
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles studies of electronic properties in lithium metasilicate (Li2SiO3)
Nguyen Thi Han*ab,
Vo Khuong Dien a,
Ngoc Thanh Thuy Tranc,
Duy Khanh Nguyend,
Wu-Pei Sue and
Ming-Fa Lin*ac
a,
Ngoc Thanh Thuy Tranc,
Duy Khanh Nguyend,
Wu-Pei Sue and
Ming-Fa Lin*ac
aDepartment of Physics, National Cheng Kung University, 701 Tainan, Taiwan. E-mail: han.nguyen.dhsptn@gmail.com; mflin@mail.ncku.edu.tw
bDepartment of Chemistry, Thai Nguyen University of Education, 20 Luong Ngoc Quyen, Quang Trung, Thai Nguyen City, Thai Nguyen Province, Vietnam
cHierarchical Green-Energy Materials (Hi-GEM) Research Center, National Cheng Kung University, Tainan, 70101, Taiwan
dInstitute of Applied Technology, Thu Dau Mot University, Binh Duong Province, Vietnam
eDepartment of Physics and Texas Center for Superconductivity, University of Houston, TX 77204, USA
First published on 29th June 2020
Abstract
Lithium metasilicate (Li2SiO3), which could serve as the electrolyte material in Li+-based batteries, exhibits unique lattice symmetry (an orthorhombic crystal), valence and conduction bands, charge density distribution, and van Hove singularities. Delicate analyses, based on reliable first-principles calculations, are utilized to identify the critical multi-orbital hybridizations in Li–O and Si–O bonds, 2s–(2s, 2px, 2py, 2pz) and (3s, 3px, 3py, 3pz)–(2s, 2px, 2py, 2pz), respectively. This system shows a huge indirect gap of 5.077 eV. Therefore, there exist many strong covalent bonds, with obvious anisotropy and non-uniformity. On the other hand, the spin-dependent magnetic configurations are thoroughly absent. The theoretical framework could be generalized to explore the essential properties of cathode and anode materials of oxide compounds.
1 Introduction
Nowadays, Li+-based batteries (LIBs) have become one of the mainstream systems in basic materials science and engineering applications,1–4 mainly owing to their diverse geometric, electronic, and transport properties. They are principally composed of the electrolyte, cathode, and anode materials (Fig. 1).5–7 In general, each component possesses unusual geometry, with a very large primitive unit cell directly reflecting the complicated chemical bonding. The greatly modulated bond length is regarded as a very important common characteristic. These lengths should be a critical condition in searching for the optimal match of three kinds of core components and determining efficient Li+-ion transport.8 Obviously, how to achieve the best LIBs with the highest performance is a universal engineering issue.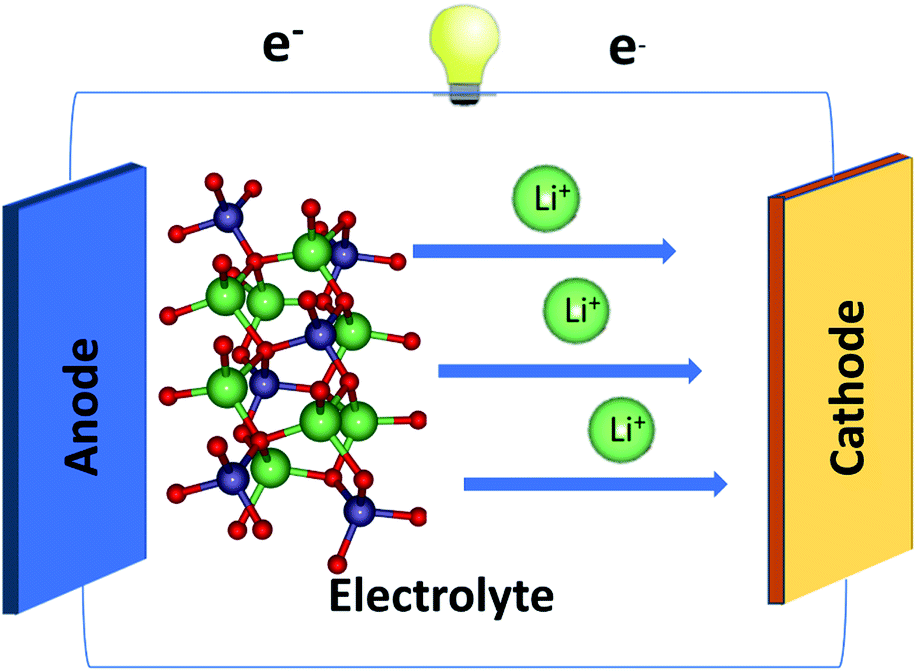 | ||
| Fig. 1 All-solid-state Li+-based battery with three-dimensional ternary Li2SiO3 electrolyte. | ||
LIBs are widely utilized in many electronic devices,9–12 e.g., cell phones, laptops, iPods, cars/buses, and radios. Their main characteristics include high capacity, large output voltage, long-term stability, and environmental friendliness.13 The crucial mechanisms of LIBs are characterized by the unique charging and discharging based on the exchange of Li+-ions. Indeed, Li+-ion transport occurs at any time during the charging/discharging process by the cathode → electrolyte → anode/anode → electrolyte → cathode path.14 Specifically, a separator membrane15 is inserted to avoid an internal short circuit and only small Li+ are accepted to freely pass between the positive and negative electrodes.15 The two electrodes are linked externally to an electric supply after the initial charging process, in which electron carriers rapidly escape from the cathode and are transported by an external wire to the anode, leading to the creation of a charge current. To keep the electric neutrality, Li+-ions are rapidly transported along the parallel direction internally from the cathode to the anode by the solid/liquid/gluon state electrolyte.16 With this efficient process, the external energy from the electrical supply is stored in the battery in the form of chemical energy. The opposite process, in which the electrons move from the anode to the cathode through the external lead and the Li+-ions move back to the cathode via the specific electrolytes, can provide electric power and thus does work in electronic devices.17
Generally, the cathode and anode systems of Li+-based batteries belong to a class of solid-state materials, such as the three-dimensional ternary LiFe/Co/NiO18–20 and LiTiO/graphite compounds,21,22 respectively. In addition, the various electrolytes can be classified into solid and liquid states.23 Compared with liquid ones, solid-state secondary LIBs may present the following merits: highly intrinsic, large energy density, safety, comparable power density, and long cycle lifetime.24 Some representative systems for solid state electrolytes are LiOCl,25 LiPS26 and LiSiO,26,27 for example. In particular, the LiSiO compound was reported as a potential electrolyte due to its low-cost synthesis,27 reliable ionic conductivity (2.5 × 10−8 S cm−1 for Li2SiO3 at 25 °C),28 and stability in a CO2 environment.27 To date, numerous theoretical and experimental studies have been conducted on the optimal electrolytes,29 in which the critical conditions are closely related to the rather high Li+-ion conductivity, the almost vanishing electron transport through them, the large electrochemical potential window, the very stable electrode–electrolyte boundary, and the optimal match of cathode and anode materials.
On the theoretical side, numerous previous predictions show that the first-principles method is very efficient and reliable in determining the rich essential properties of three-dimensional compounds, such as the electronic,30–32 lithium diffusion33 and optical properties34 of ternary LiSiO compounds. However, the extremely non-uniform chemical bonds in the large unit cell due to the critical but complicated multi-orbital hybridization, which are closely related to the high-potential functionalities of LIBs, have not been reported so far. Furthermore, they also do not deal with the unique magnetic quantization due to a very large Moire superlattice. Fortunately, the results calculated by VASP (discussed later) are sufficient to develop a theoretical framework for a thorough understanding of the diversified physical/chemical/material phenomena. Systematic studies have been successfully conducted on layered two-dimensional graphenes,35 one-dimensional graphene nano-ribbons,36 and two-dimensional silicenes with or without significant chemical modifications (absorptions and substitutions).37,38 Through delicate analyses, the critical mechanisms and a concise picture for fully comprehending the geometric, electronic, and magnetic properties can be obtained. The significant multi/single-orbital hybridizations in the various chemical bonds are obtained from the optimal lattice symmetry, the atom-created valence and conduction bands, the spatial charge densities and their changes after chemical modifications, and the atom and orbital-decomposed density of states. Furthermore, the spin distribution configurations, which are generated by the host and/or guest atoms, are accurately examined through the spin-split/spin-degenerate energy bands near the Fermi level, the spin density distributions, the net magnetic moments, and the spin-projected density of states. Therefore, these delicate calculations might be suitable for exploring the diverse fundamental properties of a lot of complicated oxide compounds, e.g., the electrolyte, cathode and anode materials, Li2SiO3, LiFe/Co/NiO,18–20 and Li4Ti5O12,21 in mainstream Li+-based batteries. Also, whether the direct combination of numerical simulations with phenomenological models is reliable needs to be clarified in independent cases, since this linking is very important in the full exploration of various properties, such as the useful combination of the VASP calculations and the generalized tight-binding models in the rich magnetic quantization.39
The main focuses of our work are the geometric symmetries and electronic properties of lithium metasilicate (Li2SiO3) – the electrolyte material of Li+-based batteries. The first-principles method is available for delicately calculating the total ground state energy, lattice symmetry, distinct Li–O and Si–O bond lengths, the atom-dominated valence and conduction bands, the spatial charge density, and the atom- and orbital-projected density of states. The spin-created phenomena, the spin-split band structure across the Fermi level, the net magnetic moment, and the spin density distributions, will be thoroughly examined as to whether they could survive in this emergent material. These physical quantities are very important in achieving the critical multi-orbital hybridizations of two kinds of chemical bonds and the spin-dependent magnetic configuration. Most of the analytical difficulties arise from the very complicated orbital-projected van Hove singularities, the existence of which is partially supported by the electronic structure and charge density distribution. The theoretical predictions of the optimal geometry, the occupied electronic states, the bandgap and whole energy spectrum can be investigated by powder X-ray diffraction (PXRD)/scanning electron microscopy (SEM)/transmission electron microscopy (TEM)/scanning tunneling microscopy (STM),40–42 optical absorption spectra,43 angle-resolved photoemission spectroscopy (ARPES),44 and scanning tunneling spectroscopy (STS),45 respectively. Besides, the close relationship between the numerical VASP calculations and the tight-binding model is discussed in detail.
2 Computational details
The rich and unique geometric structure and electronic properties of the Li2SiO3 compound were determined using density functional theory (DFT)46 implemented by the Vienna ab initio simulation package (VASP).47 The many-body exchange and correlation energies, due to the electron–electron coulomb interactions, are investigated from the Perdew–Burke–Ernzerhof functional (PBE)48 within the generalized gradient approximation (GGA). Moreover, the projector-augmented wave (PAW)49 pseudopotentials are available to characterize the significant electron–ion scatterings. Of course, those two critical interactions have no exact solutions in analytical forms. Therefore, it is difficult to carry out an accurate diagonalization of the many-particle Hamiltonian. Plane waves, with a kinetic energy cutoff of 500 eV chosen as a basic set, would make it more convenient and reliable to solve the Bloch wave functions and band structures. The first Brillouin zone is sampled by 7 × 7 × 7 and 20 × 20 × 20 k-point meshes within the Monkhorst–Pack scheme50 for geometric optimization and the electronic energy spectrum, respectively. These wave-vector points should be sufficient to calculate the suitable orbital-projected van Hove singularities, spatial charge distributions, and spin density configurations. Besides, the convergence condition of the ground-state energy is set to be 10−5 eV between two consecutive simulation steps; furthermore, the maximum Hellmann–Feynman force acting on each atom is smaller than 0.01 eV under the ionic relaxations. The accurate VASP calculations are very useful in thoroughly exploring certain physical quantities, e.g., the atom-induced valence and conduction bands, the spatial charge densities due to chemical bonding, the atom- and orbital-projected density of states, the atom-dependent spin configurations, the spin-split or degenerate states across the Fermi level, and the finite or vanishing magnetic moments.3 Results and discussions
3.1 Geometric structure
In this work, lithium metasilicate is predicted to display an extremely non-uniform chemical environment in a primitive unit cell (discussed later), so a suitable tight-binding model with the various chemical bonds might be very difficult to obtain for fitting the first-principles band structure. The ternary three-dimensional Li2SiO3 material possesses a unique lattice symmetry, according to the delicate first-principles calculations on the optimal geometric structures. We have chosen a meta-stable system to study the rich and unusual phenomena. This material, as clearly illustrated in Fig. 2, directly corresponds to an orthorhombic structure; the space group is Cmc21 (ref. 51) with a primitive unit cell containing 24 atoms (8-Li, 4-Si and 12-O atoms). The total ground state energy is −91.8 eV per unit cell, to which the spin-dependent interactions do not make any contribution. The calculated lattice constants of lithium metasilicate are 9.46 Å, 5.44 Å, and 4.72 Å for the x, y, z directions, respectively, and are very close to previously determined experimental52 and theoretical33 values. Obviously, the chemical/physical/material environments are highly anisotropic and extremely non-uniform, and other essential properties are expected to behave similarly. The diverse atomic arrangements are easily observed under distinct plane projections, e.g., the geometric structures in Fig. 3(a) (100), Fig. 3(b) (010), Fig. 3(c) (001), where the Li, Si, and O atoms are denoted by green, purple and red balls, respectively. The above mentioned real space lattice gives rise to the orthorhombic first Brillouin zone shown in Fig. 4, in which the high-symmetry points are very useful in characterizing the electronic energy spectra and states. They include Γ (0.00, 0.00, 0.00), X (0.33, 0.33, 0.00), S (0.00, 0.50, 0.00), Y (−0.50, 0.50, 0.00), T (0.50, 0.50, 0.00), Z (0.00, 0.00, 0.50), A (0.33, 0.33, 0.5), and R (0.00, 0.50, 0.00). The various chemical bonds in the unit cell dominate all the fundamental properties. Only 32 Li–O and 16 Si–O bonds exist, whereas other kinds of chemical bonds are completely absent. Most importantly, the optimal geometric parameters in Table 1 clearly illustrate that the bond lengths of Li–O and Si–O vary over a range of 1.94–2.20 Å and 1.61–1.70 Å and the fluctuation percentages Δb (%) = |bondmax − bondmin|/bondmin are over 13.3% and 5.6%, respectively, indicating possible structural transformation between two meta-stable systems with distinct geometries. This might be responsible for the outstanding electrolyte role of Li2SiO3 in Li+-ion batteries. It is well known that the bond lengths directly determine the spatial charge density distributions and the strengths of the chemical bonds (discussed later in Fig. 6), so their large changes indicate highly non-uniform multi-orbital hybridizations (the various hopping integrals in the phenomenological models). Such complicated behaviors might induce extra theoretical barriers in exploring the other fundamental properties, i.e., the rich magnetic quantization phenomena in LiSiO-related compounds.53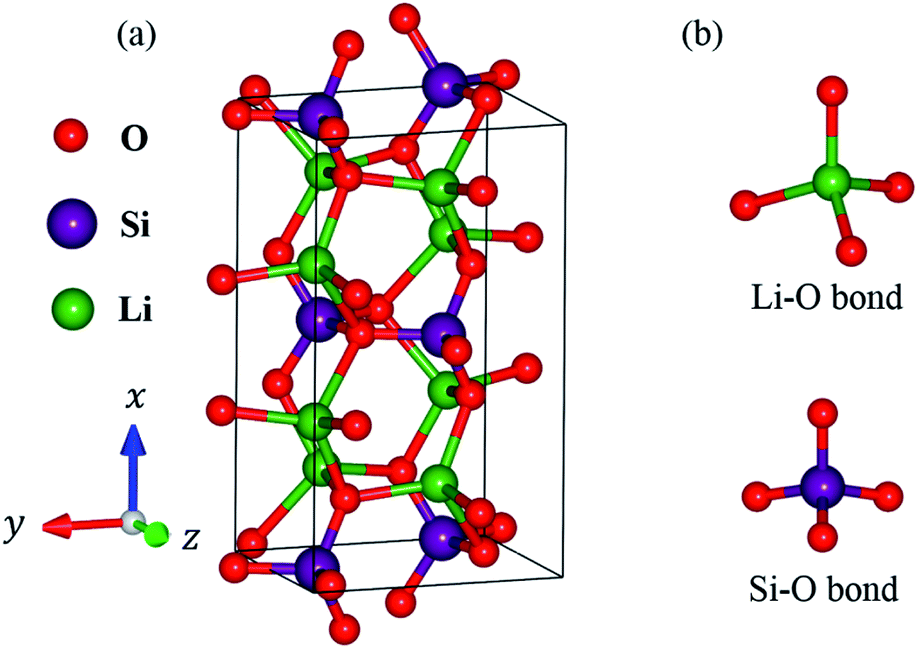 | ||
| Fig. 2 (a) The optimal lattice structure of the Li2SiO3 compound with orthorhombic symmetry with 24 atoms in a unit cell with lattice constants x = 9.46 Å, y = 5.44 Å and z = 4.72 Å. (b) Two kinds of chemical bonding: Li–O and Si–O. | ||
 | ||
| Fig. 3 Projections of Li2SiO3 onto distinct planes: (a) (100), (b) (010), (c) (001), where the green, purple and red balls correspond to Li, Si and O atoms, respectively. | ||
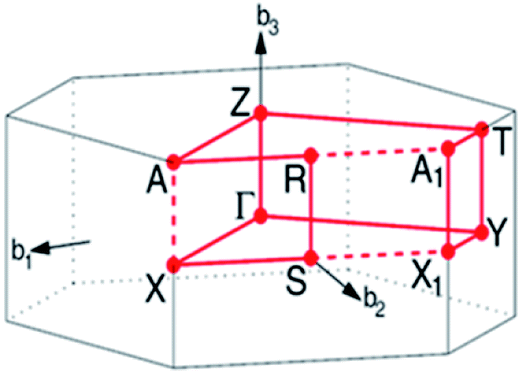 | ||
| Fig. 4 The first Brillouin zone with the high-symmetry points within the three orthogonal axes. | ||
| Atom–atom | No. of bonds | Bond length (Å) | |
|---|---|---|---|
| Experiment52 | This work | ||
| Li–O | 32 | 1.932–2.176 | 1.943–2.201 |
| Si–O | 16 | 1.591–1.680 | 1.612–1.701 |
The theoretical predictions of the essential properties of lithium metasilicate could be examined by various experimental methods. PXRD40 is the most efficient technique in identifying the lattice symmetries of three-dimensional materials. Apparently, it is very suitable to directly observe the three-dimensional ternary Li2SiO3 compound. The lattice constant and phase information of Li2SiO3 have been successfully verified by PXRD measurements.27,28,52 In addition to PXRD, the particle sizes and morphologies of samples are investigated by using higher-resolution scanning electron microscopy (SEM).40 The side- and top-view nanoscale geometries are examined by tunneling electron microscopy (TEM)41 and scanning tunneling microscopy (STM), respectively.42 They can delicately identify the complex relations within the honeycomb lattice, the finite-width quantum confinement, the flexible geometry, and the distinct chemical bonds of carbon atoms, such as the chiral and achiral hexagonal arrangements of one-dimensional planar graphene nanoribbons and carbon nanotubes, and their curved, folded, scrolled, and stacked configurations.54–59 Whether the wide modulations of the Li–O and Si–O bond lengths can be delicately identified from the measured data is worthy of systematic investigations. This is closely related to the very complex multi-orbital hybridizations in all the chemical bonds. Similar examinations could be generalized for other meta-stable or intermediate configurations, and may be very useful in understanding the transformation paths between them.
3.2 Rich electronic properties
Very interestingly, the solid-state electrolyte in Li+-based batteries, the three-dimensional ternary Li2SiO3 compound, exhibits unusual geometric structures and thus presents rich and unique band structures. The electronic energy spectrum, as clearly illustrated in Fig. 5(a)–(d), strongly depends on the wave-vector. The occupied valence bands are highly asymmetric to the unoccupied conduction bands about the Fermi level EF = 0, directly reflecting the very complicated multi-orbital hybridizations in the Li–O and Si–O bonds. The energy dispersions, which are shown along the high-symmetry point, have strongly anisotropic behavior. For example, there exist parabolic, oscillatory and partially flat dispersion relations. Furthermore, sub-band non-crossing, crossing and anti-crossing phenomena come into existence frequently. Most importantly, the highest occupied and the lowest unoccupied states, respectively, appear at the Z and Γ points (0.0, 0.0, 0.5) and (0.0, 0.0, 0.0). This indicates a very large indirect band gap of Eg = 5.077 eV (Fig. 5(a)). This band gap is close to the values of the theoretical results, which are 5.36 eV,32 4.49 eV (ref. 34) and only slightly lower than that (Eg = 5.5 eV) of diamond. Apparently, the optical threshold absorption frequency, which is measured from the reflection/absorption/transmission spectroscopies, should be higher than the band gap value. That is to say, various high-resolution optical measurements are very suitable for examining the insulating behavior. Also, spin splitting is present in any energy band, but none of the Li, Si and O atoms can create spin-up or spin-down configurations. As a result, there is no magnetic moment and the spin density distribution becomes meaningless. | ||
| Fig. 5 (a) The significant valence and conduction band of Li2SiO3 along the high-symmetry points within the first Brillouin zone for the energy range (−8.0 eV, Ec,v, 8.0 eV) with the specific (b) lithium, (c) silicon and (d) oxygen dominances (green, purple, and red balls, respectively). | ||
In addition to the main features of the band structure, the electronic wave functions for valence and conduction states can provide partial information on the chemical bonding. Each band state can be regarded as a linear superposition of different orbitals. Therefore, it can be decomposed into distinct atomic contributions. The different atom dominances, being proportional to the spheric radius, can help in understanding the important roles played by the chemical bonds in the electronic properties. The green, purple and red balls correspond to the Li, Si and O contributions, respectively. In general, the effective valence and conduction states, which are closely related to the multi-orbital hybridizations of the Li–O and Si–O bonds (discussed later in Fig. 6 and 7), lie in the energy range of −8.0 eV, Ec,v, 8 eV. All three atoms have significant contributions to the whole band structure. This unusual phenomenon may explain the wide range of modulations of the chemical bonding strength in the Li–O and Si–O bonds (the strongly modulated hopping integrals). However, it might be difficult to observe the Li-contributions (small green balls) in Fig. 5(b) because of the single 2s-orbital. On the other hand, the silicon and oxygen contributions are obvious in the electronic energy spectrum, i.e., the sufficiently large purple and red balls in Fig. 5(c) and (d). Specifically, the oxygen atoms dominate all the valence and conduction states, since they are associated with all the chemical bonds.
 | ||
| Fig. 6 The spatial charge density distribution for (a) the isolated (Li, Si, O) atoms; the shortest/medium/longest (b)/(c)/(d) Li–O and (e)/(f)/(g) Si–O bonds. | ||
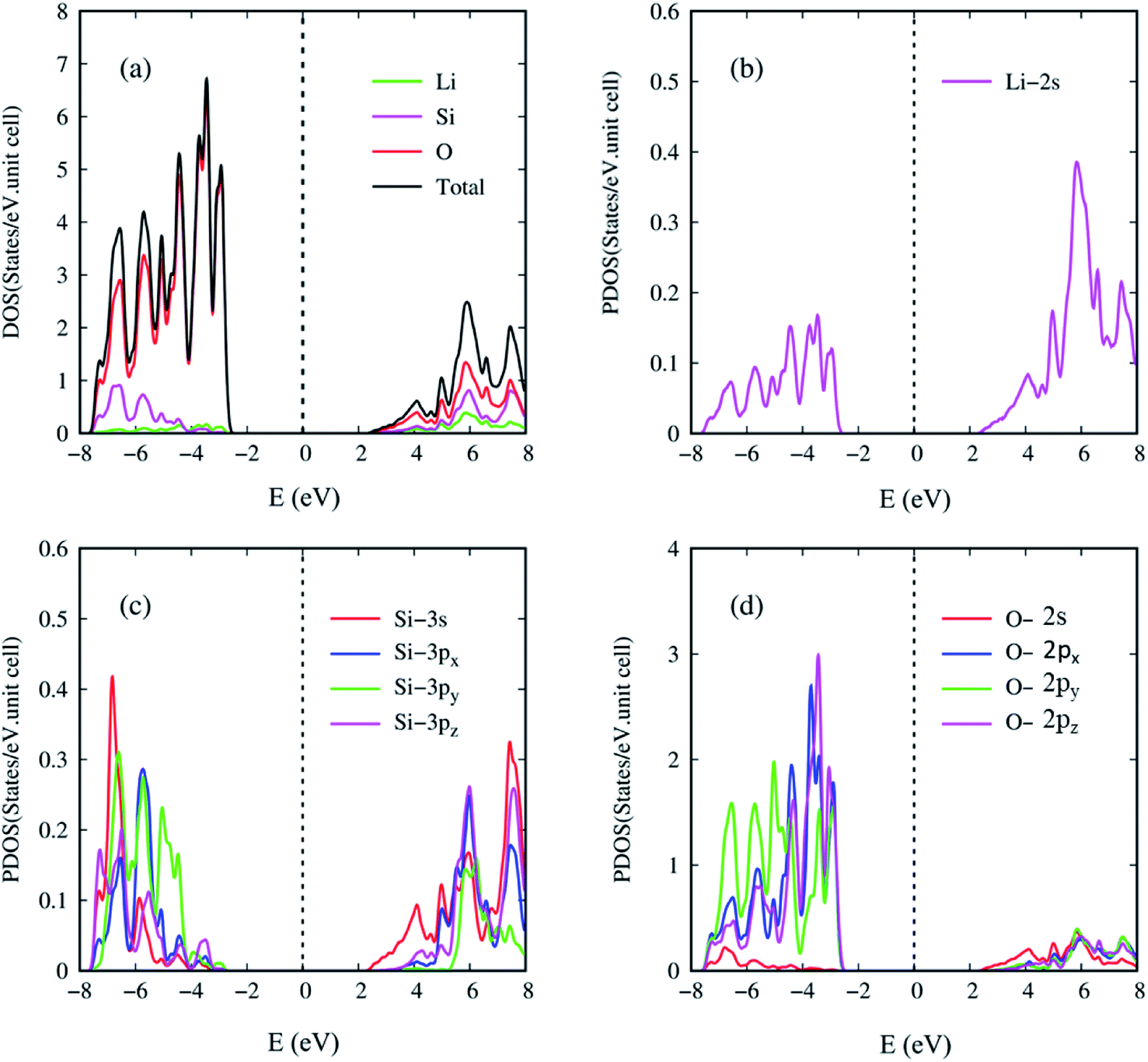 | ||
| Fig. 7 The atom- and orbital-projected density of states: those coming from (a) Li, Si, and O atoms (green, purple and red curves), (b) Li-2s orbitals (purple curve), (c) Si-(3s, 3px, 3py, 3pz) orbitals (red, blue, green and purple curves), and (d) O-(2s, 2px, 2py, 2pz) orbitals (red, blue, green and purple curves). | ||
On the experimental side, electronic energy spectra are frequently examined by optical spectroscopy methods43 and angle-resolved photoemission spectroscopy (ARPES).44 Very interestingly, the very wide energy gap in ternary lithium metasilicate, due to the extremely strong covalent bonding, is directly verified by using optical absorption spectroscopy.43 The measured excitation frequency is close to the VASP calculations. It is well known that the ARPES measurement is suitable for the direct examination of the occupied energy spectrum, with the different dispersion relations along the high-symmetry points in the first Brillouin zone. Up to now, ARPES measurements have been very successful in verifying the diverse electronic properties in graphene nanoribbons,60 carbon nanotubes,61 few-layer graphene,62 adatom-adsorbed graphene systems,63 and graphites.64 These measurements can provide sufficient information for the significant effects of the chemical bonding in these emergent materials. Nevertheless, high-resolution ARPES measurements are required for the Li2SiO3 compound in future studies.
Very interestingly, the multi-orbital hybridizations in Li–O and Si–O chemical bonds can be roughly examined via the spatial charge distributions, as clearly illustrated in Fig. 6(a)–(g). Furthermore, the delicate chemical bonds are identified from the atom- and orbital-projected density of states (later discussions) of Fig. 7(a)–(d). It is well known that the charge densities are very sensitive to a change in the bond lengths. Evidently, lithium metasilicate has highly modulated chemical bonds (Table 1), closely related to the available orbitals in different atoms. Concerning the shortest Li–O bonds of 1.943 Å in Fig. 6(b), each lithium atom only provides a single 2s-orbital, and its effective distribution range is 0.54 Å, as measured from the deep yellow region of the Li+-ion core to the light green one of the outermost orbitals. However, the two 1s orbitals do not participate in the critical orbital hybridizations with the oxygen atoms. The important oxygen orbitals, which correspond to the light green and yellow regions, are approximately predicted to be associated with the (2px, 2py, 2pz) ones. Specifically, the O-2s orbitals are relatively far away from the Li-atom, so they might play only a minor role in Li–O chemical bonding, resulting in the increase of the Li–O length to 2.201 Å. In Fig. 6(c) and (d), the orbital overlap of Li and O atoms declines quickly. As a result, the multi-orbital 2s–(2s, 2px, 2py, 2pz) hybridizations of the Li–O bonds exhibit a diverse hopping integral.
There are fewer Si–O chemical bonds, but they have a rather strong bonding strength (Fig. 6(e)–(g)) compared with the Li–O bonds. The effective charge distribution range of the Si atom is much higher than that of the Li atom because of the larger atomic number. Most importantly, both silicon and oxygen atoms can be classified into the dark red and yellow–green regions which, respectively, correspond to the 3s, 2s and (3px, 3py, 3pz), (2px, 2py, 2pz) orbitals. Very interestingly, the chemical bonding of the former is clearly illustrated by the deformed spherical distributions between the two atoms. That is to say, the Si–O chemical bonds consist of the four orbital hybridizations due to (3s, 3px, 3py, 3pz)–(2s, 2px, 2py, 2pz). The chemical bonding strength declines with the increase of bond length, as indicated by the reduced charge density between Si and O atoms. The extremely non-uniform chemical bonding even for similar bonds will induce a high calculation barrier in researching suitable hopping integrals for the phenomenological models, e.g., reliable parameters of the tight-binding model.
The density of states (DOS), in which the number of states within a very small energy range of dE is defined, allows us to fully understand the valence and conduction energy spectra simultaneously. Generally, the special structures are created by the band-edge states with vanishing group velocities. The critical points in the energy–wave-vector space cover the local extreme points (minima and maxima), the saddle points, and all the partially flat energy dispersions. Apparently, the van Hove singularities are greatly diversified under the different dimensions, e.g., the delta-function-like prominent peaks for the zero-dimensional discrete energy levels, the square-root asymmetric peaks/plateau structures of the one-dimensional parabolic/linear energy sub-bands, and the shoulder structures/V-shapes/logarithmic peaks/square-root asymmetric peaks in the two-dimensional parabolic/Dirac cone/saddle-point/constant-energy-loop band structures. Moreover, the higher dimension in three-dimensional materials makes them only show symmetric/asymmetric peaks and shoulder structures, mainly due to the absence of quantum confinement; the shoulder structures arise from the three-dimensional parabolic energy dispersions.
In the current work, the atom and orbital-decomposed van Hove singularities are very useful in directly resolving the critical multi-orbital hybridizations in the Li–O and Si–O bonds, as shown in Fig. 7(a)–(d). The three kinds of atoms have significant contributions within the whole energy spectrum between −8 eV and 8 eV, reflected in the large modulation of bond lengths (Table 1). For the wide-gap three dimensional Li2SiO3 compound, the density of states per unit cell is vanishingly small within the large energy range of Eg = 5.077 eV centered about the Fermi level (the black curve) in Fig. 7(a). The DOS is dominated by the valence states (E < 0), but not the conduction ones (E > 0). That is, the hole and electron energy spectra are highly asymmetric about the Fermi level. The van Hove singularities mainly arise from the valence and conduction band-edge states along the high-symmetry-point paths, corresponding to the local minimum, maximum, and saddle points in the reciprocal space. At least six and three special features are revealed in the valence and conduction energy spectra, respectively. As a result of the frequent crossings and anti-crossings, each feature reflects the combined phenomenon of certain neighboring band-edge states under a finite broadening effect. This indicates the experimental difficulty in examining the number of energy sub-bands and their band-edge states. Seemingly, the lithium and oxygen atoms (the green and red curves) make the weakest and strongest contributions to the total density of states, respectively. Such a result is principally determined by the number of available atom orbitals in a primitive unit cell.
Generally speaking, the important contributions, which arise from the various orbitals of the different atoms, cover (i) Li-2s orbitals (purple curve) in Fig. 7(b), (ii) Si-(3s, 3px, 3py, 3pz) orbitals (red, blue, green and purple curves) in Fig. 7(c), and (iii) O-(2s, 2px, 2py, 2pz) orbitals (red, blue, green and purple curves) in Fig. 7(d). All the orbital contributions are merged together, especially the number, energies, intensities and forms of van Hove singularities. The main reason is that the distinct chemical bonds are associated with one another through the Li–O and Si–O bonds. Consequently, there exist the multi-orbital hybridizations of 2s–(2s, 2px, 2py, 2pz) and (3s, 3px, 3py, 3pz)–(2s, 2px, 2py, 2pz). The enlarged density of states for Li-2s orbitals can provide 5 clearly visible van Hove singularities and illustrate that they are weak but significant features in Fig. 7(b). The observable contributions are obviously revealed in the half-occupied four orbitals of the silicon atoms (Fig. 7(c)), in which their differences only reflect the strong anisotropy in the primitive unit cell (Fig. 3). Most importantly, only the three orbitals of O-(2px, 2py, 2pz) make comparable and dominating contributions (Fig. 7(d)), while the 2s ones are much smaller than the others in the opposite region. This unusual behavior is consistent with the (3s, 3px, 3py, 3pz)–(2s, 2px, 2py, 2pz) and 2s–(2px, 2py, 2pz) orbital hybridizations in the Si–O and Li–O bonds, respectively. It should be noted that the O-2s orbitals are almost fully forbidden in the semiconducting Li2SiO3 compound.
High-resolution STS45 is a very efficient technique for detecting the entire valence and conduction energy spectrum through the van Hove singularities. Roughly speaking, when a quantum tunneling current (a very small I) is injected from a nanoscaled probe onto a sample surface by applying a finite potential difference, the measured differential conductance dI/dV can be regarded as the density of states. To date, STS has been successful in identifying the dimension-enriched van Hove singularities of graphene-related systems with sp2 chemical bonding. For example, there exist a lot of divergent peaks with the square-root in one-dimensional carbon nanotubes and graphene nanoribbons,65,66 a plateau structure across the Fermi level in metallic armchair tubules,67 a symmetric V-shape vanishing at EF for monolayer graphene (a zero-gap semiconductor),68 the logarithmically symmetric peaks near EF in twisted bilayer graphene systems (saddle point),69 a gate-voltage-induced energy gap for bilayer AB and tri-layer ABC stackings, a delta-function-like peak localized about EF for ABC-stacked graphenes, a sharp dip structure close to EF combined with a pair of square root peaks under tri-layer AAB stacking (a narrow-gap semiconductor with constant-energy loops), and a finite DOS at EF for the semi-metallic Bernal graphite.70 Similar STS experiments on lithium metasilicate (Li2SiO3) materials could verify a very large energy gap of Eg = 5.077 eV, six/three asymmetric/symmetric peaks and broadening shoulders in the valence/conduction spectra, and a high asymmetry about the electron and hole states and their distinct distribution widths. The experimental measurements, together with the theoretical predictions of van Hove singularities, would allow the identification of the complicated band structure and thus the multi-orbital hybridizations in the Li–O and Si–O bonds.
In current Li+-based batteries, lithium metasilicate (Li2SiO3) could serve as the solid-state electrolyte. During the charging process, many of the lithium ions are rapidly transferred from the cathode (e.g., Li/Fe/Co/NiO materials)18–20 to the electrolyte, and subsequently the anode. The opposite is true for the discharging process. Seemingly, the Li+ flow generates the dramatic transformation of materials.
4 Conclusions
We present the rich and unique properties of lithium metasilicate using density functional theory (DFT) as implemented in the VASP package. The critical multi-orbital hybridizations, which survive in Li–O and Si–O bonds, are accurately identified from the atom-dominated band structure, the charge density distributions in the greatly modulated chemical bonds, and the atom- and orbital-projected van Hove singularities. The theoretical framework could be further developed for other electrolyte, anode and cathode materials in Li+-based batteries, e.g., the important differences among various LiXO-related compounds, and the diverse phenomena driven by various components. Very importantly, highly anisotropic and non-uniform environments need to be included in the phenomenological models. For example, a suitable tight-binding model is expected to include the position-dependent hopping integrals and the orbital-created on-site coulomb potentials when simulating the VASP energy bands and the density of states. Whether the intrinsic atomic interactions of the Hamiltonian can be expressed in analytical form is the focus of future studies. Furthermore, these phenomenological models are very useful in exploring other diverse phenomena, such as the magnetic quantization in a uniform perpendicular magnetic field.The solid-state Li2SiO3 electrolyte material with 24 atoms in a primitive unit cell has an orthorhombic structure. There are 32 Li–O and 16 Si–O chemical bonds, in which each atom has four other neighboring atoms. The Li–O and Si–O bond lengths display large modulations of over 13.3% and 5.6%, respectively. Most importantly, the very strong orbital hybridizations create a very wide indirect gap of Eg = 5.077 eV, close to the largest one in diamond (5.5 eV). Its magnitude is even lower than the optical threshold absorption frequency, the high-resolution optical reflection/absorption/transmission spectroscopy71 available for detecting the obvious insulating properties of the transparent material.72 The occupied valence and the unoccupied conduction bands are highly asymmetric to each other about the Fermi level. Furthermore, there exists a very large band width (−8.0 eV, Ec,v, 8.0 eV), strong/varied energy dispersion, high anisotropy, and frequent non-crossing/crossing/anti-crossing behavior. Moreover, the van Hove singularities, which arise from the band-edge states, appear as the six/three dominating/minor special structures in the density of states of the valence/conduction energy spectra (E < 0, E > 0). They all contribute to the broadening of the asymmetric/symmetric peaks and shoulders, and play a critical role in examining the multi-orbital 2s–(2s, 2px, 2py, 2pz) and (3s, 3px, 3py, 3pz)–(2s, 2px, 2py, 2pz) hybridizations of the Li–O and Si–O bonds. The diverse covalent bonding is partially supported by the atom-dominated band structure and charge density distributions in modulated chemical bonds. The theoretical predictions of the optimal geometry, band gap, wave-vector-dependent valence bands, and valence and conduction density of states could be verified by PXRD/TEM/STM, optical spectroscopy, ARPES and STS methods, respectively.
The calculated results clearly illustrate that LiSiO-based compounds have certain meta-stable configurations, and even infinite intermediate ones during the charging and discharging processes for Li+-ion transport in batteries. Similar structural transformations between two meta-stable structures73 are expected to occur at any time. The optimal evolution paths, which might become an emergent issue, are under current investigation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Hierarchical Green-Energy Materials (Hi-GEM) Research Center, from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) and the Ministry of Science and Technology (MOST 108-3017-F-006-003) in Taiwan.References
- P. H. Maheshwari, Materials Science for Energy Technologies, 2019, 3, 490–502 CrossRef.
- Z. L. Wang, D. Xu, Y. Huang, Z. Wu, L. M. Wang and X. B. Zhang, Chem. Commun., 2012, 48, 976–978 RSC.
- I. D. Scott, Y. S. Jung, A. S. Cavanagh, Y. Yan, A. C. Dillon, S. M. George and S. H. Lee, Nano Lett., 2011, 11, 414–418 CrossRef CAS PubMed.
- B. Huang, Y. I. Jang, Y. M. Chiang and D. R. Sadoway, J. Appl. Electrochem., 1998, 28, 1365–1369 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef CAS.
- N. Kuganathan, L. H. Tsoukalas and A. Chroneos, Solid State Ionics, 2019, 335, 61–66 CrossRef CAS.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- G. Zheng, Y. Cui, E. Karabulut, L. Wågberg, H. Zhu and L. Hu, MRS Bull., 2013, 38, 320–325 CrossRef CAS.
- S. J. Andreasen, L. Ashworth, I. N. M. Remon and S. K. Kar, Int. J. Hydrogen Energy, 2008, 33, 7137–7145 CrossRef CAS.
- J. H. Wee, J. Power Sources, 2007, 173, 424–436 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135(4), 1167–1176 CrossRef CAS PubMed.
- S. Yamakawa, H. Yamasaki, T. Koyama and R. Asahi, J. Power Sources, 2013, 223, 199–205 CrossRef CAS.
- Y. J. Hwang, K. S. Nahm, T. P. Kumar and A. M. Stephan, J. Membr. Sci., 2008, 310, 349–355 CrossRef CAS.
- F. Sagane, T. Abe, Y. Iriyama and Z. Ogumi, J. Power Sources, 2005, 146, 749–752 CrossRef CAS.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- L. Liu, Z. Xiang, W. H. Li, L. Chen and X. Huang, Solid State Ionics, 2002, 152–153, 341–346 CrossRef CAS.
- H. Chen, J. A. Dawson and J. H. Harding, J. Mater. Chem. A, 2014, 2, 7988–7996 RSC.
- M. Chireh and M. Naseri, Adv. Powder Technol., 2019, 30(5), 952–960 CrossRef CAS.
- C. Lin, M. On Lai, L. Lu, H. Zhou and Y. Xin, J. Power Sources, 2013, 244, 272–279 CrossRef CAS.
- J. Rossat-Mignod, A. Wiedenmann, K. C. Woo, J. W. Milliken and J. E. Fischer, Solid State Commun., 1982, 44, 1339–1342 CrossRef CAS.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- Y. Yu, L. Gu, C. Zhu, P. A. van Aken and J. Maier, J. Am. Chem. Soc., 2009, 131, 15984–15985 CrossRef CAS PubMed.
- A. Emly, E. Kioupakis, A. Van der Ven, Y. Deng, C. Eames, J.-N. Chotard, F. Lalère, S. Vincent, S. Emge, P. Oliver, C. P. Grey, C. Masquelier and M. Saiful Islam, J. Am. Chem. Soc., 2015, 137, 9136–9145 CrossRef PubMed.
- L. Yang, G. Li, X. Jiang, T. Zhang, H. Lin and J. Y. Lee, J. Mater. Chem. A, 2017, 5, 12506–12512 RSC.
- M. Watanabe, et al., J. Ceram. Soc. Jpn., 2017, 125, 472–475 CrossRef CAS.
- A. Nakagawa, N. Kuwata, Y. Matsuda and J. Kawamura, J. Phys. Soc. Jpn., 2010, 79, 98–101 CrossRef.
- M. Cha and C. Jung, J. Appl. Electrochem., 2009, 39, 955–960 CrossRef CAS.
- J. Du and L. René Corrales, J. Phys. Chem. B, 2006, 110, 22346–22352 CrossRef CAS PubMed.
- W. Y. Ching, Y. P. Li, B. W. Veal and D. Lam, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 1203 CrossRef CAS PubMed.
- T. Tang and D. L. Luo, Journal of Atomic and Molecular Sciences, 2010, 1, 185–200 CrossRef.
- N. Kuganathan, L. H. Tsoukalas and A. Chroneos, Solid State Ionics, 2019, 335, 61–66 CrossRef CAS.
- A. Alemi, S. Khademinia and S. W. Joo, et al., Int. Nano Lett., 2013, 3, 14 CrossRef.
- T.-N. Do, C.-P. Chang, P.-H. Shih, J.-Y. Wu and M.-F. Lin, Phys. Chem. Chem. Phys., 2017, 19, 29525 RSC.
- M. Y. Han, B. Özyilmaz, Y. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef PubMed.
- C. Meier, A. Gondorf, S. Lüttjohann and A. Lorke, J. Appl. Phys., 2007, 101, 103112 CrossRef.
- X. Li, X. Pan and L. Yu, et al., Nat. Commun., 2014, 5, 3688 CrossRef CAS PubMed.
- Y. Huang, S. Chen and Y. Ho, et al., Sci. Rep., 2015, 4, 7509 CrossRef PubMed.
- S. Yang, Q. Wang, J. Miao, J. Zhang, D. Zhang, Y. Chen and H. Yang, Appl. Surf. Sci., 2018, 444, 522–529 CrossRef CAS.
- J. Garnaes, J. Vac. Sci. Technol., A, 1990, 8, 441 CrossRef CAS.
- P. K. Hansma, J. Appl. Phys., 1987, 61, R1 CrossRef CAS.
- A. Alemi, S. Khademinia and M. Sertkol, Int. Nano Lett., 2015, 5, 77–83 CrossRef CAS.
- D. Lee and A. C. Albrecht, J. Chem. Phys., 1983, 78, 3382 CrossRef CAS.
- R. MFeenstra, Surf. Sci., 1994, 299–300, 965–979 CrossRef.
- R. G. Parr, Chemistry, Springer, Dordrecht, 1980, pp. 5–15 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1998, 80, 891 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- P. Wisesa, K. A. McGill and T. Mueller, Phys. Rev. B, 2016, 93, 155109 CrossRef.
- W. Setyawan and S. Curtarolo, Comput. Mater. Sci., 2010, 49, 299–312 CrossRef.
- H. Völlenkle, Zeitschrift für Kristallographie – Crystalline Materials, 1981, 154, 77–81 Search PubMed.
- B. Dóra, K. Janik and R. Moessner, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195422 CrossRef.
- J. Van der Lit, P. H. Jacobse, D. Vanmaekelbergh and Swart, New J. Phys., 2015, 17, 053013 CrossRef.
- U. Maitra, H. S. S. Matte, P. Kumar and C. N. R. Rao, Chimia, 2012, 66, 941–948 CrossRef CAS PubMed.
- J. Zhang, J. Xiao, X. Meng, C. Monroe, Y. Huang and J.-M. Zuo, Phys. Rev. Lett., 2010, 104, 166805 CrossRef PubMed.
- K. F. Kelly, I. W. Chiang, E. T. Mickelson, R. H. Hauge, J. L. Margrave and X. Wang, et al., Chem. Phys. Lett., 1999, 313, 445–450 CrossRef CAS.
- L. B. Biedermann, M. L. Bolen, M. A. Capano, D. Zemlyanov and R. G. Reifenberger, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125411 CrossRef.
- Z. Klusek, W. Kozlowski, Z. Waqar, S. Datta, J. S. Burnell-Gray and I. V. Makarenko, et al., Appl. Surf. Sci., 2005, 252, 1221–1227 CrossRef CAS.
- H. Huang, D. Wei, J. Sun, S. L. Wong, Y. P. Feng, A. C. Neto and A. T. S. Wee, Sci. Rep., 2012, 2, 983 CrossRef PubMed.
- G. Li, A. Luican, J. L. Dos Santos, A. C. Neto, A. Reina, J. Kong and E. Andrei, Nat. Phys., 2010, 6, 109 Search PubMed.
- V. Cherkez, G. T. de Laissardière, P. Mallet and J.-Y. Veuillen, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 155428 CrossRef.
- G. Li, A. Luican and E. Y. Andrei, Phys. Rev. Lett., 2009, 102, 176804 CrossRef PubMed.
- W. Y. Ching, Y. P. Li, B. W. Veal and D. J. Lam, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 1203 CrossRef CAS PubMed.
- J. W. G. Wilder, L. C. Venema, A. G. Rinzler, R. E. Smalley and C. Dekker, Nature, 1998, 391, 59–62 CrossRef.
- L. Tapasztó, G. Dobrik and P. Lambin, et al., Nat. Nanotechnol., 2008, 3, 397–401 CrossRef PubMed.
- Y. H. Ho, C. P. Chang, F. L. Shyu, R. B. Chen, S. C. Chen and M. F. Lin, Carbon, 2004, 42, 3159–3167 CrossRef CAS.
- X. Chen, J. Tao and Y. Ban, Goos-Hänchen, Eur. Phys. J. B, 2011, 79, 203–208 CrossRef CAS.
- E. McCann and M. Koshino, Rep. Prog. Phys., 2013, 76, 056503 CrossRef PubMed.
- C.-H. Ho, Y.-H. Ho, Y.-Y. Liao, Y.-H. Chiu, C.-P. Chang and M.-F. Lin, J. Phys. Soc. Jpn., 2012, 81, 024701 CrossRef.
- T. Do, G. Gumbs and P. Shih, et al., Sci. Rep., 2019, 9, 624 CrossRef PubMed.
- H. Jia, J. Zhu, D. Kebebew Debeli, Z. Li and J. Guo, Sol. Energy Mater. Sol. Cells, 2018, 174, 140–145 CrossRef CAS.
- C. O. Saglam and K. Byl, Proc. SPIE 9467, Micro- and Nanotechnology Sensors, Systems, and Applications, 2015, vol. 7, p. 94671T Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |