
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Perfect cubic La-doped boron clusters La6&[La@B24]+/0 as the embryos of low-dimensional lanthanide boride nanomaterials†
Xiao-Qin Lu,
Mei-Zhen Ao,
Xin-Xin Tian,
Wen-Yan Zan *,
Yue-Wen Mu* and
Si-Dian Li*
*,
Yue-Wen Mu* and
Si-Dian Li*
Nanocluster Laboratory, Institute of Molecular Science, Shanxi University, Taiyuan 030006, China. E-mail: zanwy@sxu.edu.cn; ywmu@sxu.edu.cn; lisidian@sxu.edu.cn
First published on 27th March 2020
Abstract
La-doped boron nanoclusters have received considerable attention due to their unique structures and bonding. Inspired by recent experimental observations of the inverse sandwich D8h La2B8 (1) and triple-decker C2v La3B14− (2) and based on extensive global searches and first-principles theory investigations, we present herein the possibility of the perfect cubic La-doped boron clusters Oh La6&[La@B24]+ (3, 1A1g) and Oh La6&[La@B24] (4, 2A2g) which appear to be the embryos of the metallic one-dimensional La10B32 (5) nanowire, two-dimensional La3B10 (6) nanosheet, and three-dimensional LaB6 (7) nanocrystal, facilitating a bottom-up approach to build cubic lanthanide boride nanostructures from gas-phase clusters. Detailed molecular orbital and bonding analyses indicate that effective (d–p)σ, (d–p)π and (d–p)δ covalent coordination interactions exist in La6&[La@B24]+/0 (3/4) clusters, while the 1D La10B32 (5), 2D La3B10 (6), and 3D LaB6 (7) crystals exhibit mainly electrostatic interactions between the trivalent La centers and cubic B24 frameworks, with weak but discernible coordination contributions from La (5d) ← B (2p) back-donations. The IR and Raman spectra of La6&[La@B24]+/0 (3/4) and band structures of La10B32 (5) and La3B10 (6) are computationally simulated to facilitate their future characterizations.
1. Introduction
As a prototypical electron-deficient element next to carbon in the periodical table, boron has a rich chemistry characterized with delocalized multicenter-two-electron (mc-2e) bonds in both bulk allotropes and polyhedral molecules.1 Boron-based materials have found wide applications in field emissions, supercapacitors, optical absorptions, photodetectors, and etc.2–6 A bottom-up approach has received considerable attention in the past two decades to investigate the structural transitions from small boron nanoclusters to boron nanomaterials. Persistent joint photoelectron spectroscopy (PES) and first-principles theory investigations in the past two decades by Wang and coworkers have unveiled an unexpectedly rich landscape for size-selected boron clusters from planar or quasi-planar Bn−/0 (n = 3–38, 41 and 42) to cage-like borospherenes D2d B40−/0 and C3/C2 B39−. Multiple low-lying isomers appear to compete and coexist in most gas-phase boron Bn−/0 clusters, starting from n = 7.7–10 Ion-mobility measurements in combination with density functional theory (DFT) calculations, on the other hand, have shown that boron cluster monocations (Bn+) possess double-ring tubular structures in the size range between n = 16–25,11 unveiling another structural domain for boron nanoclusters. Transition-metal-doping has proven to induce dramatic structural and bonding pattern changes to boron clusters. Typical examples include the transition-metal-centered monocyclic boron wheel clusters M@Bn (Co@B8−, Ru@B9−, and Ta@B10−) with the maximum planar coordination number of CN = 10 and transition-metal-centered boron double-ring tubular clusters M@Bn− (Co@B16−, Rh@B18−, and Ta@B20−) with the record tubular coordination number of CN = 20 known in experiments.12–18 A family of inverse sandwich di-lanthanide boron complexes D7h La2B7−, D8h La2B8 (1), and D9h La2B9− with a perfect Bn ring (n = 7, 8, 9) sandwiched between two La atoms were observed in 2018 in PES measurements.19,20 The first inverse triple-decker di-lanthanide boron cluster C2v La3B14− (2) was discovered in 2019 in a joint experimental and theoretical investigation21 in which two conjoined La2B8 (1) inverse sandwiches share a B–B edge and a La vertex in the titled La–B8–La–B8–La motif. Boron clusters as typical Wankel motor molecules with fluxional bonds also exhibit structural fluxionalities where metal dopants play an important role.22–25 Our group very recently predicted the possibility of the inverse sandwich double-ring tubular molecular rotor C2h La2&[B2@B18] which possesses the smallest core–shell structure reported in boron clusters.26Bulk lanthanide hexaboride LaB6 (7) has been widely used as cathode materials due to its numerous excellent properties such as low work function, ultrahigh hardness, high chemical inertness, and high melting point.27–30 It possesses a typical cubic CaB6-type lattice (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Oh1) characterized with a 3D network constituted of B6 octahedrons, with the interstitial locations filled by trivalent La atoms.31 Interestingly, each unit cell in LaB6 (7) contains a B24 cubic framework which possesses six equivalent B8 rings on the surface sharing twelve B–B dumb-bells on the edges, with each B8 ring sandwiched between two neighboring La atoms. The strong structural similarity between the gas-phase inverse sandwich La2B8 (1) complex and cubic three-dimensional (3D) LaB6 (7) crystal has been previously noticed.31 LaB6 nanowires in cubic LaB6 (7) lattice were also recently confirmed to be active electrochemical materials for supercapacitors.32 However, a cubic embryo of the LaB6 (7) lattice still remains unknown to date in a bottom-up approach starting from gas-phase clusters. Such a cubic embryo may also be used to form one-dimensional (1D) lanthanide boride nanowires and two-dimensional (2D) lanthanide boride nanosheets.
m, Oh1) characterized with a 3D network constituted of B6 octahedrons, with the interstitial locations filled by trivalent La atoms.31 Interestingly, each unit cell in LaB6 (7) contains a B24 cubic framework which possesses six equivalent B8 rings on the surface sharing twelve B–B dumb-bells on the edges, with each B8 ring sandwiched between two neighboring La atoms. The strong structural similarity between the gas-phase inverse sandwich La2B8 (1) complex and cubic three-dimensional (3D) LaB6 (7) crystal has been previously noticed.31 LaB6 nanowires in cubic LaB6 (7) lattice were also recently confirmed to be active electrochemical materials for supercapacitors.32 However, a cubic embryo of the LaB6 (7) lattice still remains unknown to date in a bottom-up approach starting from gas-phase clusters. Such a cubic embryo may also be used to form one-dimensional (1D) lanthanide boride nanowires and two-dimensional (2D) lanthanide boride nanosheets.
Keep the inspiration in mind and based on extensive global minimum (GM) searches and first-principles theory calculations, we predict herein the highly stable perfect cubic La-doped boron clusters Oh La6&[La@B24]+ (3, 1A1g) and Oh La6&[La@B24] (4, 2A2g) which consist of six equivalent La2B8 (1) inverse sandwiches embedded in a cube sharing one La atom at center. More interestingly, La6&[La@B24]+/0 (3/4) clusters with a B24 framework turn out to be the cubic embryos of the metallic 1D La10B32 (5) nanowire, 2D La3B10 (6) nanosheet, and 3D LaB6 (7) nanocrystal in a bottom-up approach. The newly obtained ferromagnetic 1D La10B32 (5) and nonmagnetic 2D La3B10 (6) are expected to serve as efficient electronical and optical nanomaterials.
2. Theoretical procedure
Extensive GM searches were performed on La7B24+ monocation using the TGmin2 code33 at the DFT level, in combination with manual structural constructions based on the experimentally observed La2B8 (1) and La3B14− (2). Over 3000 trial structures were explored in both singlet and triplet states at PBE/TZVP. The low-lying isomers were then fully optimized at the PBE0 (ref. 34) and TPSSh35 levels with the 6-311+G* basis set36 for B and Stuttgart relativistic small-core pseudopotential for La37,38 using the Gaussian 09 program suite.39 Low-lying isomers of the open-shell neutral La7B24 were obtained from the corresponding lowest-lying structures of La7B24+.The GM search on 1D La10B32 (5) was performed with the EOU program developed by our group.40 A GM search based on the PSO technique implemented in the widely used Particle Swarm Optimization (CALYPSO)41 package was executed for 2D La3B10 (6). The calculations on 1D La10B32 (5), 2D La3B10 (6), and 3D LaB6 (7) nanostructures were performed using the Vienna ab initio simulation package (VASP),42,43 within the framework of projector augmented wave (PAW) pseudopotential method44,45 and PBE generalized gradient approximation (GGA).46,47 The Coulomb-corrected local spin-density approximation (LSDA+U) was utilized for both structural relaxation and static calculation (U = 5 eV).31,48 The cutoff energy of the plane wave basis is set to 500 eV. Atomic structures are fully relaxed using the conjugate gradient method until the maximum force on each atom was less than 0.01 eV Å−1 and the energy precision was set to 10−5 eV. Chemical bonding analyses were performed for La6&[La@B24]+ (3) using the adaptive natural density partitioning (AdNDP) approach at the PBE0 level49,50 and for 3D LaB6 (7) crystal utilizing the solid-state AdNDP (SSAdNDP) method.51,52 Natural bonding orbital (NBO) analyses were performed on La7B24+ (3) using the NBO 6.0 program.53 Born–Oppenheimer molecular dynamics (BOMD) simulations were performed on La7B24+ (3) for 30 ps at 300 K and 1000 K using the CP2K software package.54
3. Results and discussions
3.1 Structures and stabilities
Starting from the experimentally observed C2v La3B14− (2) which possesses a tilted La–B8–La–B8–La inverse triple-decker structure with two conjoined B8 rings sharing a B–B dumb-bell, we manually constructed the perfect cubic hepta-lanthanide-doped boron clusters Oh La6&[La@B24]+ (3, 1A1g) and Oh La6&[La@B24] (4, 2A2g) from two C2v La3B14− (2) clusters which share two B–B dumb-bells on the edges and one La atom at the center, with two extra La atoms added on the top and bottom, respectively. With one La atom endohedrally coordinated at the center (@) and six La atoms exohedrally coordinated on the surface (&), Oh La6&[La@B24]+/0 (3/4) can be viewed as consisting of six equivalent La2B8 (1) inverse sandwiches embedded in a cube sharing twelve B–B dumb-bells on the edges and one La vertex at the center. Interestingly and encouragingly, as shown in Fig. S1 and S2,† extensive GM searches strongly suggest that the highly stable perfect cubic La6&[La@B24]+ (3) and La6&[La@B24] (4) with a B24 framework be the well-defined GMs of the systems with the lowest vibrational frequencies of 111 cm−1 and 112 cm−1, respectively. La6&[La@B24]+/0 (3/4) possess the optimized La–B distances of rLa–B = 2.94/2.95 Å between the central La atom and its cubic B24 ligand and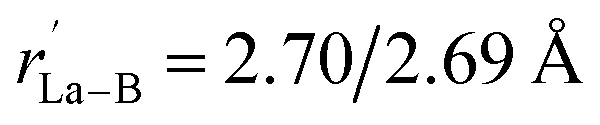 between the surface La atoms and their neighboring B8 ligands on the surface, with the B–B distances of rB–B = 1.67/1.67 Å within the B–B dumb-bells on the edges and
between the surface La atoms and their neighboring B8 ligands on the surface, with the B–B distances of rB–B = 1.67/1.67 Å within the B–B dumb-bells on the edges and 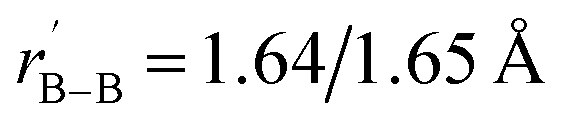 within the B3 triangles at the corners. The large HOMO–LUMO gap of ΔEgap = 2.35 eV calculated for La6&[La@B24]+ (3) well supports its high chemical stability. As shown in Fig. S1,† its second lowest-lying triplet C1 La6&[La@B24]+ (3A) which is slightly distorted (with rLa–B = 2.93–2.95 Å and
within the B3 triangles at the corners. The large HOMO–LUMO gap of ΔEgap = 2.35 eV calculated for La6&[La@B24]+ (3) well supports its high chemical stability. As shown in Fig. S1,† its second lowest-lying triplet C1 La6&[La@B24]+ (3A) which is slightly distorted (with rLa–B = 2.93–2.95 Å and 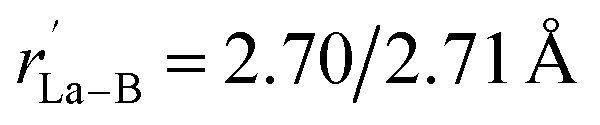 ) due to Jahn–Teller effect lies 1.30 and 1.36 eV higher than the Oh GM in energy at PBE0 and TPSSH levels, respectively. Both the distorted cubic quintet and heptet isomers with the relative energies of 2.50 eV and 3.53 eV at PBE0, respectively, appear to be much less stable (Fig. S1†). Extensive molecular dynamics simulations indicate that La6&[La@B24]+ (3) is also highly dynamically stable, with the calculated average root-mean-square-deviations of RMSD = 0.12 Å and maximum bond length deviations of MAXD = 0.38 Å at 1000 K. Similar situation exists in La6&[La@B24] (4). La6&[La@B24]+/0 (3/4) are therefore the well-defined deep-lying GMs of the systems highly stable both thermodynamically and dynamically.
) due to Jahn–Teller effect lies 1.30 and 1.36 eV higher than the Oh GM in energy at PBE0 and TPSSH levels, respectively. Both the distorted cubic quintet and heptet isomers with the relative energies of 2.50 eV and 3.53 eV at PBE0, respectively, appear to be much less stable (Fig. S1†). Extensive molecular dynamics simulations indicate that La6&[La@B24]+ (3) is also highly dynamically stable, with the calculated average root-mean-square-deviations of RMSD = 0.12 Å and maximum bond length deviations of MAXD = 0.38 Å at 1000 K. Similar situation exists in La6&[La@B24] (4). La6&[La@B24]+/0 (3/4) are therefore the well-defined deep-lying GMs of the systems highly stable both thermodynamically and dynamically.
We initially constructed a 1D La5B16 nanowire (P4/mmm) (Fig. S3†) by extending the finite La6&[La@B24]+/0 (3/4) clusters periodically in one direction (x), resulting in a 1D nanowire with one small imaginary phonon frequency at 49i cm−1. This imaginary phonon frequency corresponds to a typical Peierls phase transition which leads to the slightly more stable 1D La10B32 nanowire (5, P4/mmm) (by 0.07 eV) when the unit cell is doubled in size, with the two La atoms inside slightly off-centered by 0.22 Å (Fig. 1). Similarly, 2D La3B10 (6) (P4/mmm) and 3D LaB6 (7) (Pmm) networks can be obtained by expanding La6&[La@B24]+/0 (3/4) periodically in (x, y) and (x, y, z) directions, respectively (Fig. 1). The 1D (5), 2D (6), and 3D (7) networks possess the optimized lattice parameters of a = 8.66 Å, a = b = 4.17 Å and a = b = c = 4.16 Å at PBE level, respectively. Our calculated lattice parameters and La–B distance (rLa–B = 3.06 Å) for 3D LaB6 (7) agree well with the corresponding experimentally measured values of a = b = c = 4.16 Å and rLa–B = 3.05 Å.31 The newly obtained 2D La3B10 (6) possesses the optimized La–B distances of rLa–B(x) = rLa–B(y) = 3.05 Å and rLa–B(z) = 3.07 Å between the central La atom and its B24 cubic ligand. Similar La–B distances exist in 1D La10B32 (5). These La–B distances turn out to be slightly longer than the corresponding La–B coordination bond lengths of rLa–B = 2.94/2.95 Å in La6&[La@B24]+/0 (3/4) clusters. Interestingly, as shown in Fig. S4 and S5,† 1D La10B32 (5) has the lowest cohesive energy among the 1D structures obtained via extensive EOU global searches, while La3B10 (6) is the lowest-lying structure in all the 2D La3B10 conformations probed by extensive PSO global searches, with the second lowest-lying 1D and 2D structures lying 3.13 eV and 1.88 eV higher in cohesive energy per unit cell than La10B32 (5) and La3B10 (6), respectively. 1D La10B32 (5) and 2D La3B10 (6) are thus strongly favored in thermodynamics compared with their other low-lying counterparts.
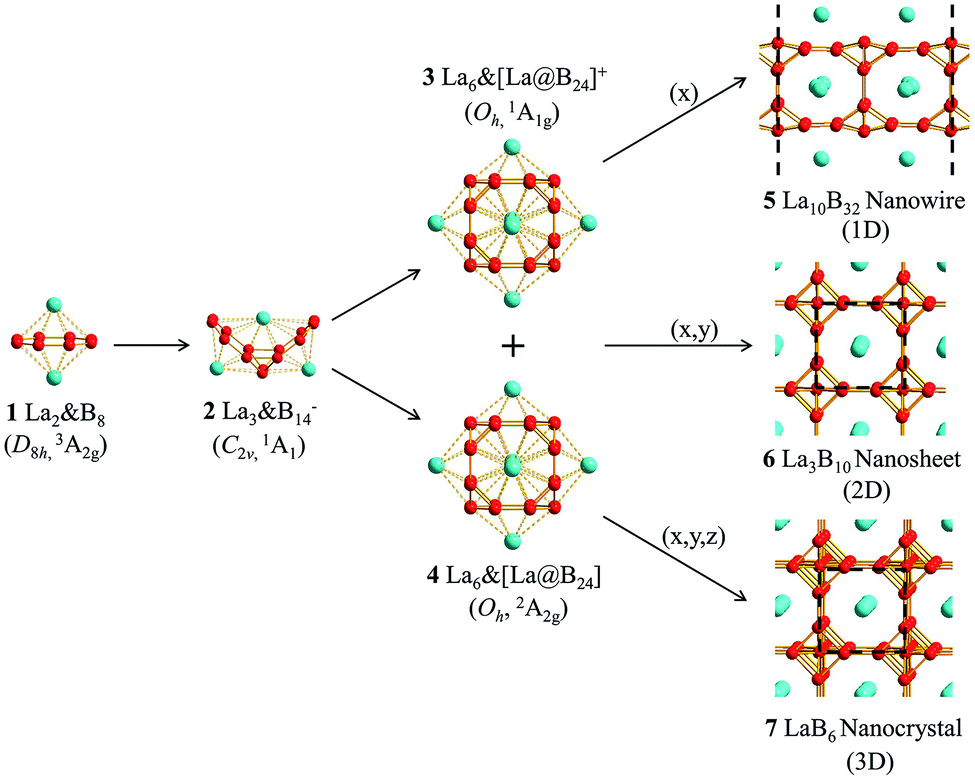 | ||
| Fig. 1 Optimized structures of inverse sandwich D8h La2&B8 (1), inverse triple-decker C2v La3@B14− (2), cubic Oh La6&[La@B24]+ (3), cubic Oh La6&[La@B24] (4), 1D La10B32 nanowire (5), 2D La3B10 nanosheet (6), and 3D LaB6 nanocrystal (7). | ||
To evaluate the relative stability of 1D La10B32 (5), 2D La3B10 (6), and 3D LaB6 (7) networks, we computed their average cohesive energy per atom
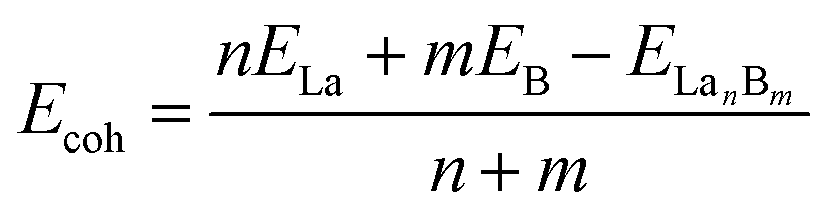
3.2 Electronic structures and bonding patterns
The high stabilities of these cubic lanthanide boride nanostructures can be traced back to their unique electronic structures and bonding patterns. Detailed NBO analyses on closed-shell La6&[La@B24]+ (3) show that the central La atom possesses the natural atomic charge of qLa = −0.50|e|, electronic configuration of La[Xe]4f0.185d3.046s0.24, and total Wiberg bond order of WBILa = 5.26, while the six surface La atoms have the natural atomic charge of qLa = +1.43|e|, electronic configuration of La[Xe]4f0.155d1.416s0.10, and total Wiberg bond order of WBILa = 2.86. Obviously, both the central and surface La atoms in La6&[La@B24]+ (3) donate their 6s2 electrons almost completely to the B24 cubic ligand and there exists an effective La 5d ← B 2p back-donation from the cubic B24 ligand to the central La atom. Especially, the central La atom with the 5d occupation of 5d3.04 has a much higher total La–B coordination bond order (5.26) than that of the partially coordinated surface La atoms (2.86) with the 5d occupation of 5d1.41. Obvious La–B coordination interaction with the average La–B bond order of WBILa–B = 0.22 exists between the central La and its 24 equivalent B ligands in La6&[La@B24]+ (3). The La coordination centers in inverse sandwich La2&B8 (1) and La3@B14− (2) possess the net atomic charges of qLa = +1.29, +1.39/1.17|e|, electronic configurations of La[Xe]4f0.175d1.496s0.10, La[Xe]4f0.13/0.165d1.37/1.636s0.11/0.11, and total Wiberg bond orders of WBILa = 2.87, 2.88/3.08, repectively, similar to the situation in the six surface La atoms in La6&[La@B24]+ (3). Detailed molecular orbital (MO) analyses indicate that all the occupied orbitals of the closed-shell La6&[La@B24]+ (3) are effective bonding MOs, while its lowest unoccupied MO (LUMO, (a2g)) is basically an anti-bonding orbital mainly originated from the 5d atomic orbitals of the six surface La atoms (Fig. S6†). With a valence electron occupying the non-degenerate LUMO (a2g) of La6&[La@B24]+ (3), neutral La6&[La@B24] (4) possesses a singly occupied highest occupied MO (SOMO (a2g)). Such an electron occupation helps maintain the Oh symmetry of the open-shell neutral which possesses the same geometry as La6&[La@B24]+ (3).To better understand the bonding nature and stabilization mechanism, we performed a detailed AdNDP bonding analysis on the closed-shell La6&[La@B24]+ (3) which recovers both the localized and delocalized bonds of the system. As clearly indicated in Fig. 2a, La6&[La@B24]+ (3) possesses 12 equivalent 2c-2e B–B σ bonds on twelve B–B dumb-bells with the occupation number of ON = 1.78|e| and 8 equivalent 3c-2e σ bonds over eight B3 triangles at the corners of the cube with ON = 1.83|e|, forming the σ-skeleton on the cubic B24 ligand. The remaining 26 delocalized bonds are mainly responsible for the La–B coordination interactions in the complex, including 6 10c-2e B8(σ)–La2(dσ) bonds, 12 10c-2e B8(σ)–La2(dπ) bonds, and 6 10c-2e B8(π)–La2(dδ) bonds with ON = 1.72–1.90|e| on six equivalent conjoined La2B8 (1) inverse sandwiches embedded in the cube and 2 31c-2e B24(π)–La7(dσ) bonds totally delocalized over the whole molecule with ON = 2.00|e| (Fig. 2a, see Fig. S7† for more details). Overall, cubic La6&[La@B24]+ (3) has similar (d–p)σ, (d–p)π, and (d–p)δ coordination interactions with that of observed inverse sandwich La2B8 (1) and triple-decker La3B14− (2).19–21 The bonding pattern presented above provides further evidence that all the occupied MOs of La6&[La@B24]+ (3) are intrinsically bonding MOs (Fig. S6†), while all its antibonding MOs remain empty. Such a unique electron occupation renders an exceptionally high stability to the monocation.
 | ||
| Fig. 2 AdNDP bonding patterns of (a) La6&[La@B24]+ (3) cluster and (b) 3D LaB6 (7) crystal, with the occupation numbers (ONs) indicated. Each 10c-2e B8–La2 bond in the second row represents six equivalent La–B8–La coordination bonds, as detailed in the ESI in Fig. S7.† | ||
We also performed a detailed SSAdNDP analysis on the periodical 3D LaB6 (7) cubic lattice in Fig. 2b. There are 3 equivalent 2c-2e B–B σ bonds on three B–B dumb-bells in the cubic unit cell with ON = 1.80|e| which correspond to the 12 2c-2e B–B σ bonds on the B24 ligand in La6&[La@B24]+ (3). The remaining 8 equivalent 4c-2c B3(σ)–La(dσ) bonds between the central La atom and the eight B3 triangles at the corners with ON = 1.77|e| represent mainly electrostatic interactions between the trivalent La center and B24 cubic framework, with weak but discernible coordination contributions from La (5d) ← B (2p) back-donations (Fig. 2b). Similar bonding patterns exist in 1D La10B32 (5) and 2D La3B10 (6). Detailed Bader charge analyses55 indicate that the central La atom in LaB6 (7) carries the net atomic charges of qLa = +1.71|e|, with its B neighbors possessing the average atomic charge of qB = −0.29|e|. The formation of rare earth (RE) boride crystals REB6 in infinite lattices involve an obvious electron transfer from the trivalent RE centers to the boron sublattice. 1D (5), 2D (6), and 3D (7) nanocrystals with the elongated La–B distances of rLa–B(z) = 3.05–3.07 Å possess obviously weaker La–B covalent coordination interactions than that in La6&[La@B24]+/0 (3/4) clusters which have the La–B coordination distances of rLa–B = 2.94–2.95 Å. The size of the trivalent RE atoms, rather than their electron configuration, is the main factor responsible for their boride structures.56
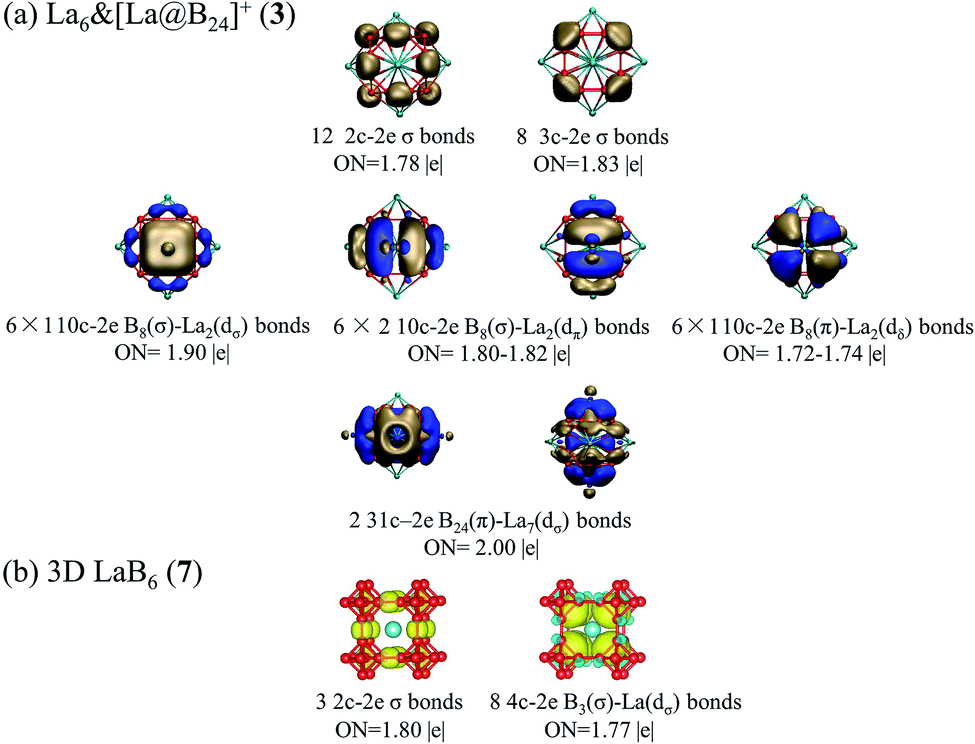
The calculated band structures and corresponding projected density of states (PDOS) of 1D La10B32 (5) and 2D La3B10 (6) nanostructures are depicted in Fig. 3. Notably, these nanostructures all appear to be metallic in nature. Both 2p orbitals from B atoms and 5d orbitals from La centers contribute to the calculated PDOS near the Fermi level, with La-5d orbitals making major contributions to the PDOS above the Fermi level, while B-2p orbitals dominating the PDOS below the Fermi level. Both spin unpolarized and spin-polarized computations were carried out to determine the ground states for these nanomaterials during the structural optimizations. 1D La10B32 (5) network turned out to be a ferromagnetic metal with the total magnetic moment of 2.03 μB per unit cell which mainly originates from the surface La atoms (with each surface La carrying the magnetic moment of 0.12 μB, while each central La carrying the magnetic moment of only 0.008 μB). In contrast, both 2D La3B10 (6) and 3D LaB6 (7) are nonmagnetic in nature with the calculated magnetic moments of zero.
 | ||
| Fig. 3 Calculated band structures and projected densities of states (PDOS) of (a) 1D La10B32 (5) and (b) 2D La3B10 (6) using the PBE functional. | ||
3.3 Simulated IR and Raman spectra of the La6&[La@B24]+ embryo
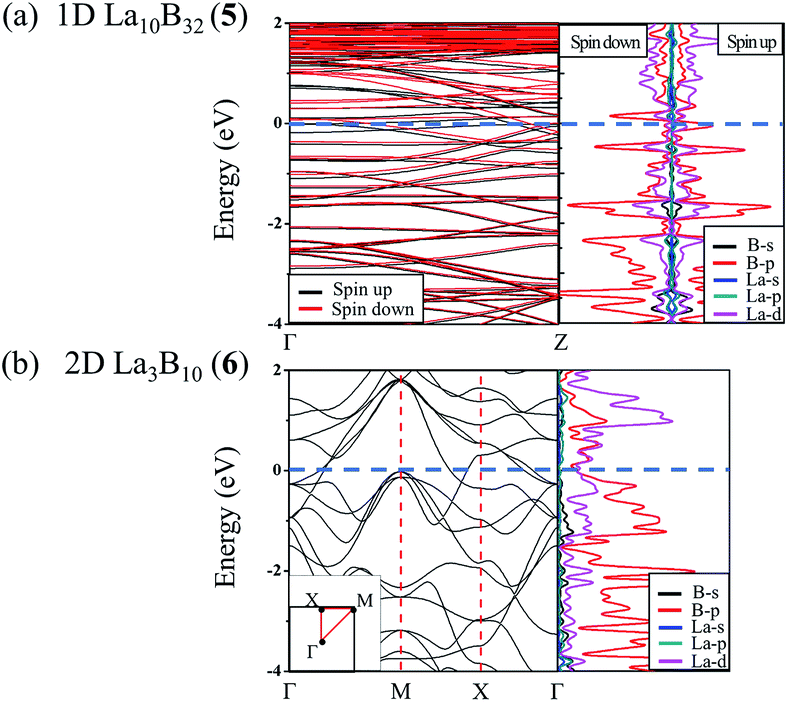
Infrared photodissociation (IR-PD) in combination with first-principles theory calculations has proven to be a powerful means to characterize novel gas-phase clusters.57,58 We computationally simulate the IR and Raman spectra of the closed-shell La6&[La@B24]+ (3) in Fig. 4a to facilitate its future experimental characterizations. The high-symmetry Oh La6&[La@B24]+ possesses relatively simple vibrational spectra, with sharp asymmetrical IR vibrations at 190 (t1u), 301 (t1u), 431 (t1u), and 1215 (t1u) cm−1 and symmetrical Raman features at 187 (a1g), 289 (t2g), 497 (a1g), 636 (t2g), 1190 (t2g) and 1298 (a1g) cm−1, respectively. The strong Raman peaks at 187 cm−1 and 497 cm−1 mainly represent the typical radial breathing modes (RBMs) (a1g) of the six evenly distributed surface La atoms and the B24 cubic framework, respectively. Such RBM spectral features can be used to characterize hollow boron structures.59 As shown in Fig. S8,† similar but slightly more complicated IR and Raman spectra are obtained for the open-shell neutral La6&[La@B24] (4). | ||
| Fig. 4 Simulated IR and Raman spectra of Oh La6&[La@B24]+ (3) at PBE0/6-311+G(d) level. | ||
4. Conclusions
Based on extensive GM searches and first-principles theory calculations, we have predicted in this work the highly stable perfect cubic Oh La6&[La@B24]+/0 (3/4) which are the embryos of the metallic 1D La10B32 (5) nanowire, 2D La3B10 (6) nanosheet, and 3D LaB6 (7) nanocrystal, building a bridge between gas-phase lanthanide boride clusters and their low-dimensional nanomaterials. La6&[La@B24]+/4 (3/4) clusters and 1D (5), 2D (6), and 3D (7) nanocrystals exhibit strong similarities in structures, with the periodical nanostructures possessing obviously weaker La (5d) ← B (2p) back-donations than their cubic embryos. Using the commercially available 3D LaB6 (7) as target, it is possible to synthesize the newly predicted 0D (3/4), 1D (5), and 2D (6) nanomaterials by laser ablations, chemical vaporizations, or arc-discharges. The construction strategy demonstrated in this work may be applicable to the design of more rare-earth boride nanomaterials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (21720102006 to S.-D. Li, 11504213 to Y.-W. Mu, and 21803037 to W.-Y. Zan).Notes and references
- F. A. Cotton, G. Wilkinson, C. A. Murrillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999 Search PubMed.
- J. Q. Xu, G. H. Hou, H. Q. Li, T. Y. Zhai, B. P. Dong, H. L. Yan, Y. R. Wang, B. H. Yu, Y. Bando and D. Golberg, NPG Asia Mater., 2013, 5, e53 CrossRef CAS.
- A. Sussardi, T. Tanaka, A. U. Khan, L. Schlapbach and T. Mori, J. Materiomics, 2015, 1, 196 CrossRef.
- G. Akopov, M. T. Yeung and R. B. Kaner, Adv. Mater., 2017, 29, 1604506 CrossRef PubMed.
- S. Carenco, D. Portehault, C. Boissière, N. Mézailles and C. Sanchez, Chem. Rev., 2013, 113, 7981 CrossRef CAS PubMed.
- Y. Tian, Z. k. Guo, T. Zhang, H. J. Lin, Z. J. Li, J. Chen, S. Z. Deng and F. Liu, Nanomaterials, 2019, 9, 538 CrossRef CAS PubMed.
- Q. Chen, W. L. Li, Y. F. Zhao, S. Y. Zhang, H. S. Hu, H. Bai, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, ACS Nano, 2015, 9, 754 CrossRef CAS PubMed.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727 CrossRef CAS PubMed.
- H. Bai, T. T. Chen, Q. Chen, X. Y. Zhao, Y. Y. Zhang, W. J. Chen, W. L. Li, L. F. Cheung, B. Bai, J. Cavanagh, W. Huang, S. D Li, J. Li and L. S. Wang, Nanoscale, 2019, 11, 23286 RSC.
- T. Jian, X. N. Chen, S. D. Li, A. I. Boldyrev, J. Li and L. S. Wang, Chem. Soc. Rev., 2019, 48, 3550 RSC.
- E. Oger, N. R. M. Crawford, R. Kelting, P. Weis, M. M. Kappes and R. Ahlrichs, Angew. Chem., Int. Ed., 2007, 46, 8503 CrossRef CAS PubMed.
- W. L. Li, C. Romanescu, T. R. Galeev, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 165 CrossRef CAS PubMed.
- T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2012, 51, 2101 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Acc. Chem. Res., 2013, 46, 350 CrossRef CAS PubMed.
- I. A. Popov, T. Jian, G. V. Lopez, A. I. Boldyrev and L. S. Wang, Nat. Commun., 2015, 6, 8654 CrossRef CAS PubMed.
- T. Jian, W. L. Li, X. Chen, T. T. Chen, G. V. Lopez, J. Li and L. S. Wang, Chem. Sci., 2016, 7, 7020 RSC.
- W. L. Li, T. Jian, X. Chen, H. R. Li, T. T. Chen, X. M. Luo, S. D. Li, J. Li and L. S. Wang, Chem. Commun., 2017, 53, 1587 RSC.
- T. Heine and G. Merino, Angew. Chem., Int. Ed., 2012, 51, 4275 CrossRef CAS PubMed.
- W. L. Li, T. T. Chen, D. H. Xing, X. Chen, J. Li and L. S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6972 CrossRef CAS PubMed.
- T. T. Chen, W. L. Li, J. Li and L. S. Wang, Chem. Sci., 2019, 10, 2534 RSC.
- T. T. Chen, W. L. Li, W. J. Chen, J. Li and L. S. Wang, Chem. Commun., 2019, 55, 7864 RSC.
- S. Pan, J. Barroso, S. Jalife, T. Heine, K. R. Asmis and G. Merino, Acc. Chem. Res., 2019, 52, 2732 CrossRef CAS PubMed.
- L. Liu, D. Moreno, E. Osorio, A. C. Castro, S. Pan, P. K. Chattaraj, T. Heinea and G. Merino, RSC Adv., 2016, 6, 27177 RSC.
- G. Martínez-Guajardo, J. L. Cabellos, A. Díaz-Celaya, S. Pan, R. Islas, P. K. Chattaraj, T. Heine and G. Merino, Sci. Rep., 2015, 5, 11287 CrossRef PubMed.
- M. Yan, H. R. Li, X. Y. Zhao, X. Q. Lu, Y. W. Mu, H. G. Lu and S. D. Li, J. Comput. Chem., 2019, 40, 966 CrossRef CAS PubMed.
- X. Q. Lu, Q. Chen, X. X. Tian, Y. W. Mu, H. G. Lu and S. D. Li, Nanoscale, 2019, 11, 21311 RSC.
- C. H. Chen, T. Aizawa, N. Iyi, A. Sato and S. Otani, J. Alloys Compd., 2004, 366, L6 CrossRef CAS.
- H. Zhang, J. Tang, J. S. Yuan, J. Ma, N. Shinya, K. Nakajima, H. Murakami, T. Ohkubo and L. C. Qin, Nano Lett., 2010, 10, 3539 CrossRef CAS PubMed.
- S. S. Ordanyan, S. V. Vikhman, D. D. Nesmelov, D. P. Danilovich and I. B. Panteleev, Adv. Sci. Technol., 2014, 89, 47 Search PubMed.
- H. Zhang, J. Tang, L. Zhang, B. An and L. C. Qin, Appl. Phys. Lett., 2008, 92, 173121 CrossRef.
- W. L. Li, C. Ertural, D. Bogdanovski, J. Li and R. Dronskowski, Inorg. Chem., 2018, 57, 12999 CrossRef CAS PubMed.
- Q. Xue, Y. Tian, S. Z. Deng, Y. Huang, M. S. Zhu, Z. X. Pei, H. F. Li, F. Liu and C. Y. Zhi, Mater. Today Energy, 2018, 10, 28 CrossRef.
- X. Chen, Y. F. Zhao, Y. Y. Zhang and J. Li, J. Comput. Chem., 2019, 40, 1105 CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- D. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS.
- L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- F.-W. Mu, A global search code based on exhaustible operations of units, 2020, to be published.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- V. I. Anisimov, I. V. Solovyev, M. A. Korotin, M. T. Czyżyk and G. A Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 16929 CrossRef CAS PubMed.
- N. V. Tkachenko and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2019, 21, 9590 RSC.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- B. D. Dunnington and J. R. Schmidt, J. Chem. Theory Comput., 2012, 8, 1902 CrossRef CAS PubMed.
- T. R. Galeev, B. D. Dunnington, J. R. Schmidt and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2013, 15, 5022 RSC.
- P. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, 2013 Search PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef CAS.
- G. Henkelman, A. Arnaldsson and H. Jonsson, Comput. Mater. Sci., 2006, 36, 354 CrossRef.
- J. Etourneau and P. Hagenmuller, Philos. Mag. B, 1985, 52, 589 CAS.
- G. Wang, M. Zhou, J. T. Goettel, G. J. Schrobilgen, J. Su, J. Li, T. Schloder and S. Riedel, Nature, 2014, 514, 475 CrossRef CAS PubMed.
- M. R. Fagiani, X. W. Song, P. Petkov, S. Debnath, S. Gewinner, W. Schcllkopf, T. Heine, A. Fielicke and K. R. Asmis, Angew. Chem., Int. Ed., 2016, 56, 501 CrossRef PubMed.
- D. Ciuparu, R. F. Klie, Y. M. Zhu and L. Pfefferle, J. Phys. Chem. B, 2004, 108, 39 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01616k |
| This journal is © The Royal Society of Chemistry 2020 |