
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of core titanium crystal dimension and crystal phase on ROS generation and tumour accumulation of transferrin coated titanium dioxide nanoaggregates
Daniel D.
Lane
ab,
Kvar C. L.
Black
a,
Ramesh
Raliya
 c,
Nathan
Reed
c,
Nalinikanth
Kotagiri
ad,
Rebecca
Gilson
ab,
Rui
Tang
a,
Pratim
Biswas
c and
Samuel
Achilefu
*abef
c,
Nathan
Reed
c,
Nalinikanth
Kotagiri
ad,
Rebecca
Gilson
ab,
Rui
Tang
a,
Pratim
Biswas
c and
Samuel
Achilefu
*abef
aOptical Radiology Laboratory, Department of Radiology, Washington University in St. Louis, St. Louis, Missouri 63110, USA. E-mail: achilefu@wustl.edu
bDepartment of Biomedical Engineering, Washington University in St. Louis, St. Louis, Missouri 63130, USA
cAerosol and Air Quality Research Laboratory, Department of Energy, Environmental, and Chemical Engineering, Washington University in St. Louis, St. Louis, MO 63130, USA
dJames L. Winkle College of Pharmacy, University of Cincinnati, Cincinnati, Ohio 45267, USA
eDepartment of Medicine, Washington University in St. Louis, St. Louis, Missouri 63110, USA
fDepartment of Biochemistry and Molecular Biophysics, Washington University in St. Louis, St. Louis, Missouri 63110, USA
First published on 23rd June 2020
Abstract
Radionuclide-stimulated therapy (RaST), which is enhanced by Cherenkov radiation, has enabled deep tissue stimulation of UV photosensitizers, providing a new path for cancer treatment. Previous reports have shown UV-active titanium dioxide (TiO2) nanoparticles (NPs) modified with transferrin inhibit tumour growth after orthogonal treatment with Cherenkov radiation-emitting radionuclides such as 18F-fluorodeoxyglucose (FDG). However, poor understanding of TiO2 NP parameters on reactive oxygen species (ROS) generation and particle distribution limits effective therapy. Here we sought to delineate the effects of crystal phase and core TiO2 crystal dimension (cTd) on ROS production and particle morphology. We prepared Transferrin (Tf)–TiO2 nanoaggregates (NAGs) using solvothermally synthesized cTd sizes from 5 to 1000 nm diameter and holo- or apo-transferrin. Holo-transferrin was unable to stabilize TiO2 NPs while apo-transferrin stabilized TiO2 into uniform nanoaggregates (NAGs), which were invariant with differing cTd, averaging 116 ± 1.04 nm for cTds below 100 nm. ROS production increased from 5 to 25 nm cTd, attaining a peak at 25 nm before decreasing with larger sizes. The supra-25 nm ROS production decrease was partially driven by a ∼1/r3 surface area decline. Additionally, amorphous TiO2 of equal core size exhibited a 2.6-fold increase in ROS production compared to anatase NAGs, although limited stability halted further use. Although both 5 and 25 nm anatase cTds formed similarly sized NAGs, 5 nm anatase showed a four-fold higher tumour-to-muscle ratio than the 25 nm NPs in tumour-bearing mice, demonstrating the intricate relationships between physical and biological properties of NAGs. The combined in vivo and ROS results demonstrate that anatase crystals and cTd size of 25 nm or less are ideal particle parameters to balance biodistribution with ROS production efficiency.
Introduction
Photosensitizing nanoparticles (NPs) are promising cancer therapy agents providing broad molecular damage via reactive oxygen species (ROS). Historically, however, limited optical light penetration in tissues, photo-bleaching, and skin sensitization confined the use of existing small molecule-based photosensitizers to surface weighted therapeutic interventions.1–6 The advent of radionuclide-stimulated therapy (RaST) has enabled the activation of UV light absorbing photosensitizers for deep tissue cancer treatment, overcoming many of the current limitations in conventional photodynamic therapy (PDT).7a,b RaST, which is also known as Cherenkov radiation induced therapy to reflect the impact of Cherenkov radiation on the therapeutic effect,7a relies on a prodrug-like colocalization of a photosensitizer and radionuclide in tumours, the timing of which can be manipulated by particle parameters and radionuclide choice. One of the earliest RaST demonstrations utilized TiO2 NPs as a nanophotosensitizer and 18F or 64Cu as the radiopharmaceutical.Titanium dioxide (TiO2) acts as a regenerative photocatalyst able to facilitate UV absorption for type I and type II photosensitization. The large bandgap of TiO2 traps higher redox potential electrons/holes than the current generation of near-infrared photosensitizers.8,9 This increased energy can generate reactive oxygen species (ROS) directly from water, reducing the demand for an oxygenated environment, which is important for cancer treatments since many tumours are characterized by hypoxic microenvironments.10,11 The most energetic of these species is the hydroxyl radical whose redox energy from water is +2.27 V, an energy level too high for near-infrared photosensitizers.8,12 Additionally, the absorbance band of TiO2 is ideal for Cherenkov radiation, as broadband UV emission can be effectively absorbed by TiO2.13–16
The efficiency of photocatalyst driven ROS generation depends on the absorption/electrical conjugation of desired species to the surface and limiting non-catalytic excitons recombination. In an aqueous environment, TiO2 rapidly traps electrons/holes in oxygen/titanium defects, respectively, limiting recombination.17–19 This trapping relies on the surface defects of TiO2, specifically acidic titanium or hydroxyl sites, with higher surface defect rates increasing the overall photoactivity.20 Furthermore, hole traps are water/hydroxyl binding sites allowing for rapid oxidation to hydroxyl radicals. The electron traps are surface oxygen defects and reduce molecular oxygen to singlet oxygen and other ROS.12
The surface defect rates for these traps has been associated with several factors, including TiO2's bulk crystal phase, core TiO2 crystal dimension (cTd), and surface coating.21 Early investigations of the natural crystal phases, anatase and rutile, revealed anatase to have a higher ROS production potential.22,23 This is attributed to the higher bandgap and surface potential of anatase, as well as increased trap density on the (0,0,1) plane.24,25 Some evidence suggests amorphous/mixed crystal phase TiO2 has a higher ROS quantum yield due to undefined surface regions with higher total surface defect rate.26 For example, P25 Degussa TiO2, the gold standard, is a mixed phase structure which is believed to increase its overall photocatalytic yield.27,28 Overall, the cTd has been heavily investigated across many TiO2 photocatalytic systems with varying particle sizes, the maximum conversion rates usually falling into a 7–40 nm range.26,29,30
In addition to ROS production, the size of an NP influences its biodistribution and tumour accumulation. Biological factors that affect NP in vivo bio-distribution include the enhanced permeability and retention (EPR) effect in tumour leaky vasculature, as well as liver, kidney or spleen filtration.31–33 These considerations cap the upper size limit for in vivo application of polystyrene NPs to about 150 nm. In general, a lower hydrodynamic diameter limit of 7 nm minimizes nephron filtration and increases circulation time, while the upper 150 nm size avoids macrophage opsonization and maintains EPR extravasation.31,34 For solid core NPs such as TiO2, the size range is modified by hydrodynamic radius increases from protein adsorption.35,36 A previous study using quantum dots showed that about 5.5 nm is the limit for hard nanostructures in renal excretion.37
In this study, we investigated how crystal size and structure influence particle geometry, ROS production, and biodistribution in tumour-bearing mice. We prepared core TiO2 NAGs in the 5–1000 nm range and stabilized the ensuing NAGs with transferrin (Tf), a protein that binds strongly to Ti4+.31–33 Our results demonstrate that, regardless of the initial TiO2 core size, all the NAGs from cTds below 100 nm have similar sizes. While the 25 nm NAGs exhibited high ROS production, the 5 nm NAGs showed a higher tumour-to-muscle ratio in tumour-bearing mice.
Experimental methods
Materials
Apo-Transferrin was purchased from Athens Research and Technology (Athens, GA), otherwise all chemicals were purchased from Sigma-Aldrich (St. Louis, MO).TiO2 core synthesis and crystal structure
TiO2 cTds of various sizes (5–1000 nm diameter) were prepared by hydrothermal reaction of titanium alkoxide (titanium isopropoxide: TTIP) stabilized in an acidic ethanol–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 1:8 v/v) solution by modifying the method previously proposed.38 The 25 nm anatase TiO2 synthesis is used as an example for the rest of the Experimental section. The pH of an ethanol and water solution was adjusted to 0.7 with 1 M nitric acid. 100 μL of 0.02 M TTIP (97%) was added drop-wise to this solution. The reaction was magnetically stirred at 400 rpm (25 °C) followed by 4 h thermal treatment at 220 °C. Synthesized crystals were washed several times with ethanol to remove un-reacted alkoxide. The materials were then either dried under vacuum or dispersed in a solvent for further use. For different crystal structures the calcination conditions were altered to 2 h at 120 °C for anatase–rutile and 250 °C for 4 h for amorphous.39
:2 to 1:8 v/v) solution by modifying the method previously proposed.38 The 25 nm anatase TiO2 synthesis is used as an example for the rest of the Experimental section. The pH of an ethanol and water solution was adjusted to 0.7 with 1 M nitric acid. 100 μL of 0.02 M TTIP (97%) was added drop-wise to this solution. The reaction was magnetically stirred at 400 rpm (25 °C) followed by 4 h thermal treatment at 220 °C. Synthesized crystals were washed several times with ethanol to remove un-reacted alkoxide. The materials were then either dried under vacuum or dispersed in a solvent for further use. For different crystal structures the calcination conditions were altered to 2 h at 120 °C for anatase–rutile and 250 °C for 4 h for amorphous.39
The size and morphology of the TiO2 cTds were investigated using transmission electron microscopy (TEM) with an 80 keV FEI Tecnai Spirit Transmission Electron Microscope. A 2 μL sample was placed on an ultrathin lacey carbon grid, 400 mesh (Ted Pella Inc.) and allowed to sit for 5 min before removal of the droplet via wicking with a Chemwipe and vacuum drying. To visualize the Tf coating a dried sample was then stained for 1 min with uranyl acetate, 3 μL drop of 4% (w/v), before blotting and vacuum drying to limit crystal contamination within the TEM. X-ray diffraction (XRD) was performed to confirm the crystal phase of TiO2 NPs.40 25 mg of dry TiO2 powder was added to a low background silicon sample holder and scanned (coupled two-theta/theta) from 5–60 degrees with a 15 rpm sample rotation and anti-scatter fins in place. The data was analysed with Bruker DIFFRAC.EVA program.
Stabilization of Tf–TiO2 NAGs
TiO2 cTd solutions (1 mg mL−1) were prepared in 1× Dulbecco's phosphate-buffered saline (DPBS) and vortexed to homogeneity. Immediately, Tf was added at a mass ratio of 1:3 (TiO2 to Tf) to the solution and mixed until dissolved. This solution was then separated into 2 mL aliquots before sonication with a small-bore probe at 3 W output for 40 seconds (kept below 50 °C from sonic heating) to form Tf–TiO2 NAGs. Post sonication, the aliquots were filtered through a Millex-HV PVDF 0.45 μm filter, unless otherwise stated. Note that due to size, >200 nm cores were not filtered, but instead centrifuged at 1k for 1 min before resuspension under sonication.
Characterization of Tf–TiO2 particles
Hydrodynamic diameter and zeta potential of the suspensions were both determined with the Malvern Zetasizer Nano ZS. Particle measurements were performed in a 2 cm path-length quartz cuvette and a folded capillary zeta cell (Malvern Instruments Ltd), respectively. A triplicate of each sample was diluted to 0.01 mg mL−1 TiO2 to produce an optically clear solution of the particles in DPBS for dynamic light scattering (DLS) and deionized water (diH2O) for zeta potential measurements. Z-Average size and polydispersity index (PDI) of the TiO2 NAGs were obtained with an average of 12 runs. TEM was performed to validate morphology and size of the coated particles. Quantification of protein coating was carried out using a Pierce BCA Protein Assay Kit to determine Tf concentration (1) before coating, (2) amount remaining in the supernatant after coating and centrifugation, and (3) the amount remaining in the centrifuged particle sample. Long-term stability was quantified using three separate 2 mL samples at 1 mg mL−1 of Sigma-Aldrich Tf–TiO2, prepared in water. These were stored at 4 °C and 100 μL samples were diluted to 0.01 mg mL−1 in water and analysed on DLS as described above.ROS quantification
Dichlorofluorescein diacetate (DCF-DA) was used to quantify general ROS production from Tf–TiO2, and hydroxyphenyl fluorescein (HPF) was used to detect hydroxyl radicals.41 DCF-DA was activated to DCF by adding DCF-DA (45 μL, 5.55 mM) in dimethyl sulfoxide (DMSO) to NaOH (5 μL, 1 N) and mixed for 10 min, producing a 5 mM stock that was refreshed for each sample run. DCF and HPF were added to TiO2 samples from DMSO with 5 mM starting concentrations. The Tf–TiO2 samples were prepared as above but not filtered to preserve the concentration more closely, relying on orbital shaking between reads to maintain homogeneity. The initial Tf–TiO2 formulation (1 and 3 mg mL−1 TiO2 and Tf, respectively) was then diluted to 0.01 mg mL−1 TiO2 in 1 mL and 5 μM HPF or DCF was added.An uncoated, black walled, flat and clear bottom 96 well plate (Greiner Bio-One) containing 150 μL sample per well was used for ROS quantification. Each plating was performed in triplicate with a well geometry that allowed an average power of 1.9 mW across each triplicate set. For comparison between runs, a bare 25 nm TiO2 DCF control was always plated to quantify variability. After loading, the plate was shaken for 20 seconds in a double orbital pattern and analysed on a plate reader (BioTek Synergy Neo2) using 487 nm excitation and 528 nm emission. Subsequently, the plate was automatically exposed to UV light for 80 seconds before being shaken again, repeating the process. This was carried out for a total of 30 min for each plate and the data was compiled into pseudo-first-order kinetic curves for reporting.
In vivo tumour model and biodistribution
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals at Washington University in St. Louis and the protocol used was approved by Washington University's Institutional Animal Care and Use Committee (IACUC). Biodistribution of different NACs was quantified on breast cancer (4T1)-bearing BALB/c mice (n = 4/group). 4T1 (1 × 106 cells) were injected subcutaneously and grown until 10 mm by calliper. Freshly prepared 5 and 25 nm Tf–TiO2 NACs in PBS were injected via tail vein (100 μL, 1 mg mL−1). The animals were euthanized 24 h post-injection. To quantify the TiO2 biodistribution, major organs and tumour tissue were harvested, homogenized, and degraded using nitric acid and H2O2 (hydrogen peroxide) and Ti content in each organ was quantified by inductively coupled plasma mass spectrometry (ICP-MS).Results and discussion
Synthesis and characterization of TiO2 core NPs
We prepared different sizes (5, 15, 25, 30, 50, 100, 200, and 1000 nm) of TiO2 cTd as described above (Fig. 1). The electrophoretic zeta potential of the cores was between −19 and −35 mV. Using the 25 nm NPs, we synthesized and characterized three different crystal phases – anatase, anatase–rutile mix, and amorphous cTds. DLS measurements showed an average PDI of 0.15 ± 0.035 at pH 5.8 in ethanol across all cores. XRD analysis shows three core crystal phases matching 26°, 37°, and 55° peaks for anatase and 36°, 42°, and 54° peaks for rutile (Fig. 1E). Amorphous particles have weaker facets of both phases due to the lack of annealing during synthesis. TEM of anatase cores match the expected pseudo-octahedral crystal shape, appearing as slight elongated, rhomboid shapes. Preferential elongation along a single axis was observed as the size of particle increased, creating NPs with a standard aspect ratio of near 2:1 for 200 nm tetragonal crystals (Fig. 2B). This was further shown in the diffraction bands (Fig. 1A) whose graph analyses exhibited two patterns with a spacing of 0.3312 ± 0.0811 nm and 0.3568 ± 0.0993 nm for each (∼3.5 Å literature).42 The different sizes and crystal phases provided diverse parameters to assess their ROS-generating properties and biodistribution.
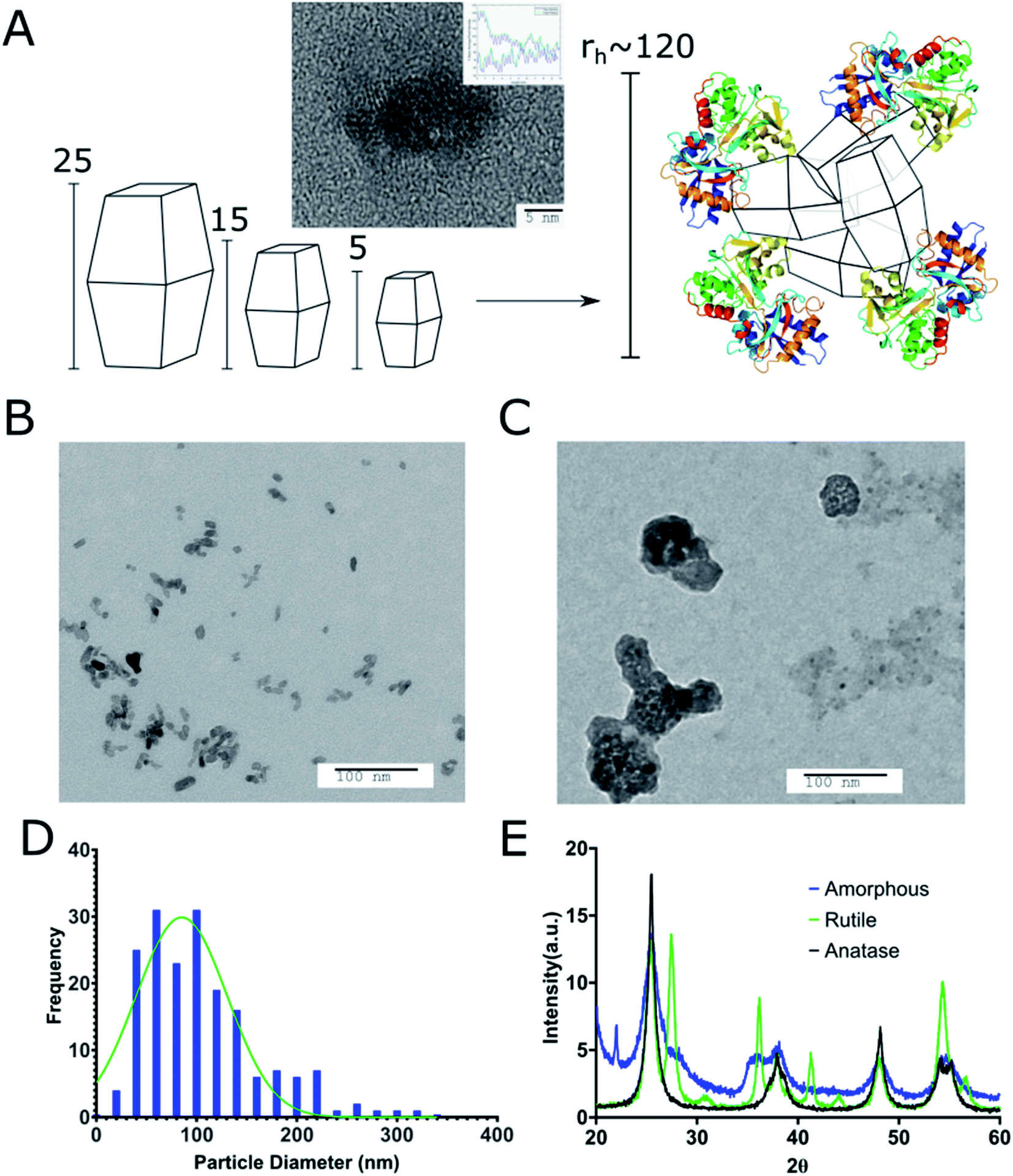 | ||
| Fig. 1 The use of differing core TiO2 crystal diameter from 5–100 nm results in NAGs of equal size. (A) A representation of the differing core sizes being coated to form regular NAGs of ∼120 nm. (B) Uncoated 25 nm anatase TiO2 compared against (C) uranyl acetate stained Tf–TiO2. (D) Histogram of the size distribution of stained 25 nm cTd NCs. (E) X-ray diffraction pattern from the TiO2 amorphous, rutile and anatase cTds. It is clear the amorphous particles are a mix of anatase, rutile and unstructured crystal domains when referenced against the JCPDS standard XRD card (88-1175 and 84-1286). | ||
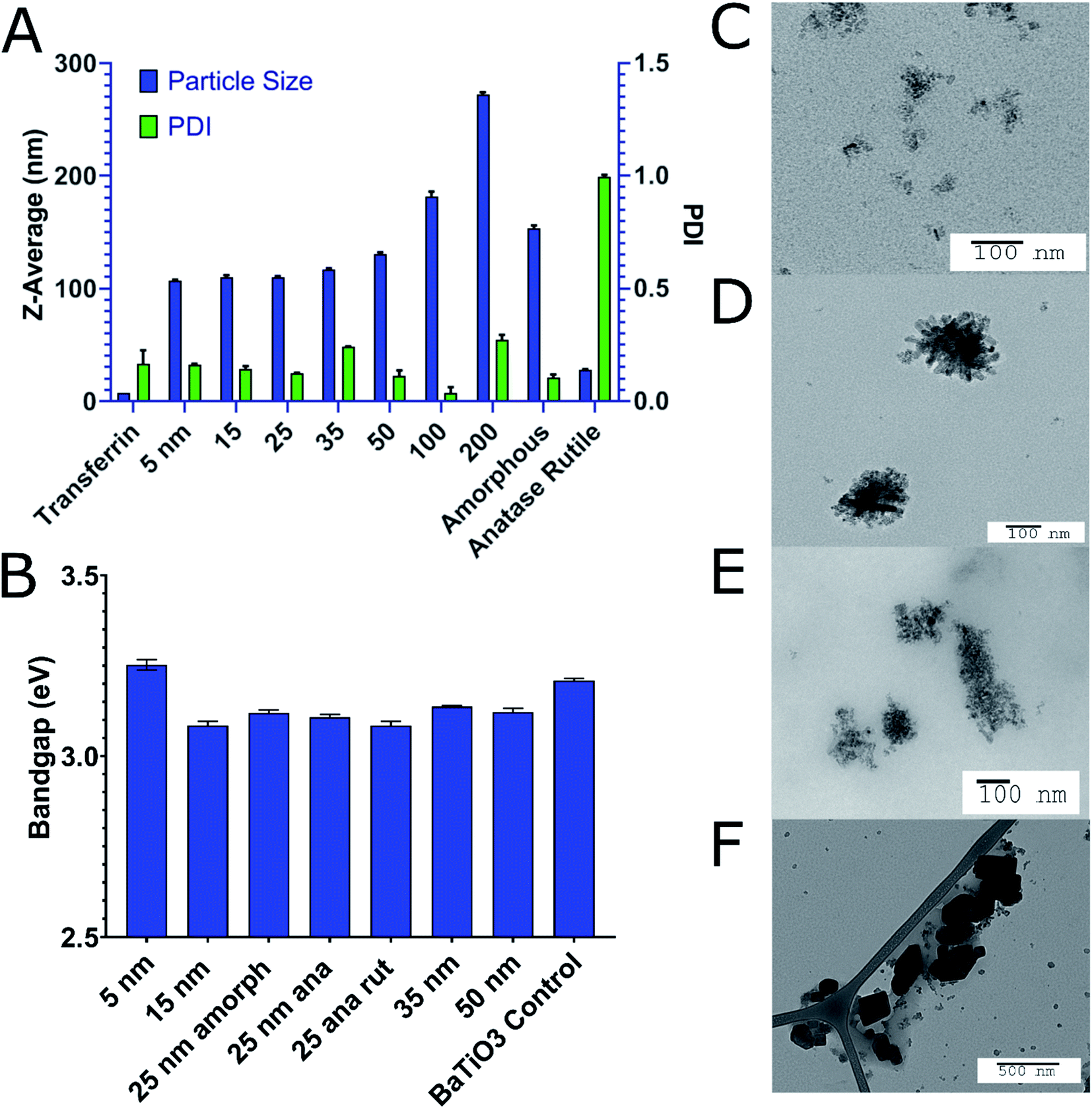 | ||
| Fig. 2 (A) Effects of cTd and crystal structure on the final Tf–TiO2 particle size and PDI by DLS. 200 nm particles were not filtered due to the proximity to our filter cutoff resulting in low concentration. (B) Band gap energies of bare cTds of TiO2 calculated from a Tauc plot (Horiba integrating quanta-phi scatter sphere). BaTiO3 sample was included as calibration, its published Eg is 2.2 eV. TEM of (C) 5, (D) 15, (E) 50 and (F) 200 nm uncoated cTds. | ||
Coating of core TiO2 NPs with Tf produces distinct NAGs
TiO2 NPs of varying size and crystal structures were coated with Tf, which served as both a dispersing and tumour-targeting agent. The 80 kDa β-globulin Tf has been shown to have the combined advantages of high affinity to Ti4+ ions, non-toxicity, and tumour targeting effects.43–45 Additionally, it is the third most common protein in serum, acting as the major systemic iron transporter, a vital resource for cancer cells. Most tumours upregulate the expression of Tf-receptor to feed their increased iron demand, which allows Tf to act also as a cancer-targeting moiety.46–48 Holo-Tf resulted in uncontrolled aggregation of TiO2 to >1 μm (PDI of 1) while apo-Tf resulted in the formation of Tf–TiO2 NAGs. DLS analysis provided an average NAG size of 108 ± 1.13 nm for 5 and 15 nm core NPs (Fig. 2A). The NAG size was consistent to 50 nm cTd before gradually increasing as the NP core size increased. It was difficult to obtain an accurate measurement of the 1000 nm NPs due to rapid settling. TEM showed consistent clustering of all Tf–TiO2 particles into NAGs. For 25 nm core NPs, they appeared as agglomerated TiO2 cores coated with Tf, with an average size of 105.1 ± 59.15 nm (Fig. 1C and D). Visual analysis suggests the number of cores in each particle decreases with increasing core size. Possibly, the NAG size is governed by a volume-restricted number of cores that are then coated with Tf, forming a stable (low PDI) NAG. BCA on 25 nm anatase Tf–TiO2 NAGs showed 95 ± 2.9 Tf per particle, indicating a monolayer of Tfs coating the surface. The size distribution, however, narrowed with increasing core size as it approached the filter cutoff, dropping from PDI 0.17 to 0.03. The crystal phase also influenced the overall NAG size. This effect was not driven by differences in buffer conditions as each NP type was synthesized through the same process, differing only in the finishing temperatures. The PBS buffer was used to maintain the formulation at pH 7 for all NPs. Amorphous particles show a higher average particle size, likely due to increased surface energy and hydroxylation in aqueous solution. This increased water affinity and lack of exposed Ti3+/4+ on the surface, characteristic of amorphous TiO2, likely lowers the affinity for Tf. Loss of the stabilizing Tf coating and drives aggregation, limiting the overall stability of amorphous NAGs.43,49 NAG size of the mixed anatase–rutile TiO2 collapsed from the anatase cTd along with a drastic increase in size heterogeneity indicated by PDI. This was driven in part by high filter retention, suggesting the Tf coating was unstable on these particles.A fundamental problem with many NP formulations is the poor shelf life. Thus, 25 nm core NAGs stored at 2 °C were monitored for over two months in diH2O. Longitudinal tracking of size and PDI via DLS show the dispersion of NPs in diH2O exhibited high stability over 60 days, with consistent PDI below 0.2 (Fig. 3). The result suggests that diH2O is useful in maintaining NAG's integrity for long term storage.
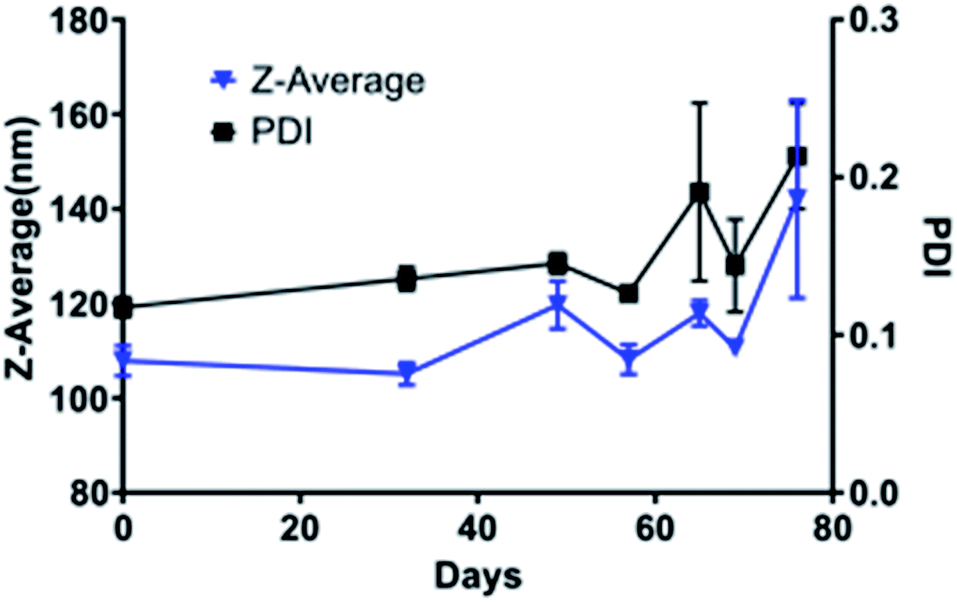 | ||
| Fig. 3 Stability of 25 nm Tf–TiO2 particles in water over the course of two months as determined by DLS. The experiment endpoint was a PDI above 0.25. | ||
ROS production exhibits nonlinear relationship with NP size
ROS producing capacity of the NPs varied with size and crystal phase. Comparison of all the NPs prepared showed that the 25 nm core NAGs produced the most ROS in both HPF and DCF measurements (Fig. 4A). This finding contrasts with standard catalytic particle theory, which suggests increased surface area per gram should show the highest ROS.50 The results here show 25 nm NPs have 180% enhanced DCF conversion rate vs. 15 nm and a 190% enhanced HPF rate over 5 nm particles, its nearest competitors for the respective reporters. Our results agree with data originally generated against non-UV exposed, bare TiO2 which suggested low cTd have decreased surface defect rate below 25 nm.26 As Ti3+ defects act as binding sites for surface hydroxyls and oxygen, they are essential for exciton separation and catalysis. Therefore, the loss of defects for 5 nm cTd TiO2 limits the ROS production.19 The drop off above 25 nm approximates the canonical loss of surface area, a 1/r3 decrease.51 Furthermore, the HPF signal increased relative to DCF at lower cTd. This phenomenon is likely driven by increased exciton confinement near the Bohr radius of the exciton (∼3.2 nm in TiO2). That confinement further increases the absorbed energy, which favours hydroxyl radical generation by holes but has little effect on oxygen catalysis.8 This shift toward hydroxyl generation may be vital in hypoxic tumour cores as water splitting is the main transfer path for hydroxyl radicals, possibly removing oxygen dependence, a vulnerability of many small molecule photosensitizers.52 Crystal type affects ROS generation in Tf–TiO2 NAGs.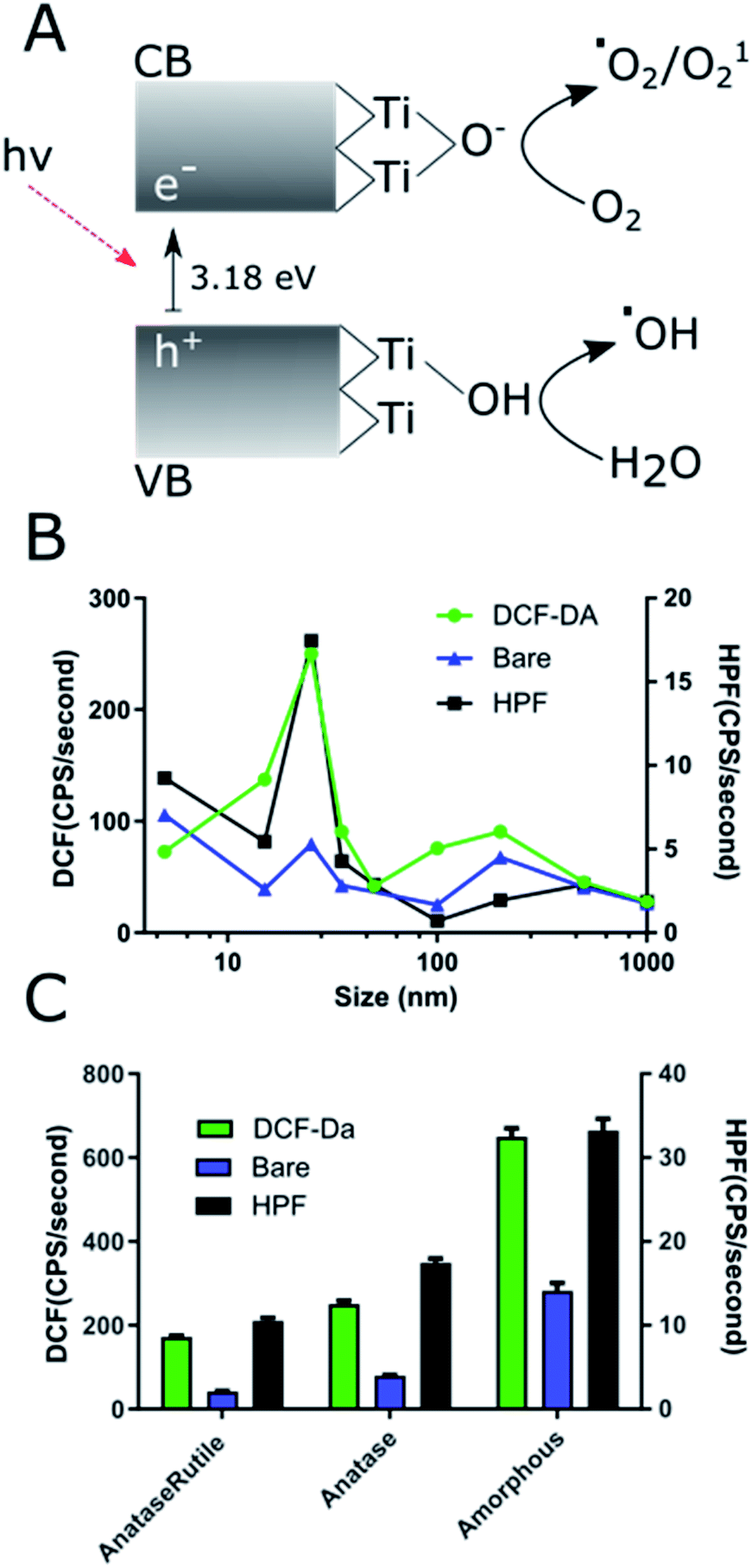 | ||
| Fig. 4 (A) Schematic representation of the primary ROS generation by TiO2 for electrons and holes. Hydroxyl radical production is the primary route sensed by HPF in this work. ROS production rate as quantified through DCF and HPF with changing size (B) and crystal structure (C). The rates were determined through pseudo-first order approximation on the fluorescent curves. | ||
Previous studies demonstrated that amorphous particles can improve ROS quantum yield.26 Here we explored whether this pattern can translate to NAGs using 25 nm core NPs. Our results show a 260% increase in ROS production of amorphous over anatase (Fig. 4B). This enhancement is likely caused by an increased surface defect rate. Since amorphous particles lack regular crystal structure (Fig. 1E), they leave grain boundaries that propagate to the surface and increase trapping. Additionally, the amorphous cTd's lack of calcination leaves a higher oxy anion concentration on the surface of particles, which has been shown to increase the catalytic rate.20 It is also apparent that the rutile content reduces overall ROS yield, as reported previously.53 This is caused by a decrease in surface energy compared to the high defect rate anatase active crystal plane (101), which, when combined with anatase–rutile's lower band gap decreases it's ROS yield compared to anatase.17 These data point to the importance of the presence of TiO2 surface defects in maximizing ROS generation.
Small TiO2 core NAGs exhibit high tumour retention
For in vivo biodistribution studies, 5 and 25 nm cTd NPs were selected for their high ROS and similar NAG size. Biodistribution of these NAGs were determined by ICP-MS measurement of total 48Ti accumulation in tissues. The analysis revealed an increase in tumour localization per unit mass for 5 nm core NAGs with a total of 2.7 μg g−1 (Fig. 5). Further, a six-fold increase in tumour to muscle ratio was observed for the 5 nm core NAGs (13.3) compared to the 25 nm core (2.83). Although the NAG sizes for the two NP cores are similar, our result suggests the in vivo biological properties are different. Probably, stripping of transferrin from the NAGs in circulation, high intravenous shear force, or other biological interactions dissociate the NAGs, thereby reducing the size to more closely match the cTd. Uptake of TiO2 NAGs in the brain was not expected because of its large size. It is possible that the elevated 48Ti could result from 48Ca interference54 or the uptake of NAGs by astrocytes via transferrin mediated internalization.55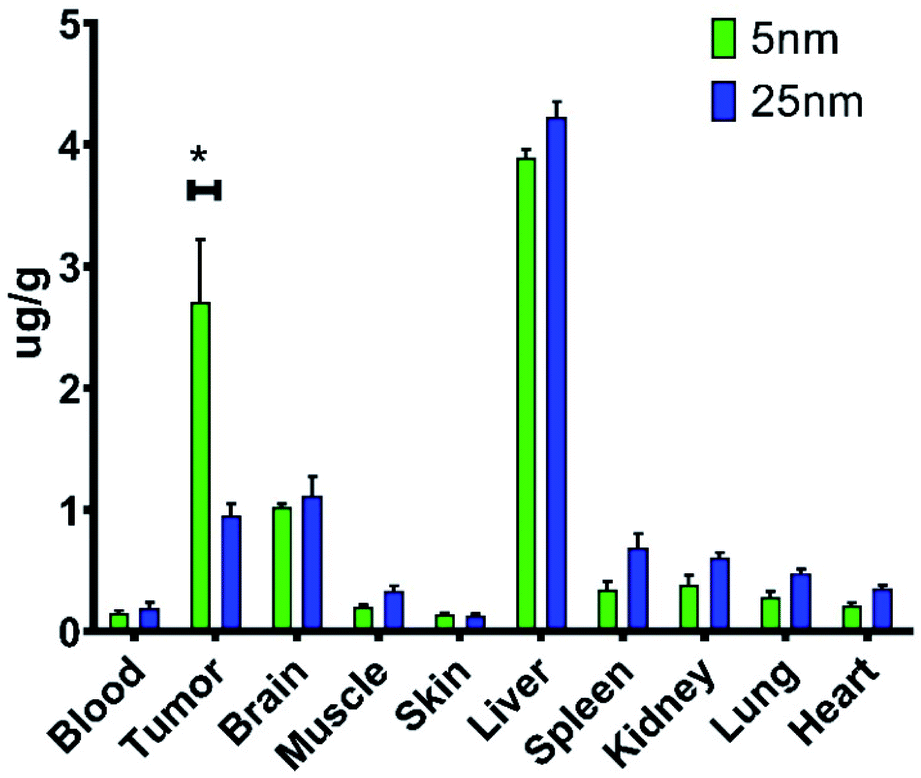 | ||
| Fig. 5 In vivo biodistribution of two core sizes of Tf–TiO2 determined by ICP-MS of homogenized tissue. This was done in 4T1 BALB/c mouse models after injecting 200 mg of TiO2. *P < 0.05. | ||
Conclusions
In summary, analysis of the TiO2 core size and crystal reveal their direct impact on ROS generation and in vivo biodistribution. First, cTd appears to have little impact on the size of the NAGs formed in aqueous suspensions, likely due to the formulation size being driven by the particle surface energy and not physical core dimensions. This is most apparent when comparing differing crystal phases against the final formulated size, with higher surface energy cores resulting in larger Tf–TiO2 NAGs. While the core size had little effect on the final NAG, it did significantly impact the ROS production. The 25 nm core NAGs clearly generated the most ROS, which held true for both bare TiO2 and Tf–TiO2 NAGs. The 5 nm NAGs produced the next largest amount of ROS at half the rate of the 25 nm NAGs. This is likely due to the 5 nm NP's large increase in the surface area over 25 nm which partially compensated for the reduction in defect sites. Furthermore, the defect hypothesis was corroborated by the highest ROS production rate from the amorphous crystal structure, having a 200% enhancement in rate. Unfortunately, amorphous NAGs sit near 200 nm which limits its utility. Future work will seek to focus on decreasing the size of NAGs. Additionally, the increase in HPF production at 5 nm implies small cTd may be used to fine-tune oxygen sensitivity of TiO2 but at the cost of absorbance range.Finally, 5 nm cTd NAGs also show improved tumour localization over 25 nm, showing the importance of cTd in tumour accumulation properties. There seems to be a paradox between the nearly 2-fold enhancement of ROS generation by the 25 nm over the 5 nm particles versus the nearly 3-fold increase in the tumour uptake of the 5 nm over the 25 nm NPs. Taken with the ROS-generating properties of various types of cTds, this points toward a balance in design parameters to synthesize the ideal TiO2-based photosensitizer. Since TiO2 photosensitizers must both generate high amounts of ROS under UV irradiation as well as significantly accumulate in tumours, a particle must be designed that can do both. Though 25 nm cTds create the most ROS, 5 nm cTds accumulate more in tumours. This implies that the ideal cTd size for depth-independent photodynamic therapy, which would both generate ROS and accumulate in tumours, is in the 5–25 nm range. Future therapy studies in mouse models of cancer will be needed to identify which of the two factors dominate therapeutic response.
Overall, RaST offers broad potential in the treatment of cancer, with TiO2 based nanophotosensitizers having already shown in vivo promise. However, the material properties play a direct role in the efficacy of the treatment. Herein, we have elucidated key parameters in the design of TiO2-based nanophotosensitizers that refine both the ROS-generating and biodistribution necessary to enhance therapeutic effect in vitro. These design features include the use of cTds that contain significant numbers of surface defect sites that generate ROS, whether in anatase or amorphous form, and the use of smaller crystals (<25 nm) with higher tumour accumulation, which should be incorporated into the future design of TiO2 photodynamic agents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded primarily by the National Institutes of Health (NIH) grants U54 CA199092 and R01 EB021048, and in part by other NIH grants (R01 CA171651, P50 CA094056, P30 CA091842, S10 OD020129, S10 OD016237, and S10 RR031625), the Department of Defense Breast Cancer Research Program (W81XWH-16-1-0286), and the Alvin J. Siteman Cancer Center Investment Program Research Development Award.Notes and references
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef.
- R. R. Allison, R. Cuenca, G. H. Downie, M. E. Randall, V. S. Bagnato and C. H. Sibata, Photodiagn. Photodyn. Ther., 2005, 2, 51–63 CAS.
- R. S. Verma and B. Bonavida, Resistance to Targeted Anti-Cancer Therapeutics, 2015, vol. 6 Search PubMed.
- H. Chen, Y. Di, D. Chen, K. Madrid, M. Zhang, C. Tian, L. Tang and Y. Gu, Nanoscale, 2015, 7, 8884–8897 RSC.
- T. S. Troutman, S. J. Leung and M. Romanowski, Adv. Mater., 2009, 21, 2334–2338 CrossRef CAS.
- T. Y. Ohulchanskyy, I. Roy, L. N. Goswami, Y. Chen, E. J. Bergey, R. K. Pandey, A. R. Oseroff and P. N. Prasad, Nano Lett., 2007, 7, 2835–2842 CrossRef CAS.
- (a) N. Kotagiri, G. P. Sudlow, W. J. Akers and S. Achilefu, Nat. Nanotechnol., 2015, 10, 370–379 CrossRef CAS; (b) N. Kotagiri, M. L. Cooper, M. Rettig, C. Egbulefu, J. Prior, G. Cui, P. Karmakar, M. Zhou, X. Yang, G. Sudlow, L. Marsala, C. Chanswangphuwana, L. Lu, L. M. Habimana-Griffin, M. Shokeen, X. Xu, K. Weilbaecher, M. Tomasson, G. Lanza, J. F. Dipersio and S. Achilefu, Nat. Commun., 2018, 9, 1–12 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- K. Xu, Y. Xie, X. Cui, J. Zhao and K. D. Glusac, J. Phys. Chem. B, 2015, 119, 4175–4187 CrossRef CAS.
- P. Vaupel, Semin. Radiat. Oncol., 2004, 14, 198–206 CrossRef.
- N. J. Mabjeesh and S. Amir, Histol. Histopathol., 2007, 22, 559–572 CAS.
- C. Turchi and D. Ollis, J. Catal., 1990, 122, 178–192 CrossRef CAS.
- J. V. Jelley, Br. J. Appl. Phys., 1955, 6, 227 CrossRef.
- J. Axelsson, S. C. Davis, D. J. Gladstone and B. W. Pogue, Med. Phys., 2011, 38, 4127–4132 CrossRef CAS.
- Z. Ouyang, B. Liu, S. Yasmin-Karim, E. Sajo and W. Ngwa, Phys. Med., 2016, 32, 944–947 CrossRef.
- R. K. Gill, G. S. Mitchell and S. R. Cherry, Phys. Med. Biol., 2015, 60, 4263–4280 Search PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- A. N. Shultz, W. Jang, W. M. Hetherington, D. R. Baer, L. Q. Wang and M. H. Engelhard, Surf. Sci., 1995, 339, 114–124 CrossRef CAS.
- T. Yamamoto and T. Ohno, Phys. Chem. Chem. Phys., 2012, 14, 589–598 RSC.
- A. Sclafani and J. M. Herrmann, J. Phys. Chem., 1996, 100, 13655–13661 CrossRef CAS.
- Z. Liu, B. H. Hu and P. B. Messersmith, Tetrahedron Lett., 2010, 51, 2403–2405 CrossRef CAS.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- A. L. Linsebigler, A. L. Linsebigler, J. T. Yates Jr, G. Lu, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- T. Luttrell, S. Halpegamage, J. Tao, A. Kramer, E. Sutter and M. Batzill, Sci. Rep., 2014, 4, 4043 CrossRef.
- C. Jin, Y. Tang, F. Guang Yang, X. Lin Li, S. Xu, X. Yan Fan, Y. Ying Huang and Y. Ji Yang, Biol. Trace Elem. Res., 2011, 141, 3–15 CrossRef CAS.
- J. Jiang, G. Oberdörster, A. Elder, R. Gelein, P. Mercer and P. Biswas, Nanotoxicology, 2008, 2, 33–42 CrossRef CAS.
- R. I. Bickley, T. Gonzalez-Carreno, J. S. Lees, L. Palmisano and R. J. D. Tilley, J. Solid State Chem., 1991, 92, 178–190 CrossRef CAS.
- B. Prasai, B. Cai, M. K. Underwood, J. P. Lewis and D. A. Drabold, J. Mater. Sci., 2012, 47, 7515–7521 CrossRef CAS.
- P. Calza, E. Pelizzetti, K. Mogyorósi, R. Kun and I. Dékány, Appl. Catal., B, 2007, 72, 314–321 CrossRef CAS.
- J. Jiang, G. Oberdörster and P. Biswas, J. Nanopart. Res., 2009, 11, 77–89 CrossRef CAS.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631–651 CrossRef CAS.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS.
- S. A. Kulkarni and S. S. Feng, Pharm. Res., 2013, 30, 2512–2522 CrossRef CAS.
- N. T. K. Thanh and L. A. W. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- Y. Zhang and A. Clapp, Sensors, 2011, 11, 11036–11055 CrossRef.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS.
- S. Y. Chae, M. K. Park, S. K. Lee, T. Y. Kim, S. K. Kim and W. I. Lee, Chem. Mater., 2003, 15, 3326–3331 CrossRef CAS.
- B. Xu, R. Tang, P. Biswas, M. Mixdorf, S. Achilefu, N. Reed and R. Raliya, ACS Appl. Bio Mater., 2019, 2, 1141–1147 CrossRef.
- P. Carpenter, Procedures for Using the Bruker D8 Advance X-ray Diffractometer, http://xraysrv.wustl.edu/web/xrd/brukerd8.html Search PubMed.
- K. ichi Setsukinai, Y. Urano, K. Kakinuma, H. J. Majima and T. Nagano, J. Biol. Chem., 2003, 278, 3170–3175 CrossRef.
- J. Anthony, R. Bideaux, K. Bladh and M. Nochols, Handbook of Mineralogy, 1990 Search PubMed.
- H. Sun, H. Li, R. A. Weir and P. J. Sadler, Angew. Chem., Int. Ed., 1998, 37, 1577–1579 CrossRef CAS.
- Y. Nuevo-Ordoñez, M. Montes-Bayón, E. Blanco González and A. Sanz-Medel, Metallomics, 2011, 3, 1297–1303 RSC.
- A. D. Tinoco and A. M. Valentine, J. Am. Chem. Soc., 2005, 127, 11218–11219 CrossRef CAS.
- F. Danhier, O. Feron and V. Preat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS.
- P. T. Tybor, C. W. Dill, J. N. Bryant and W. A. Landmann, J. Agric. Food Chem., 1970, 18, 629–631 CrossRef CAS.
- T. R. Daniels, T. Delgado, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 159–176 CrossRef CAS.
- J. J. Ramsden, J. Phys. Chem., 1994, 98, 5376–5381 CrossRef CAS.
- H. H. Hu, J. H. Xin, H. H. Hu, X. Wang, D. Miao and Y. Liu, J. Mater. Chem. A, 2015, 3, 11157–11182 RSC.
- K. Nakataa and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef.
- J. F. Reeves, S. J. Davies, N. J. F. Dodd and A. N. Jha, Mutat. Res., 2008, 640, 113–122 CrossRef CAS.
- T. Ohno, K. Sarukawa and M. Matsumura, New J. Chem., 2002, 26, 1167–1170 RSC.
- K. J. Orians and E. A. Boyle, Anal. Chim. Acta, 1993, 282, 63–74 CrossRef CAS.
- K. Tulpule, S. R. Robinson, G. M. Bishop and R. Dringen, J. Neurosci. Res., 2010, 88, 563–571 CAS.
| This journal is © The Royal Society of Chemistry 2020 |