
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu/TEMPO catalyzed dehydrogenative 1,3-dipolar cycloaddition in the synthesis of spirooxindoles as potential antidiabetic agents†
Chitrala Tejaa,
Spoorthy N. Babub,
Ayesha Noorb,
J. Arul Danielc,
S. Asha Devic and
Fazlur Rahman Nawaz Khan *a
*a
aOrganic and Medicinal Chemistry Research Laboratory, Department of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore-632014, Tamil Nadu, India. E-mail: nawaz_f@yahoo.co.in; Tel: +91-944-423-4609
bCentre for Bio Separation Technology, Vellore Institute of Technology, Vellore-632014, India
cDepartment of Biomedical Sciences, School of Bioscience and Technology, Vellore Institute of Technology, Vellore-632014, India
First published on 25th March 2020
Abstract
A series of spiro-[indoline-3,3′-pyrrolizin/pyrrolidin]-2-ones, 4, 5 and 6 were synthesized in a sequential manner from Cu–TEMPO catalyzed dehydrogenation of alkylated ketones, 1 followed by 1,3-dipolar cycloaddition of azomethine ylides via decarboxylative condensation of isatin, 2 and L-proline/sarcosine, 3 in high regioselectivities and yields. The detailed mechanistic studies were performed to identify the reaction intermediates, which revealed that the reaction proceeds via dehydrogenative cycloaddition. Additionally, the regio and stereochemistry of the synthesized derivatives were affirmed by 2D NMR spectroscopic studies. The synthesized derivatives were explored further with molecular docking, in vitro antioxidant, and anti-diabetic activities.
1. Introduction
Spirooxindoles have recently attracted significant attention towards versatile bioactivities, such as antimicrobial,1,2 antitumor,3,4 anti-tubercular,5,6 antiviral7 and related properties.8–12 Natural products, for instance, horsfiline, elacomine, alstonisine, mitraphylline,13 spirotryprostatin A,14,15 peteropodine, consist of a privileged heterocyclic skeleton, where the spiro ring is fused at the C3 position of the oxindole (Fig. 1).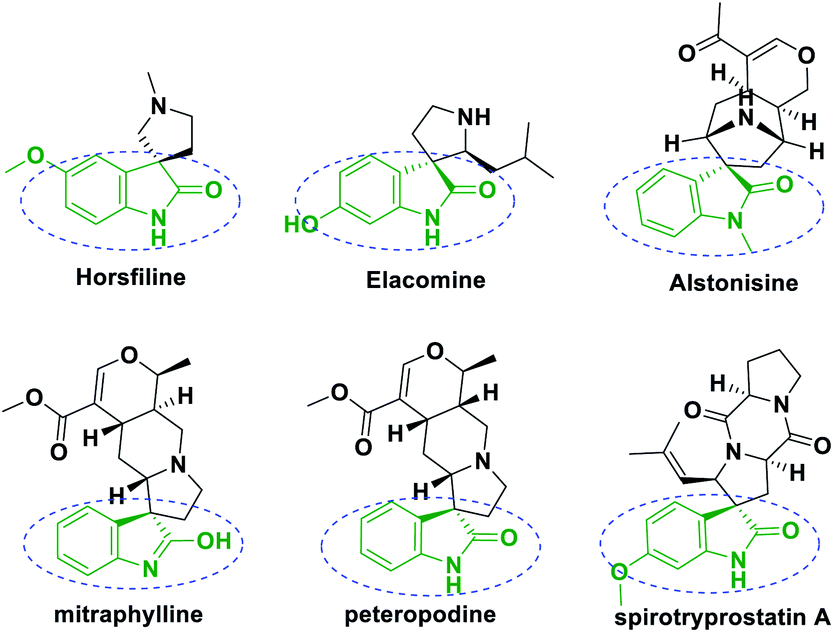 | ||
| Fig. 1 Bioactive spirooxindole natural products. | ||
The 1,3-dipolar cycloaddition16–18 of azomethine ylides is the most efficient synthetic approach towards the spirooxindoles in high regio and stereoselectivities. Moreover, these spirooxindoles were generated by the decarboxylative condensation19–21 of isatins and α-amino acids with unsaturated alkenes.22–24 A wide variation of azomethine ylides,25 including α-amino acids, and 1,3-dipolarophiles such as α,β-unsaturated carbonyl compounds,26 (chalcones) arylidene-malono-dinitriles,27 nitrostyrene,28 acrylamides29 α,β-unsaturated lactones,30 and various electron-deficient alkenes have been extensively documented.
Fokas, Lalitha and co-workers utilized α,β-unsaturated carbonyl compounds and volatile toxic solvents, alike methanol and dioxane (Scheme 1).23,31 Consequently, in the present study envisioned the α,β-unsaturated carbonyl intermediate, 1d formation from Cu–TEMPO catalyzed dehydrogenation of the saturated ketone. TEMPO is a unique free radical (oxidant); was used as co-catalyst for several mechanistic studies.32,33 In addition, several mechanistic pathways and reports were reported for dehydrogenation strategy. Nicolaou et al. developed a milder method using IBX (o-iodoxybenzoic acid) as a stoichiometric reagent,34–36 Stahl et al.,37–39 Newhouse et al.,40 and others proposed aerobic dehydrogenation of alkyl aldehydes and ketones, using palladium, and other catalysis.41–45 These reported methods were suffering from multistep reactions, expensive catalysts, and use of toxic volatile organic solvents (VOCs) Me–OH, Et–OH, 1,4-dioxane, toluene, DMF46,47 and stoichiometric reagents, the formation of unwanted by-products and so forth.
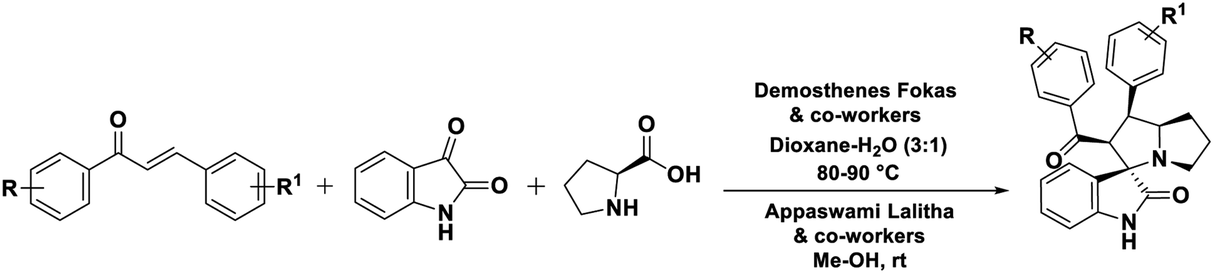 | ||
| Scheme 1 1,3-Dipolar cycloaddition of α,β-unsaturated carbonyl compounds (previous work). | ||
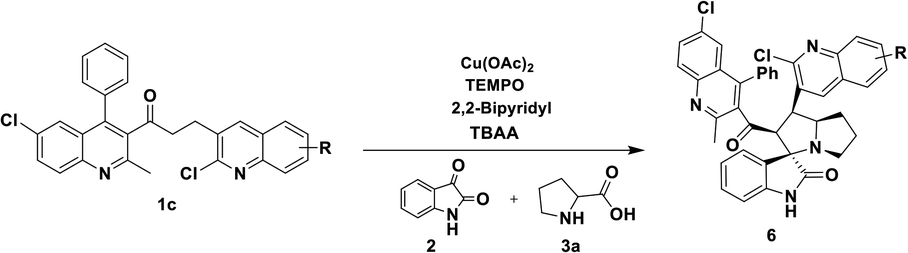
In order to overcome the above mentioned difficulties, herein, a Cu–TEMPO catalyzed dehydrogenative 1,3-dipolar cycloaddition of alkylated-ketones,48 1 utilizing isatin, 2, and L-proline/sarcosine, 348 in TBAA is reported for the spiro-indoline-3,3′-pyrrolizin/pyrrolidines (Scheme 2). Interestingly, the TBAA (tetrabutylammonium acetate) acted as a reaction medium instead of VOCs, in several C–C bond forming reactions.46,47,49–54
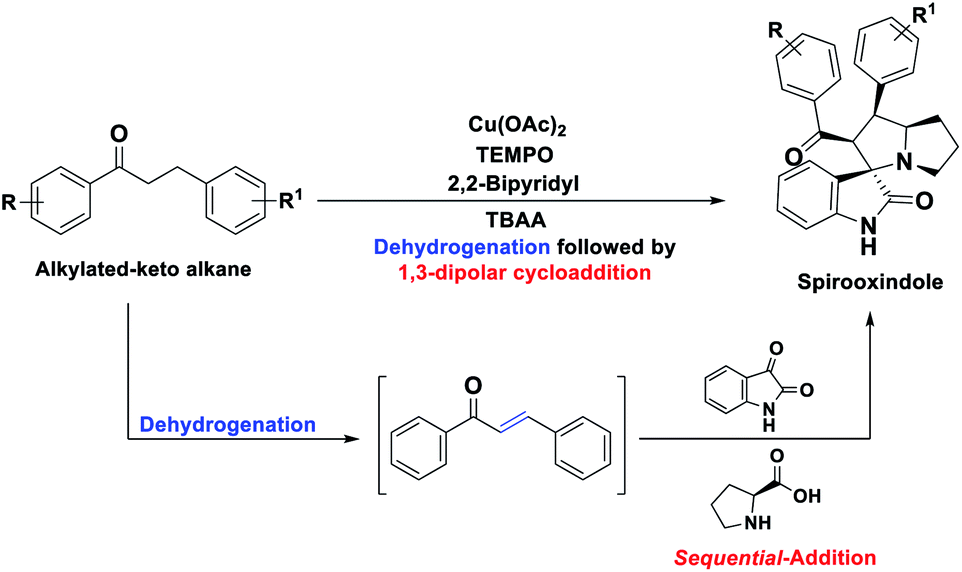 | ||
| Scheme 2 Synthesis of spirooxindoles by sequential dehydrogenative 1,3-dipolar cycloaddition of alkylated ketones (this work). | ||
Likewise, diabetes mellitus (DM) type-2, a chronic metabolic disorder, was characterized by high blood sugar, insulin resistance, and secretion. The common symptoms were continual urination, increased hunger, and thirst, tiredness, and unexpected weight loss, and so forth.55,56 According to the International Diabetes Federation (IDF) report as of 2017 approximately 425 million people were diagnosed with DM type 2.57,58 There are numerous therapeutic approaches to treat diabetes, for instances, decreasing the postprandial hyperglycemia by hydrolyzing enzyme inhibition, including α-amylase and α-glucosidase.59,60 Anti-diabetic inhibitors voglibose, Acarbose, and miglitol were utilized as drugs for years.61–63 Lately, spirooxindoles were reported as anti-diabetic inhibitors,64 therefore, the present work was envisioned to develop the spirooxindoles based anti-diabetic inhibitors.
2. Results and discussion
2.1 Chemistry
Initially, the reaction was carried out with 3-(4-fluorophenyl)-1-(p-tolyl)propan-1-one, 1ab (0.5 mmol), and optimized the reaction conditions using transition metal catalysts viz. Mn(OAc)2, Co(OAc)2, Ni(OAc)2, Cu(OAc)2, Zn(OAc)2, Pd(OAc)2 (Table 1, entries 1–6) in the presence of TEMPO (0.1 mmol), 2,2-bipyridyl (0.1 mmol) in DMF at 100 °C. Interestingly, the Cu(OAc)2 and Pd(OAc)2 provided the desired unsaturated ketone 1d within 4 h, through dehydrogenation of 1ab. Further, the sequential addition of isatin (0.5 mmol) and L-proline (0.6 mmol) leads to the formation of spiro-oxindole 4ab in 55% and 41% of yields, respectively. The TEMPO acted as a radical initiator, while the 2,2-bipyridyl as a bidentate transition metal chelating ligand for the reaction. Also, screened with Ag2O, CuSO4 CuCl2, CuBr2, and Cu(OTf)2 under TEMPO/2,2-bipyridyl conditions, a trace amount of 4ab was obtained (Table 1, entries 7–11). Likewise, the change of the ligand to 1,10-phenanthroline did not affect the yield of 4ab (43% in 8 h) (Table 1, entry 12). Other tested ligands including, DBU, DABCO, and pyridine, respectively under the same experimental conditions, provided poor yields of 4ab (Table 1, entries 13–15). Similarly, the change of the oxidant to N-hydroxyphthalimide (NHPI), tert-butyl-hydroperoxide (TBHP) using 2,2-bipyridyl in DMF at 100 °C provided a trace amount of 4ab (Table 1, entries 16 and 17). In addition, screened the effect of solvent using 1,4-dioxane, DMSO, and toluene; however, there is no significant change in yield of the desired product was observed (Table 1, entries 18–20). Here all the optimizations were carried out sequentially by in situ addition of isatin and L-proline after the formation of unsaturated ketone intermediate from dehydrogenation of the saturated ketone.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Additives | Solvent | Temp. °C | Time (h) | Yield% 4abc | |
| a Reaction conditions: saturated ketone, 1ab (0.5 mmol), Cu(OAc)2 (10 mol%), TEMPO (0.1 mmol) and 2,2-bipyridyl (0.1 mmol), in 2 mL of solvent at 100 °C after sequential addition of isatin, 2 (0.5 mmol), L-proline 3a or sarcosine 3b (0.6 mmol).b TBA-ionic-liquid (1 eq.) at 80 °C for 1 h.c Isolated yields. | |||||||
| 1 | Mn(OAc)2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 9 | — |
| 2 | Co(OAc)2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 8 | — |
| 3 | Ni(OAc)2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 12 | — |
| 4 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 6 | 55 |
| 5 | Zn(OAc)2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 10 | — |
| 6 | Pd(OAc)2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 13 | 41 |
| 7 | Ag2O | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 12 | Trace |
| 8 | CuSO4 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 20 | Trace |
| 9 | CuCl2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 18 | 13 |
| 10 | CuBr2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 18 | 19 |
| 11 | Cu(OTf)2 | TEMPO | 2,2′-Bipyridyl | DMF | 100 | 10 | 25 |
| 12 | Cu(OAc)2 | TEMPO | 1,10-Phenanthroline | DMF | 100 | 8 | 43 |
| 13 | Cu(OAc)2 | TEMPO | DBU | DMF | 100 | 15 | 10 |
| 14 | Cu(OAc)2 | TEMPO | DABCO | DMF | 100 | 18 | 22 |
| 15 | Cu(OAc)2 | TEMPO | Pyridine | DMF | 100 | 12 | 15 |
| 16 | Cu(OAc)2 | NHPI | 2,2′-Bipyridyl | DMF | 100 | 10 | 15 |
| 17 | Cu(OAc)2 | TBHP | 2,2′-Bipyridyl | DMF | 100 | 15 | 15 |
| 18 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | 1,4-Dioxane | 100 | 10 | 31 |
| 19 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | DMSO | 100 | 8 | 48 |
| 20 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | Toluene | 100 | 16 | 25 |
| 21b | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | TBAB (1 eq.) | 80 | 3 | 72 |
| 22 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | TMAB | 80 | 4 | 63 |
| 23 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | TEAB | 80 | 3 | 65 |
| 24 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | TBAA | 80 | 2 | 87 |
| 25 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | TBAI | 80 | 5 | 61 |
| 26 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | TBAA (0.5 eq.) | 80 | 2 | 82 |
| 27 | Cu(OAc)2 | TEMPO | 2,2′-Bipyridyl | TBAA (0.25 eq.) | 80 | 3 | 70 |
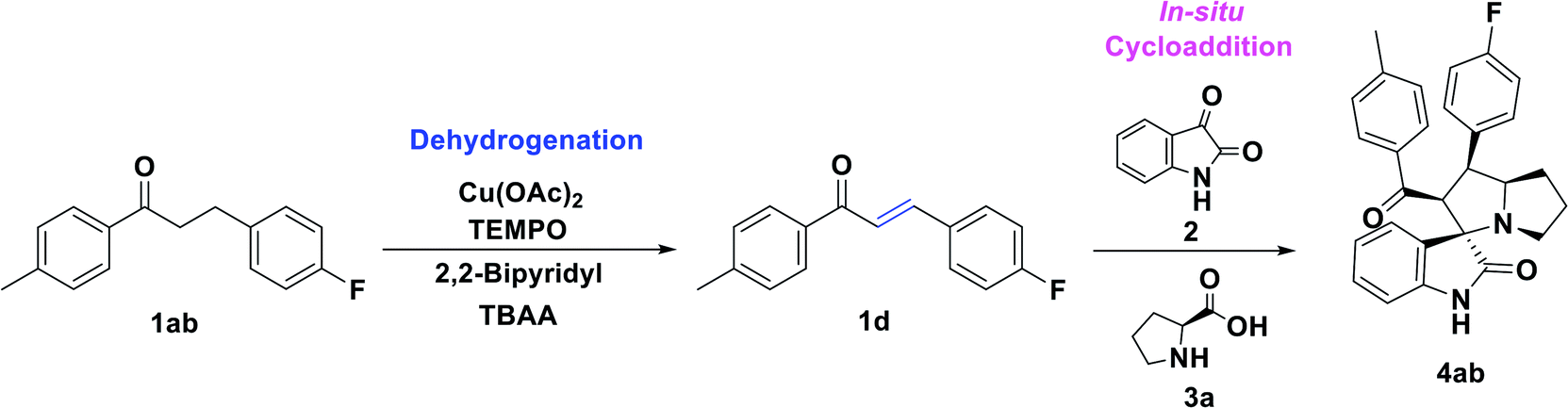
Interestingly, when a sequential dehydrogenation/1,3-dipolar cycloaddition was screened using TBA-salt as a reaction medium instead of DMF by taking 3-(4-fluorophenyl)-1-(p-tolyl)propan-1-one 1ab (0.5 mmol) TBAB (tetrabutylammonium-bromide) Cu(OAc)2 (10%), TEMPO (0.1 mmol) and 2,2-bipyridyl (0.1 mmol) at 80 °C. It provided the corresponding spirooxindole 4ab in good yield of 72% within 2 h (Table 1, entry 21). The TBAB acted as an ionic medium and was advantageous over VOCs, the TBA-salts also used in several C–C bond-forming transformations.46,47,50,52,65–67 Encouraged by this result, further screened with various tetrabutylammonium-salts, for instances, TMAB (tetra-methylammonium bromide), TEAB (tetra-ethylammonium bromide), TBAA (tetra-butylammonium acetate), TBAI (tetra-butylammonium iodide) at 80 °C carried out, here 4ab was obtained in good to excellent yields up to 87% (Table 1, entries 22–25). Among the tested TBA-salts, TBAA with 87% yield of desired product emerged out as the best-optimized condition (Table 1, entry 24). Additionally, when optimizing the loading of TBAA, for instance, 0.5 eq., 0.25 eq. (Table 1, entries 26 and 27) a less amount of product yield was observed while comparing with 1.0 eq.
Having streamlined optimized reaction condition in hand (Table 1, entry 24), various spirooxindole derivatives were obtained from different alkylated ketones by sequential dehydrogenative 1,3-cycloaddition, tabulated in Tables 2–4. Also, the scope of the reaction was explored using substrates with electron-donating groups (–Me, and –OMe), and with electron-withdrawing groups (–Cl, F and Br). Favorably, all the tested substrates transformed smoothly to provide the desired spirooxindoles.
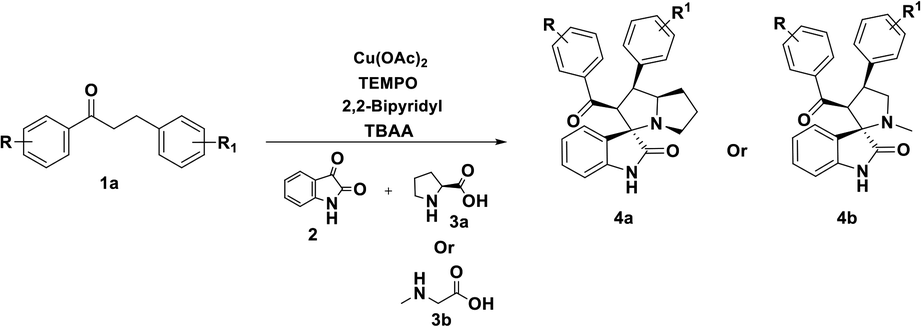
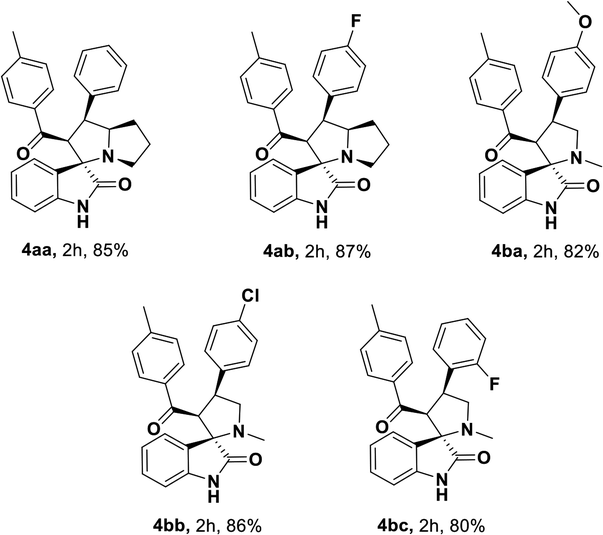
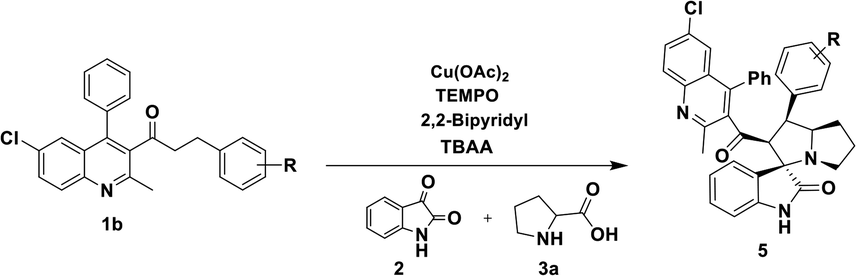
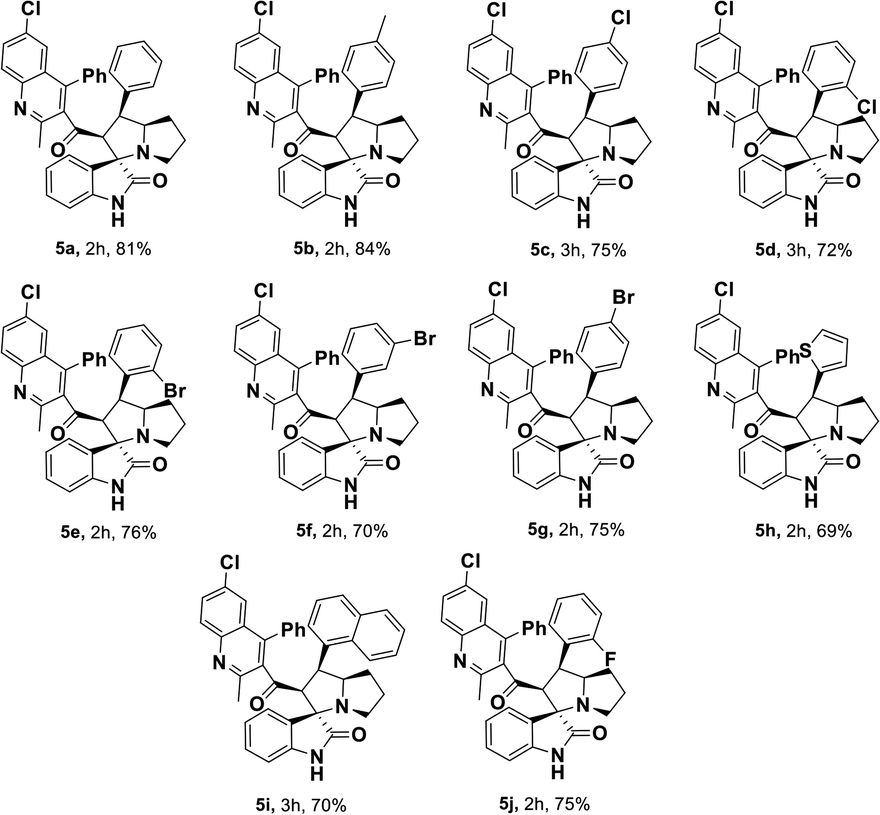
|
|---|
| a Reaction conditions: quinolinyl-alkylated ketone, 1c (0.5 mmol), Cu(OAc)2 (10 mol%), TEMPO (0.1 mmol) and 2,2′-bipyridyl (0.1 mmol), in TBAA (1 eq.) at 80 °C for 1 hour, sequential addition of isatin, 2 (0.5 mmol), L-proline 3a or sarcosine 3b (0.6 mmol) at 80 °C for another 1 h.b Isolated yields. |
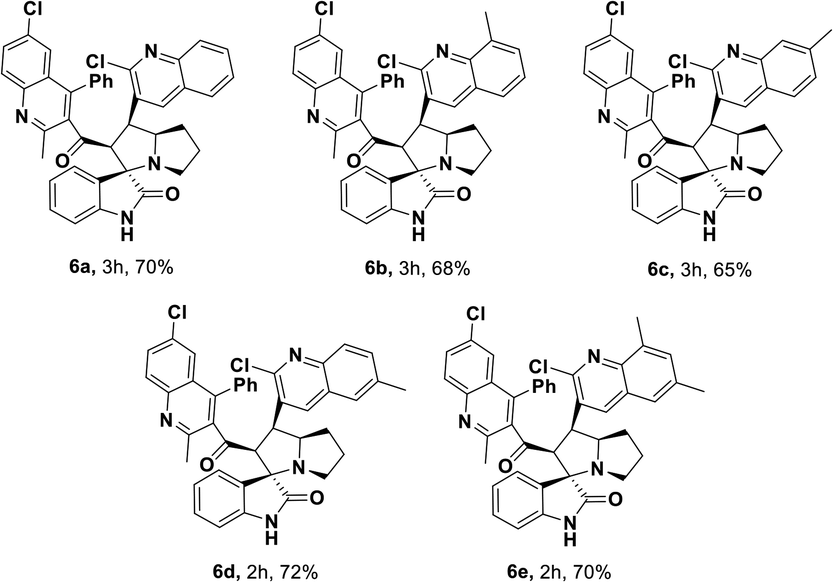 |
2.2 Proposed reaction mechanism
In addition, the control experiments and 1H NMR studies were performed to identify the reaction mechanism (results are discussed in Fig. S46 and 47 ESI Page S54–S56†). From the above mentioned mechanistic studies and previous literature reports,45,68 we proposed the plausible reaction mechanism Fig. 2. Also, Su et al. previously proposed the plausible mechanistic pathway for dehydrogenation of saturated ketones via radical addition and elimination.45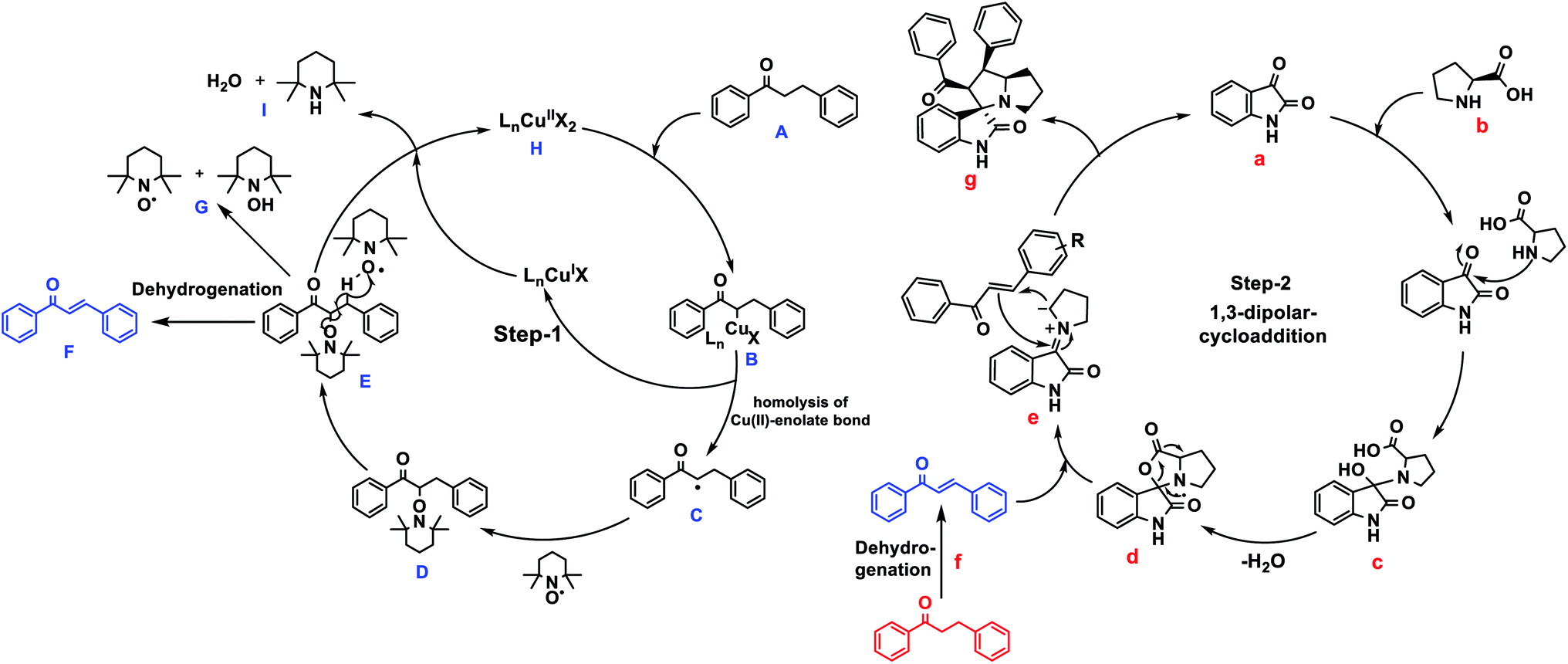 | ||
| Fig. 2 Plausible mechanism for the synthesis of spirooxindoles from keto-alkanes via Cu–TEMPO catalyzed dehydrogenation and 1,3-dipolar cycloaddition. | ||
Initially, Cu(OAc)2 gets reacted with alkylated-ketone A to provide metal-enolate complex B. Then generates Cu(I) species, α-radical intermediate C and α-TEMPO radical substituted ketone D, as confirmed by 1H NMR studies (ESI Fig. S47†).
Spectra-2 C reveals the α-TEMPO-adduct C–H proton at 4.6 ppm, where trans-ketone formation occurs, to form enones E and the formation of alkene (chalcone) via dehydrogenation F.
In (ESI Fig. S47†) C and D show the formation of unsaturated ketone (chalcone) along with saturated ketone. In the final step, oxidation occurs via Cu(I) species by TEMPO rather than TEMPO-OH G to reproduce Cu(II) H by forming 2,2,6,6-tetramethylpiperidine and H2O as by-products I.
The formation of Cu(II) species from the oxidation of Cu(I) species, was observed in literature reports.69 The reactions of Cu(OAc)2/2,2-bpy complex with TEMPO or TEMPO-OH have been demonstrated, the both TEMPO and TEMPO-OH could oxidize Cu(I) species to form Cu(II). Where the TBAA, as a basic and ionic medium, alters the reaction conditions. The sequential addition of isatin and proline to the unsaturated ketone intermediate (chalcone) resulted in the 1,3-dipolar cycloaddition leads to provide the 5-membered spirooxindoles. For instance, (Fig. 2) isatin a, L-proline b leads to carboxylative cyclic ester c, which undergoes decarboxylation d to give azomethine ylide e, a dipole. The azomethine ylide then undergoes 1,3-cycloaddition to provide the expected spirooxindole-1,3-dipolar-cycloaddition product g, water, and CO2 only the by-products. The transformation of spirooxindole from unsaturated ketone has been indicated in (ESI Page S55, Fig. S47†) NMR studies. The structural elucidation of compounds 4, 5, and 6 were identified using 1H, 13C, and 2D-NMR spectroscopic studies (see ESI, Page S53†), as mentioned in the illustrative example of 4aa.
2.3 Pharmacological studies
| S. no. | Compound name | Binding energy (kcal mol−1) | ||
|---|---|---|---|---|
| Alpha-amylase | Alpha-glucosidase | GLP-1 | ||
| 1 | 5a | −8.15 | −8.64 | −9.51 |
| 2 | 5b | −7.3 | −8.08 | −9.47 |
| 3 | 5c | −7.76 | −8.66 | −8.31 |
| 4 | 5d | −7.78 | −8.27 | −7.46 |
| 5 | 5e | −7.78 | −7.6 | −8.11 |
| 6 | 5f | −8.47 | −7.23 | −8.21 |
| 7 | 5g | −7.9 | −7.61 | −8.15 |
| 8 | 5h | −9.29 | −7.28 | −8.39 |
| 9 | 5i | −8.57 | −8.73 | −8.4 |
| 10 | 5j | −8.13 | −7.91 | −8.55 |
| 11 | 6a | −9.51 | −9.29 | −8.85 |
| 12 | 6b | −9.16 | −9.77 | −7.56 |
| 13 | 6c | −9.61 | −7.07 | −8.6 |
| 14 | 6d | −8.74 | −7.44 | −10.2 |
| 15 | 6e | −9.24 | −8.71 | −8.95 |
| 16 | 4aa | −7.81 | −7.62 | −7.65 |
| 17 | 4ab | −6.95 | −7.04 | −7.47 |
| 18 | 4ba | −6.77 | −6.85 | −6.66 |
| 19 | 4bb | −7.79 | −7.14 | −6.87 |
| 20 | 4bc | −7.78 | −6.57 | −7.31 |
| 21 | Std. drug | −0.74 | 0.32 | 37.44 |
Interaction of the synthesized compounds with α-glucosidase enzyme revealed that 6b and 6a had the best binding energy of −9.77 and −9.29 kcal mol−1, respectively. Acarbose had binding energy of 0.32 kcal mol−1 and formed 3 hydrogen bonds with PHE525 and SER626 amino acid residues of the α-glucosidase enzyme. Most of the synthesized compounds had interactions with the catalytic domain of the α-glucosidase enzyme present between residues 358 to 720.71 Molecular interactions with GLP-1 receptor revealed 6d, 5a, and 5b had the best binding energy. Previous studies have shown that exendin-4 binds with the GLU128 of the GLP-1 receptor having an agonist activity (Underwood 2010).72
Current interaction studies have imparted that 5d, 5f, 5h, 6e, 4aa, 4ab, and 4bc formed a hydrogen bond with GLU128, which implies that it could have agonist activity too as reported before.
The reaction below shows the scavenging action of DPPH due to the presence of anti-oxidants

All the synthesized compounds showed the potential to scavenge ABTS and DPPH radical with compound 6d having the highest activity at an IC50 value of 0.28 ± 0.03 mg mL−1 and 0.34 ± 0.06 mg mL−1 respectively while compound 6e had the least activity with an IC50 value of 6.04 ± 0.54 mg mL−1 and 6.98 ± 0.75 mg mL−1 respectively. The principle of metal chelating assay involves the chelation of ferrozine with ferrous ion. This forms a red-colored complex. The presence of antioxidants reduces the chelation as the antioxidants compete with ferrozine to chelate with the ferrous ion. The chelation of antioxidants with ferrous ion reduced the red color of the ferrozine ferrous ion complex.
In our study, all the compounds could chelate with ferrous ion. The compound 6e exhibited highest potential (IC50 value: 0.31 ± 0.02 mg mL−1) while compound 4ab showed least potential with an IC50 value of 6.91 ± 1.10 mg mL−1.
The H2O2 assay is based on the reduction of the tri-phenanthroline complex formed due to the reaction between ferrous ion and phenanthroline (red-orange). The addition of H2O2 reduced ferrous ion to ferric, and the tri-phenanthroline complex is not formed. The presence of compounds prevents the action of H2O2, leading to the formation of the tri-phenanthroline complex.
In our study, all compounds could scavenge the H2O2 radical with compound 5e having the highest potential, while compound 4ab had very less scavenging activity (shown in Table 6).
| S. no. | Compound | ABTS | DPPH | Metal chelating | H2O2 | α-Amylase | α-Glucosidase |
|---|---|---|---|---|---|---|---|
| 1 | 4aa | 1.62 ± 0.21 | 1.46 ± 0.21 | 0.96 ± 0.23 | 0.63 ± 0.12 | 1.47 ± 0.27 | 1.48 ± 0.32 |
| 2 | 4ab | 1.54 ± 0.30 | 1.22 ± 0.34 | 6.91 ± 1.10 | 6.15 ± 1.12 | 1.11 ± 0.41 | 1.52 ± 0.35 |
| 3 | 4ba | 2.70 ± 0.60 | 1.23 ± 0.39 | 3.21 ± 0.76 | 0.88 ± 0.06 | 0.99 ± 0.06 | 1.25 ± 0.24 |
| 4 | 4bb | 1.42 ± 0.31 | 0.80 ± 0.07 | 0.46 ± 0.08 | 0.41 ± 0.04 | 0.98 ± 0.25 | 1.46 ± 0.31 |
| 5 | 4bc | 1.50 ± 0.45 | 1.68 ± 0.34 | 0.86 ± 0.09 | 1.16 ± 0.35 | 1.52 ± 0.40 | 2.22 ± 0.57 |
| 6 | 5a | 3.25 ± 0.47 | 2.26 ± 0.13 | 0.93 ± 0.11 | 0.92 ± 0.09 | NA | 0.51 ± 0.19 |
| 7 | 5b | 0.49 ± 0.32 | 0.55 ± 0.04 | 0.81 ± 0.06 | 0.79 ± 0.02 | NA | 2.73 ± 0.52 |
| 8 | 5c | 1.57 ± 0.12 | 1.62 ± 0.43 | 0.92 ± 0.04 | 0.85 ± 0.07 | NA | 0.50 ± 0.03 |
| 9 | 5d | 1.11 ± 0.36 | 1.28 ± 0.32 | 1.16 ± 0.31 | 1.50 ± 0.35 | NA | 1.02 ± 0.34 |
| 10 | 5e | 0.79 ± 0.43 | 0.88 ± 0.09 | 6.00 ± 0.75 | 0.21 ± 0.03 | NA | 0.74 ± 0.13 |
| 11 | 5f | 0.55 ± 0.20 | 0.50 ± 0.06 | 1.40 ± 0.62 | 1.52 ± 0.32 | NA | 0.30 ± 0.05 |
| 12 | 5g | 2.72 ± 0.54 | 2.79 ± 0.32 | 0.69 ± 0.05 | 1.27 ± 0.32 | NA | 0.58 ± 0.06 |
| 13 | 5h | 0.41 ± 0.06 | 0.49 ± 0.03 | 1.04 ± 0.08 | 0.78 ± 0.06 | 0.54 ± 0.14 | 0.35 ± 0.05 |
| 14 | 5i | 0.86 ± 0.09 | 0.92 ± 0.05 | 0.77 ± 0.22 | 0.57 ± 0.09 | 0.28 ± 0.07 | 0.31 ± 0.07 |
| 15 | 5j | 3.03 ± 0.65 | 2.09 ± 0.34 | 0.38 ± 0.07 | 0.35 ± 0.04 | NA | 1.12 ± 0.15 |
| 16 | 6a | 2.75 ± 0.58 | 2.71 ± 0.43 | 0.43 ± 0.04 | 1.05 ± 0.27 | NA | 0.41 ± 0.04 |
| 17 | 6b | 0.43 ± 0.05 | 0.44 ± 0.08 | 0.46 ± 0.08 | 0.68 ± 0.06 | NA | 0.26 ± 0.06 |
| 18 | 6c | 0.34 ± 0.09 | 0.36 ± 0.02 | 0.99 ± 0.09 | 0.59 ± 0.03 | NA | 0.32 ± 0.07 |
| 19 | 6d | 0.28 ± 0.03 | 0.34 ± 0.06 | 0.37 ± 0.03 | 0.33 ± 0.04 | NA | 0.24 ± 0.05 |
| 20 | 6e | 6.04 ± 0.54 | 6.98 ± 0.75 | 0.31 ± 0.02 | 0.32 ± 0.07 | NA | 0.27 ± 0.02 |
| 21 | Ascorbic acid | 0.006 ± 0.002 | NA | 0.0073 ± 0.002 | NA | NA | NA |
| 22 | Gallic acid | NA | 0.0055 ± 0.001 | NA | 0.0045 ± 0.001 | NA | NA |
| 23 | Acarbose | NA | NA | NA | NA | 0.058 ± 0.012 | 0.046 ± 0.023 |
In our study, all the compounds showed positive results in inhibiting the α-glucosidase enzyme with compounds 5h and 5i having high activity. It was interesting to note that only 7 compounds answered positively to the α-amylase inhibition assay with compounds 5h and 5i having higher potential.
Overall, amongst the compounds, it can be inferred that compounds 5h and 5i have anti-oxidant and anti-diabetic properties with better IC50 values (shown in Table 6).
2.3.4.1 Physicochemical and pharmacokinetic properties. The in silico physicochemical properties of synthesized spiro-oxindole analogs were calculated and compared with Acarbose (standard antidiabetic drug). All the compound molecular weights are calculated according to Lipinski's rule of 5, in that 4aa, 4ab, 4ba, 4bb, and 4bc, which molecular weights are <500, remaining all the derivatives 5a series and 6a series including Acarbose are (>500) greater than its range. Polar Surface Area (PSA) is calculated based on TPSA methodology, TPSA is the essential factor that is showing molecular transport across the membranes, all the compounds showing TPSA values within satisfactory range between 50–100 except 4aa series. So the result is showing our synthesized compounds showing moderate lipophilicity across the blood-brain barrier. Other parameters, like, number of rotatable bonds (n-RotB), number of O, N atoms present and NH, OH donor/acceptor, were also in acceptable range comparing with standard drug. The results are showing excellent properties comparing with Acarbose because electronic and steric, substitution factors of the molecules, the positive as well as negative results also were recorded. 4aa series derivatives conceded 1 violation in terms of mlogPb violation, remaining all the compounds conceded 2 violations in terms of MW and mlogPb, comparing with Acarbose, which shows good results (Acarbose conceded 3 violations). So, according to Lipinski's rule of five, our compounds have no problems with bioavailability. In future new drug design and discovery point of view, a slight modification in the compounds may increase its lipophilicity and all other parameters (ESI Page S67, Table S7†).
The pharmacokinetics of the synthesized compounds were predicted using the pkCSM pharmacokinetic prediction tool to find the drug likeliness by ADMET properties (absorption, distribution, metabolism, excretion, and toxicity).73
The results show that all the synthesized molecules show excellent absorption properties including, water solubility, intestinal absorption, comparing with Acarbose as a standard drug. It shows moderate distribution properties and 4aa series few derivatives show AMES toxicity, as well as hepatotoxicity; remaining derivatives do not have any toxicity issues. Based on the above results, all the synthesized molecules show excellent ADMET properties comparing with the standard drug (ESI Page S67, Table S8†).
3. Conclusions
In summary, we have developed a solvent-free protocol in the virtue of avoiding VOCs for the synthesis of novel spirooxindoles from alkylated saturated ketones via Cu–TEMPO catalyzed dehydrogenation and sequential 1,3-dipolar cycloaddition pathway. Also, the detailed mechanistic studies revealed that the reaction proceeds via dehydrogenation followed by cycloaddition. It is an efficient protocol in-terms of one-pot sequential strategy, in high regio, stereo-selectivities and yields. All the synthesized compounds were investigated further for bio-activities, such as in silico molecular docking, in vitro antioxidant and anti-diabetic activities, most of the synthesized derivatives shows potential anti-oxidant and anti-diabetic inhibition. QSAR-studies of the synthesized derivatives show good biodegradability and bioavailability, including drug transport and ADMET properties.4. Experimental procedure
4.1 General information
All the chemicals were purchased from Sigma-Aldrich and TCI chemicals Ltd. used without further purification. All the reaction was conducted using glass reaction vials from App-tec Instrument Ltd., and performed by using in Apptec Lab Mate Manual Parallel Synthesizer. Pre-coated aluminum TLC sheets were used to check TLC (silica gel 60 F254, Merck). After completion of the reaction, the reaction mixture was allowed to warm at rt and extracted using water and CH2Cl2 as a solvent, and the organic extracts were dried over anhydrous Na2SO4, and the crude reaction mixture was passed through manual column chromatography to obtain pure product. Column chromatography performed on silica gel (100–200 mesh, Merck Ltd.) to give pure product on petroleum ether and ethyl acetate (petroleum ether/EtOAc 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v). Characterization alike, NMR-spectra was recorded using Bruker AVANCE-III (400 MHz) using CDCl3 as a solvent system. FT-IR spectra were recorded using (Shimadzu IR Tracer-100) spectrometer νmax are reported in cm−1.
:2 v/v). Characterization alike, NMR-spectra was recorded using Bruker AVANCE-III (400 MHz) using CDCl3 as a solvent system. FT-IR spectra were recorded using (Shimadzu IR Tracer-100) spectrometer νmax are reported in cm−1.
4.2 General procedure for synthesis of 1′-(4-fluorophenyl)-2′-(4-methylbenzoyl)-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (spirooxindole)
(3-(4-Fluorophenyl)-1-(p-tolyl)propan-1-one) saturated ketone, 1ab (1.0 mmol), Cu(OAc)2 (10 mol%), TEMPO (0.1 mmol) and 2,2-bipyridyl (0.1 mmol), in TBAA (1 eq.) at 80 °C for 1 h after formation of unsaturated enone intermediate 1d, confirmed by TLC, sequential addition of isatin, 2 (0.5 mmol), L-proline 3a or sarcosine 3b (0.6 mmol) at 80 °C for another 1 h led to formation of 1′-(4-fluorophenyl)-2′-(4-methylbenzoyl)-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one, as resulted spirooxindole, after confirmation of TLC, the reaction mixture was allowed to warm at rt and extracted using water and CH2Cl2 as solvent, and the organic extracts were dried over anhydrous Na2SO4, and the crude reaction mixture was passed through manual column chromatography to obtain pure product 4ab.The same procedure as follows for the synthesis of the remaining derivatives.
4.3 Molecular docking studies
Interaction studies were carried out for the synthesized derivatives with α-amylase, α-glucosidase, and glucagon-like peptide-1 (GLP-1). The structure of the compounds was obtained using ChemDraw, while the PDB structure of alpha-amylase, alpha-glucosidase, and GLP-1 were obtained from protein data bank with PDB ID 5U3A, 5NN8, and 3IOL respectively. AutoDock 4.2 was used to analyze the interactions, while LigPlot+ v1.4 was used to visualize the interactions. Interaction studies by AutoDock were carried out using different parameters for different targets. For alpha-amylase, the interacting grid points for x, y, and z-axis were set at 70, 80 and 70 of 0.9583 Å spacing, 90, 90, and 120 of 1.0 Å spacing for alpha-glucosidase and for GLP-1 it was set at 60, 90 and 80 at 0.7 Å spacing.4.4 In vitro antioxidant assay
ABTS scavenging activity was calculated as follows:
| ABTS radical-scavenging activity (%) = [(Ac − As)/Ac] × 100, |
| DPPH radical-scavenging activity (%) = [(Ac − As)/Ac] × 100, |
| H2O2 radical-scavenging activity (%) = [(Ac − As)/Ac] × 100, |
| Metal chelating activity = [(Ac − As)/Ac] × 100, |
4.5 α-Amylase inhibition assay
The α-amylase inhibition assay was performed using the 3,5-dinitrosalicylic acid (DNSA) method. The compounds were mixed with 0.25% starch azure and alpha-amylase (4 U mL−1) solution. After 3 minutes, DNS was added and boiled for 15 minutes. The absorbance was noted at 540 nm. Acarbose was used as a standard reference compound. α-Amylase inhibition was calculated as follows:| α-Amylase inhibition activity = [(Ac − As)/Ac] × 100, |
4.6 α-Glucosidase inhibition assay
α-Glucosidase inhibition of the compounds was measured with 1 mg mL−1 4-nitrophenyl-α-D-glucopyranoside and α-glucosidase (0.3 U). After incubating at 37 °C for 30 min, the reaction was stopped by the addition of 50 mM sodium hydroxide, and the absorbance was recorded at 405 nm. Acarbose was used as the standard reference compound. α-Glucosidase inhibition was calculated as follows:| α-Glucosidase inhibition activity = [(Ac − As)/Ac] × 100, |
4.7 QSAR studies
Calculation of molecular physicochemical properties from Molinspiration online database server.The pharmacokinetics of the synthesized compounds were predicted using the pkCSM pharmacokinetic prediction tool to find the drug likeliness by ADMET properties (absorption, distribution, metabolism, excretion, and toxicity).73
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are thankful to SIF-VIT for providing NMR, GC-MS and IR and other instrumentation facilities.Notes and references
- I. Gülçin, M. Elmastaşan, A. Dandia, A. K. Jain and A. K. Laxkar, RSC Adv., 2013, 3, 8422–8430 RSC.
- G. Periyasami, R. Raghunathan, G. Surendiran and N. Mathivanan, Bioorg. Med. Chem. Lett., 2008, 18, 2342–2345 CrossRef CAS PubMed.
- A. S. Girgis, Eur. J. Med. Chem., 2009, 44, 91–100 CrossRef CAS PubMed.
- B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
- S. V. Karthikeyan, B. D. Bala, V. P. A. Raja, S. Perumal, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2010, 20, 350–353 CrossRef CAS PubMed.
- P. Prasanna, K. Balamurugan, S. Perumal, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2010, 45, 5653–5661 CrossRef CAS PubMed.
- T. Jiang, K. L. Kuhen, K. Wolff, H. Yin, K. Bieza, J. Caldwell, B. Bursulaya, T. Y.-H. Wu and Y. He, Bioorg. Med. Chem. Lett., 2006, 16, 2105–2108 CrossRef CAS PubMed.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller and Y. Tomita, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS PubMed.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stuckey and K. Krajewski, J. Med. Chem., 2006, 49, 3432–3435 CrossRef CAS PubMed.
- K. Lundahl, J. Schut, J. Schlatmann, G. Paerels and A. Peters, J. Med. Chem., 1972, 15, 129–132 CrossRef CAS PubMed.
- B. K. Yeung, B. Zou, M. Rottmann, S. B. Lakshminarayana, S. H. Ang, S. Y. Leong, J. Tan, J. Wong, S. Keller-Maerki and C. Fischli, J. Med. Chem., 2010, 53, 5155–5164 CrossRef CAS PubMed.
- G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79–91 CrossRef CAS PubMed.
- E. G. Prado, M. G. Gimenez, R. De la Puerta Vázquez, J. E. Sánchez and M. S. Rodriguez, Phytomedicine, 2007, 14, 280–284 CrossRef CAS PubMed.
- C.-B. Cui, H. Kakeya and H. Osada, Tetrahedron, 1996, 52, 12651–12666 CrossRef CAS.
- P. R. Sebahar and R. M. Williams, J. Am. Chem. Soc., 2000, 122, 5666–5667 CrossRef CAS.
- A. S. Babu and R. Raghunathan, Tetrahedron, 2007, 63, 8010–8016 CrossRef.
- K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863–910 CrossRef CAS PubMed.
- W. S. Jen, J. J. Wiener and D. W. MacMillan, J. Am. Chem. Soc., 2000, 122, 9874–9875 CrossRef CAS.
- O. Tsuge, S. Kanemasa, M. Ohe and S. Takenaka, Bull. Chem. Soc. Jpn., 1987, 60, 4079–4089 CrossRef CAS.
- E. J. Corey, J. Am. Chem. Soc., 1952, 74, 5897–5905 CrossRef CAS.
- Y. Katsuyama, K.-i. Miyazono, M. Tanokura, Y. Ohnishi and S. Horinouchi, J. Biol. Chem., 2011, 286, 6659–6668 CrossRef CAS PubMed.
- J.-A. Xiao, H.-G. Zhang, S. Liang, J.-W. Ren, H. Yang and X.-Q. Chen, J. Org. Chem., 2013, 78, 11577–11583 CrossRef CAS PubMed.
- K. Revathy and A. Lalitha, RSC Adv., 2014, 4, 279–285 RSC.
- S. Haddad, S. Boudriga, F. Porzio, A. Soldera, M. Askri, M. Knorr, Y. Rousselin, M. M. Kubicki, C. Golz and C. Strohmann, J. Org. Chem., 2015, 80, 9064–9075 CrossRef CAS PubMed.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- M. Yu, G. Li, S. Wang and L. Zhang, Adv. Synth. Catal., 2007, 349, 871–875 CrossRef CAS.
- A. Dandia, A. K. Jain and D. S. Bhati, Tetrahedron Lett., 2011, 52, 5333–5337 CrossRef CAS.
- N. Mase, R. Thayumanavan, F. Tanaka and C. F. Barbas, Org. Lett., 2004, 6, 2527–2530 CrossRef CAS PubMed.
- Y. H. Bae, T. Okano and S. W. Kim, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 923–936 CrossRef CAS.
- S. A. Glickman and A. C. Cope, J. Am. Chem. Soc., 1945, 67, 1017–1020 CrossRef CAS.
- D. Fokas, W. J. Ryan, D. S. Casebier and D. L. Coffen, Tetrahedron Lett., 1998, 39, 2235–2238 CrossRef CAS.
- L. Yu, H. Cao, X. Zhang, Y. Chen and L. Yu, Sustainable Energy Fuels, 2020, 4, 730–736 RSC.
- X. Deng, H. Cao, C. Chen, H. Zhou and L. Yu, Sci. Bull., 2019, 64, 1280–1284 CrossRef CAS.
- K. Nicolaou, Y.-L. Zhong and P. Baran, J. Am. Chem. Soc., 2000, 122, 7596–7597 CrossRef CAS.
- K. Nicolaou, T. Montagnon, P. Baran and Y.-L. Zhong, J. Am. Chem. Soc., 2002, 124, 2245–2258 CrossRef CAS PubMed.
- K. Nicolaou, T. Montagnon and P. S. Baran, Angew. Chem., Int. Ed., 2002, 41, 993–996 CrossRef CAS PubMed.
- T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566–14569 CrossRef CAS PubMed.
- Y. Izawa, D. Pun and S. S. Stahl, Science, 2011, 333, 209–213 CrossRef CAS PubMed.
- T. Diao, T. J. Wadzinski and S. S. Stahl, Chem. Sci., 2012, 3, 887–891 RSC.
- Y. Chen, A. Turlik and T. R. Newhouse, J. Am. Chem. Soc., 2016, 138, 1166–1169 CrossRef CAS PubMed.
- M. Zhang, J. Zhou, J. Kan, M. Wang, W. Su and M. Hong, Chem. Commun., 2010, 46, 5455–5457 RSC.
- W. Gao, Z. He, Y. Qian, J. Zhao and Y. Huang, Chem. Sci., 2012, 3, 883–886 RSC.
- J. Zhou, G. Wu, M. Zhang, X. Jie and W. Su, Chem.–Eur. J., 2012, 18, 8032–8036 CrossRef CAS PubMed.
- Z. Wang, Z. He, L. Zhang and Y. Huang, J. Am. Chem. Soc., 2018, 140, 735–740 CrossRef CAS PubMed.
- X. Jie, Y. Shang, X. Zhang and W. Su, J. Am. Chem. Soc., 2016, 138, 5623–5633 CrossRef CAS PubMed.
- V. Calò, A. Nacci, A. Monopoli, A. Fornaro, L. Sabbatini, N. Cioffi and N. Ditaranto, Organometallics, 2004, 23, 5154–5158 CrossRef.
- V. Calo, A. Nacci, A. Monopoli, E. Ieva and N. Cioffi, Org. Lett., 2005, 7, 617–620 CrossRef CAS PubMed.
- C. Teja and F. R. Nawaz Khan, ACS Omega, 2019, 4, 8046–8055 CrossRef CAS PubMed.
- V. Caló, A. Nacci, A. Monopoli, A. Detomaso and P. Iliade, Organometallics, 2003, 22, 4193–4197 CrossRef.
- H.-J. Xu, Y.-Q. Zhao and X.-F. Zhou, J. Org. Chem., 2011, 76, 8036–8041 CrossRef CAS PubMed.
- V. Calo, A. Nacci, A. Monopoli and F. Montingelli, J. Org. Chem., 2005, 70, 6040–6044 CrossRef CAS PubMed.
- A. M. Sajith and A. Muralidharan, Tetrahedron Lett., 2012, 53, 5206–5210 CrossRef CAS.
- J. M. Khurana and S. Kumar, Tetrahedron Lett., 2009, 50, 4125–4127 CrossRef CAS.
- A. Davoodnia, S. Allameh, A. Fakhari and N. Tavakoli-Hoseini, Chin. Chem. Lett., 2010, 21, 550–553 CrossRef CAS.
- D. Meetoo, P. McGovern and R. Safadi, Br. J. Nurs., 2007, 16, 1002–1007 CrossRef PubMed.
- H. E. Lebovitz, Clin. Chem., 1999, 45, 1339–1345 CrossRef CAS.
- C. Almeida and R. Cruz, Eur. J. Publ. Health, 2019, 29(suppl. 1), ckz034.088 Search PubMed.
- A. T. Kharroubi and H. M. Darwish, World J. Diabetes, 2015, 6, 850 CrossRef PubMed.
- G. Wang, Z. Peng, Z. Gong and Y. Li, Bioorg. Chem., 2018, 78, 195–200 CrossRef CAS PubMed.
- S. Shah, K. Javaid, H. Zafar, K. M. Khan, R. Khalil, Z. Ul-Haq, S. Perveen and M. I. Choudhary, Bioorg. Chem., 2018, 78, 269–279 CrossRef CAS PubMed.
- A. Chaudhury, C. Duvoor, R. Dendi, V. Sena, S. Kraleti, A. Chada, R. Ravilla, A. Marco, N. S. Shekhawat and M. T. Montales, Front. Endocrinol., 2017, 8, 6 Search PubMed.
- P. Guillausseau, Diabetes Metabol., 2003, 29, 79–81 CrossRef CAS.
- I. W. Campbell, Drugs, 2000, 60, 1017–1028 CrossRef CAS PubMed.
- S. Mathusalini, T. Arasakumar, K. Lakshmi, C.-H. Lin, P. S. Mohan, M. G. Ramnath and R. Thirugnanasampandan, New J. Chem., 2016, 40, 5164–5169 RSC.
- T. Jeffery and M. David, Tetrahedron Lett., 1998, 39, 5751–5754 CrossRef CAS.
- V. Calo, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef CAS PubMed.
- Y. Liu, M. Li, Y. Lu, G.-H. Gao, Q. Yang and M.-Y. He, Catal. Commun., 2006, 7, 985–989 CrossRef CAS.
- C. Teja and F. R. Nawaz Khan, Org. Lett., 2020, 22, 1726–1730 CrossRef CAS PubMed.
- A. Dijksman, I. W. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232–3237 RSC.
- G. D. Brayer, G. Sidhu, R. Maurus, E. H. Rydberg, C. Braun, Y. Wang, N. T. Nguyen, C. M. Overall and S. G. Withers, Biochemistry, 2000, 39, 4778–4791 CrossRef CAS PubMed.
- V. Roig-Zamboni, B. Cobucci-Ponzano, R. Iacono, M. C. Ferrara, S. Germany, Y. Bourne, G. Parenti, M. Moracci and G. Sulzenbacher, Nat. Commun., 2017, 8, 1111 CrossRef PubMed.
- C. R. Underwood, P. Garibay, L. B. Knudsen, S. Hastrup, G. H. Peters, R. Rudolph and S. Reedtz-Runge, J. Biol. Chem., 2010, 285, 723–730 CrossRef CAS PubMed.
- D. E. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- N. S. Rajurkar and S. Hande, Indian J. Pharmaceut. Sci., 2011, 73, 146 CrossRef CAS PubMed.
- F. Nikhat, D. Satynarayana and E. Subhramanyam, Asian J. Res. Chem., 2009, 2, 218–221 CAS.
- D. Mukhopadhyay, P. Dasgupta, D. S. Roy, S. Palchoudhuri, I. Chatterjee, S. Ali and S. G. Dastidar, Free Radic. Antioxidants, 2016, 6, 124–132 CrossRef CAS.
- I. Gülçin, M. Elmastaş and H. Y. Aboul-Enein, Phytother Res., 2007, 21, 354–361 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR, HRMS and FT-IR spectra of all compounds and 2D-NMR spectra for selected compounds. See DOI: 10.1039/d0ra01553a |
| This journal is © The Royal Society of Chemistry 2020 |