J. Alves, P. A. S. Júnior, A. J. Romanha, S. M. F. Murta, J. H. McKerrow, C. R. Caffrey, R. B. de Oliveira and R. S. Ferreira, Antimicrob. Agents Chemother., 2015, 59, 2666–2677 CrossRef CAS PubMed
- N. Raghav and R. Kaur, Med. Chem. Res., 2014, 23, 4669–4679 CrossRef CAS
- R. W. W. Hooft, PLoS One, 1998, 8, e77460 Search PubMed
- M. A. Alves, A. C. De Queiroz, M. S. Alexandre-Moreira, J. Varela, H. Cerecetto, M. González, A. C. Doriguetto, I. M. Landre, E. J. Barreiro and L. M. Lima, Eur. J. Med. Chem., 2015, 100, 24–33 CrossRef CAS PubMed
- P. Yogeeswari, D. Sriram, R. Thirumurugan, J. V. Raghavendran, K. Sudhan, R. K. Pavana and J. Stables, J. Med. Chem., 2005, 48, 6202–6211 CrossRef CAS PubMed
- P. C. Lima, L. M. Lima, K. C. da Silva, P. H. Léda, A. L. de Miranda, C. A. Fraga and E. J. Barreiro, Eur. J. Med. Chem., 2000, 35, 187–203 CrossRef CAS PubMed
- L. I. Camara Coelho, M. Paes, J. A. Guerra, M. das Graças Barbosa, C. Coelho, B. Lima, M. E. Brito and S. P. Brandão Filho, Parasitol. Res., 2011, 108, 671–677 CrossRef PubMed
- R. de Oliveira
Lopes, M. A. Alves, R. O. Pinheiro, C. S. Freitas, F. Q. Cunha, E. J. Barreiro and L. M. Lima, Lett. Drug Des. Discov., 2018, 15, 766–777 CrossRef
- L. Di, E. H. Kerns, Y. Hong and H. Chen, Int. J. Pharm., 2005, 297, 110–119 CrossRef CAS PubMed
- R. W. W. Hooft, Collect data collection software, 1998 Search PubMed
- J. T. Granato, J. A. Santos, S. L. Calixto, N. P. Silva, J. S. Martins, A. D. Silva and E. S. Coimbra, Biomed. Pharmacother., 2018, 106, 1082–1090 CrossRef PubMed
- D. L. Sacks and P. C. Melby, Curr. Protoc. Immunol., 2015, 108, 19.2.1–19.2.24 Search PubMed
- C. W. Carter and R. M. Sweet, Methods in Enzymology: macromolecular crystallography, Academic Press, 1997 Search PubMed
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 1985, 189, 189–190 Search PubMed
- M. P. Nunes, L. Cysne-Finkelstein, B. C. Monteiro, D. M. de Souza, N. A. Gomes and G. A. DosReis, Microbes Infect., 2005, 7, 78–85 CrossRef CAS
- J. C. Melo Pereira, V. Carregaro, D. L. Costa, J. Santana da Silva, F. Q. Cunha and D. W. Franco, Eur. J. Med. Chem., 2010, 45, 4180–4187 CrossRef CAS PubMed
- C. Taswell, in Cell Separation, Elsevier, 1987, pp. 109–145 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1824217. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra00287a |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2020 |