
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual roles of 3-aminopropyltriethoxysilane in preparing molecularly imprinted silica particles for specific recognition of target molecules†
Fenying Wang*a,
Baoping Ling b,
Qianjin Li*c and
Rahma Abouhanyd
b,
Qianjin Li*c and
Rahma Abouhanyd
aCollege of Chemistry, Nanchang University, Nanchang, Jiangxi 330031, China. E-mail: wangfenying@ncu.edu.cn
bSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, Shandong 273165, China
cDepartment of Food Science and Engineering, School of Food Science and Pharmaceutical Engineering, Nanjing Normal University, Nanjing 210023, China. E-mail: liqianjinnju@163.com
dDepartment of Biomedical Sciences, Faculty of Health and Society, Malmö University, Malmö 20506, Sweden
First published on 27th May 2020
Abstract
3-Aminopropyltriethoxysilane (APTES) is a silane widely used to supply amino groups for further modifications on various materials, but it is less studied as a catalyst to catalyze sol–gel silica polymerization. Here, by using APTES as the catalyst instead of the conventional basic catalysts, a novel strategy was developed to prepare silica-based molecularly imprinted polymers (MIPs). Meanwhile, APTES was employed as the functional monomer to create imprinted nanocavities for specific recognition of target molecules. The as-synthesized MIP exhibited ultra-high recognition capability due to the elimination of the detrimental effect on the imprinting performance caused by the additional catalysts. The preparation process, specificity, pH effect, binding capacity and affinity of the MIP were studied in detail. The MIP microparticles could be packed into a solid phase extraction column for removing the target molecule in water efficiently, and the molecule could easily be enriched by 40 times. The interaction of the functional monomer and template was studied by the calculation method, giving a more clear understanding of the recognition behaviours of the imprinted polymers. The strategy could be extended not only to prepare highly specific MIPs for other small phosphoric molecules, but also for biomolecules e.g. phosphorylated peptides or proteins.
1. Introduction
Molecularly imprinted polymers (MIPs) are special molecular recognition materials with template-created cavities, which are complementary to the template shape and have functional ligands inside to interact with the template.1–3 MIPs are widely used in the fields of separation, catalysis, bioassay, sensing, imaging, drug delivery and protein crystallization, because of their specificity, low cost, high stability, long shelf-life and possibility of mass production.4–12 Normally, MIPs are prepared by following steps: (1) preparation of a pre-polymerization mixture containing functional monomer, template, crosslinker and solvent, in which the functional monomer and the template will assemble into complexes; (2) addition of initiator or catalyst to initiate the polymerization; (3) removal of the template from the polymers by suitable washing solution to obtain MIPs. Many polymerization methods were developed to prepare MIPs, for example, thermal, UV, oxide/reduce reaction, or enzyme initiated radical polymerizations,13–17 which are used to prepare organic MIPs; and bases or acids initiated sol–gel polymerizations, which are usually employed to synthesize inorganic MIPs, particularly silica-based MIPs.18–22 Comparing with organic MIPs, silica-based MIPs are friendly to environment, easy to degrade after usage, and economical to produce at room temperature which is beneficial to save thermal energy, simplify equipment and improve producing safety.3-Aminopropyltriethoxysilane (APTES) is a well-known silane reagent as a linker for immobilization of functional molecules on various materials,23 and it could also act as a functional monomer for making silica-based MIPs. There were several papers reporting that APTES was employed as the functional monomer for the preparation of silica-based MIPs targeting molecules which have acidic groups such as carboxylic acid group,24–27 phenolic hydroxyl group,28,29 or alcoholic hydroxyl group.30 However, all the reported sol–gel silica imprinting systems used bases (e.g. NH3 or NaOH) or acids (e.g. acetic acid) as the catalyst to initiate the hydrolysis and condensation reaction, resulting in poor imprinting performance and low selectivity of the MIPs, because the catalysts would interrupt the interactions between APTES and the template.
If the additional catalysts could be avoided to use in the sol–gel silica imprinting systems, their detrimental effect on the imprinting performance would be eliminated. It has been reported that APTES was successfully employed to catalyse the sol–gel silica polymerization to produce silica aerogels with excellent adsorption and elasticity properties,31 as well as metal embedded silica particles,32,33 but to the best of our knowledge, there is no report to synthesize silica-based MIPs by using its self-catalytic property.34,35
In this paper, a novel strategy was proposed by employing APTES as the catalyst to prepare silica-based MIPs. In the sol–gel silica imprinting system, APTES also acted as the functional monomer, an acidic molecule of 1-naphthyl phosphate (NPA) and tetraethoxysilane (TEOS) were used as the template and the crosslinker, respectively. No additional catalysts were added into the imprinting system. Silica-based MIPs were produced successfully with high specificity by comparing with their corresponding non-imprinted polymers (NIPs) and the control MIPs which were synthesized in the presence of additional basic catalysts. Furthermore, the recognition behaviours of the MIPs were studied in detail with the assistance of calculation, showing the nature of the interactions between the functional ligand and the template in imprinted cavities of the MIP was electrostatic attraction interaction.
2. Results and discussion
2.1. Synthesis and characterization of the silica-based MIPs
To study the availability of the proposed strategy, an acidic molecule 1-naphthyl phosphate (NPA) was selected as the template, tetraethyl orthosilicate (TEOS) was used as the crosslinker, and APTES was expected to act as both functional monomer and catalyst. As shown in Scheme 1, APTES firstly assembled with NPA into APTES–NPA complex in the co-solvent of ethanol and water. As NPA is a diprotic acid that can interact with two APTES to form a neutral complex, so we set the ratio of NPA/APTES to be 1/4 to make sure there were enough free APTES to catalyse the sol–gel polymerization. After introduction of TEOS into the mixture, polymerization began in less than 5 min. After two days, silica particles with embedded templates precipitated and were collected by centrifugation. After template removal, MIP silica particles (MIP1) were obtained and expected to possess the NPA imprinted nanocavities having amino ligands inside (Scheme 1). To investigate the self-catalytic behaviour of the APTES in the imprinting mixture, another ratio of NPA/APTES was set to be 1/2 to prepare the MIP silica particles (MIP2). Meanwhile, two basic catalysts of NH3 and NaOH were employed as the additional catalysts to prepare MIP–NH3 and MIP–NaOH, respectively. Non-imprinted polymers (NIPs) were prepared under the same experimental conditions of their corresponding MIPs except for the omission of NPA.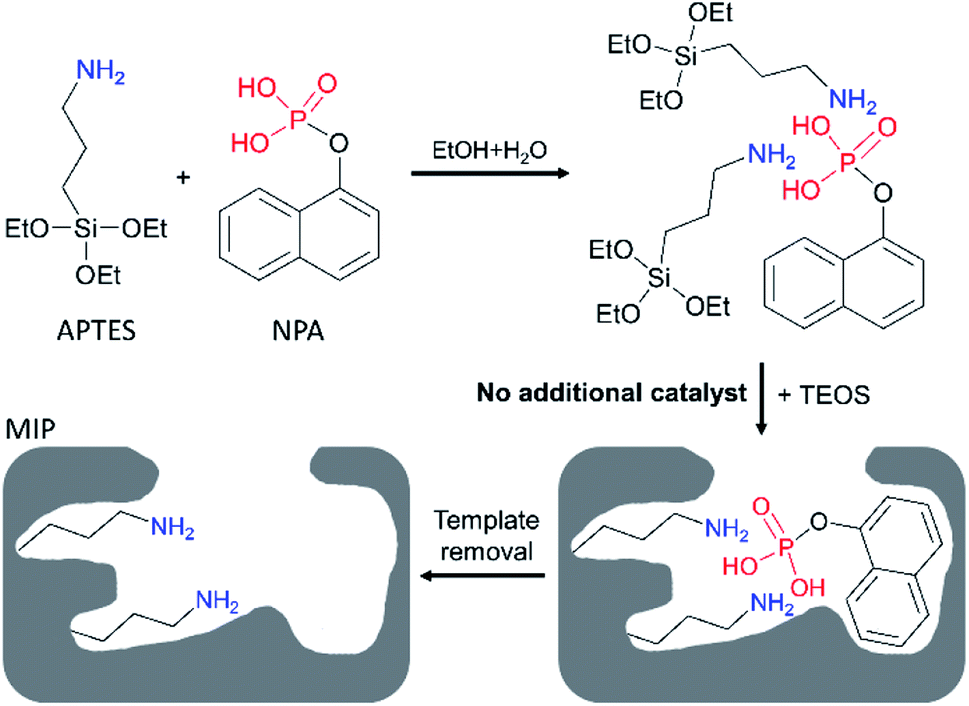 | ||
| Scheme 1 Preparation of 1-naphthyl phosphate (NPA) imprinted polymer by using 3-aminopropyltriethoxysilane (APTES) as both catalyst and functional monomer in the co-solvent of ethanol and water. | ||
FT-IR measurements were performed on the as-synthesized MIPs and NIPs. The peak at 1389 cm−1 in NPA (Fig. 1a) was also observed in MIP1 and MIP2 before template removal (Fig. 1b), demonstrating the template was successfully polymerized into them. But the peak signal of 1389 cm−1 was very weak in both MIP–NH3 and MIP–NaOH, indicating less template was polymerized into them, due to their low template per polymer weight because of their high polymer yields (Table 1). After template removal, the peak at 1389 cm−1 in MIP1 and MIP2 disappeared (Fig. 1c), and all the MIPs showed the same spectra as their corresponding NIPs (Fig. 1d), suggesting the template could be removed successfully by the washing solution.
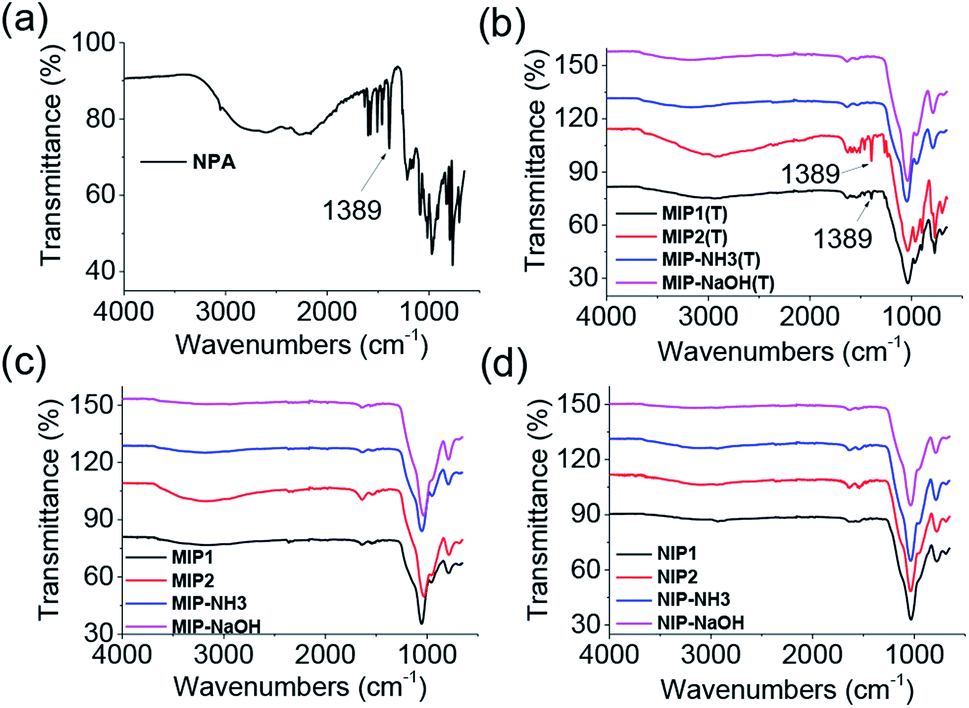 | ||
| Fig. 1 FT-IR spectra of NPA (a), MIPs before template removal (b), MIPs after template removal (c) and NIPs (d). | ||
| Polymer name | APTES, mmol | NPA/APTES/TEOS, mol/mol/mol | Template utilization, % | Yield, % |
|---|---|---|---|---|
| a With catalyst of NH3 (1.6 mmol).b With catalyst of NaOH (1.6 mmol). | ||||
| MIP1 | 1.0 | 1/4/18 | 80 | 12 |
| NIP1 | 1.0 | 0/4/18 | — | 20 |
| MIP2 | 0.5 | 1/2/18 | 88 | 11 |
| NIP2 | 0.5 | 0/2/18 | — | 13 |
| MIP–NH3a | 0.5 | 1/2/18 | 39 | 30 |
| NIP–NH3a | 0.5 | 0/2/18 | — | 26 |
| MIP–NaOHb | 0.5 | 1/2/18 | 27 | 28 |
| NIP–NaOHb | 0.5 | 0/2/18 | — | 26 |
In view of the polymer yields of NIP1 (20%), NIP2 (13%), NIP–NH3 (26%) and NIP–NaOH (26%), it is clear to see that the more basic group in the polymerization mixture (Table 1), the higher yield of the NIP. However, for the MIPs without using additional catalysts, the yields of MIP1 (12%) and MIP2 (11%) were lower than their corresponding NIPs, due to the acidic template NPA inhibited part of the self-catalytic activity of APTES by forming the complex of NPA–APTES in the mixture. But for the MIPs using additional basic catalysts, the yields of MIP–NH3 (30%) and MIP–NaOH (28%) were not affected by the acidic template, as the amount of the basic catalysts (1.6 mmol) was much more than the acidic template (0.25 mmol).
To find out how many template molecules were polymerized into the MIPs, template utilization was calculated. As shown in Table 1, the template utilization of MIP1, MIP2, MIP–NH3 and MIP–NaOH was measured to be 80%, 88%, 39% and 27%, respectively. It is clear to see the template utilization of MIP2 > MIP1 ≫ MIP–NH3 > MIP–NaOH, suggesting the additional basic catalysts present in the imprinting mixture was detrimental to let the template polymerized into the polymers for creating imprinted cavities, because the basic catalysts could destroy the NPA–APTES complex. In addition, it is interesting to find that the template utilization in the NH3 catalytic system was higher than that in the NaOH catalytic system, indicating the stronger basic catalyst would result in less template molecules polymerized into the polymer. Therefore, it can be predicted that the selectivity of the MIPs would be in the order of MIP2 > MIP1 > MIP–NH3 > MIP–NaOH.
2.2. Specificity evaluation
Equilibrium binding experiments were performed on investigation of the selectivity of the MIPs. As shown in Fig. 2a and b, MIP1, MIP2 and MIP–NH3 bound more NPA than their corresponding NIPs in both ethanol and water, indicating the three MIPs all had selectivity towards the template molecule. However, MIP–NaOH bound less NPA than NIP–NaOH in both solvents, meaning it had no selectivity, due to its rather low template utilization. Furthermore, both of MIP1 and MIP2 bound at least twice more NPA than MIP–NH3, demonstrating the MIPs prepared using APTES as the catalyst had more imprinted cavities, and the additional basic catalysts were detrimental to the MIPs selectivity. MIP1 exhibited the highest selectivity comparing with other MIPs (Table S1†), giving imprinting factors (IFs) of 32.2 and 15.2 in ethanol and water, respectively, which were also much higher than the IF values (1.8–5.6) of the reported MIPs produced by using APTES as the functional monomer.24–30 As MIP1 gave the best selectivity among the MIPs, it was selected to perform the remaining experiments.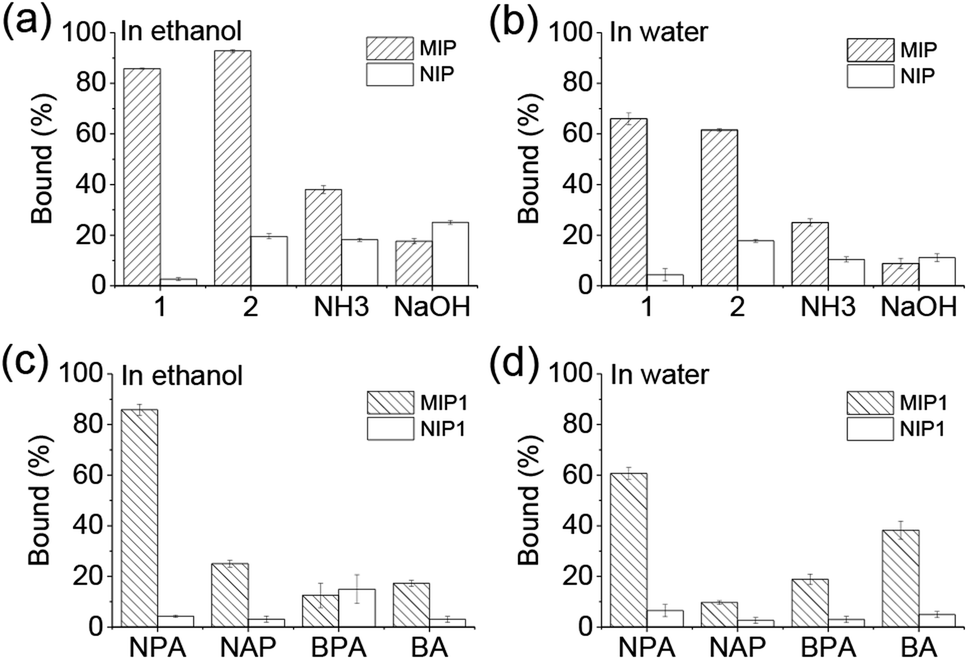 | ||
| Fig. 2 Uptakes of 1-naphthyl phosphate (NPA) by MIPs and NIPs in ethanol (a) and water (b), respectively; as well as uptakes of NPA, naproxen (NAP), benzylphosphonic acid (BPA) and benzoic acid (BA) by MIP1 in ethanol (c) and water (d), respectively. Polymer: 5 mg mL−1; testing molecule concentration: 0.5 mM. | ||
Several acidic molecules including benzylphosphonic acid (BPA) having phosphonic acid group, naproxen (NAP) and benzoic acid (BA) both having carboxylic acid group, were selected to investigate the specificity of MIP1 to NPA. As shown in Fig. 2c and d, MIP1 bound more NPA than NAP, BPA and BA in both ethanol and water, demonstrating its good specific recognition capability towards the template molecule of NPA.
2.3. Binding behaviours under different environmental conditions
It is interesting to find the binding behaviours of MIP1 to NPA in ethanol (Fig. 3a) and water (Fig. 3b) was very different. In ethanol, when the concentration of NPA was lower than 300 μM, the binding amount of NPA on MIP1 displayed a linear relationship to its doses, and reached to the maximum binding capacity of ∼320 μmol g−1 when the NPA concentration was higher than 400 μM. However, in water the binding behaviour of MIP1 exhibited a non-linear relationship between the binding amount and the NPA doses, giving a binding capacity of ∼200 μmol g−1, lower than that in ethanol. However, at low concentration (1–20 μM) of NPA, MIP1 can adsorb all of them in both ethanol and water. By analysis of the equilibrium binding data using the Scatchard equation (Fig. S1†),36 the dissociation constant (Kd) of MIP1 to NPA was calculated to be 12.6 and 13.7 μM in ethanol and water, respectively, meaning the binding affinity of MIP1 to NPA was slightly stronger in ethanol than in water, in agreement with the binding capacity was higher in ethanol than in water. Furthermore, the Kd between APTES and NPA was determined to be 1516 μM in ethanol (Fig. S2†), ∼120 times more than that of MIP1 to NPA (12.6 μM), demonstrating MIP1 had a much stronger binding affinity to NPA due to its imprinted nanocavities, and further confirming the imprinting strategy based on the self-catalytic ability of APTES was successful.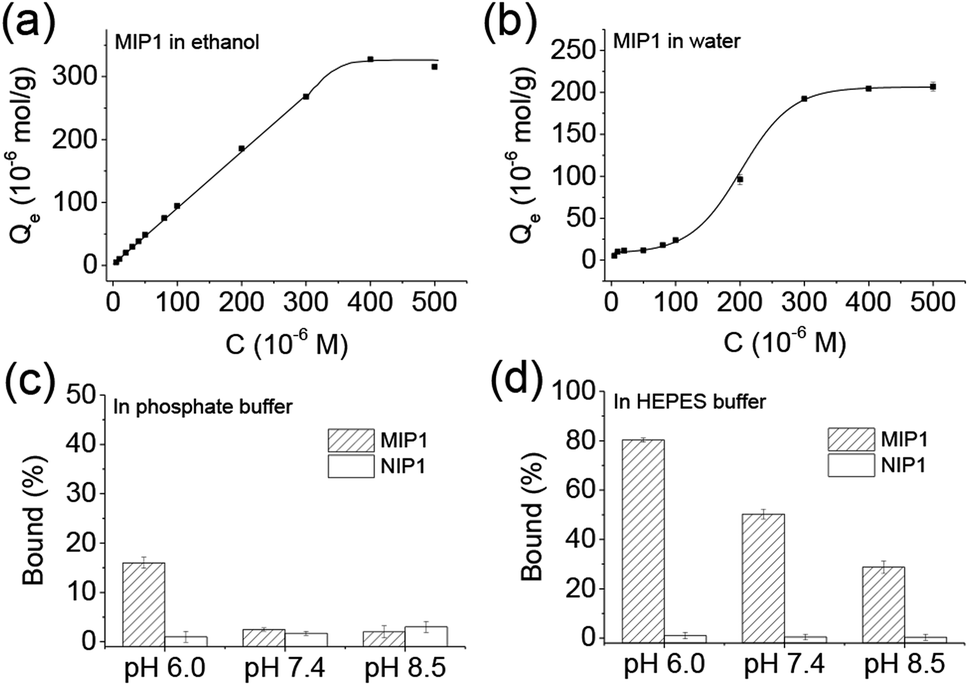 | ||
| Fig. 3 Binding behaviours of MIP1 to NPA in ethanol (a) and water (b), respectively, and its selective recognition ability in phosphate buffer (c, 10 mM) and HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer (d, 10 mM) under different pH environments. Polymer: 5 mg mL−1; NPA: 0.5 mM. | ||
As the recognition interaction in the imprinted nanocavities was mainly from the hydrogen bonding interaction between the functional amino ligand and the phosphoric group (Scheme 1), environmental pH would obviously affect the binding behaviour of the MIP. As shown in Fig. 3c, it is clear to see MIP1 lost its binding ability to NPA in the phosphate buffers at pH 7.5 and 8.5, while at pH 6.0 it still could selectively bind certain amount of NPA (∼15%, Fig. 3c), much less than the binding amount in water (∼60%, Fig. 2b), which could be explained by the following two reasons: (1) competition interaction between the inorganic phosphate and NPA to the functional amino ligands; (2) electrostatic repelling interaction between the less negatively charged amino group in MIP1 and the negatively charged NPA (pKa1 = 1.2, pKa2 = 5.9) under high pH environment. To eliminate the competition interaction effect, we used HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer instead of the phosphate buffer under the same pH conditions to perform the experiments. As shown in Fig. 3d, MIP1 bound much more NPA in HEPES buffer than that in phosphate buffer (Fig. 3c) under the same pH condition, further confirming the phosphate did compete with NPA to interact with the amino ligand in the imprinted cavities. Without the competition interaction effect, we can see MIP1 bound less NPA by increasing the environmental pH values (Fig. 3d), due to the increasing electrostatic pushing interactions between the amino group in MIP1 and the phosphoric group in NPA.
2.4. Recognition mechanism
To further investigate the recognition mechanism in MIP1, calculations were carried out to study the interactions between the functional monomer and the template by using the DFT method.37,38 As shown in Fig. 4, after calculation and model, the most stable complexes of NPA–APTES(I) and NPA–APTES(II) were obtained by optimization under each independent condition. The complex NPA–APTES(I) showed lower free energy than that of the complex NPA–APTES(II), indicating the complex was more stable in ethanol than in water, which agreed with the binding affinities of MIP1 in the two solvents (Fig. S1†). Moreover, the amino group in APTES is protonated and the phosphoric acid group in NPA is deprotonated in both complexes (Fig. 4a and b), suggesting the hydrogen bonding interaction does date from electrostatic attraction interactions. In addition, both of the NPA–APTES(I) and NPA–APTES(II) complexes are linked together through four hydrogen bonds, and each amino group in APTES can form a three-dimensional (3D) six-member ring with the phosphoric group in NPA, which could make the complex more stable than the single hydrogen bond linked complex. Based on the calculation results, it is clear to understand the pH effect mechanism on the binding behaviours of MIP1 (Fig. 4c) in view of the electrostatic attraction and repulsion interactions, as well as the recognition mechanism of such MIPs.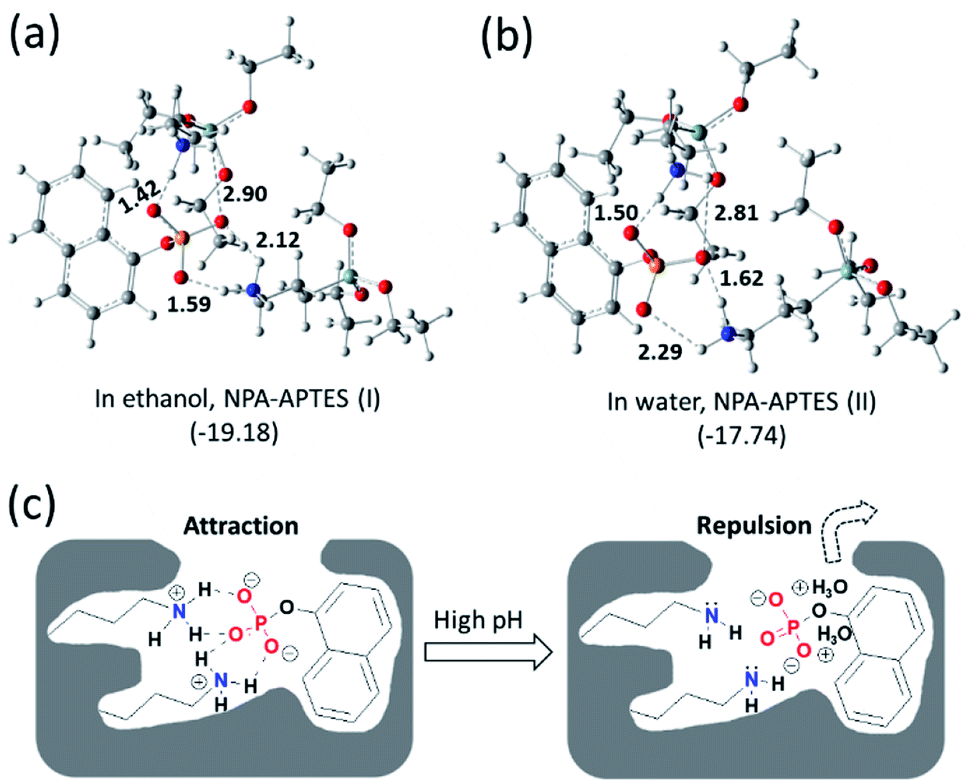 | ||
| Fig. 4 Interactions between APTES and NPA in ethanol (a) and water (b), respectively, calculated by the DFT method. Proposed recognition mechanisms of MIP1 to NPA under low and high pH environments (c). The values inserted into the molecular structures mean hydrogen bond length (Å); the values in the brackets mean calculated free energy (kcal mol−1). | ||
2.5. Removal and enrichment of target molecule in water
As shown in Fig. 5, the particle size distribution of MIP1 was measured by dynamic light scattering (DLS) method in water, it had a relatively narrow particle size distribution with polydispersity index (PDI) of 0.22 and an average diameter size of 1.5 μm. The morphology of MIP1 was characterized by scanning electronic microscope (SEM) and displayed spherical shapes although most of them preferred to aggregate together. The MIP1 microparticles were then employed to prepare solid phase extraction (SPE) column for the separation of NPA in tap water (Fig. 5), in which the spiked NPA concentration was set at 1 μM. After the tap water sample was treated by the SPE column, no NPA could be detected in the effluent, suggesting its high efficiency for contamination removal in water purification. Furthermore, the adsorbed NPA molecules could be desorbed completely by the phosphate buffer (10 mM, pH 8.5), giving a recycle rate of 100 ± 2% (Fig. S3 and Table S2†), and the enrichment factor reached to 40 times, which was beneficial for detection of the trace target molecules in samples.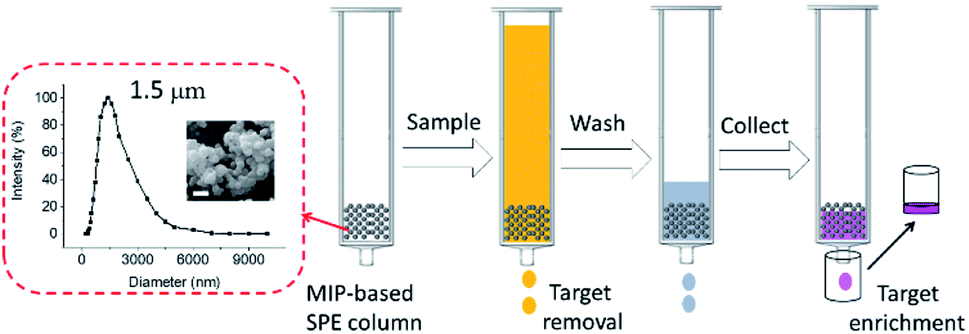 | ||
| Fig. 5 Particle size distribution and scanning electronic microscope (SEM) images of MIP1 particles which were employed to make solid phase extraction (SPE) column for target molecules removal and enrichment. The scale bar inserted in the SEM image is 2 μm. | ||
3. Conclusions
In this paper, by using APTES as both catalyst and functional monomer, a novel imprinting strategy was developed for the preparation of silica-based MIP. The MIP exhibited much higher selectivity than both of its NIPs and control MIPs synthesized using conventional basic catalysts, which would lead to low imprinting efficiency by destroying the functional monomer–template complex in the pre-polymerization mixture. Binding environments including buffer types and pH values would affect the binding behaviours of the MIP, whose recognition mechanism was based on the electrostatic attraction interactions between the functional ligand and the template which were linked into 3D ring complex. The MIP particles displayed great performance on target molecule removal and enrichment. In summary, this novel imprinting strategy could produce silica-based MIPs successfully with ultra-high selectivity, and reduce waste water treatment when the MIPs were in mass production. The strategy also provides a general approach for preparing high selective MIPs targeting other phosphoric biomolecules e.g. phosphorylated peptides or proteins.4. Experimental
4.1. Chemicals and equipment
3-Aminopropyltriethoxysilane (APTES, 99%) and tetraethoxysilane (TEOS, 99%) were bought from Shanghai Aladdin Bio-Chem Technology Co., LTD (Shanghai, China). Ethanol (99.7%) were purchased from Tianjin Damao Chemical Reagent Factory (Tianjin, China). 1-Naphthyl phosphate (NPA), benzylphosphonic acid (BPA), naproxen (NAP, racemate) and benzoic acid (BA) were obtained from J&K Scientific, LTD (Shanghai, China). Pure water used here is double-distilled water. Other chemicals were all in analytical grade.UV measurements were performed on Varian Cary 50 UV-vis spectrophotometer (Agilent Technologies, USA). IR spectra were recorded on a FTIR-Tensor27 spectrophotometer (Bruker, Germany). Scanning electron microscopy (SEM) imaging was carried out on a JEOL JSM-6701F field emission scanning electron microscope (Tokyo, Japan). Particle size distribution was measured on a Zetasizer Nano ZS90 dynamic light scattering instrument in pure water (Malvern Instruments Ltd., Worcestershire, UK).
4.2. Synthesis of molecularly imprinted polymers
At first, the template of NPA (56 mg, 0.25 mmol) was dissolved in the co-solvent of H2O/EtOH (20 mL/12 mL) under magnetic stirring at a speed of 500 rpm; then, APTES (234 μL, 1.0 mmol) was added and the stirring was kept for 30 min; finally, TEOS (1.0 mL, 4.5 mmol) was added into the mixture to obtain the pre-polymerization mixture solution. The reaction mixture was stirred at room temperature for 48 h. The synthesized MIPs were collected by centrifugation at a speed of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The supernatants were also collected for measuring the amount of the template left in the solution. The template in the MIPs was removed by washing with phosphate buffer (50 mM, pH 7.4). After template removal, the MIPs were washed with pure water and methanol three times separately before drying. The control MIPs was also prepared under the same experimental conditions as the MIPs except for the addition of the catalysts of NH3 or NaOH. Non-imprinted polymers (NIPs) were prepared under the same conditions of MIPs except for the omission of the template. All the MIPs and their NIPs were characterized by FT-IR. The yields and template utilization of all the polymers were measured and summarized in Table 1.
000 rpm. The supernatants were also collected for measuring the amount of the template left in the solution. The template in the MIPs was removed by washing with phosphate buffer (50 mM, pH 7.4). After template removal, the MIPs were washed with pure water and methanol three times separately before drying. The control MIPs was also prepared under the same experimental conditions as the MIPs except for the addition of the catalysts of NH3 or NaOH. Non-imprinted polymers (NIPs) were prepared under the same conditions of MIPs except for the omission of the template. All the MIPs and their NIPs were characterized by FT-IR. The yields and template utilization of all the polymers were measured and summarized in Table 1.
4.3. Equilibrium binding experiments
Polymers (5 mg or 1 mg) and test compounds at certain concentrations were mixed in 1 mL solvent. After shaking for 12 h, the supernatant was obtained by centrifugation and measured using a UV spectrophotometer at the wavelength of 282 nm for NPA, 272 nm for both NAP and BA, and 260 nm for BPA.4.4. Dissociation constants
Dissociation constant (Kd) of the polymer to the testing molecule was calculated using the Scatchard equation:(36)where Qmax is the saturated adsorption capacity, Qe is the amount of molecule bound on the polymer, Cs is the free molecule concentration.
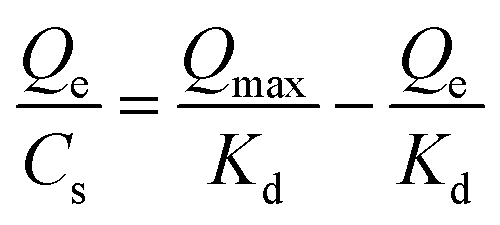
The Kd between APTES and NPA was determined based on the fluorescence changes of NPA (100 μM) by titration with APTES in ethanol using the following equation:39
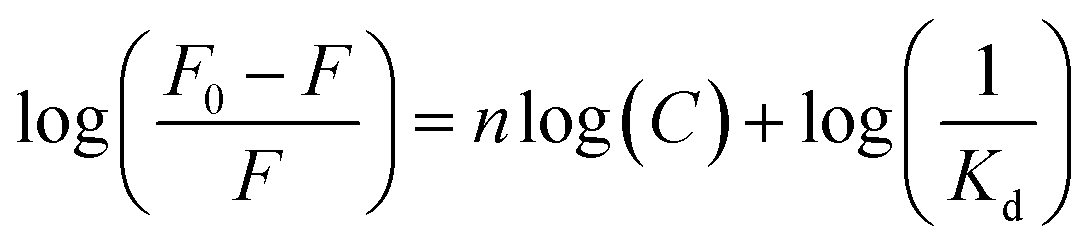
4.5. Computational details
All calculations were conducted by using density functional theory (DFT) method with the Gaussian 09 program.40 The geometry optimizations were performed with the M06-2x functional,37 and the 6-31G(d,p) basis set was used for all the atoms. Vibrational frequencies were computed at the same level of theory to obtain the free energy correction. The solvent effect was considered by a self-consistent reaction field (SCRF) using the SMD implicit solvent model,38 and ethanol or water was applied as the solvent separately. The free energy of the complex was calculated at the M06-2x/6-311++G(d,p) level. Each complex's energy was described by the Gibbs free energy in solution including the free energy correction from gas phase calculations, and the final energy was obtained by comparing the complex energy with the energy of reference state.4.6. SPE extraction
MIP1 microspheres (20 mg) were packed into a column to make a MIP-based solid phase extraction (SPE) column for the removal and enrichment of the target molecule NPA in tap water. The tap water spiked with NPA (40 mL, 1 μM, NPA 40 nmol) was pushed through the SPE column. The effluent was measured by UV to check if the spiked NPA were completely removed from the tap water. After the SPE column was washed by the pure water (500 μL), the adsorbed NPA was collected by using the phosphate buffer (10 mM, pH 8.5) for three times, each time 400 μL. The quantification result of the NPA in the desorbed solution were shown in Table S2.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21705073, 21603116 and 51406074).References
- G. Wulff and A. Sarhan, Angew. Chem., Int. Ed., 1972, 11, 341 CAS.
- G. Vlatakis, L. I. Andersson, R. Muller and K. Mosbach, Nature, 1993, 361, 645 CrossRef CAS.
- T. Takeuchi and H. Sunayama, Chem. Commun., 2018, 54, 6243 RSC.
- B. Sellergren and K. J. Shea, J. Chromatogr., 1993, 635, 31 CrossRef CAS.
- L. Ye, O. Ramstrom, M. O. Mansson and K. Mosbach, J. Mol. Recognit., 1998, 11, 75 CrossRef CAS.
- G. Wulff, Chem. Rev., 2002, 102, 1 CrossRef CAS PubMed.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859 CrossRef CAS.
- M. J. Whitcombe, I. Chianella, L. Larcombe, S. A. Piletsky, J. Noble, R. Porter and A. Horgan, Chem. Soc. Rev., 2011, 40, 1547 RSC.
- L. X. Chen, X. Y. Wang, W. H. Lu, X. Q. Wu and J. H. Li, Chem. Soc. Rev., 2016, 45, 2137 RSC.
- L. Ye, Anal. Bioanal. Chem., 2016, 408, 1727 CrossRef CAS PubMed.
- Z. Liu and H. He, Acc. Chem. Res., 2017, 50, 2185 CrossRef CAS PubMed.
- J. M. Pan, W. Chen, Y. Ma and G. Q. Pan, Chem. Soc. Rev., 2018, 47, 5574 RSC.
- G. Q. Pan, Y. Zhang, X. Z. Guo, C. X. Li and H. Q. Zhang, Biosens. Bioelectron., 2010, 26, 976 CrossRef CAS PubMed.
- L. Li, Y. Lu, Z. J. Bie, H. Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 7451 CrossRef CAS PubMed.
- Q. J. Li, T. Kamra and L. Ye, Chem. Commun., 2016, 52, 12237 RSC.
- M. D. Attieh, Y. Zhao, A. Elkak, A. Falcimaigne-Cordin and K. Haupt, Angew. Chem., Int. Ed., 2017, 56, 3339 CrossRef PubMed.
- Q. J. Li, L. D. Jiang, T. Kamra and L. Ye, Polymer, 2018, 138, 352 CrossRef CAS.
- A. Katz and M. E. Davis, Nature, 2000, 403, 286 CrossRef CAS PubMed.
- M. E. Diaz-Garcia and R. B. Laino, Microchim. Acta, 2005, 149, 19 CrossRef CAS.
- J. E. Lofgreen and G. A. Ozin, Chem. Soc. Rev., 2014, 43, 911 RSC.
- Z. J. Bie, Y. Chen, J. Ye, S. S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2015, 54, 10211 CrossRef CAS PubMed.
- X. H. Pan, X. P. He and Z. Liu, Anal. Chim. Acta, 2018, 1019, 65 CrossRef CAS PubMed.
- S. K. Vashist, E. Lam, S. Hrapovic, K. B. Male and J. H. T. Luong, Chem. Rev., 2014, 114, 11083 CrossRef CAS PubMed.
- Z. J. Duan, L. P. Fan, G. Z. Fang, J. H. Yi and S. Wang, Anal. Bioanal. Chem., 2011, 401, 2291 CrossRef CAS PubMed.
- S. F. A. Raof, S. Mohamad and M. R. Abas, Int. J. Mol. Sci., 2013, 14, 5952 CrossRef PubMed.
- D. Zhang, Y. K. Lv, R. Chen and C. C. Shi, Asian J. Chem., 2013, 25, 3922 CrossRef CAS.
- B. Gao, X. P. He, Y. Jiang, J. T. Wei, H. Suo and C. Zhao, J. Sep. Sci., 2014, 37, 3753 CrossRef CAS PubMed.
- Y. Zhou, Z. B. Qu, Y. B. Zeng, T. S. Zhou and G. Y. Shi, Biosens. Bioelectron., 2014, 52, 317 CrossRef CAS PubMed.
- K. K. Zhi, L. L. Wang, Y. G. Zhang, Y. F. Jiang, L. T. Zhang and A. Yasin, Materials, 2018, 11, 777 CrossRef PubMed.
- S. Sadeghi and M. Jahani, Food Anal. Methods, 2014, 7, 2084 CrossRef.
- Y. Wei, J. Wang, Y. L. Zhang, L. Wang and X. T. Zhang, RSC Adv., 2015, 5, 91407 RSC.
- L. X. Xu, F. Cui, J. J. Zhang, Y. J. Hao, Y. Wang and T. Y. Cui, Nanoscale, 2017, 9, 899 RSC.
- L. X. Xu, F. Cui, J. J. Zhang, X. Zhang, Y. Wang and T. Y. Cui, Nanoscale, 2018, 10, 9460 RSC.
- B. V. Zhmud and J. Sonnefeld, J. Non-Cryst. Solids, 1996, 195, 16 CrossRef CAS.
- S. G. Coombs, S. Khodjaniyazova and F. V. Bright, Talanta, 2018, 177, 26 CrossRef CAS PubMed.
- Q. J. Li, X. Y. Tu, J. Ye, Z. J. Bie, X. D. Bi and Z. Liu, Chem. Sci., 2014, 5, 4065 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian Inc., Wallingford CT, 2009.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01684e |
| This journal is © The Royal Society of Chemistry 2020 |